

Abstract
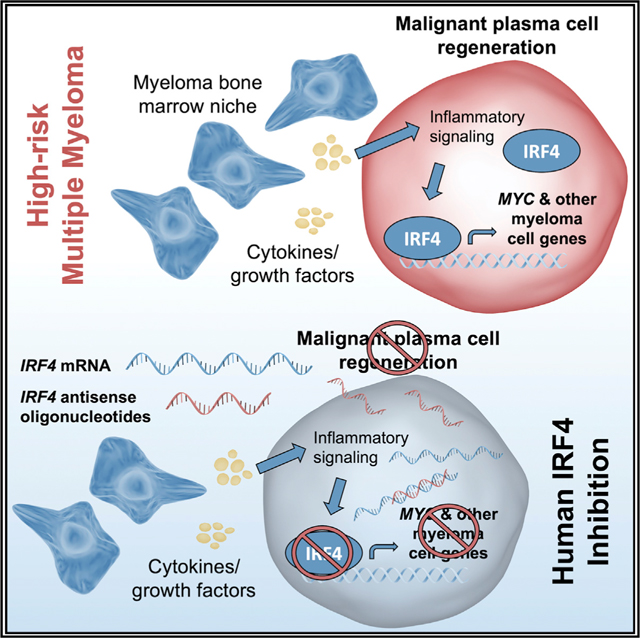
SUMMARY
In multiple myeloma, inflammatory and anti-viral pathways promote disease progression and cancer stem cell generation. Using diverse pre-clinical models, we investigated the role of interferon regulatory factor 4 (IRF4) in myeloma progenitor regeneration. In a patient-derived xenograft model that recapitulates IRF4 pathway activation in human myeloma, we test the effects of IRF4 antisense oligonucleotides (ASOs) and identify a lead agent for clinical development (ION251). IRF4 overexpression expands myeloma progenitors, while IRF4 ASOs impair myeloma cell survival and reduce IRF4 and c-MYC expression. IRF4 ASO monotherapy impedes tumor formation and myeloma dissemination in xenograft models, improving animal survival. Moreover, IRF4 ASOs eradicate myeloma progenitors and malignant plasma cells while sparing normal human hematopoietic stem cell development. Mechanistically, IRF4 inhibition disrupts cell cycle progression, downregulates stem cell and cell adhesion transcript expression, and promotes sensitivity to myeloma drugs. These findings will enable rapid clinical development of selective IRF4 inhibitors to prevent myeloma progenitor-driven relapse.
In Brief
Crews and colleagues demonstrate that selective antisense oligonucleotides targeting the plasma cell transcription factor, IRF4, reduce disease burden and myeloma regeneration in human-relevant pre-clinical models. Mechanistically, IRF4 overexpression expands a myeloma progenitor population, while IRF4 inhibition impairs cell survival via cell cycle arrest and sensitization to clinical myeloma drugs.
Graphical Abstract
INTRODUCTION
Multiple myeloma (MM) is the second most common blood cancer in the United States, with over 32,000 new cases predicted in 2020 and a 5-year survival of only 53.9 percent (https://seer.cancer.gov/statfacts/html/mulmy.html). Characterized by abnormal expansion of malignant clonal progenitors and their immature antibody-producing plasma cell progeny, MM generally recurs and is refractory to further treatment within 5 years. Despite a plethora of novel therapies (Costello and Mikhael, 2018; Kumar et al., 2019; Stewart et al., 2015), treatment toxicities have presented new challenges for patients and clinicians (Paner et al., 2018). Moreover, disease relapse rates remain high, in part due to acquisition of drug resistance (Siegel et al., 2020), along with malignant regeneration of MM cells in inflammatory microenvironments (Bianchi and Ghobrial, 2014; Yaccoby, 2018). More selective and effective therapies that prevent malignant regeneration represent an unmet medical need for MM.
In addition to mutational evolution and epigenetic alterations, inflammatory cytokine-responsive RNA modifications contribute to cancer stem cell (CSC) generation and maintenance, which govern cancer progression and drug resistance (Crews and Jamieson, 2013; Jiang et al., 2017). In MM, pro-inflammatory cytokine and anti-viral interferon-responsive signals derived from the bone marrow microenvironment play a pivotal role in disease progression (Lazzari et al., 2017; Mantovani and Garlanda, 2006). These cytokine drivers include activation of interleukin-6 (IL-6) signaling with downstream interferon regulatory factor (IRF) (Claudio et al., 2002) and signal transducer and activator of transcription (STAT)-dependent signaling (Fuhler et al., 2010), along with acquired mutations in pro-inflammatory genes (Kortüm et al., 2016). Cumulative evidence supports a role for aberrant activation of the key B cell progenitor fate determinant (Klein et al., 2006) and essential MM cell survival factor (Low et al., 2019; Shaffer et al., 2008), IRF4, in the pathogenesis and progression of MM (Claudio et al., 2002; Lopez-Girona et al., 2011).
Induction of vital stem cell self-renewal pathways, such as NOTCH1 (Colombo et al., 2016) and interferon-responsive RNA editase adenosine deaminase acting on RNA (ADAR1) activation, promote progression and therapeutic resistance of a broad array of malignancies, including MM (Crews et al., 2015; Jiang et al., 2013; Lazzari et al., 2017; Mirandola et al., 2013; Zipeto et al., 2016). In addition, acquisition of mutations in pro-inflammatory pathways and the emergence of a relatively rare (<4%) CD138−CD20+CD27+ myeloma-initiating progenitor population (Matsui et al., 2004, 2008) have been implicated as drivers of therapeutic resistance and MM progression. Interestingly, IRF4 drives expression of the stem cell reprogramming genes, MYC and KLF4 (Cheng et al., 2017; Shaffer et al., 2008). However, the role of IRF pathway activation in the maintenance of malignant progenitors in MM has not been explored. Thus, we hypothesized that IRF4 governs MM progenitor regeneration, thereby promoting disease progression. Using an array of in vitro and in vivo assays, we tested the effects of selective IRF4 inhibition on MM progenitors and normal hematopoietic stem and progenitor cell (HSPC) development.
Because transcription factors are notoriously difficult to target with traditional small-molecule strategies, we utilized an antisense oligonucleotide (ASO)-based inhibitory platform to directly reduce stability of IRF4 transcripts. We evaluated the effects of in vitro ASO-mediated inhibition of IRF4 on MM cell viability, IRF4 expression, cell cycle status, and sensitization to standard-of-care drug treatment. Subcutaneous tumor formation, MM dissemination and survival assays, along with IRF4 and c-MYC expression analyses were performed to assess the therapeutic efficacy of IRF4 ASOs in human myeloma cell xenograft models. We then examined the burden of malignant plasma cells and myeloma-initiating progenitor cells in the hematopoietic tissues of engrafted mice in a patient-derived xenograft (PDX) model of high-risk disease (Lazzari et al., 2017). The pre-clinical results supported the identification of a lead ASO agent for clinical development, ION251 (NCT: NCT04398485), providing a mechanistic framework for imminent human trials targeting IRF4. Together, inhibition of MM regeneration through direct modulation of IRF4 expression may provide a potent therapeutic strategy for preventing disease progression driven by therapeutically recalcitrant progenitors in inflammatory microenvironments.
RESULTS AND DISCUSSION
Human IRF4 is highly expressed in patient-derived in vivo models of high-risk MM
We previously established PDX models from primary patients with high-risk plasma cell neoplasms (Lazzari et al., 2017). These serially transplantable PDX models recapitulate multiple features of each patient’s original disease, including clonality, plasma cell burden in the bone marrow, and plasmacytoma formation (Lazzari et al., 2017). We utilized the most aggressive model, MM9-PDX (Figures 1A, 1B, S1A, and S1B), as an in vivo platform for evaluating selective IRF4 inhibitors for high-risk MM.
Figure 1. Characterization of IRF4 pathway activation in a patient-derived pre-clinical MM model.

(A) Representative live animal bioluminescence imaging of serial transplant recipients of MM9-PDX-derived cells (1:1 ratio of human cells isolated from the bone marrow and plasmacytomas of MM PDX mice; 106 cells transplanted per mouse). Image shows mice harboring representative average engraftment levels across all transplanted animals, compared with a non-transplanted control mouse.
(B) Quantification of human kappa light chain levels in MM9-PDX mouse plasma (n = 17).
(C) IRF4 gene expression levels (transcripts per million [TPM]) by RNA sequencing in newly diagnosed (n = 2) and high-risk (n = 3) primary MM samples.
(D) Gene expression heatmap showing 117 differentially expressed IRF4 target genes in high-risk disease (plasma cell leukemia [PCL]) versus newly diagnosed MM samples.
(E) Intranuclear flow cytometry histograms showing human IRF4 protein expression in total CD38+ cells from primary MM9 MNCs and MM9-PDX bone marrow (BM), compared with a representative normal age-matched bone marrow control and negative (no antibody) control. IRF4 protein expression quantification is shown as median fluorescence intensity (MFI) in the CD38++ fractions of primary MM9 versus MM9-PDX BM.
(F) Flow-cytometry-based quantification of intranuclear IRF4 expression in MM9-PDX tissues. MFI values were quantified in the live, CD38++ population of cells from engrafted mouse BM, peripheral blood (PB), and plasmacytomas (PCs) 9 weeks after transplant (n = 4). Graph shows means ± SEM.
(G) Double-labeling immunohistochemical analysis (IHC) of human IRF4 and human CD138 expression in tissue sections from the BM of serially transplantedMM9-PDX mice. Scale bar represents 100 μm.
(H) Gene set enrichment analysis (GSEA) of top 10 Reactome pathways (false discovery rate [FDR] q-values < 0.03) that were significantly over-represented within the differentially expressed IRF4 target genes in (D).
Human cells isolated from the bone marrow of transplanted mice established serially transplantable disease in immunocompromised mice (Lazzari et al., 2017). We further observed that transplantation of human cells isolated from plasmacytomas also facilitated rapid MM engraftment (Figures S1A and S1B). Thus, in some experiments, we transplanted equal numbers of human cells derived from both MM PDX bone marrow and plasmacytomas, which propagated disease in a rapid and reproducible manner in recipient mice (Figures 1A and 1B). To examine IRF4 pathway activation in high-risk MM samples and our humanized MM mouse models, we first generated a molecular profile of IRF4 and downstream target gene expression by whole-transcriptome RNA sequencing (RNA-seq) of cells from the primary patient sample used to generate this in vivo model. For comparison, additional plasma cell leukemia (PCL), relapsed/refractory MM, and newly diagnosed primary MM samples (Table S1) were also analyzed. High levels of IRF4 mRNA were detected by RNA-seq and qRT-PCR in high-risk primary patient-derived cells, in particular in primary cells from MM9 cells prior to in vivo transplantation (Figures 1C and S1C). Moreover, pathway analysis demonstrated that, of 306 IRF4 target genes (Shaffer et al., 2008), 117 were differentially expressed in PCL samples compared with newly diagnosed MM samples (Figure 1D). The majority of these IRF4 target genes were upregulated, including three key plasma cell surface markers (CD38, CD319, and TNF receptor superfamily member 17 [TNFRSF17]/B cell maturation antigen [BCMA]), suggesting that the IRF4 pathway is broadly activated in high-risk MM samples capable of engrafting into immunocompromised mice (Lazzari et al., 2017).
We further verified IRF4 expression at the protein level in primary MM samples and engrafted tissues from MM9-PDX mice (Figure S1D). In previous MM immunohistochemistry studies, protein levels of IRF4 were higher in Multiple Myeloma International Staging System (ISS) stage III patients compared with stage I and II samples (Bai et al., 2017). In keeping with these observations, we found that, in CD38-high (CD38++) cells, which represent the malignant plasma cell population (Leo et al., 1992), IRF4 protein levels were variable but higher on average in untreated PCL samples compared with low-risk MM samples and normal bone marrow controls (Figure S1E; Table S1). Strikingly, the high levels of IRF4 protein expression detected in primary MM9 cells were conserved in engrafted MM9-PDX bone marrow (Figures 1E and S1E). Moreover, a population of IRF4-high cells was enriched in MM9-PDX mouse tissues (Figure 1E), suggesting that the CD38++, IRF4-high cells represent a regenerative population of MM cells in vivo. Overall, IRF4 expression was consistently high in engrafted mouse tissues (Figure 1F). Bone marrow tissue sections from MM9-PDX mice also displayed abundant human IRF4 protein expression, both within CD138low/negative progenitors and CD138+ plasma cells (Figure 1G). This highlights the persistence of IRF4-expressing, CD138low/negative MM-initiating progenitors in serial murine transplant recipients. In plasmacytoma sections, CD138 immunoreactivity was more prominent (Figure S1F), supporting the notion that niche-dependent signals might promote the maintenance of an IRF4-enriched, regenerating MM progenitor population.
Because tumor-initiating cells have the capacity to become dormant in protective microenvironments, and several key cell cycle regulators are IRF4 target genes (Shaffer et al., 2008), we performed a gene set enrichment analysis on the IRF4 target genes that were differentially expressed in the high-risk MM samples. The top enriched gene sets were predominantly cell-cycle-associated pathways, including G1 and G1/S regulatory genes (Figure 1H), suggesting that the model could recapitulate IRF4-driven cell cycle deregulation in vivo. Thus, this serially transplantable MM9-PDX model captures many features of human myeloma, including robust human IRF4 expression, thereby providing a relevant and reproducible platform to study IRF4 as a vital therapeutic target in MM.
