

Abstract
Gene-environment interaction is implicated in the majority of idiopathic Parkinson’s disease (PD) risk, and some of the most widespread environmental contaminants are selectively toxic to dopaminergic neurons. Pesticides have long been connected to PD incidence, however, it has become increasingly apparent that other industrial byproducts likely influence neurodegeneration. For example, organic solvents, which are used in chemical, machining, and dry-cleaning industries, are of growing concern, as decades of solvent use and their effluence into the environment has contaminated much of the world’s groundwater and soil. Like some pesticides, certain organic solvents, such as the chlorinated halocarbon trichloroethylene (TCE), are mitochondrial toxicants, which are collectively implicated in the pathogenesis of dopaminergic neurodegeneration. Recently, we hypothesized a possible gene-environment interaction may occur between environmental mitochondrial toxicants and the protein kinase LRRK2, mutations of which are the most common genetic cause of familial and sporadic PD. In addition, emerging data suggests that elevated wildtype LRRK2 kinase activity also contributes to the pathogenesis of idiopathic PD. To this end, we investigated whether chronic, systemic TCE exposure (200 mg/kg) in aged rats produced wildtype LRRK2 activation and caused nigrostriatal dopaminergic dysfunction. Interestingly, we found that TCE not only induced LRRK2 kinase activity in the brain, but produced a significant dopaminergic lesion in the nigrostriatal tract, elevated oxidative stress, and caused endolysosomal dysfunction and α-synuclein accumulation. Together, these data suggest that TCE-induced LRRK2 kinase activity contributed to the selective toxicity of dopaminergic neurons. We conclude that gene-environment interactions between certain industrial contaminants and LRRK2 likely influence PD risk.
Keywords: Trichloroethylene, LRRK2, Neurodegeneration, α-Synuclein, Oxidative stress
1. Introduction
Halogenated organic solvents are heavily used chemicals throughout the world and represent widespread industrial byproducts that contaminate the environment (Bridbord et al., 1975; Matysik et al., 2010; Savage, 1989; Simonetti et al., 2020). The chlorinated solvent trichloroethylene (TCE) is a common degreasing product and chemical feedstock, which was widely used during the 20th century and remains in regulated use today (Doherty, 2000; Wu et al., 2019). Due to its chlorinated structure, TCE does not readily break down in the environment, resulting in lasting contamination of the solvent, particularly in ground water (Morrison et al., 2005). In recent decades TCE has been detected in up to 34% of drinking water, and is currently estimated to contaminate the treated water supply of 14–20 million individuals within the United States (Stoiber and Naidenko, 2020). Despite its continued use, TCE is a well-known human toxicant, implicated in renal cancer, liver cancer, non-Hodgkin’s lymphoma, autoimmune disease, and neurotoxicity (Agency for Toxic Substances and Disease Registry (ATSDR)., 2019). TCE is also implicated in the development of the most common neurodegenerative movement disorder, Parkinson’s disease (PD), as some evidence suggests occupational exposure to TCE is associated with increased PD risk (Bove et al., 2014; Goldman et al., 2011). Similarly, proof-of-principal neurotoxicity studies describe the degeneration of dopaminergic neurons following high levels (800–1000 mg/kg) of TCE treatment in rodents (Liu et al., 2018; Liu et al., 2010). Thus, TCE represents a candidate environmental or occupational toxicant that could increase risk for PD, however, it is unclear what mechanisms are responsible for its selective neurotoxicity to dopaminergic neurons.
The vast majority – about 85% – of PD is considered idiopathic (iPD), and represents a spectrum of disease sequelae among patients, including motor and non-motor symptoms that progressively worsen (Jankovic, 2008). For most, gene-environment interaction (GxE) likely drives the majority of iPD risk, however, quantifiable GxE may be difficult to assess (Marras et al., 2019). The most commonly inherited genetic mutations associated with elevated risk for PD are within the leucine rich repeat kinase 2 (LRRK2) gene, and account for approximately 1–2% of PD cases (Alessi and Sammler, 2018). Recently, we and others have observed that wildtype LRRK2 may play a role in iPD (Kluss et al., 2019; Di Maio et al., 2018), as elevated LRRK2 protein kinase activity was observed in dopaminergic neurons of postmortem brain tissue (Maio et al., 2018a), peripheral blood cells (Dzamko et al., 2013), and urinary exosomes of iPD patients (Fraser et al., 2016). While the cause of aberrant LRRK2 kinase activity in iPD is unknown, activity of endogenous, wildtype LRRK2 appears to be induced, either directly or indirectly by oxidative stress (Mamais et al., 2014). A potential source for which is the cellular oxidative damage produced by mitochondrial toxicants (e.g. pesticides), which are implicated as environmental factors in PD risk (Martinez and Greenamyre, 2012; De Miranda et al., 2016; Smith et al., 1997).
LRRK2 is a master regulator of protein and vesicular trafficking; its kinase substrates, Rab GTPases, drive the direction and fusion of endosomal vesicles important to the autophagy-lysosomal pathway (ALP; Pfeffer, 2018). Increases in LRRK2 kinase activity, whether the result of inherited mutations (e.g. G2019S) or overactivation of the wildtype protein, results in ALP dysfunction (Madureira et al., 2020), and is implicated in the aberrant accumulation of cytotoxic protein (e.g. α-synuclein; Bae et al., 2018; Hu et al., 2018; Volpicelli-Daley et al., 2016). As such, endolysosomal impairment mediated by LRRK2 appears to be an early and important pathological factor in dopaminergic cell survival. As a mitochondrial toxicant (Elkin et al., 2019), TCE represents a putative environmental source for LRRK2 kinase activation that could induce a cascade of pathology within dopaminergic neurons, ultimately resulting in neurodegeneration over time.
To this end, we have investigated whether a gene-environment interaction exists between the ubiquitous environmental contaminant TCE and LRRK2, which could provide a mechanism for the observed dopaminergic pathology following TCE exposure. To identify TCE-induced LRRK2 kinase activity and associated neuropathology, we developed a rat model of TCE exposure (200 mg/kg) supplied in a daily gavage over a chronic dosing period (6 weeks) in aged Lewis rats. In the United States, occupational daily exposure limits for TCE are set at 100 ppm, and short-term (5-min) exposure limits at 300 ppm (OSHA, trichloroethylene), therefore, we chose 200 mg/kg of to rep TCE to represent a moderate exposure. As this dose is considerably lower than previously reported studies, we examined multiple cohorts of aged (10–15 month old) Lewis rats to identify pathology within nigrostriatal dopaminergic neurons, potentially as a result of TCE-induced LRRK2 kinase activity. Aged animals displayed a marked loss of dopaminergic neurons and their striatal terminal projections, corresponding with a reduction in motor behavior following 6 weeks of TCE exposure. Within surviving dopaminergic neurons, TCE caused a clear increase in oxidative stress, produced dysfunction in endolysosomal and protein degradation pathways, and resulted in the accumulation of endogenous α-synuclein. TCE also caused LRRK2 kinase activation in the nigrostriatal tract, detected by protein auto- and substrate phosphorylation, which occurred prior to any significant dopaminergic neurodegeneration. Collectively, these data show that a moderate, systemic TCE exposure resulted in elevated LRRK2 kinase activity in the brain that mirrors pathological activation observed in PD patients, providing an avenue for PD risk from exposure to this industrial contaminant.
2. Materials and methods
2.1. Chemical reagents and supplies
Trichloroethylene (CAS Number 79–01-6) and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Antibody information for immunohistochemistry (IHC), immunocytochemistry (ICC) and western blot is listed in Table 1.
Table 1.
Antibody information.
| Antigen | Antibody Catalog Information | Company | IHC or Western Blot Concentration |
|---|---|---|---|
| Tyrosine hydroxylase 3-Nitrotyrosine |
AB1542 06-284 |
EMD Millipore / Sigma (St. Louis, MO) |
1:2000 1:500 |
|
IHC Ser129-α-Synuclein Lamp1 GAD67 DARPP32 4-HNE |
ab51253 ab24170 ab26116 ab40801 ab48506 |
Abcam (Cambridge, MA) |
1:500 1:500 1:500 1:500 1:500 |
|
Western Blot pThr73-Rab10 pSer935-LRRK2 pSer1292-LRRK2 LRRK2 c41-2 |
ab231707 ab133450 ab203181 ab133474 |
1:2000 1:2000 1:2000 1:2000 |
|
|
ICC pThr73-Rab10 |
ab241060 | 1:1000 | |
| Iba1 | 019-19741 | Wako Chemicals USA (Irvine, CA) | 1:500 |
| CD68 (ED1) | MCA341 | BioRad (Hercules, CA) | 1:500 |
| α-Synuclein | 610787 | BD Biosciences (San Jose, CA) | 1:500 |
| p62/SQSTM1 (2C11) | H00008878-M01 | Abnova (Tapei, Taiwan) | 1:500 |
|
IHC pThr73-Rab10 |
10th bleed | MRC Protein Unit (Dr. Dario Alessi, University of Dundee) | 1:500 |
|
IHC Total LRRK2 |
N241A/34 | NeuroMab UC Davis (Davis, CA) | 1:500 |
|
IHC 14-3-3 |
NBP2-67831 | Novus Biologics (Centennial, CO) | 1:500 |
2.2. Animals and TCE administration
Three separate cohorts of rats were administered TCE in this study (Supplemental Fig. 1). Adult, male and female Lewis rats were obtained through a retired breeding program from Envigo (Indianapolis, IN). Upon arrival, rats were separated into single-housing two weeks prior to the onset of treatment and handled daily to prevent stress upon study commencement. Conventional diet and water were available to rats ad libitum, and animals were maintained under standard temperature-controlled conditions with a 12-h light-dark cycle. TCE was dissolved into premium olive oil (Trader Joe’s, Monrovia, CA) for a final concentration of 200 mg/kg based on individual rat weight (cohorts 1 and 2), or 50, 100, 200, 400 or 800 mg/kg (Cohort 3). TCE and vehicle (olive oil) groups were randomly divided, and each animal was administered a single daily oral gavage for one, three, or six weeks corresponding to each cohort endpoint. Animal weight was recorded daily, and rats were observed twice a day for signs of overt toxicity or animal morbidity (Supplemental Fig. 1). Following TCE exposure, animals were euthanized using a lethal dose of pentobarbital, followed by transcardial perfusion with PBS and fixation with 4% paraformaldehyde (cohorts 1 and 2). Cohort 3 rat brain tissue was collected, flash frozen, and stored at —80 °C. Following euthanasia and tissue collection, animals were assigned a four-digit code and researchers were blind to treatment groups during all subsequent analyses.
