

Abstract
Tropomyosin and troponin regulate muscle contraction by participating in a macromolecular scale steric-mechanism to control myosin-crossbridge – actin interactions and consequently contraction. At low-Ca2+, the C-terminal 30% of troponin subunit-I (TnI) is proposed to trap tropomyosin in a position on thin filaments that sterically interferes with myosin-binding, thus causing muscle relaxation. In contrast, at high-Ca2+, inhibition is released after the C-terminal domains dissociate from F-actin-tropomyosin as its component switch-peptide domain binds to the N-lobe of troponin-C (TnC). Recent, paradigm-shifting, cryo-EM reconstructions by the Namba group have revealed density attributed to TnI along cardiac muscle thin filaments at both low- and high-Ca2+ concentration. Modeling the reconstructions showed expected high-Ca2+ hydrophobic interactions of the TnI switch-peptide and TnC. However, under low-Ca2+ conditions, sparse interactions of TnI and tropomyosin, and in particular juxtaposition of non-polar switch-peptide residues and charged tropomyosin amino acids in the published model seem difficult to reconcile with an expected steric-blocking conformation. This anomaly is likely due to inaccurate fitting of tropomyosin into the cryo-EM volume. In the current study, the low-Ca2+ cryo-EM volume was fitted with a more accurate tropomyosin model and representation of cardiac TnI. Our results show that at low-Ca2+ a cluster of hydrophobic residues at the TnI switch-peptide and adjacent H4 helix (Ala149, Ala151, Met 154, Leu159, Gly160, Ala161, Ala163, Leu167, Leu169, Ala171, Leu173) draw-in tropomyosin surface residues (Ile143, Ile146, Ala151, Ile154), presumably attracting the entire tropomyosin cable to its myosin-blocking position on actin. The modeling confirms that neighboring TnI “inhibitory domain” residues (Arg145, Arg148) bind to thin filaments at actin residue Asp25, as previously suggested. ClusPro docking of TnI residues 137–184 to actin-tropomyosin, including the TnI inhibitory-domain, switch-peptide and Helix H4, verified the modeled configuration. Our residue-to-residue contact-mapping of the TnI-tropomyosin association lends itself to experimental validation and functional localization of disease-bearing mutations.
Keywords: Actin, tropomyosin, troponin, cryo-electron microscopy, molecular modeling, protein-protein docking
Graphical Abstract

Introduction
Troponin regulates muscle thin filament activity in a Ca2+-dependent manner by controlling tropomyosin configuration on actin. Here, tropomyosin coiled coils linked head-to-tail into continuous cables shift between blocked, B-state and Ca2+-activated, C-state tropomyosin positions, respectively obstructing or exposing much of the myosin-binding site on actin-subunits along the thin filament [1–3]. Such tropomyosin movement is therefore responsible for either interfering with or initiating binding of myosin-heads and the crossbridge cycle on actin that powers contractility. Thus troponin controls the Ca2+-dependent on-off switching mechanism, which then, coupled to additional myosin-induced movement of tropomyosin, fully activates the muscle thin filaments [4–6]. Each step of this 3-state steric-model of muscle regulation [2,4,9,10] is obligatory, and defects result in cardiomyopathy and skeletal muscle disease [7,8]. Still, tropomyosin transitions between states have not been observed directly nor have tropomyosin or troponin binding sites on actin been unambiguously established. Moreover, the structural mechanism by which TnI, the inhibitory subunit of troponin, traps tropomyosin in the myosin-blocking B-state remains controversial, despite being identified 50 years ago [11]. Nonetheless, recent cryo-EM reconstructions of thin filaments by Yamada et al. [12] have provided a platform to enhance understanding steric-regulation of thin filament activity, and, in this context, we have reevaluated TnI-binding to actin-tropomyosin at low-Ca2+ in order to better understand tropomyosin positioning on thin filaments.
Yamada et al. [12] and Pavadai et al. [13,14] offer support for the troponin-tropomyosin-based steric-model, but with a paradigm-shifting, mechanistic view of tropomyosin regulatory movement and therefore steric-regulation. Tropomyosin, in fact, does not move uniformly over actin-subunits between its low- and high-Ca2+ states in large azimuthal steps or as a continuous unit as previously surmised. Rather, the ends of each tropomyosin molecule are stabilized on actin by their head-to-tail overlap domain augmented by the tail-domain of troponin subunit-T (TnT1), while the mid-piece of tropomyosin molecules, covering six successive actin subunits between the molecule’s ends, pivot side-to-side over electrostatically defined sites on actin residues Lys326 and Lys328, thereby blocking or exposing myosin-binding sites on actin. This tropomyosin rocking is controlled by C-terminal domain residues of TnI extending from the troponin core, where the so-called TnI inhibitory-peptide (residues 137–148), switch-peptide (residues 149–164), H4-helix (residues 165–184) and C-terminal tail (residues 185–210) all likely play critical roles in biasing tropomyosin to its blocked-state pivot [12–15]. However, while the overall high-Ca2+ and myosin-induced states of the filament are well-characterized at atomic resolution [12,14,16,17], the low-Ca2+ filament remains less well defined, and this includes contacts between TnI and actin-tropomyosin evaluated in the current work.