IRF4 overexpression in an inflammatory niche enhances myeloma progenitor survival
To characterize IRF4 expression levels in various myeloma in vitro models, we evaluated a publicly available RNA-seq dataset from 66 human myeloma cell lines (https://www.keatslab.org/data-repository; Keats et al., 2007). A spectrum of IRF4 mRNA expression was observed (Figure S2A). We selected a representative subset of cell lines with relatively high (e.g., H929 and EJM), low (RPMI-8226 and U266), and moderate (KMS-11, MM.1R, L363, JJN3, and AMO1) IRF4 levels for further study and functional modulation of IRF4. Lentivirally enforced expression of human IRF4 in MM cells with low endogenous levels of IRF4 (RPMI-8226) promoted expansion of a CD138− fraction (Figure S2B), a subpopulation that has been linked to clinical drug resistance and progenitor-like phenotypes (Chaidos et al., 2013). Similarly, in our in vitro model of acquired lenalidomide resistance (Lazzari et al., 2017), MM cells with high endogenous IRF4 expression (H929) demonstrated a significant expansion of the CD138− population compared with lenalidomide-sensitive controls (Figure S2C). Together, these data suggest that modulation of the inflammatory response and IRF4 promotes the survival or proliferation of progenitor-like, drug-resistant MM cells.
Furthermore, co-culture of wild-type MM cells with human bone marrow stromal cell lines or primary MM stroma resulted in expansion of CD138− MM cells (Figure S2D). In contrast, mouse OP9 stromal cells did not elicit this effect (Figure S2D), suggesting that species-specific factors support emergence of a stem-like phenotype in myeloma cells grown in co-culture models that mirror key aspects of the human bone marrow microenvironment.
The bone marrow niche can modulate cell cycle progression associated with CSC quiescence (Forsberg et al., 2010; Goff and Jamieson, 2010; Goff et al., 2013; Jiang et al., 2019; Yaccoby, 2005, 2018), and IRF4 governs the expression of powerful cell cycle regulatory genes (Lopez-Girona et al., 2011). To investigate a functional link between MM progenitor generation, microenvironmental inflammatory cues, response to drug treatment, and cell cycle status, we established a stable MM cell cycle reporter line using H929 cells expressing lentiviral fluorescence ubiquitination cell cycle indicator (FUCCI) vectors (H929-FUCCI) (Pineda et al., 2016; Sakaue-Sawano et al., 2008). MM cell cycle status was evaluated in H929-FUCCI cells grown in suspension culture or in co-culture with adherent bone marrow stromal cells (Video S1). Although exposure to human stromal cell lines and primary MM stromal cells significantly reduced the fraction of MM cells in G1, mouse bone marrow stromal co-culture was similar to suspension culture alone (Figure S2E). In H929-FUCCI cells treated with lenalidomide, there was an induction of G1 cell cycle arrest, which was reversed by co-culture with human bone marrow stromal cells (Figure S2F). Thus, microenvironmental signals may play a key role in protecting MM cells from drug-induced cell cycle arrest.
IRF4 antisense oligonucleotides impair MM cell survival and induce cell cycle arrest
Although immunomodulatory drugs (IMiDs) (e.g., lenalidomide) represent an important first line of therapy for MM patients, alternative strategies that target inflammation mediators will be vital in order to overcome drug resistance mediated by the protective effects of the human bone marrow microenvironment. To selectively inhibit human (h)IRF4 using an ASO strategy in MM models, we tested a panel of human-specific ASO agents targeting IRF4 and profiled their effects on MM cell viability and IRF4 expression. Treatment of genetically diverse MM cell lines with IRF4 ASO agents dramatically reduced MM cell survival concomitant with decreased human IRF4 mRNA and protein expression (Figures 2A–2C and S3A–S3I). Growth inhibition and half-maximal inhibitory concentration (IC50) values were in the nanomolar to low micromolar range (Table S2), irrespective of genetic alterations. Conversely, negative control (non-targeting) and non-IRF4 targeted ASOs did not affect IRF4 expression in cells with high endogenous IRF4 levels (H929; Figure 2D).
Figure 2. Selective antisense oligonucleotides targeting IRF4 reduce MM cell viability commensurate with reduced IRF4 and c-MYC expression and induction of cell cycle arrest.

(A) Luminescence-based cell viability (CellTiter Glo) assays showing representative dose response curves of H929 and RPMI-8226 myeloma cells (left) and quantification of biological replicate assays performed in H929 cells (right) after treatment with increasing doses of human (h)IRF4-targeted ASOs for 5 days (n = 3 to 4 biological replicates analyzed from separate passages of cells).
(B) Quantitative RT-PCR analyses showing human IRF4 expression in representative dose response curves of H929 and RPMI-8226 myeloma cells (left) and quantification of biological replicate assays performed in H929 cells (right) treated with increasing doses of hIRF4-targeted ASOs for 48 h (n = 3 to 4 biological replicates analyzed in separate passages of cells).
(C) Flow-cytometry-based quantification of intranuclear human IRF4 protein expression in H929 and RPMI-8226 cells treated with hIRF4 ASO-4 (left) and hIRF4 ASO-Lead (right). MFI values quantified within the live, CD38+ population of cells after treatment with ASOs for 72 h (n = 3 biological replicates analyzed from separate passages of cells).
(D) IRF4 mRNA expression in H929 cells treated with control ASO agents, including 2–10 μM Ctrl ASO and ASO specific for a non-IRF4 target (human MALAT1).
(E) Quantitative RT-PCR analysis of MYC expression in hIRF4 ASO-treated H929 cells (n = 3 biological replicates analyzed from separate passages of cells).
(F) Colony formation assay of H929 cells after treatment with hIRF4-targeted ASOs (n = 3 individual wells analyzed per condition).
(G and H) Flow cytometric gating strategy (G) and quantification (H) of cell cycle status in H929-FUCCI cells treated with hIRF4 ASO (n = 3 individual wells analyzed per condition).
Bar graphs show means ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001 compared to vehicle (PBS)-treated cells by unpaired, two-tailed Student’s t test. See also Figures S2, S3, and S6, Table S2, and Video S1.
In vitro IRF4 ASO treatment also reduced mRNA and protein levels of the IRF4 target gene MYC (Figures 2E and S3G–S3I), which is a well-known stem cell reprogramming factor and cell cycle regulator. As a surrogate measure of cell death pathway activation, poly (ADP-ribose) polymerase (PARP) cleavage rates increased in cells treated with IRF4 ASOs for 3 days (Figures S3G–S3I). Overt apoptosis was detected after 6 days of hIRF4 ASO treatment (Figures S3J–S3L). Consistent with a role for IRF4 in MM cell survival, colony formation was significantly reduced after hIRF4 ASO treatment of H929 cells (Figure 2F). To explore the mechanisms of cytotoxicity associated with IRF4 ASOs, we turned to the H929-FUCCI model and evaluated cell cycle changes following ASO-mediated IRF4 inhibition. hIRF4 ASO treatment resulted in a significant and dose-dependent cell cycle arrest in G1, with a concomitant reduction in the proportion of cells in G2/M phase (Figures 2G and 2H). This effect was observed at 48 h post-treatment and preceded significant changes in overall cell viability.
To verify the MM cell line results in additional clinically relevant assays, ex vivo treatments were performed using MM9-PDX-derived human myeloma cells isolated from fresh bone marrow (BM) or plasmacytoma (PC) tissues and grown in the presence of stem-cell-supportive extracellular matrix components. MM9-PDX cells were exquisitely sensitive to ASO-mediated IRF4 inhibition, with significant reductions in cell viability and IRF4 mRNA expression observed after treatment with concentrations of less than 1 μM (Figures 3A–3D). Because IRF4 has been implicated in the function of other immune cell types (Mittrücker et al., 1997), we also sought to establish a therapeutic index for hIRF4 ASO treatment compared with normal hematopoietic cell types. In mononuclear cells (MNCs) from primary MM patient samples and the bone marrow of healthy age-matched donors (Table S1), we observed relatively low levels of hIRF4 protein expression in all normal immune subsets analyzed (Figure 3E). In contrast, levels of IRF4 protein in CD38++ cells from MM patients were approximately 5- to 10-fold higher than in normal immune cell populations (Figure 3E). Notably, ex vivo hIRF4 ASO treatment of primary normal bone marrow samples resulted in moderate on-target reductions in hIRF4 protein expression, although normal CD19+ B cell frequencies remained unaltered (Figures 3F and 3G).
Figure 3. IRF4 inhibition reduces primary patient-derived MM cell viability ex vivo while sparing normal B cell populations.

(A–D) Luminescence-based ex vivo MM9-PDX cell viability assays (A and B) and quantitative RT-PCR analyses (C and D) of BM and PC-derived human cells treated with increasing doses of hIRF4 ASOs, vehicle (PBS) control, or control ASO (up to 2 μM) for 5 days (viability) or 2 days (qPCR; n = 3 individual wells analyzed per tissue for each assay).
(E) Flow cytometry quantification of intranuclear IRF4 expression in primary MNCs from normal bone marrow (NBM) compared with low-risk (smoldering and newly diagnosed MM) and high-risk MM (PCL). MFI was quantified within the live, CD19+, CD3+, CD14+, or CD38++ populations of cells (n = 3–5 samples per group).
(F) Reduced hIRF4 protein expression in NBM samples treated with hIRF4 ASO-Lead (2 μM) compared with PBS control for 3 days.
(G) Unchanged CD19+ B cell frequency after treatment with hIRF4 ASOs as in (F).
Graphs show means ± SEM; *p < 0.05 and **p < 0.01 compared to vehicle or Ctrl ASO-treated cells by unpaired, two-tailed Student’s t test. See also Table S1.
IRF4 inhibition reduces myeloma burden and improves survival in MM xenograft models
To evaluate the pre-clinical therapeutic efficacy of selective IRF4-targeted ASO agents, we tested a panel of hIRF4 ASO tool compounds in MM cell line xenograft models. In MM.1R subcutaneous xenograft assays, hIRF4 ASOs were well tolerated in myeloma tumor-bearing mice (Figure S4A). Treatment with hIRF4 ASOs significantly reduced tumor growth and IRF4 mRNA expression in tumor tissues in a dose-responsive manner (Figures 4A and 4B). Confirming on-target pharmacodynamic activity of IRF4 ASOs, in short-term in vivo dose-response assays, reduced hIRF4 protein expression was detected in tumors from MM.1R subcutaneous xenograft models (Figure S4B). Similarly, in KMS-11 subcutaneous xenograft assays, hIRF4 ASO treatment significantly reduced tumor growth, circulating kappa light chain levels, and hIRF4 protein expression compared to PBS-treated and non-targeting negative control ASOs (Figures S4C–S4E). In both subcutaneous xenograft models, hIRF4 ASO-4 completely halted further tumor growth after the initiation of treatment (Figures 4A and S4C).
Figure 4. Human IRF4-targeted ASOs reduce tumor burden and IRF4 expression in xenograft models and improve overall survival.

(A and B) For human (h)IRF4 ASO treatment in a subcutaneous xenograft MM model using MM.1R cells, tumor-bearing non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice were injected subcutaneously with control (Ctrl) ASOs or IRF4 ASOs (25 or 50 mg/kg [mpk]), or PBS, 5 times per week for 3 weeks (n = 12 mice per group). Average tumor size (A) and quantitative RT-PCR analysis (B) of human IRF4 mRNA expression in tumors from ASO or control mice are shown.
(C–E) In a MM.1R dissemination model, MM cells were transplanted into NSG mice intravenously, followed by treatment with IRF4 ASOs or controls.
(C and D) Tumor burden estimated by qPCR of relative human β-actin levels (C) and human IRF4 mRNA expression (D) in the bone marrow of hIRF4 ASO-treated MM.1R disseminated mice that received three daily doses of ASOs at 50 mg/kg (n = 4 per group).
(E) Overall survival analysis in the MM.1R dissemination model.
Graphs with errors bars show means ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to PBS-treated controls by ordinary one-way ANOVA for (A)–(D). Significance was assessed using log rank (Mantel-Cox) test for (E). See also Figure S4.
In a MM.1R in vivo dissemination model with significant bone marrow involvement, hIRF4 ASO treatment significantly decreased myeloma burden and IRF4 gene expression, which was undetectable in the bone marrow of some treated animals, leading to increased overall animal survival (Figures 4C–4E). Similarly, in a KMS-11 disease dissemination model where cells home to the bone marrow and can also be detected in the peripheral blood, hIRF4 ASO treatment reduced IRF4 and MYC gene expression and significantly improved overall animal survival (Figures S4F–S4H).
In support of a favorable in vivo therapeutic index for IRF4 inhibition, in immune-competent murine ASO assays, high-dose treatment with murine-specific (m)Irf4 ASOs for 6 weeks moderately reduced mIrf4 expression in murine splenic B cells (Figure S4I), suggesting that higher concentrations of IRF4-targeted ASOs may be required to achieve similar levels of knockdown in normal B cells as compared with malignant MM cells. In tolerability assays, mice treated with mIrf4 ASOs for 4 weeks also showed on-target reduction of Irf4 expression in macrophages but no significant differences in normal hematopoietic cell frequencies compared with controls (Figures S4J and S4K). Together, these results indicate that hIRF4 ASO treatment can significantly reduce human myeloma cell burden in MM xenograft models, and mouse-specific ASOs maintain normal hematopoietic cell frequencies in immune-competent mice.