Wildtype and LRRK2-G2019S (C57BL/6-LRRK2tm4.1Arte) mice were purchased from Taconic Biosciences (Germantown, NY) as breeding pairs. Adult male and female retired breeding mice (10–12 months) were euthanized using a lethal dose of pentobarbital, followed by rapid collection and freezing of brain tissue (N = 3/group). Mice were not treated with TCE during this study. Brain tissue collected from wildtype and LRRK2-G2019S knockin mice was used as a comparison of endogenous LRRK2 kinase activity and expression. All experiments involving animal treatment and euthanasia were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. TCE handling and disposal was carried out following University of Pittsburgh Environmental Health and Safety procedures.
2.3. Motor behavior
Open field behavior was monitored after 6 weeks of TCE treatment (200 mg/kg) in Cohort 1, aged male Lewis rats. Following a period of acclimation, each rat was placed in an open field chamber and allowed to freely explore for 20 min while tracking software (Noldus EthoVision XT, Leesburg, VA) recorded a standard array of open field parameters. Motor behavior analyses were carried out following all protocols set by the University of Pittsburgh Rodent Behavioral Analysis Core (RBAC), including ambient noise, light, and animal handling parameters to limit rodent stress and erroneous stimuli that could affect behavioral outcomes.
2.4. Striatal terminal intensity
A series of brain sections (35 μm) encompassing the volume of the rat striatum (1/6 sampling fraction, approximately 10 sections per animal) were stained for tyrosine hydroxylase (TH) and detected using an infrared secondary antibody (IRDye® 680, LiCor Biosciences). Striatal tissue sections were analyzed using near-infrared imaging for density of dopamine neuron terminals (LiCor Odyssey), and quantified using LiCor Odyssey software (V3.0; Licor Biosciences, Lincoln, NE) with consistent background subtraction. Results are reported as striatal TH intensity in arbitrary fluorescence units.
2.5. Stereology
Stereological analysis of dopamine neuron number in the SN was achieved using an adapted protocol from Tapias et al. (2013; 2014) as reported in De Miranda et al. (2019, 2018) employing an unbiased, automated system. Briefly, nigral tissue sections were stained for TH and counterstained with DAPI and Nissl NeuroTrace Dye (640; Life Technologies) then imaged using a Nikon 90i upright fluorescence microscope equipped with high N.A. plan fluor/apochromat objectives, Renishaw linear encoded microscope stage (Prior Electronics) and Q-imaging Retiga cooled CCD camera (Center for Biological Imaging, University of Pittsburgh). Images were processed using Nikon NIS-Elements Advanced Research software (Version 4.5, Nikon, Melville, NY), and quantitative analysis was performed on fluorescent images colocalizing DAPI, TH, and Nissl-positive stains. Results are reported as the number of TH-positive cell bodies (whole neurons) within the SN.
2.6. Immunohistochemistry and immunopathology
Brain sections (35 μm) were maintained at —20 °C in cryoprotectant, stained while free-floating, and mounted to glass slides for imaging, using a “primary antibody delete” (secondary antibody only) stained section to subtract background fluorescence. The proximity ligation assay (PLA) was performed using the DuoLink (Millipore Sigma, St. Louis, MO) protocol, and as reported in Di Maio et al. (2018). Briefly, fixed rat brain tissue was stained for TH (Ab1542, Millipore) followed by staining with total LRRK2 (N241A/34, NeuroMab, UC Davis), and 14–3-3 (NBP2–67831, Novus Biologicals) mouse and rabbit primary antibodies, respectively, overnight at 4 °C. DuoLink rabbit plus and mouse minus probes conjugated to oligonucleotides (Step 1) were incubated for 1 h at 37 °C in a humid chamber. Ligation of the probes using DuoLink Ligase Solution (Step 2) occurred for 45 min at 37 °C in a humid chamber. Amplification of the probes using DuoLink Green (488) Detection Reagents (Step 3) was achieved by a 100 min incubation at 37 °C in a humid chamber. Following PLA amplification, tissue sections were washed and treated with the autofluoresence quenching reagent TrueBlack (Biotium, Fremont, CA), coverslipped, and imaged 24-h later.
Fluorescent immunohistochemical images were collected using an Olympus BX61 confocal microscope and Fluoview 1000 software (Melville, NY). Quantitative fluorescence measurements were thoroughly monitored using standard operating imaging parameters to ensure that images contained no saturated pixels. For quantitative comparisons, all imaging parameters (e.g., laser power, exposure, pinhole) were held constant across specimens. Confocal images were analyzed using Nikon NIS-Elements Advanced Research software (Version 4.5, Nikon, Melville, NY). Background subtraction was achieved by using a primary antibody delete tissue section containing only corresponding secondary antibodies. A minimum of 6 images per tissue slice were analyzed per animal, averaging 9–15 neurons per 60-100× image (approximately 120–180 cells per animal, per histological stain depending on magnification). 20× magnification was used to generate montage imaging of the ventral midbrain or striatum, for representative images or analysis per image using anatomical region of interest (ROI) boundaries. Results are reported as a measure of puncta within TH-positive cells, either number of objects (# of objects), area (μm2), or fluorescence intensity (arbitrary units).
2.7. Western blots
Brain tissue sections from Cohort 3 rats (3-week TCE treatment and dose response) were microdissected and flash frozen. Tissue lysates were prepared by homogenizing each brain section in a Dounce tissue grinder using 20% weight/volume lysis buffer (see below for recipe). Tissue was ground using 20 loose strokes and 20 tight strokes, then left on ice for 30 min to lyse. Homogenized tissue was centrifuged at 15,000 ×g for 10 min at 4 °C, and supernatant was collected and centrifuged for an additional 15,000 ×g for 10 min at 4 °C to pellet remaining debris. Protein was quantified using a Pierce Bovine Serum Albumin Standard (23,208, ThermoFisher Scientific). Lysis buffer was generated from 10× Cell Signaling Lysis Buffer (9803, Danvers, MA) diluted to 1 × using ultra pure water, followed by the addition of a protease inhibitor cocktail (78430) and phosphatase inhibitor cocktail (788427) from ThermoFisher Scientific (Waltham, MA).
Samples were incubated with NuPage LDS 4× sample buffer (NP0007, ThermoFisher Scientific) and boiled for 10 min at 95 °C. A total of 100 μg brain lysate was run on a Bio-Rad Mini-Protean TGX 4–20% polyacrylamide gel (4,561,094, Bio-Rad, Hercules, CA) at 200 V for approximately 40 min. Gels were transferred to a nitrocellulose membrane (1,704,159, Bio-Rad) using the Bio-Rad Trans-Blot Turbo system for 7 min (Midi-Gel setting). Nitrocellulose membranes were blocked for 1 h in 50% TBS (20 mM Tris, 0.5 M NaCl, pH 7.5) 50% Odussey blocking buffer (LiCor, Lincoln, NE), then incubated overnight with primary antibodies in 50% TBS-T (20mM Tris, 0.5 M NaCl, pH 7.5, 0.1% Tween 20), 50% Odyssey blocking buffer at 4 °C. Membranes were washed 3 × 10 min in TBS-T followed by secondary incubation with Odyssey IRDye® 800 (926–32,213) and IRDye® 680 (926–68,072) for 1 h at room temperature. Following 3 × 10 min wash in TBS-T, membranes were scanned using the Odyssey CLx imaging system, and analyzed using LiCor Image Studio Software. Quantified intensity is reported as phosphorylated protein in relation to total LRRK2, in relation to β-actin.
2.8. Blood cell analysis
Rodent blood was collected via cardiac stick in Cohorts 1–3. Approximately 1000 μl blood was extracted and transferred to K2 EDTA tubes (ThermoFisher Scientific, Waltham MA). Samples were spun at 2500 g for 20 min and the plasma was removed and stored. The buffy coat enriched fraction was collected and diluted in 1000 μl OptiMEM reduced serum medium (Thermo Fisher). Samples were then plated at 100 μl per well in 16-well chamber slides, which were pre-coated with Cell-Tak (Corning Inc., Corning NY). Chamber slides were centrifuged at 500 g for 5 min to promote cell adherence. Post centrifuge, media was removed, and cells were fixed with 4% PFA for 20 min at RT. Samples containing white blood cells (WBC) were stained using pThr73-Rab10 (ab241060, Abcam) overnight at 4 °C, and analyzed via confocal microscopy as described above.