Understanding of the troponin-tropomyosin regulatory scheme requires complete elucidation of thin filament switching interactions in “off” and “on” B- and C-states at near atomic resolution. Indeed, some of the most functionally significant thin filament domains are very poorly resolved by cryo-EM and by other techniques [12,18]. These include the TnT-linker that connects the troponin core-domain to the tropomyosin overlap nexus [12,18] as well as the aforementioned TnI C-terminal domains, including the switch-peptide, H4-helix and C-terminal tail bound to actin-tropomyosin. In solution, many of the TnI domains are unstructured, while at low-Ca2+on actin, the domains may adopt metastable conformations [19–21].
In the current study, we focus on the structure of the low-Ca2+ switch-peptide region, the neighboring H4-helix and C-terminal tail of TnI when bound to actin-tropomyosin in order to determine their potential influence on thin filament B-state conformation. Since cryo-EM densities of this stretch of TnI [12,15,18] are insufficiently resolved for direct characterization at a residue-to-residue scale, we have undertaken alternative computational approaches to account for cryo-EM densities at an atomic level. Here we describe polar and non-polar interactions between cardiac TnI, actin and tropomyosin likely to trap tropomyosin in the blocked B-state configuration at low-Ca2+. Our results suggest that the TnI inhibitory-peptide acts as an anchor on actin-tropomyosin to position the TnI’s C-terminal domain on actin and prevent off-target promiscuous binding along actin-tropomyosin filaments. Once precisely positioned on actin, the C-terminal domains are primed to bind to both actin and tropomyosin specifically, trapping them in the steric-blocking configurations under muscle’s low-Ca2+ relaxed conditions.
MATERIALS AND METHODS
Thin filament structures were generated from component atomic models by flexible-fitting into the experimentally determined Yamada et al. cryo-EM volume (EMDB-0728) [12] using the program MDFF [23,24]. The F-actin structure used was taken from von der Ecken et al. [25] (PDB ID:5JLF), troponin from PDB ID:1YV0 as modified by Yamada et al. (PDB ID:6KN7) [12], and tropomyosin from a library of structures in Pavadai et al. [14] previously fitted to Yamada et al. EM volumes. The composite map (see PDB coordinates, Supplementary Material) was then subjected to energy minimization and molecular dynamics as a means of further refinement [14]. Interaction energy determination between residue domains was carried out in NAMD [24]. TnI segments, excised from PDB ID:6KN7, were docked onto actin-tropomyosin using the programs PIPER and ClusPro [26,27], as previously [14]. The programs, UCSF-Chimera and VMD [28,29], were used for molecular graphics.
RESULTS and DISCUSSION
Premise
Our premise is that the C-terminal domain of TnI must interact closely with the tropomyosin coiled coil in order for it to trap tropomyosin in its low-Ca2+ configuration on actin, thereby hindering myosin-head binding to the blocked-B-state thin filament. In fact, the Yamada et al. reconstructions show convincing, although weak, TnI density abutting tropomyosin, supporting the logic of this supposition [12]. In addition, their modeling indicates intimate electrostatic contact between the TnI inhibitory peptide and actin between arginine residues 145, 146 and 148 of TnI and Asp 24 and 25 on actin. However, densities corresponding to the TnI switch peptide and the rest of C-terminal TnI are insufficiently distinct to identify and therefore precisely align respective TnI and tropomyosin residues one-on-one in order to provide convincing evidence of such interaction. Indeed, we were surprised to note the paucity of appropriate interactions between the region of TnI neighboring tropomyosin in the Yamada PDB (ID: 6KN7) modeling of the low-Ca2+ thin filament reconstruction [12] (detailed in Supplementary Material Fig. S1). PDB 6KN7 shows a hydrophobic patch on TnI beginning at switch peptide residue Leu155 and extending to Leu173 approaches tropomyosin closely but unexpectedly abuts charged tropomyosin residues Arg133, Lys136, Glu138, Lys140 and Glu145, suggesting possible modeling error. In addition, a second interaction in the 6KN7 model quite distal to the switch peptide appears to consist of an attractive pairing of Ser199 on TnI and Asp84 on tropomyosin but repulsion between neighboring Glu202 and Asp80 as well as between Lys178 and Arg125, all leading to extremely unfavorable van der Waals interactions (measured to be >7900 kcal/mol between tropomyosin and TnI). Possible modeling error is compounded by an inappropriate choice of tropomyosin structure fitted into the 6KN7 (and to 6KN8) maps, that was previously shown to be compressed by about 10 percent to fit to the EM density [14], thus corrupting the native TnI-actin-tropomyosin register. Given the lack of obvious connectivity between the C-terminal region of TnI and actin-tropomyosin described by 6KN7, it is difficult to envision cardiac thin filaments displaying residual Ca2+-regulatory function following cardiac stunning and removal of 17 C-terminal residues from TnI [30].