IRF4 inhibition reduces myeloma cell regeneration in patient-derived in vivo assays
To evaluate IRF4 ASOs in patient-derived pre-clinical models, we established a cohort of MM9-PDX mice (Lazzari et al., 2017). Due to the rapid progression of the disease features in this model once engraftment is detected, we selected a 2-week daily dosing regimen (7× per week) with animals distributed among treatment groups, including two unique human IRF4-targeted agents, vehicle (PBS), or non-targeting ASO control conditions (Figures S5A and S5B). In MM9-PDX mice treated by subcutaneous injection of IRF4 ASOs, live animal bioluminescent imaging showed a reduction in total body luminescence, with a significant decrease in animals receiving hIRF4 ASO-Lead (Figures 5A and 5B). This particularly aggressive model also develops plasmacytomas, and tumor formation was significantly reduced in animals treated with hIRF4 ASO-Lead (Figures 5C, S5C, and S5D).
Figure 5. Human IRF4-targeted ASOs reduce overall tumor burden and myeloma engraftment in a PDX model of high-risk MM.

MM9-PDX mice were treated with human (h)IRF4 ASOs, non-targeting control (Ctrl) ASO, or PBS for 2 weeks (n = 4 to 5 animals per group).
(A and B) Representative live animal bioluminescence images (A) and quantification of normalized (background-subtracted) luminescent signal (B) in a subset of mice from each treatment group (n = 3 per condition). Mice housed together were imaged together (up to 5 mice per image), and individual mouse images were cropped and shown in groups according to treatment condition.
(C) Average tumor numbers in treated MM9-PDX mice.
(D–F) Flow cytometric analyses of live, CD38+ (D), CD138+ (E), and CD319+ (F) cell frequencies in the BM and peripheral blood (PB) of MM9-PDX mice treated with hIRF4 ASOs (n = 5 for hIRF4 ASO-4; n = 4 for hIRF4 ASO-Lead), Ctrl ASO (n = 4), or PBS (vehicle; n = 4). Tissues from a non-transplanted (no tp) control animal are shown for comparison.
Graphs with error bars show means ± SEM; *p < 0.05 compared to vehicle-treated controls by unpaired, two-tailed Student’s t test; n.s., not significant. See also Figure S5.
Analysis of three different plasma cell markers—two of which are direct IRF4 target genes (CD38 and CD319)—showed a significant reduction in the peripheral blood of animals that received hIRF4 ASO-Lead compared to PBS controls, with levels of CD319+ cells also being significantly reduced in the bone marrow of treated animals (Figures 5D–5F). Levels of all plasma cell markers in the peripheral blood and bone marrow of hIRF4 ASO-treated animals were similar to minimal background levels detected in a no-transplant control (Figures 5D–5F), with significant reductions also detected in the liver and plasmacytomas compared with PBS-treated controls (Figures S5E–S5G).
Sensitive intranuclear flow cytometric analyses of IRF4 protein levels revealed a significant reduction of IRF4 protein expression in CD38+ and CD138+ plasma cells in hematopoietic tissues of mice treated with hIRF4 ASO-Lead (Figure 6A). Similarly, there was a striking decrease in IRF4 protein immunoreactivity in bone marrow sections from hIRF4 ASO-treated mice compared with PBS and negative control animals (Figure 6B), suggesting a potent on-target effect of this agent in degrading IRF4 transcripts and reducing IRF4 protein production in myeloma-relevant tissues. Human IRF4 mRNA expression in the bone marrow also correlated with the frequency of plasma cell markers in all study animals, regardless of treatment group, as animals with lower burden of CD38+ plasma cells in particular harbored lower levels of human IRF4 by qRT-PCR (R2 = 0.65; p < 0.0001; Figure S5H).
Figure 6. IRF4 ASOs reduce IRF4 expression and pathway activation in MM9-PDX mice and spare hematopoietic stem cell development in normal human immune cell xenografts.
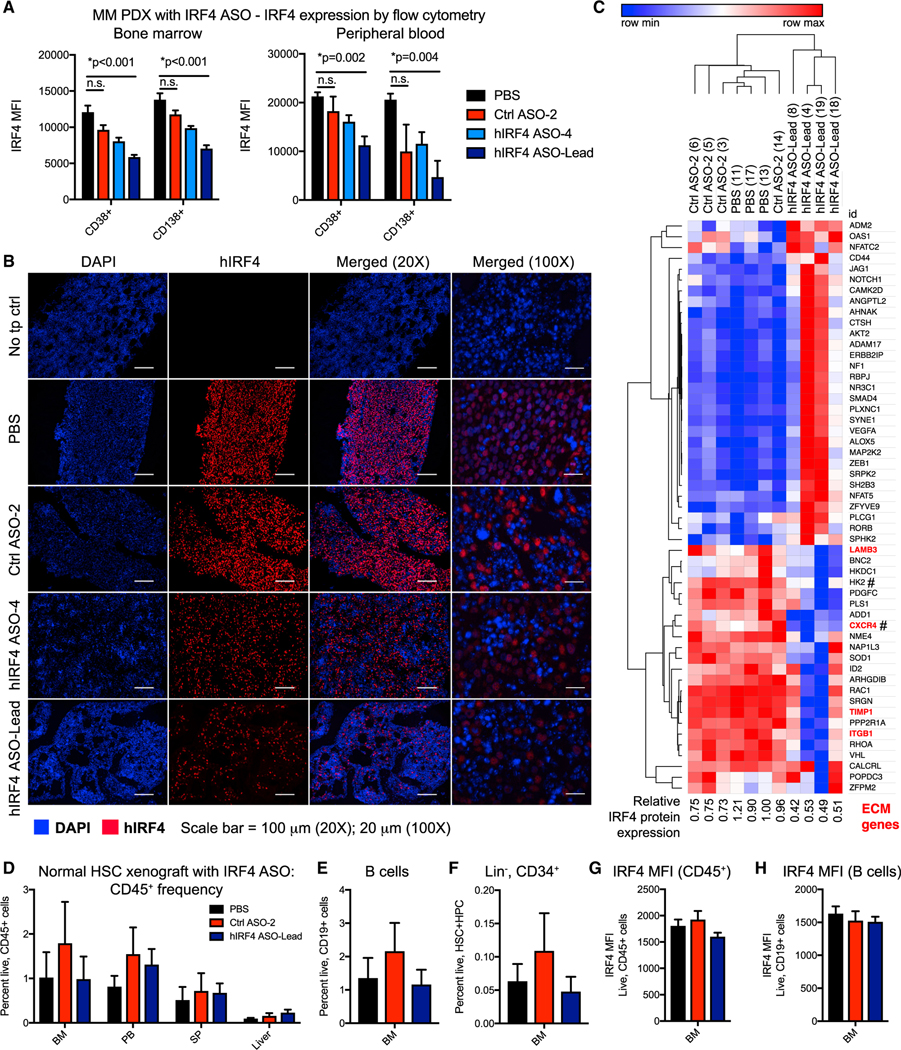
(A) Intranuclear flow cytometric analyses showing MFI of IRF4 protein expression in CD38+ and CD138+ cell populations in the bone marrow and peripheral blood of MM9-PDX mice treated with human-specific (h)IRF4 ASOs (n = 5 hIRF4 ASO-4 and n = 4 hIRF4 ASO-Lead), control (Ctrl) ASO (n = 4), or PBS (vehicle, n = 4) as in Figure 5.
(B) Immunofluorescence analyses of human IRF4 protein, with DAPI staining both mouse and human nuclei, in the bone marrow of control or ASO-treated MM9-PDX mice. Sections from a non-transplanted (no tp) control animal show no IRF4 immunoreactivity. Scale bars represent 100 μm (20×); 20 μm (100×).
(C) NanoString analysis of top differentially expressed human transcripts in total RNA from the bone marrow of treated mice. # represents IRF4 target genes (Shaffer et al., 2008).
(D–F) Human-cord-blood-engrafted mice were treated with hIRF4 ASO-Lead (n = 5), Ctrl ASO (n = 4), or PBS (n = 5). Frequencies of total human hematopoietic cells (CD45+; D), B cells (CD19+; E), and lineage-negative hematopoietic stem and progenitor cells (Lin−, CD34+; F) were determined by flow cytometry.
(G and H) IRF MFI in total hematopoietic (G) and B cell populations (H).
Graphs show means ± SEM; *p < 0.01 compared to PBS vehicle-treated controls by unpaired, two-tailed Student’s t test.
In vivo ASO inhibition of IRF4 impairs myeloma progenitor cell regeneration and spares normal human hematopoietic stem cells and progeny
We further analyzed subpopulations of the CD38+ plasma cell compartment to quantify the frequencies of CD138+ (bulk tumor) and CD138− (progenitor) cells after ASO treatment. In mice treated with hIRF4 ASO-Lead, engraftment levels of human CD38+CD138− MM progenitors and their malignant plasma cell progeny (CD38+CD138+) in the bone marrow were comparable to a no-transplant control mouse (Figures S5I and S5J). Then, to investigate the extent to which hIRF4 ASO knockdown elicits gene expression changes consistent with IRF4 pathway downregulation and cancer burden reduction, a pan-cancer progression NanoString analysis of over 700 transcripts was performed in bone marrow samples from treated MM9-PDX mice. Analysis of human-specific transcripts revealed reduced expression of IRF4 target genes (C-X-C motif chemokine receptor 4 [CXCR4] and hexokinase 2 [HK2]; Johnson et al., 2008; Shaffer et al., 2008, 2009) as well as other microenvironment-responsive and cell cycle regulatory transcripts, including LAMB3, TIMP1, and ITGB1 (Figure 6C). These results suggest that IRF4 ASO treatment provides a highly specific strategy to inhibit IRF4 pathway activation and selectively target MM cells by influencing their cell-cycle-responsive interactions with the bone marrow microenvironment. In addition, genes that respond to ASO treatment in vivo represent potentially valuable functional biomarkers of response to IRF4 inhibition with clinical relevance to planned human trials with these agents.
In a normal human HSPC xenograft model where cord blood CD34+ cells engraft into immunocompromised mice (Crews et al., 2016), in vivo treatment with hIRF4 ASO-Lead spared normal hematopoietic cells (Figure 6D). Comprehensive flow cytometric analyses of hematopoietic populations showed no significant alterations in B cell progeny (CD19+) as well as stem and progenitor populations (CD34+Lineage−; Figures 6E and 6F). Overall expression of endogenous human IRF4 protein was relatively low in engrafted cord blood-derived cells (Figures 6G and 6H) compared with MM9-PDX cells (Figure 6A), suggesting that potential mechanisms underlying a favorable therapeutic index for IRF4 inhibition may include low baseline expression of IRF4 in normal hematopoietic cells and a reduced reliance on the IRF4 pathway for normal immune cell survival.
IRF4 ASO inhibition sensitizes human MM cells to standard-of-care drug treatment
In the clinical setting, selective inhibition of IRF4 might be most useful in combination with other standard-of-care agents. Because lenalidomide and IRF4 ASO treatment both induced G1 arrest, we evaluated whether these two agents may act synergistically to impair human MM cell survival. Indeed, in dose-response assays, H929 cells treated in vitro with IRF4 ASO agents were sensitized to lenalidomide treatment (Figure S6A). Combined IRF4 ASO and lenalidomide treatment also resulted in greater reductions in human IRF4 expression compared with either condition alone (Figure S6B). H929 cells are reported to be more sensitive to lenalidomide than some other MM cell lines (Gandhi et al., 2014), so we then tested whether IRF4 ASOs could sensitize more resistant cell lines to lenalidomide treatment. Combination treatment of KMS-11 cells with IRF4 ASOs and lenalidomide demonstrated a synergistic effect of these two agents even with low doses of lenalidomide (Figure S6C). Moreover, the effects of another standard-of-care agent—the proteasome inhibitor bortezomib—were also potentiated by combination treatment with IRF4 ASO agents in dexamethasone-resistant MM.1R cells (Figure S6D). Together, ASO-mediated IRF4 inhibition enhances sensitivity to lenalidomide and other clinical MM drugs, providing a strong rationale for future combination therapy trials and evaluation of IRF4 ASOs in the relapsed/refractory setting.
Therapeutic mechanisms and translational implications of IRF4 inhibition
In MM patients, high IRF4 expression is associated with lower overall survival rates (Lopez-Girona et al., 2011). Although a recent study in cell lines found that indirect inhibition of IRF4 in vitro could provide a viable strategy to overcome lenalidomide resistance (Zhu et al., 2019), no pre-clinical studies to date have successfully inhibited expression or activity of this transcription factor directly. Notably, the mechanism of action of standard-of-care IMiD therapies for MM involves targeting the cereblon-Ikaros family zinc finger proteins-IRF4 (CRBN-IKZF-IRF4) protein degradation axis (Krönke et al., 2014). Because mutations in these factors have also been implicated in IMiD resistance, disruption of IRF4 via CRBN-independent methods may be necessary to effectively treat disease in drug-resistant cases. Together, IRF4 represents a potentially valuable therapeutic target for MM, particularly for patients who are refractory to standard-of-care drugs.