2.9. Statistical analyses
All data are expressed as mean values ± standard error of the mean (SEM). For statistical comparison between vehicle and TCE groups, a non-parametric t-test was used. Statistical significance between male and female rats was evaluated for normally distributed means by two-way analysis of variance (two-way ANOVA) with a Sidak multiple comparisons test to correct for mean comparison between multiple groups, where source of variation was defined as “sex” or “TCE” groups. P-value and F-statistic are reported for the interaction comparison between “vehicle” and “TCE” variables as F (DFn, DFd) = x within each figure legend. Prior to study onset, a-priori power analyses were conducted using G*power software (Heinrich-Heine-University Düsseldorf) to determine the sample size required for a 20–40% difference between mean, with a 95% power at α = 0.05. Statistical significance between treatment groups is represented in each figure as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, unless otherwise specified on graph. Statistical outliers from each data set were determined using the extreme studentized deviate (Grubbs’ test, α = 0.05). Statistical analyses were carried out using GraphPad Prism software (V. 5.01).
3. Results
3.1. Aged rats exposed to TCE display selective nigrostriatal neurodegeneration
Aged adult (15 month), male Lewis rats were given a daily oral gavage of 200 mg/kg TCE or vehicle (olive oil) for 6-weeks (Cohort 1, Fig. 1a). Following 6-weeks of TCE exposure, locomotor behavior was assayed using the Open Field test, which revealed a significant reduction in total Distance Traveled, as well as Ambulatory Time in rats receiving TCE treatment (Fig. 1b-d). The reduction in spontaneous locomotor activity was also evident from trace plots of TCE-treated animals from open field monitoring (Fig. 1e).
Fig. 1. Aged rats exposed to TCE display selective nigrostriatal neurodegeneration.
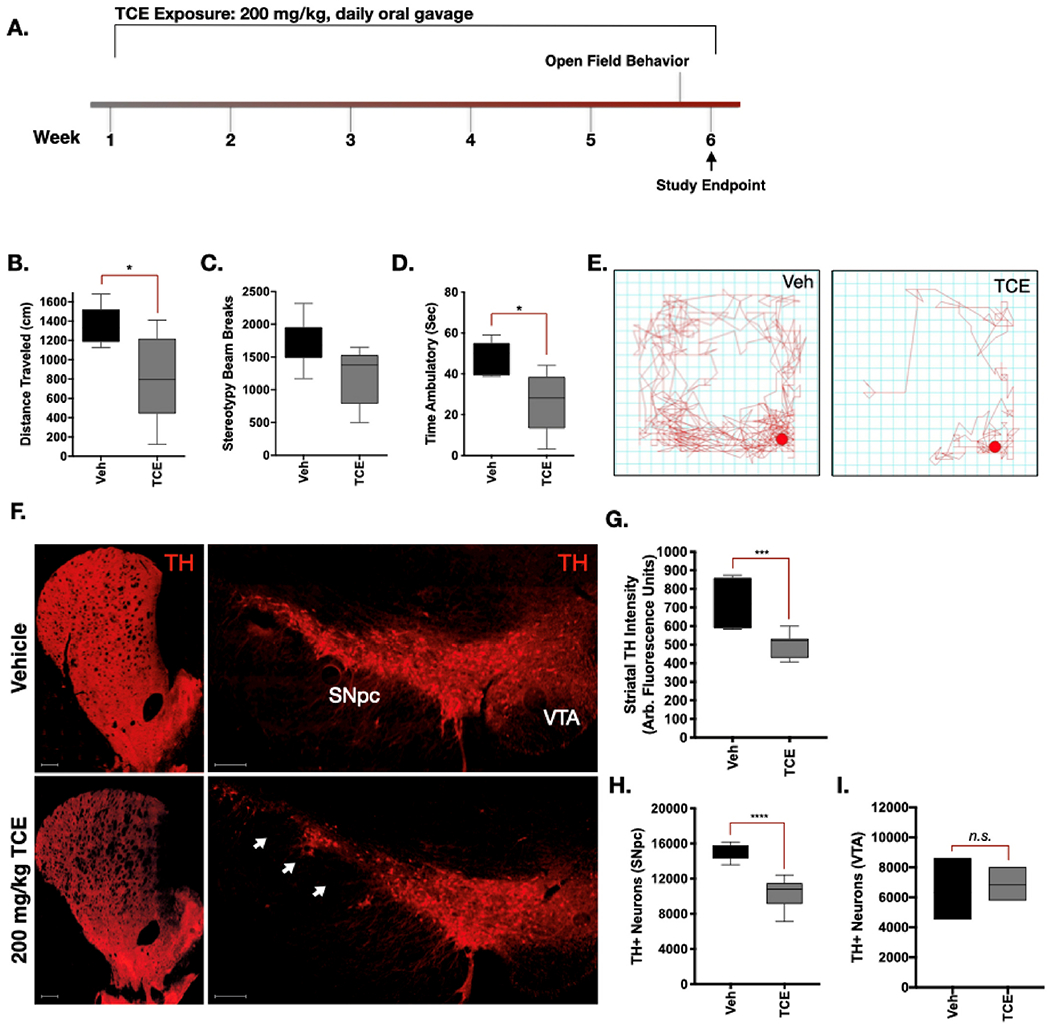
A. 15-Month-old, male Lewis rats were exposed to 200 mg/kg TCE or vehicle (olive oil) via daily oral gavage for 6 weeks. B—D. Quantitative parameters measured from open-field chamber motor behavior analysis showed significant differences in “Distance Traveled” (F(4, 5 = 5.267), p = 0.015) and “Time Ambulatory” (F(4, 4 = 3.182), p = 0.03) but not “Beam Breaks” (F(4, 5 = 1.408), p = 0.06). E. Depiction of a trace plot from free movement of TCE or vehicle treated rats after 6 weeks. F. Representative 20× montage images of tyrosine hydroxylase (TH; red) positive cells in the striatum, substantia nigra pars compacta (SNpc), and ventral tegmental area (VTA). G-I. Quantification of dopaminergic cell loss from the striatum (F(5, 11 = 3.567), p = 0.0001) and SNpc (F(8, 4 = 2.959), p < 0.0001) but not the VTA (F (2, 2 = 3.46) (p = 0.73) following 6 weeks of TCE exposure. Statistical analysis unpaired t-test, error bars represent SEM, (N = 7 vehicle, 10 TCE).
Immunohistochemistry of striatal brain sections from TCE-treated rats revealed approximately a 30% reduction in tyrosine hydroxylase (TH) intensity within the striatum compared to vehicle (p = 0.0001), indicative of a loss of dopaminergic neuron terminals (Fig. 1f-g). Assessment of neuronal subtypes within the striatum, such as DARPP32-positive medium spiny neurons and GAD67-positive neurons, showed no substantive change in protein expression. Similarly, total neuronal cell bodies within the striatum visualized using NeuN did not appear altered between vehicle and TCE treated rats (Supplemental Fig. 2a).
TCE caused approximately 32% loss of dopaminergic neurons in the substantia nigra (SN) assessed by stereology, compared to vehicle (p < 0.0001; Fig. 1f,h). Dopaminergic neurons within the ventral tegmental area (VTA) were not significantly altered by TCE exposure in these animals (p = 0.73; Fig. 1f,i). Stereological TH counts were colocalized with Nissl stain for dopaminergic cell bodies within the SN, which showed loss of cells following TCE treatment (Supplemental Fig. 2b).
3.2. TCE induced oxidative stress within dopaminergic neurons
Evidence of oxidative stress was measured using markers for oxidative protein modification within the dopaminergic neurons of the SN from 6-week, 200 mg/kg TCE or vehicle treated rats. Significantly elevated levels of 3-nitrotyrosine (3-NT; p < 0.001), a marker for peroxynitrite (ONOO−) protein adducts on tyrosine residues were observed in dopaminergic (TH-positive) neurons of TCE-exposed aged rats (Fig. 2a,b). Similarly, elevated levels of the protein 4-hydroxynonenal (4-HNE; p < 0.01), a marker for lipid peroxidation caused by oxygen radicals, were observed in dopaminergic neurons of the SN within TCE treated rats (Fig. 2a,c). Together, these data indicate surviving dopaminergic neurons within TCE treated animals display evidence of elevated levels of oxidative damage compared to age-matched, vehicle treated rats.
Fig. 2. Evidence of oxidative damage and neuroinflammation in the ventral midbrain of TCE exposed rats.