Refining PDB 6KN7
A first step in amending the alignment of regulatory components in the low-Ca2+ thin filaments required replacing the tropomyosin structure in the Yamada PDB (ID: 6KN7) model with one that is less conjectural. We therefore fitted the well-vetted Pavadai et al. tropomyosin structure based on crystal structures [14] to match corresponding densities in the Yamada et al. low-Ca2+ EM reconstruction (EMDB-0728) [12] (see Supplementary Material Fig. S2). Indeed, cross-correlation metrics confirmed the excellent correspondence of the tropomyosin structure to the cryo-EM densities (0.88 value vs 0.84 for Yamada’s tropomyosin as calculated by VMD [29]). We then completed building the thin filament by refitting F-actin and troponin models to their respective positions in the EM volume, as done previously by Yamada [12]. Thus, the only significant difference to distinguish the thin filament coordinates and those in the Yamada et al. is the tropomyosin alignment along actin and TnI. Other than the tropomyosin fitting, the relative positioning of troponin on actin, including the troponin core-domain, inhibitory and switch-peptides as well as the H4-helix and remaining C-terminal tail TnI are essentially the same as in 6KN7. The amended structure now based on the corrected tropomyosin alignment yields typical polar and non-polar protein-protein interactions between TnI and actin-tropomyosin (Fig. 1). While the alignment of residues in the TnI inhibitory-peptide and actin is the same as shown by Yamada et al., clustered hydrophobic residues in the switch-peptide and in the neighboring H4-helix of TnI (Ala149, Ala151, Ala153, Met 154, Met155, Ala157, Leu159, Gly160, Ala161, Ala163, Leu167, Leu169, Ala171, Leu173) are now grouped among nonpolar Ala155, Ile154, Ala151, Leu148, Ile146, Ile143 on tropomyosin; i.e. they do not fall interspersed with charged residues on tropomyosin as in 6KN7. In fact, the nonpolar stretch of TnI interweaves with exceedingly rare solvent facing hydrophobic residue pairs at helical wheel c/f positions (I143, I146) on tropomyosin (Fig. 1C, D arrows). The nonpolar TnI domains are flanked on one end by salt-bridges linking the inhibitory-peptide with actin and on the other end by Lys170 and Lys174 on TnI helix H4 contacting Glu139 on tropomyosin. In addition, Arg192/Glu114, Asp196/Arg105, Ser199/Glu98 and Glu202/Arg91 TnI/tropomyosin links are observed on the tail end of TnI, along with a possible secondary TnI contact (Leu169) with the hydrophobic D-Loop on an adjoining actin subunit (Fig. 1). Thus all of the TnI segments participate in multivalent interactions with tropomyosin and actin (contacts listed in Supplementary Material Fig. S3). Electrostatic interactions dominate the binding of the TnI inhibitory domain and actin and outweigh those between helix-H4 and C-terminal tail domain of TnI and tropomyosin (Fig. 2). Little electrostatic interaction is noted between the TnI switch-peptide and actin-tropomyosin, which as mentioned is a region replete with non-polar residues (Fig. 2). Molecular dynamics simulation of the new structure showed that the average residue-by-residue alignment of TnI along the thin filament components is stable.
Figure 1.