In comprehensive pre-clinical studies, we show that blocking human IRF4 gene and protein expression with selective ASO agents potently inhibits malignant plasma cell survival and regeneration while sparing normal HSPC and lymphoid cell development. The cell growth inhibitory and cytotoxic effects of IRF4 ASOs were observed across an array of different MM cell lines, in primary patient-derived ex vivo cultures, in subcutaneous tumor xenograft and myeloma cell dissemination assays, and in MM PDX mouse models of high-risk, aggressive disease. Decreased IRF4 mRNA and protein expression corresponded with reduced expression of stem cell regulatory and microenvironment-responsive IRF4 target genes, such as MYC and CXCR4. Together, IRF4 ASOs selectively reduce IRF4 pathway activation and attenuate MM cell survival in diverse pre-clinical models of MM.
RNA-based drug development strategies have been studied for over 40 years (Zamecnik and Stephenson, 1978), and recent advancements in ASO medicinal chemistry have significantly improved their potency and bioavailability. Other ASO strategies that have shown promising pre-clinical or clinical activity include targeting STAT3 in solid tumors (Hong et al., 2015; ClinicalTrials.-gov NCT: NCT02983578), androgen receptor (AR) in prostate cancer (NCT: NCT02144051 and NCT: NCT03300505), and KRAS in solid tumors (Ross et al., 2017; NCT: NCT03101839). In addition, ASOs can be delivered into cells by free uptake, harnessing natural endocytic mechanisms. This enables rapid gene expression modulation in myeloma cells, which are relatively resistant to other gene delivery strategies.
IRF4 plays a role in normal immune cell development, governing B cell differentiation into plasma cells, as well as T cell and macrophage functions (Mittrücker et al., 1997). However, a 50% loss in Irf4 expression in mouse knockout models was well tolerated (Mittrücker et al., 1997), but a 50% knockdown in IRF4 expression is cytotoxic to MM cells (Shaffer et al., 2008), suggesting a potential therapeutic window where normal immune cells may tolerate some reduced IRF4 levels, but MM cells would be more sensitive at the same doses. In addition, although IRF4 RNA expression was detectable in normal plasma cells from healthy donors, it is expressed at significantly higher levels in plasma cells from MM patients (Bai et al., 2017).
In striking contrast to the cytotoxic effects observed on malignant plasma cells in MM models, in pre-clinical normal stem cell developmental in vivo models, we found that IRF4 ASO treatment did not alter the engraftment frequency of total hematopoietic cells, B cell progeny, or lineage-negative HSC and progenitors (CD34+). Similarly, normal B cell frequencies were stable in in vitro normal bone marrow ASO treatment assays. These results are further supported by ASO in vivo pharmacodynamic and tolerability assays, where murine-specific IRF4 ASOs in immune-competent mice maintained normal hematopoietic cell frequencies in peripheral blood. Together, these results suggest a favorable therapeutic index for IRF4 ASO treatment in MM, whereby IRF4 ASOs impair MM cell regeneration while sparing normal HSPC development.
Previous reports suggest that a subpopulation of drug-resistant MM primary cells that are negative for the plasma cell surface marker CD138 display stem-like behavior with enhanced survival and self-renewal capacity, which can be recapitulated in cell line models of MM (Chaidos et al., 2013; Ghosh and Matsui, 2009; Matsui et al., 2004; Van Valckenborgh et al., 2012). Although specific positive cell surface markers for the MM CSC population remain poorly defined, a functional self-regenerating population of MM cells clearly exists and can serially transplant disease in immunocompromised mice (Lazzari et al., 2017)—the gold standard indicator of CSC self-renewal capacity (Kreso and Dick, 2014). We have previously shown that serially transplantable MM cells express the CD319 surface marker (encoded by the IRF4 target gene, SLAMF7; Lazzari et al., 2017), and in the present study, the engraftment of this population was significantly reduced in the bone marrow after treatment with IRF4 ASOs, suggesting that IRF4 inhibition may be able to target a MM-regenerating cell population.
The bone marrow microenvironment provides numerous pro-inflammatory and growth factors that support stem cell maintenance (Pinho et al., 2013). Moreover, intercellular interactions via stem cell regulatory factors, such as NOTCH and CXCR4, can promote microenvironment reprogramming (Colombo et al., 2013) in the inflammatory milieu of the bone marrow in MM (Colombo et al., 2016; Mirandola et al., 2013). We found that recapitulating the bone marrow microenvironment in a stromal co-culture assay enriched for CD138− MM cells and reduced the frequency of cells in the G1 phase. These effects were associated with stromal cell-mediated protection of H929-FUCCI cells against G1 cell cycle arrest induced by lenalidomide treatment. Because G1 phase length has been reported to govern the capacity of multipotent stem cells to remain quiescent versus differentiating in vivo (Calder et al., 2013; Nakamura-Ishizu et al., 2014; Pauklin and Vallier, 2013), a reduced G1 phase length of myeloma cells in the bone marrow microenvironment may support emergence of quiescent progenitor populations.
In keeping with previous findings in myeloid hematologic malignancies where clinically relevant ASOs targeting CSC-enriched transcription factors, such as STAT3, could impair leukemic growth in vivo (Shastri et al., 2018), the results of the present study suggest that overall survival outcomes in MM could be improved by IRF4 ASO-mediated clearance of malignant myeloma cells with self-renewal capacity. Notably, Shastri et al. also demonstrated that STAT3-specific ASOs were detected at a 2-fold higher frequency in myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML) CSC compared with human-cord-blood-derived HSPC, thus supporting the notion that malignant progenitors are more receptive to uptake of ASOs than their healthy blood stem cell counterparts.
Considering that lenalidomide acts in part by downregulating IRF4 expression (Lopez-Girona et al., 2011) and myeloma cytotoxic cell death is enhanced by G1 arrest following inhibition of the cell cycle regulatory cyclin-dependent kinases (CDKs) CDK4/CDK6 via a mechanism involving cell-cycle-coupled loss of IRF4 (Huang et al., 2012), the present results provide new evidence that direct inhibition of IRF4 using selective ASO agents could be a strong potentiator of lenalidomide therapy. Our results further demonstrate that IRF4 ASO treatment induced G1 cell cycle arrest and reduced cell adhesion molecule gene expression, thus providing a potential mechanism of action along with functional biomarkers of response to selective IRF4 inhibition in vivo. In the bone marrow microenvironment, myeloma cell interactions with stromal cells are amplified by CXCR4 (Mirandola et al., 2013; Shaffer et al., 2009; Tokoyoda et al., 2004), the expression of which is induced by IRF4 (Shaffer et al., 2009). Notably, recent clinical studies have tested a CXCR4-directed imaging probe, 68Ga-Pentixafor, for diagnostic receptor targeting in MM patients (Herrmann et al., 2015), suggesting that targeting the IRF4/CXCR4 pathway may have a range of clinical applications.
Together, the results of the present study suggest that the pro-inflammatory bone marrow niche may contribute to drug resistance through cell-cycle-associated deregulation of microenvironment interactions between MM progenitor cells and stromal cells. IRF4 downmodulation may mobilize bone-marrow-resident quiescent myeloma cells away from the bone marrow niche through disruption of CXCR4 and other extracellular matrix components. Notably, a clinical candidate hIRF4 ASO-Lead (ION251) has been identified and is advancing into human clinical development. Thus, antisense targeting of IRF4 represents a clinically tractable strategy that may be effective for relapsed/refractory MM patients and could be monitored longitudinally in patients through biomarker testing as well as CXCR4-directed molecular imaging modalities. Selective detection and inhibition of this key viral-response transcription factor pathway using antisense therapies could prevent disease relapse driven by malignant regeneration in inflammatory microenvironments.
Limitations of study
The RNA-seq studies of primary patient samples were designed to characterize the IRF4 pathway in the sample used for PDX model development, with additional primary samples shown for comparison. A potential caveat of this is the relatively small sample sizes. Therefore, we also evaluated IRF4 RNA and protein expression in a second cohort of primary samples. Our results provide new data in support of increased IRF4 expression in untreated, high-risk MM, including PCL, with relatively low protein expression in normal B, T, and macrophage/monocyte populations. In addition, in MM9-PDX assays with IRF4 ASO treatment, myeloma progenitor and malignant plasma cell engraftment reached the lower limit of detection by flow cytometry, precluding analyses such as serial transplantation after treatment. Therefore, several different subcutaneous and dissemination-based xenograft mouse models were tested. In these complementary assays, alternative endpoint measurements (e.g., tumor growth and overall animal survival) were used to confirm the therapeutic efficacy of IRF4 ASO treatment. Future investigations could also address whether a selective IRF4 inhibitory strategy could lead to deep remissions if used in monotherapy or combination therapeutic approaches at the disease initiation stage. We anticipate that clinically relevant efficacy and mechanistic data on hIRF4 antisense therapy will be generated through ancillary studies to forthcoming clinical trials.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Leslie A. Crews (lcrews@ucsd.edu).
Materials availability
The study did not generate new unique biological reagents. There are restrictions to the availability of the ASO reagents described here due to their current development as part of an industry-sponsored clinical trial.
Data and code availability
The accession number for the RNA-sequencing data reported in this paper is dbGaP: phs002291.v1.p1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Experiments
All animal studies were performed in accordance with UCSD and NIH-equivalent ethical guidelines and were approved by the university institutional animal care and use committee (IACUC). Newborn (1–3 days) BALB/c Rag2−/− IL2Rγc−/− mice of both genders (sample size depending on litter survival rates) were intrahepatically injected with a 30-gauge Hamilton syringe (Hamilton Company). Depending on the experiment and number of viable cells available for transplant, animals were injected with 105-106 cells (constant numbers of cells were used in all mice for each individual experiment) from human cell-enriched bone marrow (BM) or collagenase-digested plasmacytoma (PC) tissues processed using a mouse cell depletion kit (Miltenyi). We found that engraftment of cells derived from either PCs or BM was detectable in immunocompromised mice in as few as 5–7 weeks post-transplant. While cells derived from PCs alone showed somewhat more rapid but variable engraftment levels in a small cohort of mice (Figures S1A and S1B), the mixture of BM and PC-derived cells showed consistent engraftment rates at weeks 6–8 post-transplant (Figures 1A and 1B). Mixing of the MM9-PDX bone marrow-derived and PC-derived cells was performed for some serial transplantation studies to promote rapid but consistent engraftment without selecting for a specific niche, as well as for technical reasons associated with insufficient numbers of human-enriched cells recoverable from the bone marrow alone. The mouse cell depletion strategy allows the engrafted human cells to remain minimally manipulated (no positive antigen selection), and ensures that transplanted cells contain both CD138+ fractions as well as CD138− progenitor populations.
Transplanted animals were weaned at 3 weeks of age and monitored regularly by health status assessment. In vivo bioluminescence imaging (IVIS 200) and peripheral blood screening of kappa chain levels using the human kappa ELISA quantitation set (Bethyl Laboratories, Inc) were regularly performed until clinical signs of disease were observed, including significant loss of weight, limited mobility and/or presence of palpable tumors.
Human Subjects
Patient samples were obtained from consenting patients at the University of California, San Diego or the University of Toronto in accordance with approved human research protections program Institutional Review Board approved protocols that meet the requirements as stated in 45 CFR 46.404 and 21 CFR 50.51. Human cord blood samples were purchased as purified CD34+ cells from AllCells Inc or StemCell Technologies Inc. Detailed patient information can be found in Table S1.
Cell Lines
The human myeloma cell lines H929 (RRID:CVCL_1600) and RPMI-8226 (RRID:CVCL_0014) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured under the supplier’s recommended conditions. Briefly, H929 cells were cultured in RPMI 1640 containing 10% Fetal Bovine Serum, 1% Penicillin/Streptomycin, 1% Non-Essential Amino Acids and 0.05mM β-Mercaptoethanol, and RPMI-8226 cells were cultured in RPMI-1640 supplemented with 10% Fetal Bovine Serum (FBS). The human bone marrow stromal cell lines (HS5, RRID:CVCL_3720 and HS27a, RRID:CVCL_3719) were obtained from ATCC and cultured in DMEM or RPMI-1640 media supplemented with 10% FBS as previously described (Crews et al., 2016). The murine bone marrow stromal cell line OP9 was generously provided by Dr. Raffaella Chiaramonte (University of Milan, Italy), and cultured in Alpha Minimum Essential Medium without ribonucleosides and deoxyribonucleosides and with 2.2 g/L sodium bicarbonate, supplemented with 20% FBS. All other human myeloma cell lines were obtained from the Japanese Collection of Research Bio-resources (JCRB), ATCC, or the German Collection of Microorganisms and Cell Cultures (DMSZ) and cultured under the suppliers’ recommended conditions: KMS-11 (JCRB), MM.1R (ATCC), U266 (ATCC), JJN3 (DSMZ), EJM (DSMZ), L363 (DSMZ), AMO1 (DSMZ).