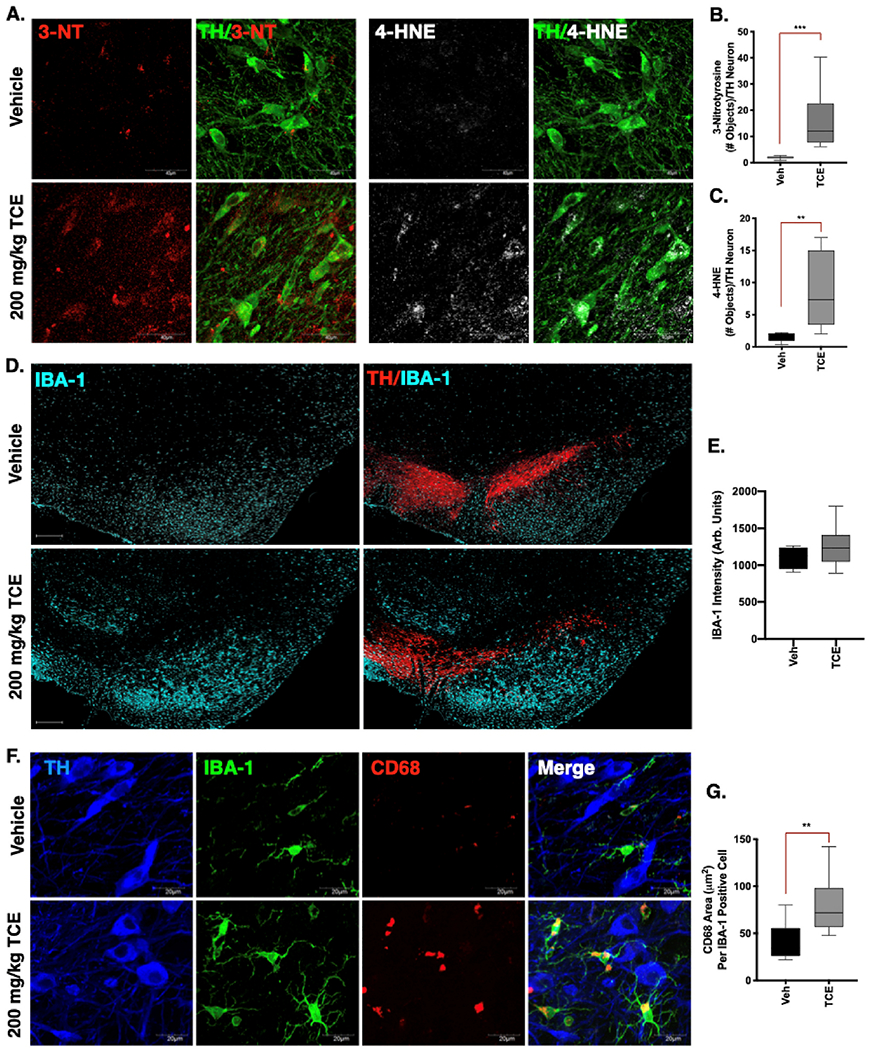
Immunohistochemistry was conducted in brain tissue from 15-month old male Lewis rats exposed to 200 mg/kg TCE or vehicle (olive oil) for 6 weeks. A. Representative confocal images (60×) of the peroxynitrite adduct 3-nitrotyrosine (3-NT, red) and the lipid peroxidation maker 4-hydroxynonenal (4-HNE, white) in dopaminergic neurons of the SNpc (TH, green). B—C. Quantification of 3-NT (F(8, 10 = 517.6), p = 0.0006) and 4-HNE (F(8, 6 = 65.19), p = 0.0061) within dopaminergic neurons indicates elevated oxidative damage following 6 weeks of TCE exposure. D. Representative images (20× montage) of microglia (IBA-1, cyan) and dopaminergic neurons (TH, red) in the SNpc of vehicle and TCE treated rats. E. Quantification of IBA-1 intensity is not significantly different (F(13, 3 = 2.82), p = 0.309) between treatment groups. F. Representative confocal images (60×) of IBA-1 (green) and the myeloid lysosomal protein CD68 (red), surrounding dopaminergic neurons (TH, blue) in the SNpc of TCE or vehicle treated rats. G. Significantly elevated CD68 area in the SNpc of TCE treated rats compared to vehicle (F(13, 6 = 1.59), p = 0.005). Statistical analysis unpaired t-test, error bars represent SEM, (N = 7 vehicle, 10 TCE).
3.3. Moderate TCE exposure results in a mildly activated microglial phenotype
Previous studies using high doses of TCE exposure in rats (up to 1000 mg/kg) reported a hypertrophic microglial phenotype consistent with activated microglia (Liu et al., 2010). Despite the lower dose used here (200 mg/kg), we observed a similar trend in hypertrophic microglia throughout the ventral midbrain and surrounding the SN in TCE treated rats compared to vehicle (Fig. 2d). Observational differences in IBA-1 positive cell volume, however, were not statistically different between treatment groups (p = 0.309; Fig. 2c). On the other hand, higher-magnification analysis of lysosomal protein CD68 area within IBA-1 positive cells, indicative of increased ongoing phagocytic activity, was significantly elevated (p < 0.001) in the microglia of TCE treated rats (Fig. 2e-f).
3.4. Endolysosomal dysfunction and protein accumulation in the SN of TCE-treated rats
Within nigral dopaminergic neurons, we observed a marked increase of Rab5 positive puncta in rats exposed to TCE, an indication that early endosomes were accumulating within these cells (p < 0.01; Fig. 3a,b). TCE exposure also caused a significant accumulation of protein p62/Sequestosome 1 within dopaminergic neurons of the SN, the ubiquitin-like protein that tags cargo destined for lysosomal degradation (p < 0.05; Fig. 3a,c). In conjunction, a robust decrease of the late endosome and lysosomal membrane protein Lamp1 was observed in dopaminergic neurons of the SN in TCE treated rats (p < 0.0001; Fig. 3a,d). Collectively, these data suggest TCE exposure caused impairment of ALP within the surviving nigral dopaminergic neuron population. These changes also correlated with accumulation of both phosphorylated (pSer129-αSyn; p = 0.0076) and total α-synuclein (p = 0.0003) within the soma of dopaminergic neurons of TCE treated animals (Fig. 3e-g).
Fig. 3. Endolysosomal dysfunction and protein accumulation occurs in dopaminergic neurons of TCE-treated rats.
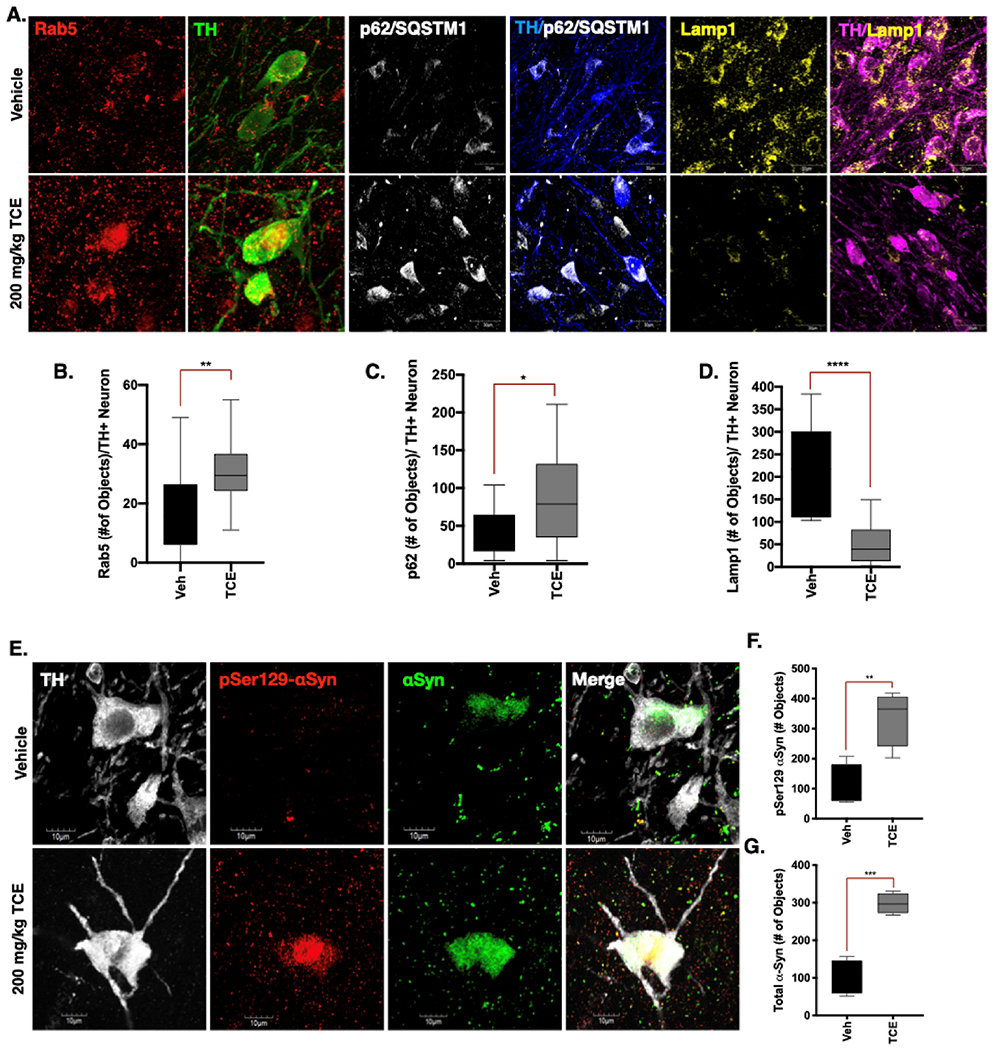
Immunohistochemistry was conducted in brain tissue from 15-month old male Lewis rats exposed to 200 mg/kg TCE or vehicle (olive oil) for 6 weeks. A. Representative confocal images of Rab5 (200×, red), p62/SQSTM1 (100×, white), and Lamp1 (100×, yellow) in the SNpc of vehicle or TCE treated rats (counterstained for TH, green, blue, magenta, respectively). B. The early endosome marker Rab5 is significantly elevated in dopaminergic neurons in the SNpc of rats exposed to TCE (F (12, 23 = 1.62), p = 0.0087). C. The ubiquitin like protein p62/SQSTM1 is also significantly elevated in TCE treated rats (F(14, 9 = 3.976), p = 0.035). D. The lysosomal membrane protein Lamp1 is significantly reduced in dopaminergic neurons of rats treated with TCE (F(9, 14 = 4.84), p < 0.0001). E-G. Phosphorylated (pSer1 29-αSyn, red) and total αSyn (green) protein was accumulated in dopaminergic neurons (TH, white) of rats exposed to TCE; pSer129-αSyn (F(3, 3 = 1.87), p = 0.0076), total αSyn (F(3, 3 = 2.82), p = 0.0003). Statistical analysis unpaired t-test, error bars represent SEM, (N = 7 vehicle, 10 TCE).