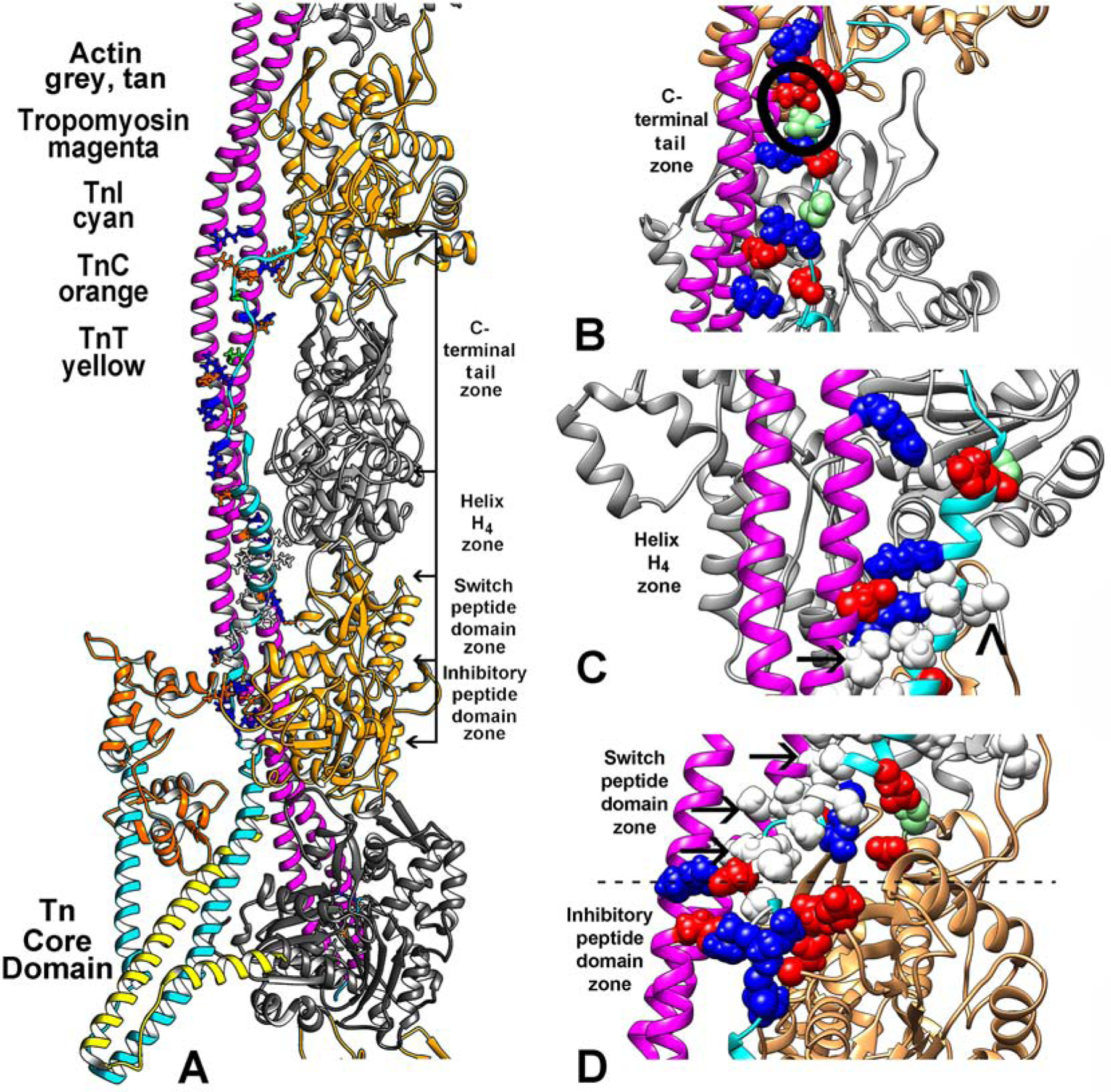
The low-Ca2+ thin filament structure determined in this study. (A) Ribbon diagram focused on the region of the filament over which TnI (cyan) spans actin (grey, tan) and tropomyosin (magenta). The uppermost region of the C-terminal chain of TnI may require zooming to readily locate. The troponin core-domain points (TnI cyan, TnC, orange, TnT, yellow) toward the barbed-end side of actin filament. Only one long-pitch strand of the filament helix is shown for clarity. Selected residues that interface TnI and actin-tropomyosin are highlighted, electrostatic (colored red–acidic, blue-basic), polar (light green) and hydrophobic (white) residues (contacts also noted in (E-G)). (B-D) enlargements of (A) with regions of the filament rotated to focus on segments of TnI at the C-terminal tail end of TnI (B), at its H4–helix region (C), and over the switch-peptide and inhibitory-domains (D). Here, sidechains of interacting resides noted in (A) are drawn in sphere format for emphasis. The inhibitory-peptide seen in (D) makes robust salt-bridge contacts with actin and tropomyosin as first suggested by the Yamada et al. model (see Supplementary Material Fig. S1). In the current study, hydrophobic residues in the TnI switch-peptide and in H4-helix cluster together to abut nonpolar residues on tropomyosin (arrows in (C) indicate examples; a caret points to potential interaction of L369 on TnI and Met47 on the D-loop of actin). In (B), S199, a potential site of TnI phosphorylation, is circled. In addition, salt-bridges link TnI to both actin and tropomyosin here and throughout the structure (see Table 1).
Figure 2.

Interaction energetics between TnI and actin and tropomyosin. Electrostatic and van der Waals interactions measured over different segments of TnI. Note considerable electrostatic interaction energy between actin and the inhibitory-domain, but little between actin and the rest of TnI, while alternatively tropomyosin interactions with TnI domains increase in magnitude toward distal TnI C-terminal domains.