For stromal co-culture experiments, HS or OP9 cells were irradiated at 4000 Rads and stained with CellTrace Violet dye (Life Technologies) prior to co-culture with human myeloma cells. Stromal cells were plated as confluent monolayers, and then myeloma cells were added on top of stromal cells for 24 h. Drug treatment (lenalidomide 10μM) was added to co-cultures or MM cells grown in suspension culture alone, for an additional 24 h, followed by CD138 and cell cycle quantification by flow cytometry. H929-FUCCI cells were co-cultured with human or mouse bone marrow stromal cell lines (HS or OP9), along with primary MM stromal cells (prepared as adherent monolayers established from the CD34-negative fraction of primary MM patient bone marrow samples).
Primary Cell Cultures
For ex vivo studies using primary normal bone marrow samples, cryopreserved MNCs from the bone marrow of healthy donors were plated directly into 96-well plates and cultured for up to 3 days in StemPro serum-free media (ThermoFisher Scientific). For ex vivo studies using primary patient-derived MM cells, fresh cells isolated from the bone marrow or collagenase-digested plasmacytomas of MM9-PDX mice were washed in HBSS containing 1% FBS and plated directly into flat 96-well plates pre-coated with Matrigel (ThermoFisher Scientific, Carlsbad, CA). Cells were cultured for up to 5 days in StemPro serum-free media (ThermoFisher Scientific).
METHOD DETAILS
Primary patient samples and in vivo patient-derived xenograft model maintenance
All primary patient samples were collected according to local institutional review board (IRB) guidelines under approved protocols and patient consent. Primary MM samples were processed as previously described (Crews et al., 2016; Lazzari et al., 2017) by Ficoll density centrifugation to isolate mononuclear cell (MNC) fractions from bone marrow or peripheral blood samples. Viably cryopreserved cells (in 90% FBS supplemented with 10% DMSO) were stored in liquid nitrogen until use in experimental studies. For in vivo transplantation of primary samples, total MNCs were thawed and plated in cytokine-supplemented StemPro serum-free media (ThermoFisher Scientific, Carlsbad, CA) containing lentiviral vectors encoding GFP-luciferase or control vector constructs (Abrahamsson et al., 2009) for up to 48 h. Aliquots of transduced cells were reserved for RNA-sequencing (RNA-seq) analyses to confirm gene pathway activation in cells used to establish patient-derived xenograft (PDX) models of MM such as MM9.
RNA-sequencing and analyses
For whole transcriptome analyses, total RNA was isolated from primary MM sample cell aliquots reserved from transduction and transplantation experiments, quantified by Nanodrop, and submitted to the Scripps Research Next Generation Sequencing (NGS) Core (San Diego, CA) for analysis as previously described (Crews et al., 2016). Briefly, RNA quality was assessed using a Bioanalyzer instrument, and only samples with RNA integrity (RIN) values > 7 were subjected to library preparation for RNA-seq. The SMARTer v4 cDNA amplification kit was used along with library construction by the NEB Ultra DNA kit for Illumina. Sequencing was performed on Illumina NextSeq instruments (Scripps NGS Core), with all samples run over two separate flow-cells (to generate additional sequencing reads); > 95M reads were generated in total for each sample.
Data were analyzed by Rosalind (https://rosalind.onramp.bio/), with a HyperScale architecture developed by OnRamp BioInformatics, Inc. (San Diego, CA). Reads were trimmed using cutadapt (Martin, 2011). Quality scores were assessed using FastQC (Andrews, 2010). Reads were aligned to the Homo sapiens genome build GRCh38 using STAR (Dobin et al., 2013). Individual sample reads were quantified using HTseq (Anders and Huber, 2010) and normalized via Relative Log Expression (RLE) using DESeq2 R library (Love et al., 2014). Read Distribution percentages, violin plots, identity heatmaps, and sample MDS plots were generated as part of the QC step using RSeQC (Wang et al., 2012). DEseq2 was also used to calculate fold changes and p values and perform optional covariate correction. Clustering of genes for the final heatmap of differentially expressed genes was done using the PAM (Partitioning Around Medoids) method using the CRAN: fpc R library (Hennig, 2013). Hypergeometric distribution was used to analyze the enrichment of pathways, gene ontology, domain structure, and other ontologies. The topGO R library (Alexa and Rahnenfuhrer, 2019), was used to determine local similarities and dependencies between GO terms in order to perform Elim pruning correction. Several database sources were referenced for enrichment analysis, including Interpro (Mitchell et al., 2019), NCBI (Geer et al., 2010), MSigDB (Liberzon et al., 2011; Subramanian et al., 2005), REACTOME (Fabregat et al., 2018), and WikiPathways (Slenter et al., 2018). Enrichment was calculated relative to a set of background genes relevant for the experiment. Functional enrichment analysis of pathways, gene ontology, domain structure and other ontologies was performed using HOMER (Heinz et al., 2010).
For IRF4 pathway-specific analyses on the Rosalind platform, a custom gene list was generated based on putative IRF4 target genes that have been previously reported (Shaffer et al., 2008). Differentially expressed genes from this gene set were calculated in Rosalind and displayed in a heatmap. The differentially expressed IRF4 target genes were further tested for pathway enrichment using GSEA (https://www.gsea-msigdb.org/gsea/index.jsp) and the number of represented genes within the top 10 Reactome pathways were plotted in graphical format using Prism (GraphPad Software, San Diego, CA).
An additional publicly-available RNA-sequencing dataset was also utilized to evaluate IRF4 expression levels from 66 human myeloma cell lines (Keats et al., 2007). Relative mRNA expression values of IRF4 from these lines were extracted from the available processed transcript expression data, in Fragments Per Kilobase of transcript, per Million mapped reads (FPKM), downloaded from the Keats Lab repository (https://www.keatslab.org/data-repository).
Lentiviral vectors and generation of a stable fluorescent ubiquitination cell cycle indicator (FUCCI) myeloma cell line for stromal co-culture assays
Lentiviral vector constructs expressing FUCCI components (mCherry-hCdt1 and mVenus-hGeminin) were generously provided by Dr. Atsushi Miyawaki (Riken, Japan) (Sakaue-Sawano et al., 2008), and lentiviral vectors expressing human IRF4 (GFP) or GFP control were obtained from Genecopoeia (Rockville, MD). Infectious lentiviral particles were produced and purified as previously described (Crews et al., 2016; Lazzari et al., 2017; Pineda et al., 2016). The FUCCI system is comprised of two vectors expressing fluorescent markers that are detectable during the G0-G1/S or S/G2/M phases of the cell cycle. Red (mCherry) or green (mVenus) fluorescent proteins are fused to Cdt1 and Geminin, respectively, to allow live tracking of individual cells and quantification of proportions in G1 or S/G2/M-phase. The MM cell line NCI-H929, which harbors a CD138− subpopulation, underwent sequential rounds of lentiviral transduction with these two vectors. For production of a stably-transduced H929-FUCCI cell line, wild-type H929 cells were cultured under standard conditions. Freshly-passaged H929 cells were transduced sequentially with lentiviral-FUCCI-Cdt1-mCherry followed by lentiviral-FUCCI-Geminin-mVenus with FACS purification to isolate the transduced cells. Briefly, H929 cells were plated into a 96-well plate and transduced with lentiviral-FUCCI-Cdt1-mCherry at a multiplicity of infection (MOI) of 5–20. Cultures were expanded to a minimum of 1×106 cells followed by FACS purification on a HEPA filter-equipped BD Influx cell sorter (BD Biosciences) of mCherry-positive cells. Then, the positive cells were further expanded and the H929-FUCCI-Cdt1 line was plated into a 96-well plate and transduced with lentiviral-FUCCI-Geminin-mVenus (MOI = 20) followed by FACS purification of mVenus-positive or mCherry/mVenus double-positive cells. The dual-transduced stable cell line (H929-FUCCI) was then expanded for cryopreservation and in vitro experiments.
For stromal co-culture assays using the H929-FUCCI model, stromal cells were plated as a confluent monolayer, and irradiated and stained with CellTrace Violet as described above. Then wild-type H929 cells or H929-FUCCI cells were added on top of stromal cells for 24 h. Drug treatment (lenalidomide 10 μM) was added for an additional 24 h, and then cells were collected for cell cycle quantification by flow cytometry.
For lentiviral overexpression of human IRF4, cells were incubated with concentrated lentiviral particles (MOI ranging from 10–100) for 48 h before harvesting and processing for flow cytometry and qRT-PCR. Overexpression of IRF4 was verified using Taqman qPCR assay (ThermoFisher) and lentiviral transduction was confirmed by using flow cytometry to confirm presence of GFP positive cells before analysis of the CD138 marker.
In vitro treatments, viability assays, and apoptosis analyses
Antisense oligonucleotides and standard-of-care drug treatment conditions
All antisense oligonucleotides (ASOs) were designed and synthesized by Ionis Pharmaceuticals. The ASOs, or PBS vehicle, were diluted in the previously described cell culture media and for most experiments cells were plated into multiwell plates and treated in triplicate with 2-fold serial dilutions of targeted ASO agents, or at the concentrations specified in each figure legend. As an additional control, negative non-targeting ASO agents were used at the same concentrations, or at concentrations that result in > 50% IRF4 knockdown in quantitative RT-PCR studies. For all in vitro assays (Figure 2), cells were incubated with human (h)IRF4-targeted ASOs at doses ranging from 0.1 nM-10 μM for 2–5 days, compared with non-targeting control (Ctrl) ASOs (1–5 μM). For viability assays, cells were incubated for 5–6 days, while for RNA and protein knockdown assessments, cells were incubated for 48 or 72 h, respectively. Since it is difficult to capture true biological variability with assays performed in a single cell line, we performed all assays across a panel of different myeloma cell lines, and with multiple unique ASOs that each targets a different sequence of human IRF4 mRNA. For all viability, qPCR, and colony assays, assays for each cell line were performed in at least three individual experimental wells treated with ASO agents separately. For verification of the results, a subset of assays in H929 and RPMI-8226 cells was repeated in a minimum of three experimental (biological) replicates analyzed in separate passages of cells on separate days.
For single agent or combination treatment studies with standard-of-care myeloma drugs, MM cell lines were pre-incubated with ASO agents for 24 h, followed by the addition of lenalidomide (ThermoFisher Scientific) at doses of 0.4–10 μM or bortezomib (Sigma) at doses of 1.25–2.5 nM, with PBS or DMSO used as vehicle controls. Cells were incubated under these combination treatment conditions for a further 1–2 days (for RNA analyses) or 4–5 days (for viability assays).
Cell viability and apoptosis assays
For human myeloma cell lines and ex vivo PDX cells treated with ASO agents alone or in combination with standard-of-care drugs, cells were cultured in 96 well plates in triplicate and then analyzed using the CellTiter Glo 2.0 (Promega) cell growth and viability assay or the Caspase Glo 3/7 apoptosis assay system (Promega). Luminescence readings were collected on a GloMax (Promega) luminometer and relative luminescence values were quantified after background subtraction of media-only containing wells.
Colony assay
50,000 H929 cells were plated into wells of a 96 well plate and treated with increasing concentrations of IRF4 ASO, non-targeting control ASOs or PBS for 48 h. 100–200 cells were then plated in 1ml of methylcellulose (HSC-CFU basic media, Miltenyi) in separate wells of a 12-well plate and incubated at 37°C for 14 days. Total colony numbers in each well were counted and averaged across a minimum of 3 separate wells for each condition.
Flow cytometry analyses in primary patient samples and in vitro cultures
For flow cytometric analyses of IRF4 protein levels in primary samples and in vitro culture assays, MNCs were stained with a live/dead NearIR stain (ThermoFisher Scientific) and then incubated with antibodies against normal immune cell markers (CD3 AF488, CD19 PE, CD14 BV605) and MM cell markers (CD38 PE-Cy7, CD138 VioBlue, CD319 PerCP-Cy5.5). For IRF4 protein analyses in myeloma cell lines treated with IRF4 ASOs, only the MM cell markers were included. Cells were then fixed and permeabilized (BioLegend True-Nuclear Kit) according to the manufacturer’s protocol to allow for intranuclear staining with an AF647-labeled antibody against human IRF4 (BioLegend). Samples were analyzed on BD LSR Fortessa (BD Biosciences) or MACSQuant instruments (Miltenyi), with cell frequencies and IRF4 median fluorescence intensities (MFI) quantified on FlowJo (Treestar, Inc.). For intranuclear flow cytometry-based protein analyses of IRF4, we focused on the CD38-high (CD38++) fraction (Figure 1E), as this surface marker is a direct IRF4 target gene, and malignant plasma cells are uniformly high in CD38 expression (Leo et al., 1992).
For flow cytometric analyses of cell cycle status in H929-FUCCI cells, stably-transduced cells were treated with lenalidomide or serial dilutions of IRF4 ASO in 96-well plates, with non-targeting ASOs, DMSO, or PBS as controls. For IRF4 ASO studies, after 72 h of incubation the cells were collected and stained with CD138 APC-conjugated antibody (Clone 44F9, Miltenyi) and DAPI (1 μg/mL, ThermoFisher Scientific) and analyzed using the BD LSR Fortessa (BD Biosciences). Live (DAPI-negative) cells were evaluated for CD138+ cells as well as fluorescence in the Texas Red and FITC channels to determine the proportion of cells in each phase of the cell cycle, based on the fluorescence of the FUCCI vectors; mCherry (red) is expressed during G1 phase, an intermediate mCherry+/mVenus+ (yellow) detected during G1/S phase, and mVenus (green) during G2/M phase (Pineda et al., 2016; Sakaue-Sawano et al., 2008). All analyses were performed using FlowJo (Treestar, Inc.) and plotted using Prism (GraphPad, San Diego, CA). Similar analyses were performed for CD138+/− populations in lentiviral-IRF4-transduced cells. Briefly, cells were stained with CD138 APC antibodies (Miltenyi) and DAPI, with analysis on a MACSQuant instrument (Miltenyi).