3.5. TCE exposure influences LRRK2 protein-protein interactions associated with elevated kinase activity
A proximity ligation (PL) assay was performed in rat brain tissue between LRRK2 and its regulatory protein 14–3-3, which is inversely correlated with LRRK2 kinase activity when 14–3-3 is bound at phosphorylation sites Ser910 and Ser953 (N-terminal) or Ser1444 (Roc domain) (Lavalley et al., 2015; Li et al., 2011; Nichols et al., 2010; Stevers et al., 2017) Within dopaminergic neurons, PL signal between LRRK2 and 14–3-3 was significantly lower (p < 0.008) in TCE treated rats than vehicle, providing evidence that LRRK2 kinase activity was elevated in these cells (Fig. 4a-b). In conjunction, Rab10, a kinase substrate of LRRK2, was highly phosphorylated (pThr73) in dopaminergic neurons (p < 0.05) in the SN of TCE treated rats Fig. 4c-d, f-g). Six weeks of TCE exposure also resulted in elevated pThr73-Rab10 signal compared to vehicle in white blood cells (WBC) cultured from rat blood samples collected at study termination (p < 0.05; Fig. 4e).
Fig. 4. TCE exposure influences LRRK2 protein-protein interactions associated with elevated kinase activity.
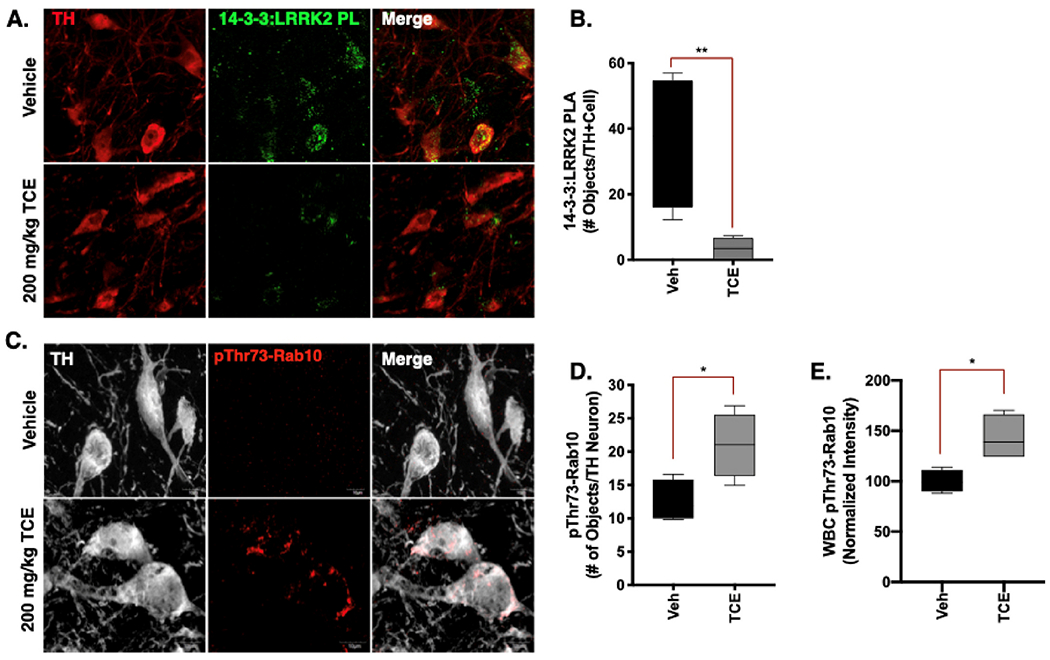
Immunohistochemistry was conducted in brain tissue from 15-month old male Lewis rats exposed to 200 mg/kg TCE or vehicle (olive oil) for 6 weeks. A-B. Proximity ligation assay (PLA) between LRRK2 (N241A) and its regulatory protein 14–3-3 (PL; green) in dopaminergic neurons (TH, red) of TCE treated animals shows significant reduction in signal compared to vehicle (F(3, 4 = 35.43), p = 0.0084). C-E. An antibody generated toward pThr73-Rab10 (red) indicates a significant accumulation of the LRRK2 kinase substrate in dopaminergic neurons (TH, white; F(3, 3 = 1.87), p = 0.0076), and in white blood cells (WBC) cultured from Lewis rats after 6 weeks of TCE exposure (F(3, 3 = 4.20), p = 0.0238). Statistical analysis unpaired t-test, error bars represent SEM, (N = 7 vehicle, 10 TCE).
3.6. TCE-induced LRRK2 kinase activity is elevated prior to dopaminergic neuron loss
To assess the temporal loss of dopaminergic neurons in response to TCE exposure, adult male and female rats were treated for one, three, or six weeks with 200 mg/kg TCE (or vehicle, olive oil) via daily oral gavage (Cohort 2, Fig. 5a). Analysis of striatal TH intensity over the time course, revealed progressive dopaminergic terminal loss from three-weeks to six-weeks of continuous TCE exposure in both male (p < 0.001) and female animals (p < 0.0001; Fig. 5b). Striatal terminal loss induced by TCE preceded dopaminergic neuron death, as no significant neurodegeneration was observed until six weeks of TCE exposure (p = 0.005; Fig. 5c). In contrast to other neurotoxicant models, such as rotenone (De Miranda et al., 2019), it did not appear that sex influenced a robust difference in dopaminergic neurodegeneration in TCE treated rats (p = 0.6795, two-way ANOVA, α = 0.05). Representative images of the SN are provided in Supplemetnal Fig. 3.
Fig. 5. TCE-induced LRRK2 kinase activity is elevated prior to dopaminergic neuron loss.
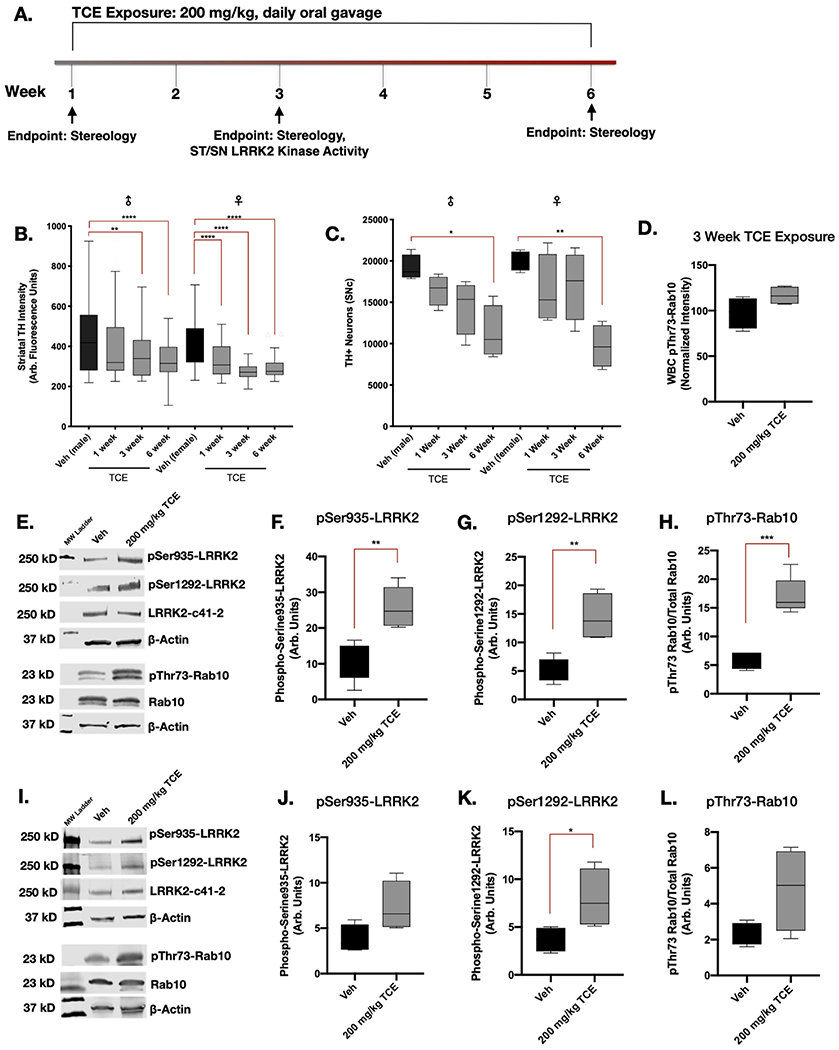
A. Aged adult (10 month) male and female Lewis rats were treated with 200 mg/kg TCE or vehicle (olive oil) via daily oral gavage for 1, 3, or 6 weeks. B—C. Dopaminergic neuron terminals in the striatum were significantly reduced in male (p = 0.0054) and female (p < 0.0001) animals after 3 weeks of exposure (F(7, 705 = 20.33), p < 0.0001), however, loss of dopaminergic neurons measured using stereology did not reach significance in male (p = 0.005) and female (p = 0.0039) rats until 6 weeks of continuous TCE exposure (F(3, 24 = 0.639), p < 0.0001); N = 5. Tukey post-hoc comparisons between vehicle and TCE-treated animals are indicated on graphs. D. pThr73-Rab10 accumulation was not present in cultured WBC of male Lewis rats following 3 weeks of TCE treatment (F(3, 3 = 2.808), p = 0.11). E-H. Western blot quantification of LRRK2 phosphorylation in the striatum of aged adult (10 month) male Lewis rats after 3 weeks of 200 mg/kg TCE exposure revealed significant elevation in pSer935 (F(4, 4 = 1.168), p = 0.0028), pSer1292 (F(3, 4 = 3.924), p = 0.003), and pThr73-Rab10 (F(4, 3 = 4.488), p = 0.0004). I-L. Western blot quantification of LRRK2 phosphorylation in the SN following 3 weeks of TCE exposure shows a significant elevation in pSer1 292-LRRK2 (F(3, 3 = 5.820), p = 0.0439), but not pSer935 (F(3, 3 = 3.213), p = 0.0671) nor pThr73-Rab10 (F(3, 3 = 13.93), p = 0.0822); unpaired T-test (N = 5), error bars represent SEM.