Model validation
The aim of the above approach was to generate an atomic model structure of C-terminal domains of TnI and tropomyosin to match cryo-EM volumes. Nevertheless, the model constructed may have been predisposed to densities in the EM images compiled for analysis by Yamada et al. or by the cryo-EM conditions chosen [12]. We therefore elected to investigate the structure of the regulatory segment of TnI on actin-tropomyosin ab initio without reference to the TnI volume in the cryo-EM data. Our rationale was to gauge TnI binding to TnI-free actin-tropomyosin filaments based on unbiased computation of surface complementarity and the interaction energetics. Here we used a combination of protein-protein docking and molecular dynamics as a means to assemble the TnI segments computationally onto these TnI-free actin-tropomyosin filaments. To be most credible, the computational docking and the model generated from the cryo-EM data above should cross-validate; in other words, to be most meaningful, the computational docking and experimental work shown in Figure 1 should match each other.
Docking regulatory domains of TnI to actin-tropomyosin filaments
To begin, three C-terminal TnI segments were excised from the crystal structure of troponin (PDB ID:1YV0 as represented in reference [12] containing the TnI inhibitory-domain (residues 137–148), the switch-peptide (residues 149–164) and the H4-helix (residues 165–184). The C-terminal tail of TnI (residues 188–210) is not resolved in the crystal structure and therefore was not studied by this methodology. The latter is intrinsically disordered in solution, and not appropriate for standard rigid-body protein docking.
Docking the switch-peptide and H4-helix, as individual domains to F-actin using the programs PIPER and ClusPro [26,27] was unsatisfactory, yielding variously aberrant cross-filament interactions with little correspondence to positions or polarity of TnI on F-actin. The very basic inhibitory-domain docked to acidic residues 24 and 25 on actin but without a single precise orientation. Much the same result was obtained when other combinations of two of the three domains were tested. In contrast, docking longer TnI segments that consisted of a contiguous set of the three TnI fragments (i.e. the inhibitory-domain, switch-peptide and helix-H4) succeeded in targeting this peptide to actin-tropomyosin modules consisting of two actin subunits and tropomyosin pseudo-repeats 4 and 5. The ClusPro docking yielded structures (Fig. 3) with polarity and positioning corresponding to those observed in our molecular fitting of high resolution models to the EM volumes above. Since virtually the same residue-to-residue contacts between tropomyosin, the switch-peptide and helix-H4 were observed by each of the two methods, these two different approaches are mutually supportive. When based on combined electrostatic and van der Waals criteria, ClusPro docking studies ranked the TnI-actin-tropomyosin pose represented in Figure 3 with the top score. When ranking was measured by electrostatic interaction criteria alone, the same pose ranked second but was preceded by one with non-native TnI linkages at the uncapped ends of the actin-tropomyosin module. Other less high ranking poses crisscross actin-tropomyosin with no relationship to the native structure and were not considered further.