RNA extraction and quantitative RT-PCR
For analysis of gene expression by qRT-PCR, cells or tissue fragments were harvested in RNA lysis buffer and total RNA was extracted using RNeasy mini, micro, or 96 extraction kits (QIAGEN, Germantown, MD). Taqman one-step qRT-PCR assays were performed using human IRF4-specific (FW-5′-GGCAAAGAAAGCTCATCACAG-3′; REV-5′-GGATTGCTGATGTGTTCTGGTA-3′; Probe: 5′FAM-TAGCCCCTCAGGAAATGTCCACTG-IOWA-BLACK-3′ (with internal ZEN) and human β-actin (loading control; FW-5′CGGACTATGACTTAGTTGCGTTACA-3′; REV-5′-GCCATGCCAATCTCATCTTGT-3′; Probe: 5′FAM-CCTTTCTTGACAAAACCTAA CTTGCGCAGA-TAMRA-3′) primer/probe sets, or murine IRF4 (FW-5′TCAGAGACAGAGGAAGCTCAT-3′; REV-5′-GTGGTAATCTGG AGTGGTAACG-3′; Probe: 5′−6-FAM-HA-TGGCTAGCAGAGGTTCCACATGAG-Iowa Black-3′ (with internal ZEN) or murine cyclophilin A (PPIA) (FW-5′-TCGCCGCTTGCTGCA-3′; REV-5′-ATCGGCCGTGATGTCGA-3′; Probe: 5′-FAM-CCATGGTCAACCCC ACCGTGTTC-TAMRA-3′) as a loading control. For this purpose, 10 ng of RNA per sample and primer/probe sets were mixed with one-step RT-qPCR reagents using the EXPRESS One Step SuperScript® RT-PCR Kit (ThermoFisher Scientific) with quantification and analysis carried out on a StepOnePlus real-time PCR system. Transcript levels were quantified using gene-specific primer-probe sets and were normalized to human β-Actin/ACTB mRNA, or murine Ppia levels. For two-step SYBR-green based assays, as previously described (Crews et al., 2016; Lazzari et al., 2017), 100–1000 ng of RNA were subjected to cDNA synthesis using the SuperScript III (ThermoFisher Scientific) kit followed by qRT-PCR using SYBR GreenER (ThermoFisher Scientific) master mix according to the manufacturer’s recommended cycling conditions on BioRad iQ5 or BioRad CFX384 instruments using primers specific for human IRF4 that correspond to the same primer sequences utilized in the Taqman assays (FW-5′-GGCAAAGAAAGCTCATCA CAG-3′, REV-5′-GGATTGCTGATGTGTTCTGGTA-3′). Some validation assays were also performed using previously published primers specific for human IRF4 (Lazzari et al., 2017), and human HPRT was utilized as a loading control as previously described (Crews et al., 2016; Lazzari et al., 2017). Additional Taqman assays were also performed using cDNA and primer/probe sets specific for human MYC or human β-actin (loading control) along with Taqman Fast Advanced master mix (all from ThermoFisher Scientific), with qRT-PCR performed according to the supplier’s recommended cycling conditions on a ThermoFisher QuantStudio3 instrument. Relative mRNA expression values were calculated using the 2-ΔCT method (Bustin et al., 2009), with normalization to untreated or vehicle-treated controls.
In vivo ASO treatments and analyses
For all in vivo ASO efficacy studies, target doses and treatment regimens were selected based on the optimal therapeutic dose observed in potency assays (at least 50 mg/kg), extensive previous pharmacokinetic studies of ASO agents (Crooke et al., 2019), and the rate of disease progression in each in vivo model, with a goal of achieving a similar total exposure to the ASO agents over each study period (ranging from 750–1000 mg delivered). In pharmacodynamic and tolerability studies in healthy animals, less frequent dosing regimens were used because the ASO half-life in post mitotic tissues is approximately 2 weeks. In addition, in higher disease burden models such as the PDX assays, ASOs were delivered with a higher bolus loading dose to promote rapid uptake into the plasmacytomas and achieve tissue (tumor) levels sufficient to reduce target RNA, and up to 7X per week to maintain consistent exposure to IRF4 ASO agents in rapidly-dividing tumor cell populations. For in vivo treatments in xenograft assays using human myeloma cell lines, subcutaneous tumor formation and dissemination strategies were used. For subcutaneous transplantations of KMS-11 and MM.1R cells, 5×106 cells were implanted subcutaneously into the flanks of female 5-week old NOD/SCID mice. Dosing was initiated when tumors reached an average of ~150 mm3. For in vivo treatments, animals received subcutaneous injections of ASO agents (IRF4 targeted tool compounds or negative non-targeting ASO controls) at 25 or 50 mg/kg, or PBS vehicle control in equivalent volumes. For in vivo potency assays, animals were dosed 5 times weekly for up to 30 days and for in vivo target knockdown assays (IRF4 protein analyses), animals received 5 (KMS-11 model) or 3 (MM.1R model) consecutive daily doses at 50 mg/kg. At the end of each study animals were euthanized 48 h after the last dose and peripheral blood samples were collected for light chain analyses, and tumor tissues were removed and processed for RNA (qRT-PCR) and protein (western blot) analyses.
For in vivo treatments and survival analyses in a KMS-11 transplantation model where MM cells distribute mostly to bone marrow, 107 cells were transplanted intravenously into cyclophosphamide-pretreated (50 mg/kg) 5-week old NOD/SCID mice. Mice were treated with PBS control or ASO agents by subcutaneous injection (50 mg/kg/dose, 3 times per week) for up to 5.5 weeks. Bone marrow and peripheral blood were collected for evaluation of IRF4 pathway gene expression by qRT-PCR. Similar studies were also performed in a systemically disseminated MM.1R model with significant bone marrow involvement. For these studies, NOD-scid IL2Rγ−/− (NSG) mice were given a single 50 mg/kg dose of cyclophosphamide, and the next day, 107 MM.1R cells were transplanted intravenously. Two weeks after transplantation, serum light chain levels (Igλ chain) were quantified to monitor engraftment levels and randomize animals for treatment. Animals in the efficacy (survival) cohort received a loading dose regimen of PBS control or ASO treatments (IRF4 targeted tool or lead compounds or negative non-targeting ASO controls) at 50 mg/kg injected daily, subcutaneously, for one week, followed by a maintenance dose regimen of 3 doses per week at 50 mg/kg, until body weight loss exceeded 20% or clinical symptoms required euthanasia. Animals in the pharmacodynamic cohort received 3 daily consecutive doses of ASO agents or PBS, and were euthanized 48 h after the last dose to quantify IRF4 expression in the bone marrow and relative tumor burden by qRT-PCR.
For in vivo pharmacodynamic studies in an immune-competent mouse strain, C57BL/6 mice were treated with ASO agents targeting murine Irf4 at a dose of 120 mg/kg delivered once weekly by subcutaneous injection for six weeks (total ASO dose = 720 mg). At the end of the study, murine splenic B cells were analyzed by qRT-PCR for mIrf4expression. For in vivo tolerability studies in an immune-competent mouse strain, C57BL/6 mice were treated with ASO agents targeting murine Irf4 at a dose of 100 mg/kg delivered once weekly by intraperitoneal injection for four weeks (total ASO dose = 400 mg). At the end of the study, peritoneal macrophages and peripheral blood samples were collected for qRT-PCR of mouse IRF4 and evaluation of hematology parameters. For this purpose, approximately 0.5 mL blood was collected into K2-EDTA-coated tubes (ThermoFisher Scientific) from all study animals. Hematology endpoints, including lymphocyte, white blood cell (WBC), monocyte, hemoglobin (HGB), and mean corpuscular volume (MCV) were analyzed by IDEXX Laboratories (Westbrook, ME).
For in vivo treatments in a patient-derived pre-clinical model, MM9-PDX-engrafted mice were established by intrahepatic injection of individual Rag2−/−IL2Rγ−/− neonates with 106 human-enriched cells from plasmacytomas of MM9-PDX mice (processed using a mouse cell depletion kit, Miltenyi). Human myeloma cell engraftment levels were monitored as described above by bioluminescence imaging (IVIS 200) and peripheral blood screening of kappa chain levels using the human kappa ELISA quantitation set (Bethyl Laboratories, Inc). For MM9-PDX efficacy studies with IRF4-targeted ASOs, starting 7 weeks after transplant, engrafted mice were treated daily by subcutaneous injection for a two-week period with IRF4 tool or lead ASO agents, non-targeting control, or PBS vehicle. ASO-treated animals received a loading dose regimen of 3 initial doses at 100mg/kg followed by 11 additional consecutive daily doses at 50mg/kg. A subset of treated animals were imaged by IVIS 24 h before euthanasia. For bioluminescence imaging, mice housed together were imaged together (up to 5 mice per image), and individual mouse images were cropped and shown in groups according to treatment condition. Quantitative bioluminescence signals were normalized by subtracting background flux levels for each image. Animals were injected subcutaneously and sacrificed 24–48 h after the last dose. Mice were killed by CO2 inhalation. Peripheral blood was collected by cardiac puncture immediately after sacrifice. Bones, spleen, liver, and plasmacytomas were collected in ice cold HBSS containing 2% FBS. Tissues were processed to a single-cell suspension. After manual mincing, plasmacytomas were digested with 1 mg/mL collagenase IV (GIBCO) for 30 minutes-1hr for each sample. Tissue samples were processed for flow cytometry and RNA analyses. In addition, one femur from each animal, along with one plasmacytoma for animals with more than one tumor, were also preserved by fixation in 10% neutral-buffered formalin (NBF) followed by 70% ethanol, then paraffin-embedding and sectioning for immunohistochemical and immunofluorescence analyses (Moores Cancer Center Tissue Technology Shared Resource).
For in vivo treatments in a normal hematopoietic stem and progenitor cell xenograft model, CD34+ cells from human umbilical cord blood samples (AllCells) were transplanted (105 cells per mouse) as previously described (Crews et al., 2016). Animals were weaned at 3 weeks of age and monitored regularly by health status assessment; peripheral blood screenings using human CD45 BB515 (BD) were regularly performed until a population of human CD45+ cells was detected in the blood (average of 1% positive cells in the peripheral blood). Eight weeks after transplant, mice were distributed among treatment groups on the basis of human cell engraftment in their peripheral blood, and treated with the same ASO dosing regimen as MM9-PDX mice. Normal hematopoietic tissues were collected and processed for flow cytometry analyses.
Western blot analyses
For analysis of human IRF4 protein expression, other downstream targets, and markers of cell death, cell lysates were prepared from cells treated with ASO agents or controls for 3 days and processed for western blot analyses, and tumor tissue lysates were prepared from animals treated as described above. For western blots, cells and tissue fragments were collected in RIPA buffer and 20 μg per sample were analyzed by gel electrophoresis and immunoblot using antibodies against IRF4 (Abcam ab133590, or Dako M7259 or GA64461-2), c-MYC (Abcam), cleaved PARP (Abcam), and β-actin (loading control, Abcam). After incubation with species-specific secondary antibodies, blots were imaged using enhanced chemiluminescence reagents and X-ray film. For western blot analyses, the band corresponding to the molecular weight of each protein is shown as a single cropped bar for each membrane image. Some antibodies were analyzed on separate blots to avoid stripping membranes repeatedly.
In vivo flow cytometry analyses
Tissues from MM PDX mice were stained with CD138 VioBlue (Miltenyi), CD38 PE-Cy7 (BD Biosciences), CD319 PE (Miltenyi), and IRF4 AF647 (BioLegend). Tissues from cord blood-transplanted mice were stained with two separate antibody panels: a stem and progenitor panel including CD45 APC (Invitrogen), Lineage PE-Cy5 (BD Biosciences), CD34 BV421, and CD38 PE-Cy7 (BD Biosciences) and a differentiated panel including CD45 APC (Invitrogen), CD3 FITC (BioLegend), CD19 PE (BioLegend), CD14 PE-Cy7 (ThermoFisher Scientific, and IRF4 AF647 (BioLegend). The differentiated panel was fixed and permeabilized (BioLegend True-Nuclear Kit) after surface antibody staining according to the manufacturer’s protocol to allow for IRF4 AF647 intranuclear staining. Samples were analyzed on a MACSQuant instrument (Miltenyi), with cell frequencies quantified on FlowJo (Treestar, Inc.).
Immunohistochemical and immunofluorescence analyses
Tissues (femurs and plasmacytomas) from MM9-PDX mice were fixed in 10% neutral buffered formalin for 4 days. Tissues were transferred to 70% EtOH and stored at room temperature until being processed for paraffin embedding and sectioning by the Moores Cancer Center Tissue Technology Shared Resource.