Quantification of LRRK2 phosphorylation at Ser935, Ser1292, and its kinase substrate Rab10 at pThr73 were measured via western blot in the striatum and SN of male rats after three weeks of daily 200 mg/kg TCE exposure. In the striatum, significantly elevated levels of phosphorylated pSer935-LRRK2 (p = 0.0028), pSer1292-LRRK2 (p = 0.003), and pThr73-Rab10 (p = 0.0004) were observed in TCE treated rats compared to vehicle (Fig. 5d-g). Within the SN, three weeks of TCE exposure resulted in elevated pSer1292-LRRK2 (p = 0.0439), but not pSer935-LRRK2 (p = 0.0671), nor pThr73-Rab10 (p = 0.0822; Fig. 5h-k). Thus, LRRK2 kinase activity appears to be elevated in nigrostriatal tissue prior to measurable dopaminergic neuron loss. The elevation of pThr73-Rab10 observed in striatal brain tissue after three weeks of TCE treatment did not correlate with elevated pThr73-Rab10 in blood cells at the same timepoint (p < 0.34; Fig 5d).
3.7. Systemic TCE exposure dose-dependently elevates LRRK2 kinase activity in the rat brain
To further understand the extent of wildtype LRRK2 kinase activity in the brain as a response to systemic TCE exposure, we administered increasing doses of TCE (50, 100, 200,400, or 800 mg/kg) or vehicle (olive oil) for three weeks, via daily oral gavage to separate groups of adult male Lewis rats (Cohort 3). Western blot analysis for pSer935-LRRK2, pSer1292-LRRK2, and pThr73-Rab10 in the striatum and SN of rat brain lysates showed that TCE dose-dependently elevated LRRK2 phosphorylation (Fig. 6a-h). As toxicant-induced, wildtype LRRK2 kinase activity in the brain has not been extensively characterized, we compared LRRK2 kinase activity levels from TCE treated rats to whole brain lysates of untreated LRRK2-G2019S knock-in or wildtype mice (Fig. 6i-l). As previously reported (Kluss et al., 2018), LRRK2-G2019S mice displayed significant elevation in pSer1292-LRRK2 (p = 0.04) and pThr73-Rab10 (p = 0.017), but not pSer935-LRRK2 (p 0.314). Together these data indicate that elevated phosphorylation of wildtype LRRK2, as a measure of increased LRRK2 kinase activity in the brain, is influenced by the relative amount of systemic TCE exposure sustained over a three-week time period.
Fig. 6. Systemic TCE exposure dose-dependently elevates LRRK2 kinase activity in the rat brain.
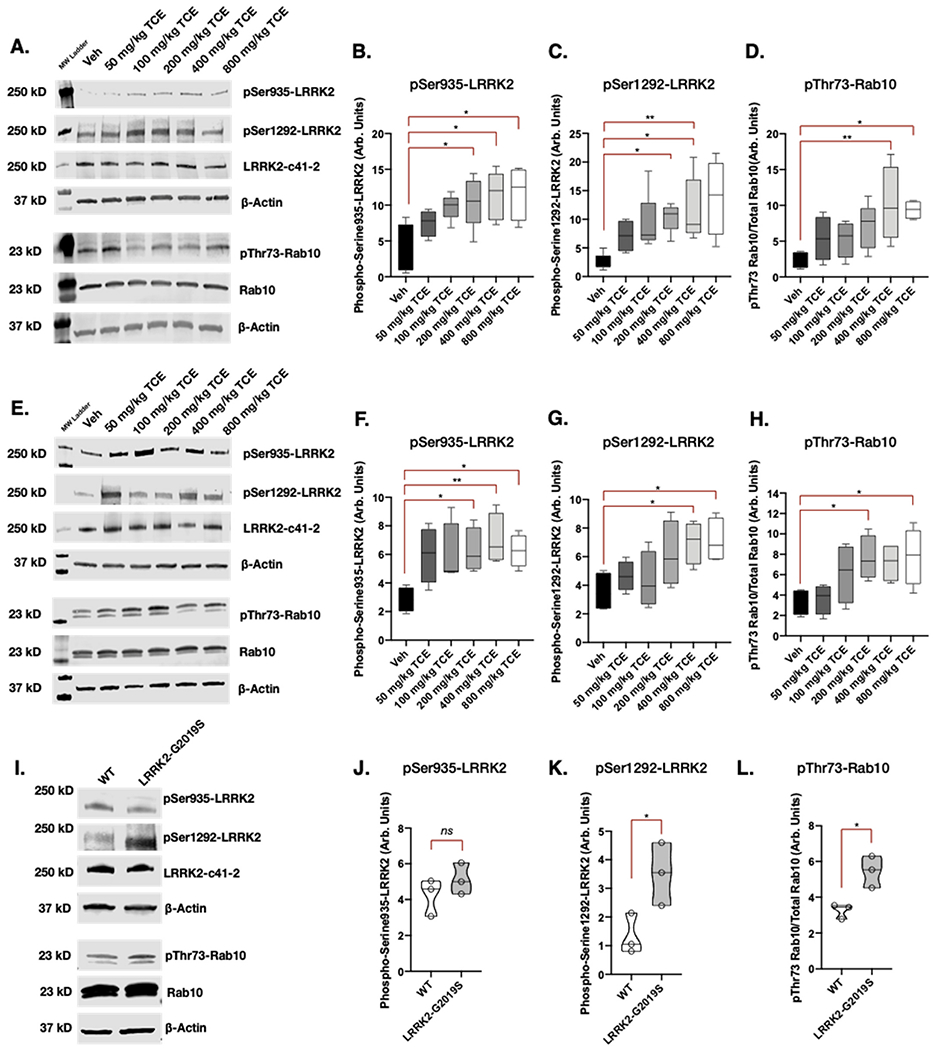
A-D. Western blot analysis of LRRK2 kinase activity in striatal tissue in response to increasing concentrations of TCE (50–800 mg/kg) or vehicle (olive oil) given to aged adult (10 month) male Lewis rats for 3 weeks via daily oral gavage; pSer935 (F(5, 18 = 3.806), p = 0.0158), pSer1292 (F(5, 22 = 3.3692), p = 0.0141), pThr73-Rab10 (F(5, 21 = 4.111), p = 0.0093). E-H. Corresponding western blot analysis of LRRK2 kinase activity in the SN; pSer935 (F(5, 18 = 3.098), p = 0.0344), pSer1292 (F(5, 18 = 3.201), p = 0.0306), pThr73-Rab10 (F(5, 18 = 3.385), p = 0.0249); one-way ANOVA, N = 5, error bars represent SEM. I-L. A comparison of LRRK2 kinase activity in untreated LRRK2-G2019S knock-in mouse brain tissue; pSer935 (F(2, 2 = 1.371), p = 0.314), pSer1292 (F(2, 2 = 2.421), p = 0.0449), pThr73-Rab10 (F(2, 2 = 4.590), p = 0.0176); unpaired T-test, N = 3, error bars represent SEM.
4. Discussion
There is growing concern for environmental exposures in the etiology for idiopathic PD, as industrialization around the world, particularly in developing countries, is paralleled by the rise of PD incidence (Dorsey and Bloem, 2018). While the link between certain pesticides and PD risk has been extensively characterized(Ascherio et al., 2006; Baltazar et al., 2014; Dick et al., 2007; Marras et al., 2019), many industrial byproducts with established neurotoxic properties have not been thoroughly assessed in the context of PD. TCE is a prototypical compound in the class of halogenated solvents, which is comprised of chlorinated, fluorocarbon, brominated or iodinated solvents and their structural derivatives (Humans, I.W.G. on the E. of C.R. to, 2014). Thus, while TCE represents one of many industrial solvents, its mechanisms of dopaminergic neurotoxicity may be relevant to other structurally similar, ubiquitous environmental toxicants.