Figure 3.
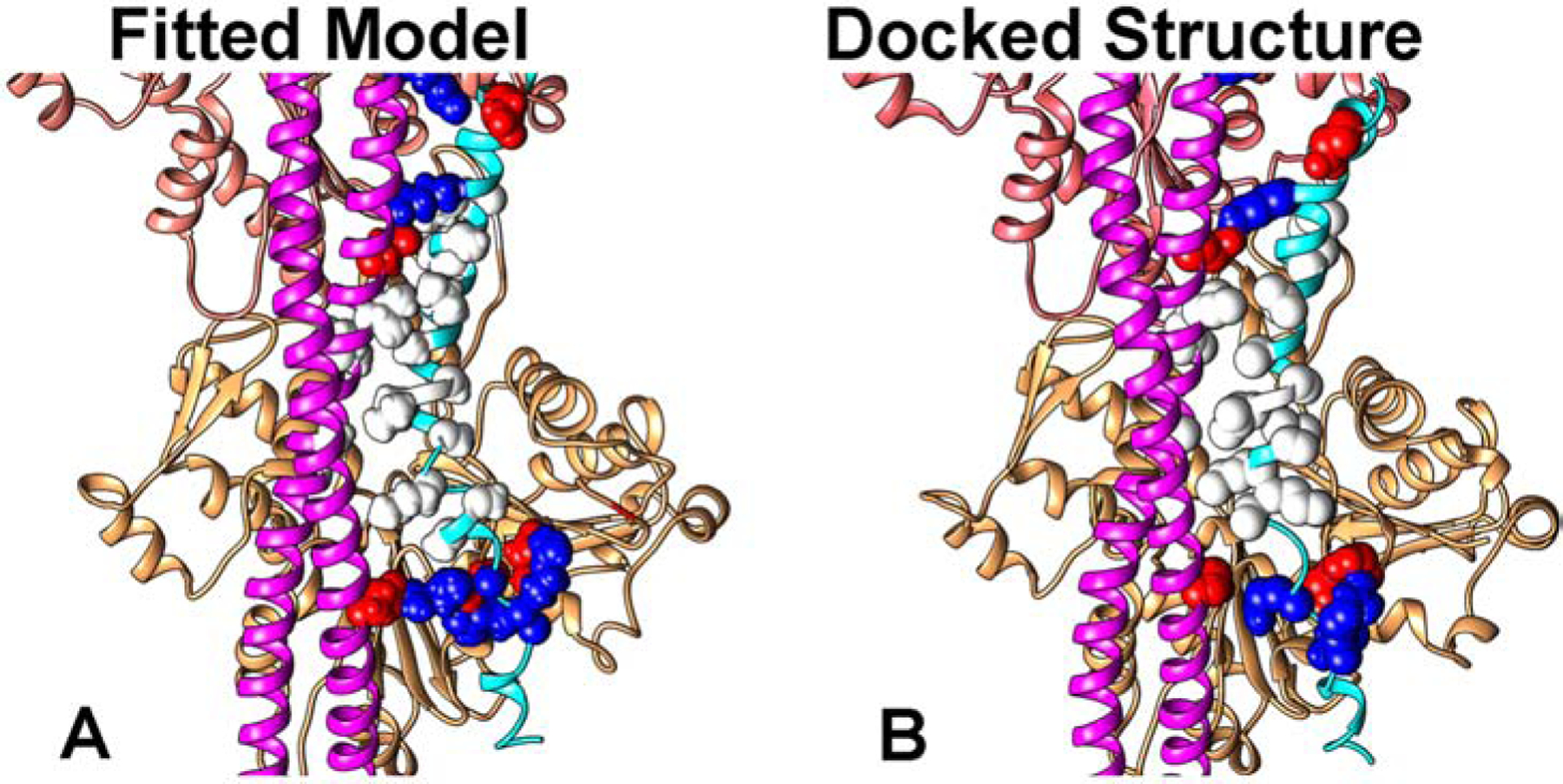
Docking of TnI to actin-tropomyosin. Comparison of (A) atomic models flexibly-fitted to cryo-EM densities shown in Figure 2 and (B) the docked structure. Note the good alignment of TnI residue sidechains in the two.
The extended TnT1 tail of troponin bridges between the tropomyosin overlapping domain on actin and TnT2 of troponin core domain, and thus, acts as a ruler locating the core-domain over tropomyosin pseudo-repeat 5. In turn, this arrangement defines the TnI inhibitory-peptide position and hence the neighboring switch-peptide, H4-helix and final C-terminal tail TnI binding sites to unique actin subunits and tropomyosin residues along thin filaments. The docking of TnI shown (Fig. 3) was performed on a two-actin module containing a stretch of tropomyosin ranging from residues 120 to 184, namely that segment of the tropomyosin coiled coil representing its pseudo-repeats 4 and 5, thereby targeting the TnI segments to the region of thin filament that they occupy in situ. When the TnI fragment was docked to longer stretches of actin-tropomyosin, for example, a four actin-subunit long actin-tropomyosin module, inhibitory-domain of the fragment docked without preference to residues 24 and 25 on one or another neighboring actin subunit, while the switch-peptide and H4 located against cognate but not specific tropomyosin pseudo-repeats. Here TnI-to-tropomyosin pseudo-repeat binding specificity was lost once freed of constraints imposed by the rest of troponin.
Conclusion and perspective
The analysis presented here reveals the residue-to-residue contacts occurring between the C-terminal domains of TnI and actin-tropomyosin which are responsible for trapping tropomyosin in its off-state pivot over actin. The so-called TnI inhibitory-peptide is linked statically to actin-tropomyosin, binding actin almost identically both at low- and high-Ca2+ [12,18]. By connecting the troponin core-domain to the switch-peptide, the inhibitory-peptide acts at the fulcrum of Ca2+-induced C-terminal TnI translocation. As beautifully shown by Yamada et al. [12], high-Ca2+ is accompanied by (1) release of C-terminal TnI domains from the actin-tropomyosin surface and relocation of the switch-peptide away from tropomyosin and onto the Ca2+-laden N-lobe of TnC of the troponin core-domain; and then (2) following C-state tropomyosin pivoting, myosin-binding itself shunts tropomyosin further to the fully active M-state position on actin [6,16,17], resulting in contraction. The regulatory switching mechanism no doubt involves a competition between TnC and actin-tropomyosin for C-terminal TnI domains. Here a dynamic equilibration between multiple weak binding interactions is likely to be in play during these transitions [2,31].