Immunostaining was performed according to standard and optimized protocols at the Moores Cancer Center Tissue Technology Shared Resource. Tissues were baked at 60°C for 1 hour, cleared and rehydrated through successive alcohols, and underwent antigen retrieval in sodium citrate buffer (pH 6) at 110°C/15 min (Intellipath Automated IHC Stainer, Biocare). For immunohistochemical (IHC) double-labeling, tissues were stained with the following primary antibodies for 1 h: Anti-MUM1 primary antibody (Rabbit, Abcam, Cat# ab133590, 1:2000). Anti-Syndecan-1 (CD138) primary antibody (Rat, R&D Systems, MAB2780, 1:1000). Tissues were then stained with the corresponding secondary Abs for 30 min: IHC - Biotinylated Anti-Rabbit IgG (1:200, Vector, Cat# BA-1100), Biotinylated Anti-Rat IgG (Vector, Cat# BA-9401). For immunofluorescence (IF), Anti-Rabbit HRP Secondary Antibody (Jackson, Cat# 711-036-152) was used. Tissues were then incubated with corresponding reagents for 30 mins: IHC - ABC-HRP (Vector, Cat# PK-6100), ABC-AP (Vector, Cat# AK-5000). For visualization, tissues were incubated with corresponding reagents for 5–10 mins: IHC - Deep Space Black Chromogen (Biocare, Cat# BRI4015), Warp Red Chromogen (Biocare, Cat# WR806), IF - Alexa Fluor 647 Tyramide Reagent (Thermo, Cat# B40958) and Vectashield Hardset with DAPI (Vector, H-1500). Tissues were mounted and imaged at 20X and 100X magnification on a Keyence BZ-X710 (Moores Cancer Center Microscopy Shared Resource).
NanoString analysis
Total bone marrow cells from control (non-transplanted) mice, or MM9-PDX mice treated with hIRF4 ASO-Lead, control ASO, or PBS (vehicle) were collected and RNA was extracted as for qRT-PCR assays. 50 ng of RNA per sample was prepared for analysis with a NanoString PanCancer Progression chip. The assay was performed on an nCounter MAX Analysis System (Sanford Consortium for Regenerative Medicine Genomics Core) according to the manufacturer’s instructions. The nSolver software by NanoString was used to normalize, analyze, and interpret targeted gene expression data. Transcripts that were noted to cross-react with mouse genes were excluded from the analysis based on genes that were detected at a 2 fold or higher level in the no transplant control sample than in the PBS-treated samples. Expression of human-specific genes were then compared between the hIRF4 ASO-treated and PBS treated samples. Genes that were expressed at significantly different levels between the two groups were then used to create heatmaps using Morpheus software.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed with at least three biological or experimental replicates, with specific number of replicates and animal numbers stated in the figure legends. Unless otherwise stated, statistical analyses of non-RNA-seq data were performed using Microsoft Excel or GraphPad Prism (v7.0) and differences were considered significant at p value < 0.05, with specific statistical tests and p values stated in the figure legends.
ADDITIONAL RESOURCES
A first-in-human clinical trial to test the lead agent identified in this study has been approved by the FDA (ClinicalTrials.gov registry number NCT: NCT04398485).
Description: https://clinicaltrials.gov/ct2/show/NCT04398485
Supplementary Material
{kind=link}
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
Antibodies | ||
| CD138 APC (Clone 44F9) | Miltenyi | Cat# 130-117-395; RRID: AB_2751386 |
| CD45 BB515 (Clone HI30) | BD | Cat# 564586; RRID: AB_2869588 |
| CD38 PE-Cy7 | BD | Cat# 335790; RRID: AB_399969 |
| CD45 APC | Invitrogen | Cat# MHCD4505; RRID: AB_10372216 |
| CD34 BV421 (Clone 581) | BD | Cat# 562577; RRID: AB_2687922 |
| CD3 FITC | BioLegend | Cat# 300306; RRID: AB_314042 |
| CD19 PE | BioLegend | Cat# 302208; RRID: AB_314238 |
| Rabbit monoclonal Anti-MUM1 (IRF4) | Abcam | Cat# ab133590 |
| Rat monoclonal Anti-Syndecan-1 (CD138) | R&D Systems | Cat# MAB2780; RRID: AB_2182840 |
| Anti-Rabbit IgG (H+L) | Vector | Cat# BA-1100; RRID: AB_2336201 |
| Anti-Rat IgG (H+L) | Vector | Cat# BA-9401; RRID: AB_2336208 |
| Anti-Rabbit HRP Secondary Antibody | Jackson | Cat# 711-036-152; RRID: AB_2340590 |
| CD138 VioBlue (discontinued product) | Miltenyi | Cat# 130-098-202; RRID: AB_2655034 |
| CD319 PE | Miltenyi | Cat# 130-099-573; RRID: AB_2657428 |
| IRF4 AF647 | BioLegend | Cat# 646408, RRID: AB_2564048 |
| CD14 PE-Cy7 | BD Biosciences | Cat# 562698; RRID: AB_2737729 |
| Mouse monoclonal Anti-Human MUM1 | Agilent Dako | Cat# M7259, RRID: AB_2127157 |
| CD3 AF488 (Clone HIT3a) | Biolegend | Cat# 300320; RRID: AB_493691 |
| CD14 BV605 (Clone M5E2) | Biolegend | Cat# 301834; RRID: AB_2563798 |
| CD138 VioBlue (Clone 44F9) | Miltenyi | Cat# 130-119-843; RRID: AB_2751882 |
| CD319 PerCP-Cy5.5 (Clone CRACC) | Biolegend | Cat# 331814; RRID: AB_2565235 |
|
Biological samples | ||
| CD34+ Cord Blood | AllCells | Cat# CB, CD34+, Single donor, 1M cells |
| MM patient samples | Dr. Caitlin Costello (UC San Diego Moores Cancer Center) |
N/A |
| PCL patient samples | Dr. Caitlin Costello and Dr. Mark Minden (Univ. of Toronto) |
N/A |
|
Chemicals peptides and recombinant proteins | ||
| Lenalidomide | ThermoFisher Scientific | Cat# NC1025152 |
| Collagenase IV | StemCell Technologies | Cat# 07909 |
| Matrigel | ThermoFisher Scientific | Cat# CB40230C |
| 10% neutral buffered formalin | EMD Millipore | Cat# MR0458682 |
| Antisense oligonucleotides | Ionis Pharmaceuticals | N/A |
| Luciferin | Caliper Life Sciences | Cat# 119222 |
|
Critical commercial assays | ||
| VECTASTAIN Elite ABC-Peroxidase Kit | Vector | Cat# PK-6100; RRID: AB_2336819 |
| VECTASTAIN ABC-ALKALINE PHOSPHATASE KIT |
Vector | Cat# AK-5000, RRID: AB_2336792 |
| Deep Space Black Chromogen Kit | Biocare | Cat# BRI4015 |
| Warp Red Chromogen Kit | Biocare | Cat# WR806 |
| Vectashield Hardset with DAPI | Vector | Cat# H-1500; RRID: AB_2336788 |
| Mouse Cell Depletion Kit | Miltenyi | Cat# 130-104-694 |
| Human Kappa ELISA Quantitation Set | Bethyl Laboratories, Inc. | Cat# E80-115 |
| CellTrace Violet Cell Proliferation Kit | Life Technologies | Cat# C34557 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit | Takara Bio USA, Inc. | Cat# 635025 |
| NEB Ultra DNA kit | Illumina | Cat# E7370 |
| CellTiter Glo 2.0 | Promega | Cat# G9241 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| EXPRESS One-Step SuperScript qRT-PCR Kits | ThermoFisher Scientific | Cat# 11781200 |
| SuperScript III | ThermoFisher Scientific | Cat# 11752250 |
| SYBR GreenER | ThermoFisher Scientific | Cat# 11761500 |
| Taqman Fast Advanced master mix | ThermoFisher Scientific | Cat# 44 445 57 |
| Biolegend True Nuclear Kit | Biolegend | Cat# 424401 |
| NanoString PanCancer Progression Panel | NanoString | Cat# XT-CSO-PROG1-12 |
| Live/Dead Fixable NearIR Dead Cell Stain Kit |
ThermoFisher Scientific | Cat# L10119 |
|
Deposited data | ||
| Whole transcriptome RNA-sequencing data | dbGaP: https://dbgap.ncbi.nlm.nih.gov/ | Accession# phs002291.v1.p1 |
|
Experimental models: cell lines | ||
| Human: H929 | ATCC | Cat# CRL-9068; RRID: CVCL_1600 |
| Human: RPMI-8226 | ATCC | Cat# CCL-155; RRID:CVCL_0014 |
| Human: HS5 | ATCC | Cat# CRL-11882; RRID: CVCL_3720 |
| Human: HS27a | ATCC | Cat# CRL-2496; RRID: CVCL_3719 |
| Mouse: OP9 | Dr. Raffaella Chiaramonte | N/A |
| Human: KMS-11 | JCRB | Cat# JCRB1179; RRID: CVCL_2989 |
| Human: U266 | ATCC | Cat# TIB-196; RRID: CVCL_0566 |
| Human: MM.1R | ATCC | Cat# CRL-2975; RRID: CRL-2975 |
| Human: JJN3 | DSMZ | Cat# ACC 541; RRID: CVCL_2078 |
| Human: EJM | DSMZ | Cat# ACC 560; RRID: CVCL_2030 |
| Human: L363 | DSMZ | Cat# ACC 49; RRID: CVCL_1357 |
| Human: AMO1 | DSMZ | Cat# ACC-538; RRID: CVCL_1806 |
|
Experimental models: organisms/strains | ||
| Mouse: Rag2− /− IL2Rγc− /− | Jamieson laboratory (UC San Diego) | N/A |
| Mouse: NOD/SCID | Jackson Labs | Cat# 001303; RRID: IMSR_JAX:001303 |
| Mouse: NOD-scid IL2Rγ− /− | Jackson Labs | Cat# 005557; RRID: IMSR_JAX:005557 |
|
Oligonucleotides | ||
| Human IRF4 (FW-5′ GGCAAAGAAA GCTCATCACAG 3′; REV-5′ GGATTG CTGATGTGTTCTGGTA 3′; Probe: 5′F AM- TAGCCCCTCAGGAAATGTCCAC TG 3′ IOWA-BLACK (w/ internal ZEN) |
IDT | Custom primer/probe set |
| Human Actin FW-5′- CGGACTATGA CTTAGTTGCGTTACA 3′; REV-5′ -GC CATGCCAATCTCATCTTGT-3′; Probe: 5′ FAM- CCTTTCTTGACAAAACCTA ACTTGCGCAGA -TAMRA-3′ |
IDT | Custom primer/probe set |
| Human IRF4 FW: 5′-GACATCTCAGAC CCGTACAAAG-3′, REV: 5′ -GAAG GGTAAGGCGTTGTCAT-3′ |
Lazzari et al., 2017 | N/A |
| Human HPRT primers | Crews et al., 2015 | N/A |
| Human MYC Taqman primer/probe set | ThermoFisher Scientific | Cat# Hs00153408_m1 |
| Human β-actin Taqman primer/probe set | ThermoFisher Scientific | Cat# Hs99999903_m1 |
|
Recombinant DNA | ||
| pHR2-GLF | Abrahamsson et al., 2009 | N/A |
| mCherry-hCdt1 | Dr. Atsushi Miyawaki (Sakaue-Sawano et al., 2008) | N/A |
| mVenus-hGeminin | Dr. Atsushi Miyawaki (Sakaue-Sawano et al., 2008) | N/A |
| pReceiver pEZ-Lv225 lentiviral vector | Genecopoeia | Cat# EX-NEG-Lv225 |
| ORF expression clone for IRF4 (NM_002460.3) (Purified plasmid) |
Genecopoeia | Cat# EX-Z3214-Lv225 |
|
Software and algorithms | ||
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| FastQC | Andrews, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTseq | Anders and Huber, 2010 | https://htseq.readthedocs.io/en/master/ |
| Interpro | Mitchell et al., 2019 | https://www.ebi.ac.uk/interpro/ |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Rosalind | https://rosalind.onramp.bio/ | |
|
Other | ||
| DESeq2 R library | Love et al., 2014 | N/A |
| fpc R library | Hennig, 2013 | N/A |
| topGO R library | Alexa and Rahnenfuhrer, 2019 | N/A |
| NCBI | Geer et al., 2010 | https://www.ncbi.nlm.nih.gov |
| MSigDB | Liberzon et al., 2011; Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| WikiPathways | Slenter et al., 2018 | https://www.wikipathways.org/index.php/WikiPathways |
| GSEA | https://www.gsea-msigdb.org/gsea/index.jsp | |
| HMCL66_Gene_Expression_FPKM | Keats Lab | https://www.keatslab.org/data-repository |
Highlights.