Previous studies found that high doses of TCE (800–1000 mg/kg) in young adult rats or mice produced selective neurodegeneration of dopaminergic neurons, providing proof-of-concept evidence for dopaminergic neurotoxicity from TCE exposure (Liu et al., 2018; Liu et al., 2018). The US Occupational Safety and Health Administration (OSHA) sets daily exposure limits to TCE at 100 ppm over 8 h, or 300 ppm for five-minute intervals (OSHA, 2019). In contrast, the US Environmental Protection Agency (EPA) sets water limits for TCE at just 5 ppb, however as TCE is classified as a Group 1 carcinogen, there is probably no safe exposure level for TCE in drinking water (Humans, I.W.G. on the E. of C. R. to, 2014). We utilized a dose of 200 mg/kg in this study to model a more moderate level of chronic TCE exposure over 6 weeks.
As aging represents the most significant risk for PD development (Reeve et al., 2014), we incorporated this factor by using 15-month-old male Lewis rats for the majority of pathological analyses in this study. Indeed, despite receiving a much lower dose of TCE than in previous reports Liu et al., 2010), aged rats used here displayed significant and selective loss of dopaminergic neurons and their striatal terminal projections (Fig. 1). Likewise, the death of nigrostriatal dopaminergic neurons appeared to be a progressive, slowly developing retrograde lesion; 10-month-old female and male Lewis rats showed loss of striatal dopaminergic terminals three weeks prior to dopaminergic soma death within the SN during continuous TCE exposure (Fig. 5). The corresponding reduction in motor behavior (Fig. 1), likely as a result of dopaminergic neurodegeneration, should also be considered in the contex of aging, as previous studies examining low-dose TCE exposure in juvenile rodents reported an increase in spontaneous locomotor activity (Blossom et al., 2013). These opposing behavioral outcomes indicate that both the amount of TCE and age of exposure ultimately contribute to the type of neurotoxocity, and resultant behavioral pathology induced by the solvent. In contrast to other neurotoxicant models (Gezer et al., 2020; De Miranda et al., 2019), no significant sexual dimorphism relating to nigrostriatal neurodegeneration was apparent, perhaps due to the chronic time course over which dopaminergic neurodegeneration occurred. Together, these data demonstrate age, in combination with TCE dose, is an important co-factor in dopaminergic neurodegeneration, and should be considered in the translation to human risk for PD stemming from past or ongoing solvent exposures.
The predominant risk factor evaluated by this study, however, was the elevation of endogenous, wildtype LRRK2 kinase activity in the brain following systemic exposure to TCE. We demonstrated that TCE treatment induced LRRK2 kinase activation in the nigrostriatal tract, with temporal relevance to a developing dopaminergic lesion (Fig. 5). In addition, TCE dose-dependently elevated wildtype, endogenous LRRK2 kinase activity, indicating that LRRK2 activation might correlate with the severity of neurotoxicity elicited by exogenous chemicals (Fig. 6). Other mitochondrial toxicants, such as the pesticide rotenone, similarly activate LRRK2 in dopaminergic neurons (Rocha et al., 2019; Di Maio et al., 2018), indicating that LRRK2 activation is a common theme of PD-associated environmental contaminants. In opposition, reducing LRRK2 kinase activity, either by genetic ablation (LRRK2 knockout) or pharmacological inhibition, prevented or reduced dopaminergic neuropathology caused by pesticides and other environmental toxicants (Chen et al., 2018; Kim et al., 2019; Rocha et al., 2019; Rudyk et al., 2019). While we suspect LRRK2 inhibition would be similarly protective against TCE-induced neurodegeneration, this remains to be assessed. Nonetheless, accumulating data support that PD risk could be influenced by gene-environment interactions between LRRK2 and environmental mitochondrial toxicants like TCE.
The mechanism of LRRK2 kinase activation by TCE is less clear. As oxidative stress appears to influence LRRK2 kinase activity (Di Maio et al., 2018; Mamais et al., 2014), the robust oxidative response observed in surviving dopaminergic neurons of TCE treated rats (Fig. 2) provides a putative source for LRRK2 activation in these cells. For example, Mamais et al. (2014) propose that oxygen radicals indirectly activate LRRK2 by modulating protein phosphatases (e.g. PP1) that dephosphorylate serine residues (Ser910/935) required for 14–3-3 binding, resulting in LRRK2 complex formation at membranes. In this same vein, as LRRK2 membrane association appears to drive its activation status (Gomez et al., 2019; Hatano et al., 2007; Schapansky and LaVoie, 2013), ongoing membrane lipid peroxidation caused by TCE, measured here by 4-hydroxynonenal (Fig. 2), positions membrane-bound LRRK2 in close proximity to reactive species. Membrane dynamics may be of particular importance in TCE exposure and PD risk, as its solvent properties impart physical changes to the lipid bilayer of cellular membranes, thus providing the anesthetic effects of TCE (Huang et al., 1995). Whether TCE interaction at the membrane plays a specific role in LRRK2 kinase activation is yet unknown, but could delineate distinct neurotoxic mechanisms of solvents like TCE from other environmental PD toxicants, such as pesticides.
In addition to oxidative stress, there is a high degree of concordance between toxicant models of PD and the loss of endolysosomal function as predegenerative pathology within dopaminergic neurons (De Miranda et al., 2018; Rocha et al., 2019). Most phenotypes of iPD exhibit dysfunctional ALP (Nixon, 2005), as well as protein accumulation, suggesting loss of protein degradation pathways are central to dopaminergic neuron injury (Hou et al., 2020). Likewise, as the kinase activity of LRRK2 regulates vesicular trafficking and influences the endolysosomal system, mutations (e.g. G2019S) or activation of wildtype LRRK2 lead to disruptions in ALP (Manzoni et al., 2013a, 2013b; Park et al., 2016; Saha et al., 2015). TCE treated rats displayed an accumulation of Rab5-positive early endosomes as well as a total reduction in LAMP1-positive late endosomes / lysosomes in dopaminergic neurons of the SN (Fig. 3). Coupled with an accumulation of lysosomal cargo (p62/SQSTM1), this suggests comprehensive endosomal dysfunction that ranges from endocytic trafficking at the cell membrane (Gorvel et al., 1991) or trans-Golgi (Nagano et al., 2019), to maturation of endosomes and lysosomes responsible for macroautophagy (Lie and Nixon, 2018). As such, the impairment of endolysosomal function observed in dopaminergic neurons following six weeks of TCE treatment (Fig. 3), may be a direct result of LRRK2 kinase activation (Kuwahara et al., 2016; Kuwahara and Iwatsubo, 2020), ongoing oxidative damage to cellular macromolecules (Ramirez-Moreno et al., 2019), a byproduct of mitochondrial dysfunction (Rocha et al., 2017), or more likely, a combination thereof. However, as we recently demonstrated, pharmacological kinase inhibition of LRRK2 by the small molecule PF-360 (Pfizer) limited endolysosmal dysfunction in dopaminergic neurons induced by rotenone, which was ultimately neuroprotective against cell death (Rocha et al., 2019). In consideration, we hypothesize that selective toxicity of nigral dopaminergic neurons to TCE is a result of mitochondrial dysfunction and oxidative stress that converge upon LRRK2-regulated trafficking and endolysosomal pathways important to neuron survival (Fig 7).
Fig. 7. A pathway to LRRK2-mediated pathology in dopaminergic neurons following TCE exposure.
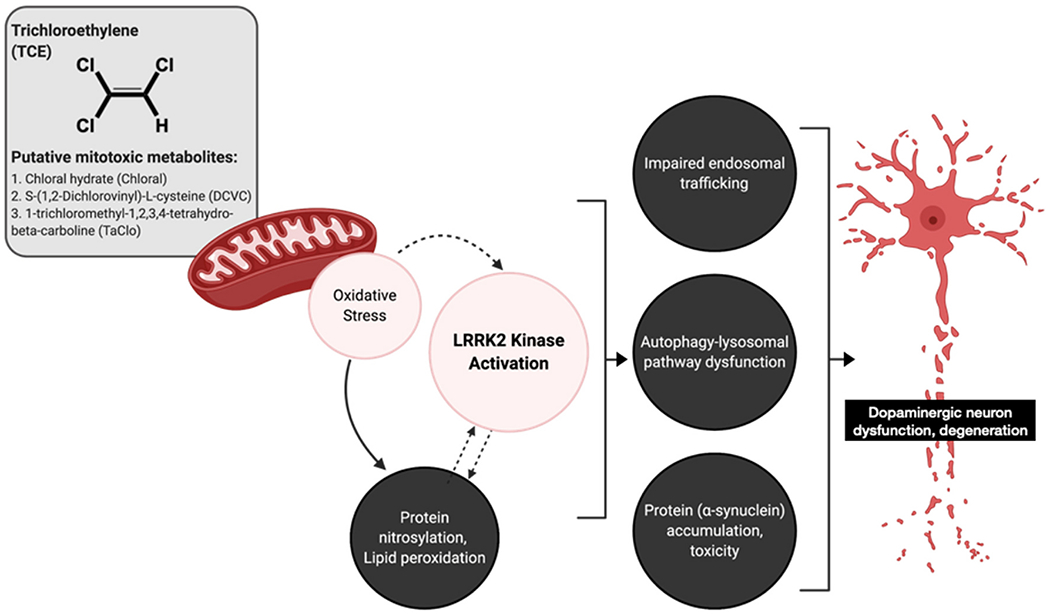
Trichloroethylene (TCE) undergoes extensive metabolism which generates several putative mitochondrial toxicants. Mitochondrial damage following TCE exposure causes cellular oxidative stress, which could induce LRRK2 kinase activity and cause downstream cellular dysfunction in dopaminergic neurons. In the current study, TCE-treated rats displayed quantifiable detection of oxidative protein modification (3-nitrotyrosine), lipid peroxidation (4-hydroxynonenal), accumulation of Rab5-positive early endosomes, reduction in autophagy (Lamp1, p62), and accumulation of endogenous α-synuclein (total, pSer129). Dashed arrows indicate hypothesized mechanisms. Image created in part with Biorender.com.