We expect that our current analysis of low-Ca2+ TnI contacts with actin-tropomyosin will offer insights linking TnI mutations to cardiomyopathy and skeletal muscle disease development (32). For example, an HCM-linked mutation hotspot is located at cardiac TnI inhibitory domain-residue Arg145 [32,33]. Preliminary modeling of point mutations in Arg145 and neighboring Arg148 suggest mutation-induced disruption of salt bridges to Asp24 and Asp25 on actin and Glu163 on tropomyosin potentially perturb thin filament blocking state fidelity. In contrast, myopathies linked to mutant hydrophobic switch-peptide residues seem infrequent, and some may be lethal [32]. However, HCM-linked mutants of polar C-terminal residues Asp196 and Arg204 may interfere with tropomyosin interaction [32–34]. Furthermore, our results suggest that changing the charge on TnI Ser199 by for example S199R mutation, will likely attract of tropomyosin residue Glu98, causing B-state stabilization, while phosphorylation will repel Glu98, inducing a C-state bias as found in situ [35]. The functional and structural impact of these and other mutations on TnI conformation and interaction with actin or tropomyosin should be assessed using results determined in the current study as a guide as well as a platform to develop small molecules that reverse their pathological effects.
Supplementary Material
Table 1.
Salt-bridge distances between TnI and actin-tropomyosin at less than 5 Å separation. Distances were measured between NH3+ hydrogen atoms on lysine and arginine sidechains and neighboring COO− oxygen atoms of aspartate and glutamate.
| Domain | Tnl Residue | Actin Residue | Tropomyosin Residue | Salt Bridge Distance in Å |
|---|---|---|---|---|
| Inhibitory peptide | LYS 138 | GLU 167 | 1.74 | |
| ARG 145 | ASP 25 | 1.97 | ||
| ASP 24 | 1.92 | |||
| GLU 163 | 1.96 | |||
| Switch peptide | ASP 152 | ARG 160 | 1.86 | |
| ASP 56 | 4.55 | |||
| Helix H4 | GLU 165 | LYS 61 | 3.26 | |
| ARG 170 | GLU 139 | 1.86 | ||
| LYS 174 | GLU 139 | 1.63 | ||
| LYS 177 | ASP 25 | 1.75 | ||
| GLU 182 | ARG 125 | 3.84 | ||
| GLU 184 | ARG 28 | 1.91 | ||
| C-terminal | ARG 186 | GLU 2 | 4.67 | |
| GLU 187 | ARG 28 | 4.29 | ||
| ARG 192 | GLU 114 | 1.84 | ||
| ARG 105 | 1.87 | |||
| GLU 202 | ARG 91 | 1.81 | ||
| ARG 204 | ASP 25 | 4.02 | ||
| LYS 205 | GLU 97 | 1.84 |
Highlights.
The C-terminal 30% of TnI traps tropomyosin in the actin filament blocked-state.
This C-terminal TnI linkage to tropomyosin and actin relaxes muscle at low-Ca2+.
Our atomic model describes residue-to-residue TnI-actin-tropomyosin interaction.
The model provides a baseline map to understand TnI mutation-based imbalances.
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01HL036153 (to W.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Gordon AM, Homsher E, Regnier M Regulation of contraction in striated muscle Physiol. Rev, 80 (2000), pp. 853–924 [DOI] [PubMed] [Google Scholar]
- [2].Geeves MA Thin Filament Regulation In: Comprehensive Biophysics, vol. 4, Molecular Motors and Motility, Egelman EH, Goldman YE, Ostap EM editors. Academic Press, Oxford: (2012), pp. 251–267 [Google Scholar]
- [3].Lehman W Thin filament structure and the steric blocking model Comp. Physiol, 6 (2016), pp. 1043–1069 [DOI] [PubMed] [Google Scholar]
- [4].Vibert P, Craig R, Lehman W Steric-model for activation of muscle thin filaments J. Mol. Biol, 266 (1997), pp. 8–14 [DOI] [PubMed] [Google Scholar]
- [5].Poole KJ, Lorenz M, Evans G, Rosenbaum G, Pirani A, Tobacman LS, Lehman W, Holmes KC A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle J. Struct. Biol, 155 (2006), pp. 273–284 [DOI] [PubMed] [Google Scholar]
- [6].Behrmann E, Müller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S Structure of the rigor actin–tropomyosin–myosin complex Cell, 150 (2012), pp. 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marston SB How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res, 4 (2011), pp. 245–255 [DOI] [PubMed] [Google Scholar]
- [8].Tardiff JC Thin filament mutations: developing an integrative approach to a complex disorder Circ. Res, 108 (2011), pp. 765–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McKillop DFA, Geeves MA Regulation of the interaction between actin and myosin subfragment-1: Evidence for three states of the thin filament Biophys. J, 65 (1993), pp. 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lehman W Switching muscles on and off in steps: The McKillop-Geeves three-state model of muscle regulation Biophys. J, 112 (2017), pp. 2459–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Greaser ML, Gergely J Reconstitution of troponin activity from three protein components J. Biol. Chem, 246 (1971), pp. 4226–4233 [PubMed] [Google Scholar]