Myeloma progenitors are enriched in protective niches and with IRF4 overexpression
IRF4 antisense agents impair myeloma cell survival through cell cycle disruption
Selective IRF4 inhibition reduces myeloma regeneration in pre-clinical models
IRF4 inhibitors sensitize myeloma cells to clinical drugs and spare normal immune cells
ACKNOWLEDGMENTS
We thank Mark Minden (University of Toronto, Princess Margaret Cancer Center) for providing PCL patient material and Atsushi Miyawaki (Riken) for generously providing FUCCI lentiviral vectors. We also thank Cayla Mason, Ida Deichaite, Leisa Sutton, and the Tissue Technology and Microscopy Shared Resources at the Moores Cancer Center for technical assistance. We appreciate the vital support from funding agencies, including NIH (NCI 1R21CA194679, R01CA205944, and UCSD Specialized Cancer Center Support Grant 2P30CA023100-33; NIDDK R01DK114468), CIRM (TRAN1-10540), LLS Blood Cancer Discoveries, NASA (NRA NNJ13ZBG001N), and pilot grant funding from the UC San Diego Health Sciences Academic Senate and the Altman Clinical and Translational Research Institute (UCSD NIH CTSA Grant UL1TR001442). This work was also supported in part by Ionis Pharmaceuticals. Finally, we appreciate the generous support of the Moores Family Foundation, the Strauss Family Foundation, the Koman Family Foundation, the Sanford Stem Cell Clinical Center, the International Myeloma Foundation in collaboration with the San Diego Multiple Myeloma Support Group, and the UC San Diego Moores Cancer Center.
Footnotes
DECLARATION OF INTERESTS
T.Z., X.L., Y.K., and A.R.M. are current or past employees of Ionis Pharmaceuticals and Ionis has filed an international patent application related to this work. All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2020.12.017.
REFERENCES
- Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, Giles FJ, Durocher J, Creusot RS, Karimi M, et al. (2009). Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc. Natl. Acad. Sci. USA 106, 3925–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, and Rahnenfuhrer J (2019). topGO: enrichment analysis for gene ontology, R package version 1.38.1 (Bioconductor). [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data (Babraham Bioinformatics). [Google Scholar]
- Bai H, Wu S, Wang R, Xu J, and Chen L (2017). Bone marrow IRF4 level in multiple myeloma: an indicator of peripheral blood Th17 and disease. Oncotarget 8, 85392–85400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi G, and Ghobrial IM (2014). Biological and clinical implications of clonal heterogeneity and clonal evolution in multiple myeloma. Curr. Cancer Ther. Rev 10, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Calder A, Roth-Albin I, Bhatia S, Pilquil C, Lee JH, Bhatia M, Levadoux-Martin M, McNicol J, Russell J, Collins T, and Draper JS (2013). Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells Dev. 22, 279–295. [DOI] [PubMed] [Google Scholar]
- Chaidos A, Barnes CP, Cowan G, May PC, Melo V, Hatjiharissi E, Papaioannou M, Harrington H, Doolittle H, Terpos E, et al. (2013). Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 121, 318–328. [DOI] [PubMed] [Google Scholar]
- Cheng WL, She ZG, Qin JJ, Guo JH, Gong FH, Zhang P, Fang C, Tian S, Zhu XY, Gong J, et al. (2017). Interferon regulatory factor 4 inhibits neointima formation by engaging Kruppel-like factor 4 signaling. Circulatio€ n 136, 1412–1433. [DOI] [PubMed] [Google Scholar]
- Claudio JO, Masih-Khan E, Tang H, Gonçalves J, Voralia M, Li ZH, Nadeem V, Cukerman E, Francisco-Pabalan O, Liew CC, et al. (2002). A molecular compendium of genes expressed in multiple myeloma. Blood 100, 2175–2186. [DOI] [PubMed] [Google Scholar]
- Colombo M, Mirandola L, Platonova N, Apicella L, Basile A, Figueroa AJ, Cobos E, Chiriva-Internati M, and Chiaramonte R (2013). Notch-directed microenvironment reprogramming in myeloma: a single path to multiple outcomes. Leukemia 27, 1009–1018. [DOI] [PubMed] [Google Scholar]
- Colombo M, Galletti S, Bulfamante G, Falleni M, Tosi D, Todoerti K, Lazzari E, Crews LA, Jamieson CH, Ravaioli S, et al. (2016). Multiple myeloma-derived Jagged ligands increases autocrine and paracrine interleukin-6 expression in bone marrow niche. Oncotarget 7, 56013–56029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello C, and Mikhael JR (2018). Therapy sequencing strategies in multiple myeloma: who, what and why? Future Oncol. 14, 95–99. [DOI] [PubMed] [Google Scholar]
- Crews LA, and Jamieson CH (2013). Molecular evolution of leukemia stem cells. In Stem Cells Handbook, Sell S, ed. (Springer Science+Business Media; ). [Google Scholar]
- Crews LA, Jiang Q, Zipeto MA, Lazzari E, Court AC, Ali S, Barrett CL, Frazer KA, and Jamieson CH (2015). An RNA editing fingerprint of cancer stem cell reprogramming. J. Transl. Med 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, Sadarangani A, Zipeto MA, La Clair JJ, Villa R, et al. (2016). RNA splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell 19, 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST, Baker BF, Xia S, Yu RZ, Viney NJ, Wang Y, Tsimikas S, and Geary RS (2019). Integrated assessment of the clinical performance of GalNAc3-conjugated 2′-O-methoxyethyl chimeric antisense oligonucleotides: I. human volunteer experience. Nucleic Acid Ther. 29, 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46 (D1), D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg EC, Passegué E, Prohaska SS, Wagers AJ, Koeva M, Stuart JM, and Weissman IL (2010). Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS ONE 5, e8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhler GM, Baanstra M, Chesik D, Somasundaram R, Seckinger A, Hose D, Peppelenbosch MP, and Bos NA (2010). Bone marrow stromal cell interaction reduces syndecan-1 expression and induces kinomic changes in myeloma cells. Exp. Cell Res 316, 1816–1828. [DOI] [PubMed] [Google Scholar]
- Gandhi AK, Mendy D, Waldman M, Chen G, Rychak E, Miller K, Gaidarova S, Ren Y, Wang M, Breider M, et al. (2014). Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. Br. J. Haematol 164, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, and Bryant SH (2010). The NCBI BioSystems database. Nucleic Acids Res. 38, D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh N, and Matsui W (2009). Cancer stem cells in multiple myeloma. Cancer Lett. 277, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff D, and Jamieson C (2010). Cycling toward elimination of leukemic stem cells. Cell Stem Cell 6, 296–297. [DOI] [PubMed] [Google Scholar]
- Goff DJ, Court Recart A, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, Leu H, Low-Marchelli J, Ma W, Shih AY, et al. (2013). A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 12, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig C (2013). Cran-package fpc. https://cran.r-project.org/web/packages/fpc/index.html. [Google Scholar]
- Herrmann K, Lapa C, Wester H-J, Schottelius M, Schiepers C, Eberlein U, Bluemel C, Keller U, Knop S, Kropf S, et al. (2015). Biodistribution and radiation dosimetry for the chemokine receptor CXCR4-targeting probe 68Ga-pentixafor. J. Nucl. Med 56, 410–416. [DOI] [PubMed] [Google Scholar]
- Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M, et al. (2015). AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med 7, 314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, Shaffer AL 3rd, Louie T, Chen I, Randolph S, et al. (2012). Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood 120, 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, et al. (2013). ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 110, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Holm F, and Jamieson CHM (2017). RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat. Rev. Cancer 17, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Isquith J, Zipeto MA, Diep RH, Pham J, Delos Santos N, Reynoso E, Chau J, Leu H, Lazzari E, et al. (2019). Hyper-editing of cell-cycle regulatory and tumor suppressor RNA promotes malignant progenitor propagation. Cancer Cell 35, 81–94.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, and Singh H (2008). Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity 28, 335–345. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al. (2007). Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 12, 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, and Dalla-Favera R (2006). Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol 7, 773–782. [DOI] [PubMed] [Google Scholar]
- Kortüm KM, Mai EK, Hanafiah NH, Shi CX, Zhu YX, Bruins L, Barrio S, Jedlowski P, Merz M, Xu J, et al. (2016). Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 128, 1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, and Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. [DOI] [PubMed] [Google Scholar]
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SK, Callander NS, Hillengass J, Liedtke M, Baljevic M, Campagnaro E, Castillo JJ, Chandler JC, Cornell RF, Costello C, et al. (2019). NCCN guidelines insights: multiple myeloma, Version 1.2020. J. Natl. Compr. Canc. Netw 17, 1154–1165. [DOI] [PubMed] [Google Scholar]
- Lazzari E, Mondala PK, Santos ND, Miller AC, Pineda G, Jiang Q, Leu H, Ali SA, Ganesan A-P, Wu CN, et al. (2017). Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun 8, 1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo R, Boeker M, Peest D, Hein R, Bartl R, Gessner JE, Selbach J, Wacker G, and Deicher H (1992). Multiparameter analyses of normal and malignant human plasma cells: CD38++, CD56+, CD54+, cIg+ is the common phenotype of myeloma cells. Ann. Hematol 64, 132–139. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Heintel D, Zhang LH, Mendy D, Gaidarova S, Brady H, Bartlett JB, Schafer PH, Schreder M, Bolomsky A, et al. (2011). Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br. J. Haematol 154, 325–336. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low MSY, Brodie EJ, Fedele PL, Liao Y, Grigoriadis G, Strasser A, Kallies A, Willis SN, Tellier J, Shi W, et al. (2019). IRF4 activity is required in established plasma cells to regulate gene transcription and mitochondrial homeostasis. Cell Rep. 29, 2634–2645.e5. [DOI] [PubMed] [Google Scholar]
- Mantovani A, and Garlanda C (2006). Inflammation and multiple myeloma: the Toll connection. Leukemia 20, 937–938. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. [Google Scholar]
- Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, and Jones RJ (2004). Characterization of clonogenic multiple myeloma cells. Blood 103, 2332–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, et al. (2008). Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 68, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirandola L, Apicella L, Colombo M, Yu Y, Berta DG, Platonova N, Lazzari E, Lancellotti M, Bulfamante G, Cobos E, et al. (2013). Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia 27, 1558–1566. [DOI] [PubMed] [Google Scholar]
- Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, et al. (2019). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47 (D1), D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittrücker HW, Matsuyama T, Grossman A, Kündig TM, Potter J, Shahinian A, Wakeham A, Patterson B, Ohashi PS, and Mak TW (1997). Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275, 540–543. [DOI] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Takizawa H, and Suda T (2014). The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 141, 4656–4666. [DOI] [PubMed] [Google Scholar]
- Paner A, Okwuosa TM, Richardson KJ, and Libby EN (2018). Triplet therapies - the new standard of care for multiple myeloma: how to manage common toxicities. Expert Rev. Hematol 11, 957–973. [DOI] [PubMed] [Google Scholar]
- Pauklin S, and Vallier L (2013). The cell-cycle state of stem cells determines cell fate propensity. Cell 155, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda G, Lennon KM, Delos Santos NP, Lambert-Fliszar F, Riso GL, Lazzari E, Marra MA, Morris S, Sakaue-Sawano A, Miyawaki A, and Jamieson CH (2016). Tracking of normal and malignant progenitor cell cycle transit in a defined niche. Sci. Rep 6, 23885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, and Frenette PS (2013). PDGFRa and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med 210, 1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, Pandey SK, Revill M, Rooney C, Buckett LK, et al. (2017). Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med 9, eaal5253. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. (2008). IRF4 addiction in multiple myeloma. Nature 454, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Emre NC, Romesser PB, and Staudt LM (2009). IRF4: immunity. Malignancy! Therapy? Clin. Cancer Res 15, 2954–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri A, Choudhary G, Teixeira M, Gordon-Mitchell S, Ramachandra N, Bernard L, Bhattacharyya S, Lopez R, Pradhan K, Giricz O, et al. (2018). Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J. Clin. Invest 128, 5479–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel DS, Schiller GJ, Samaras C, Sebag M, Berdeja J, Ganguly S, Matous J, Song K, Seet CS, Talamo G, et al. (2020). Pomalidomide, dexamethasone, and daratumumab in relapsed refractory multiple myeloma after lenalidomide treatment. Leukemia 34, 3286–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slenter DN, Kutmon M, Hanspers K, Riutta A, Windsor J, Nunes N, Mélius J, Cirillo E, Coort SL, Digles D, et al. (2018). WikiPathways: a multi-faceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 46 (D1), D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, Hájek R, Rosiñol L, Siegel DS, Mihaylov GG, et al. ; ASPIRE Investigators (2015). Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med 372, 142–152. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokoyoda K, Egawa T, Sugiyama T, Choi BI, and Nagasawa T (2004). Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20, 707–718. [DOI] [PubMed] [Google Scholar]
- Van Valckenborgh E, Matsui W, Agarwal P, Lub S, Dehui X, De Bruyne E, Menu E, Empsen C, van Grunsven L, Agarwal J, et al. (2012). Tumor-initiating capacity of CD138- and CD138+ tumor cells in the 5T33 multiple myeloma model. Leukemia 26, 1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Yaccoby S (2005). The phenotypic plasticity of myeloma plasma cells as expressed by dedifferentiation into an immature, resilient, and apoptosis-resistant phenotype. Clin. Cancer Res 11, 7599–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaccoby S (2018). Two states of myeloma stem cells. Clin. Lymphoma Myeloma Leuk 18, 38–43. [DOI] [PubMed] [Google Scholar]
- Zamecnik PC, and Stephenson ML (1978). Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 75, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YX, Shi C-X, Bruins LA, Wang X, Riggs DL, Porter B, Ahmann JM, de Campos CB, Braggio E, Bergsagel PL, and Stewart AK (2019). Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 9, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun H-J, Pineda G, Morris SR, Mason CN, Geron I, et al. (2016). ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 19, 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-sequencing data reported in this paper is dbGaP: phs002291.v1.p1.