- [12].Yamada Y, Namba K, Fujii T Cardiac muscle thin filament structures reveal calcium regulatory mechanism Nature Commun, 11 (2020), pp. 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pavadai E, Rynkiewicz MJ, Ghosh A Lehman W. Docking troponin T onto the tropomyosin overlapping domain of thin filaments Biophys. J, 118 (2020), pp. 325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pavadai E, Lehman W, Rynkiewicz MJ Protein-protein docking reveals dynamic interactions of tropomyosin on actin filaments Biophys. J, 119 (2020), pp. 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tobacman Troponin revealed LS: Uncovering the structure of the thin filament on-off switch in striated muscle Biophys. J, 120 (2021), pp. 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Doran MH, Pavadai E, Rynkiewicz MJ, Walklate J, Bullitt E, Moore JR, Regnier M, Geeves MA, Lehman Cryo-EM W and molecular docking shows myosin Loop 4 contacts actin and tropomyosin on thin filaments Biophys. J, 119 (2020), pp. 821–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Risi C, Schӓfer LU, Belknap B, White HD, Schröder GF, Galkin VE High-resolution cryo-EM of the cardiac actomyosin complex Structure, 29 (2021), pp. 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oda T, Yanagisawa H, Wakabayashi T Cryo-EM structures of cardiac thin filaments reveal the 3D architecture of troponin J. Struct. Biol, 209 (2020), pp. 107450. [DOI] [PubMed] [Google Scholar]
- [19].Marston S, Zamora JE Troponin structure and function: a view of recent progress J. Muscle Res. Cell Motil, 41 (2020), pp. 71–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Metskas LA, Rhoades E Conformation and dynamics of the troponin I C-terminal domain: Combining single-molecule and computational approaches for a disordered protein region J. Am. Chem. Soc, 137 (2015), pp. 11962–11969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Metskas LA, Rhoades E Order-disorder transitions in the cardiac troponin complex J. Mol. Biol, 428 (2016), pp. 2965–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Levine BA, Moir AJ, Perry SV The interaction of troponin-I with the N-terminal region of actin Eur. J. Biochem, 172 (1988), pp. 389–397 [DOI] [PubMed] [Google Scholar]
- [23].Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K Molecular dynamics flexible fitting: A practical guide to combine cryo-electron microscopy and x-ray crystallography Methods, 49 (2009), pp. 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Phillips JC, Braun R, Wang W W, Gumbart J J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K Scalable molecular dynamics with NAMD J. Comput. Chem, 26 (2005), pp. 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].von der Ecken J, Heissler SM, Pathan-Chhatbar S, Manstein DJ, Raunser S Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution Nature, 534 (2016), pp. 724–728 [DOI] [PubMed] [Google Scholar]
- [26].Kozakov D, Hall DR, Beglov D, Brenke R, Comeau SR, Shen Y, Li K, Zheng J, Vakili P, Paschalidis IC, Vajda S Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19 Proteins, 78 (2010), pp. 3124–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S The ClusPro web server for protein-protein docking Nature Protoc, 12 (2017), pp. 255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE UCSF Chimera—a visualization system for exploratory research and analysis J. Comput. Chem, 25 (2004), pp. 1605–1612 [DOI] [PubMed] [Google Scholar]
- [29].Humphrey W, Dalke A, Schulten K VMD: visual molecular dynamics J. Mol. Graphics, 14 (1996), pp. 33–38 [DOI] [PubMed] [Google Scholar]
- [30].Murphy AM, Kögler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marbán E Transgenic mouse model of stunned myocardium Science, 287 (2000), pp. 488–491 [DOI] [PubMed] [Google Scholar]
- [31].Lehrer The SS 3-state model of muscle regulation revisited: Is a fourth-state involved? J. Muscle Res. Cell Motility, 32 (2011), pp. 203–208 [DOI] [PubMed] [Google Scholar]
- [32].Mogensen J, Hey T, Lambrecht S A systematic review of phenotypic features associated with cardiac troponin I mutations in hereditary cardiomyopathies Can. J. Cardiol, 31 (2015), pp. 1377–1385 [DOI] [PubMed] [Google Scholar]
- [33].Gilda JE, , Xu Q, Martinez ME, Nguyen ST, Chase PB, Gomes AV The functional significance of the last 5 residues of the C-terminus of cardiac troponin I Arch. Biochem. Biophys, 601 (2016), pp. 88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu X, Zhang L, Pacciulli D, Zhao J, Nan C, Shen W, Quan J, Tian J, Huang X Restrictive cardiomyopathy caused by troponin mutations: Application of disease animal models in translational studies Front. Physiol, 7 (2016), pp. 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Winkler PJM, Li Y, Zhang P, Foster DB, dos Remedios C, Van Eyk JE, Steinen GJM, Murphy AM, van J der Velden A novel phosphorylation site, serine 199 in the C-terminus of cardiac troponin I regulates calcium sensitivity and susceptibility to calpain-induced proteolysis J. Mol. Cell Cardiol, 82 (2015), pp. 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.