The chronic dosing paradigm presented here offers the potential to limit ALP dysfunction as a neuroprotective mechanism against TCE exposure, as elevated LRRK2 kinase activity and progressive dopaminergic cell loss from three to six weeks of TCE treatment (Fig. 5) provides a post-lesion model for therapeutic intervention. While outside the scope of the current study, a thorough examination of ALP dysfunction over the protracted TCE time course, in conjunction with LRRK2 kinase inhibition (e.g. MLi-2, Merck), will be an important future direction. Collectively, these data indicate a central role for wildtype LRRK2 and endolysosmal dysfunction in toxicant-induced neurodegeneration, and suggest that LRRK2 inhibition might be a protective strategy against exposure to a broad spectrum of environmental toxicants that cause selective dopaminergic neurodegeneration.
As LRRK2 mutations and quantitative trait loci (QTL) are the most common genetic risk factors for PD (Rudenko and Cookson, 2014), environmental toxicant exposures that induce aberrant LRRK2 kinase activity could ‘tip the balance’ toward a PD phenotype in mutation carriers. Toward this end, we observed that TCE induces wildtype LRRK2 autophosphorylation (pSer1292) to a similar extent as brain tissue from untreated G2019S-LRRK2 knock-in mice (Fig. 6). In contrast, TCE highly elevated pSer935-LRRK2 phosphorylation in rat brain tissue, which was not observed in untreated G2019S-LRRK2 knock-in mice. These data are consistent with the observation that oxidative stress derived from TCE exposure influences upstream signaling kinase (e.g IκB) and phosphatase (e.g. PP1) proteins that regulate wildtype LRRK2 phosphorylation at Ser910/935 (Mamais et al., 2014). In this sense, aberrant activation of wildtype LRRK2 and its downstream consequences may be somewhat distinct from the G2019S-LRRK2 mutation. Whether toxicant-induced LRRK2 activation has an additive or synergistic effect for LRRK2 mutation carriers is unclear. Only about 50% of individuals with a LRRK2 mutation are clinically diagnosed with PD, suggesting that additional endogenous or exogenous factors, such as TCE, could contribute to overall PD risk in these populations. Studies to determine the relative susceptibility of the dopaminergic system to a combination of LRRK2 mutation and environmental toxicant exposure are currently underway in our lab.
Supplementary Material
Acknowledgments
This work was supported by research grants from the National Institutes of Health (K99ES029986 to BRD, and NS100744, NS095387 to JTG), the American Parkinson’s disease Association (JTG), the Parkinson’s Foundation (BRD), the Michael J Fox Foundation, and the friends and family of Sean Logan (JTG).
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2021.105312.
References
- Agency for Toxic Substances and Disease Registry (ATSDR)., 2019. Toxicological Profile for Trichloroethylene (TCE). U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA. [PubMed] [Google Scholar]
- Alessi DR, Sammler E, 2018. LRRK2 kinase in Parkinson’s disease. Science 360, 36–37. 10.1126/science.aar5683. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ, 2006. Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol 60, 197–203. 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- Bae E-J, Kim D-K, Kim C, Mante M, Adame A, Rockenstein E, Ulusoy A, Klinkenberg M, Jeong GR, Bae JR, Lee C, Lee H-J, Lee B-D, Monte DAD, Masliah E, Lee S-J, 2018. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun 9, 3465. 10.1038/s41467-018-05958-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltazar MT, Dinis-Oliveira RJ, de Bastos ML, Tsatsakis AM, Duarte JA, Carvalho F, 2014. Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases—a mechanistic approach. Toxicol. Lett 230, 85–103. 10.1016/j.toxlet.2014.01.039. [DOI] [PubMed] [Google Scholar]
- Blossom SJ, Cooney CA, Melnyk SB, Rau JL, Swearingen CJ, Wessinger WD, 2013. Metabolic changes and DNA hypomethylation in cerebellum are associated with behavioral alterations in mice exposed to trichloroethylene postnatally. Toxicol Appl Pharm 269, 263–269. 10.1016/j.taap.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove FJ, Ruckart PZ, Maslia M, Larson TC, 2014. Mortality study of civilian employees exposed to contaminated drinking water at USMC Base camp Lejeune: a retrospective cohort study. Environmental health : a global access science source 13. 10.1186/1476-069x-13-68, 68–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridbord K, Brubaker PE Jr., Gay B, French JG, 1975. Exposure to halogenated hydrocarbons in the indoor environment. Environ Health Persp 11, 215–220. 10.1289/ehp.7511215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Jiayan, Su P, Luo W, Chen Jingyuan, 2018. Role of LRRK2 in manganese-induced neuroinflammation and microglial autophagy. Biochem. Biophys. Res. Commun 10.1016/j.bbrc.2018.02.007. [DOI] [PubMed] [Google Scholar]
- De Miranda BR, Houten BV, Sanders LH, 2016. Toxin-mediated complex I inhibition and Parkinson’s Disease. In: Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease, pp. 115–137. 10.1007/978-3-319-42139-1_6. [DOI] [Google Scholar]
- De Miranda BR, Rocha EM, Bai Q, Ayadi AE, Hinkle D, Burton EA, Greenamyre JT, 2018. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol. Dis 115, 101–114. 10.1016/j.nbd.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda BR, Fazzari M, Rocha EM, Castro S, Greenamyre JT, 2019. Sex differences in rotenone sensitivity reflect the male-to-female ratio in human Parkinson’s disease incidence. Toxicological sciences : an official journal of the Society of Toxicology 354, 319. 10.1093/toxsci/kfz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Laar AV, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG, Greenamyre JT, 2018. LRRK2 activation in idiopathic Parkinson’s disease. Science translational medicine 10. 10.1126/scitranslmed.aar5429eaar5429 eaar5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FD, Palma GD, Ahmadi A, Osborne A, Scott NW, Prescott GJ, Bennett J, Semple S, Dick S, Mozzoni P, Haites N, Wettinger SB, Mutti A, Otelea M, Seaton A, Soderkvist P, Felice A, Group GS, 2007. Gene-environment interactions in parkinsonism and Parkinson’s disease: the Geoparkinson study. Occup. Environ. Med 64, 673–680. 10.1136/oem.2006.032078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty RE, 2000. A history of the production and use of carbon tetrachloride, Tetrachloroethylene, trichloroethylene and 1,1,1-Trichloroethane in the United States: part 1—historical background; carbon tetrachloride and Tetrachloroethylene. Environ. Forensic 1, 69–81. 10.1006/enfo.2000.0010. [DOI] [Google Scholar]
- Dorsey ER, Bloem BR, 2018. The Parkinson pandemic-a call to action. JAMA neurology 75, 9–10. 10.1001/jamaneurol.2017.3299. [DOI] [PubMed] [Google Scholar]
- Dzamko N, Chua G, Ranola M, Rowe DB, Halliday GM, 2013. Measurement of LRRK2 and Ser910/935 phosphorylated LRRK2 in peripheral blood mononuclear cells from idiopathic Parkinson’s Disease patients. J. Parkinsons Dis 3, 145–152. 10.3233/jpd-130174. [DOI] [PubMed] [Google Scholar]
- Elkin ER, Bridges D, Loch-Caruso R, 2019. The trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine induces progressive mitochondrial dysfunction in HTR-8/SVneo trophoblasts. Toxicology 427, 152283. 10.1016/j.tox.2019.152283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser KB, Rawlins AB, Clark RG, Alcalay RN, Standaert DG, Liu N, Consortium PDBP, West AB, 2016. Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov Disord Official J Mov Disord Soc 31, 1543–1550. 10.1002/mds.26686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gezer AO, Kochmanski J, VanOeveren SE, Cole-Strauss A, Kemp CJ, Patterson JR, Miller KM, Kuhn NC, Herman DE, McIntire A, Lipton JW, Luk KC, Fleming SM, Sortwell CE, Bernstein AI, 2020. Developmental exposure to the organochlorine pesticide dieldrin causes male-specific exacerbation of α-synuclein-preformed fibril-induced toxicity and motor deficits. Neurobiol. Dis 141, 104947. 10.1016/j.nbd.2020.104947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SM, Quinlan PJ, Ross GW, Marras C, Meng C, Bhudhikanok GS, Comyns K, Korell M, Chade AR, Kasten M, Priestley B, Chou KL, Fernandez HH, Cambi F, Langston JW, Tanner CM, 2011. Solvent exposures and parkinson disease risk in twins. Ann. Neurol 71, 776–784. 10.1002/ana.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez RC, Wawro P, Lis P, Alessi DR, Pfeffer SR, 2019. Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPasesRab GTPase phosphorylation by LRRK2. J. Cell Biol 218, 4157–4170. 10.1083/jcb.201902184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel J-P, Chavrier P, Zerial M, Gruenberg J, 1991. rab5 controls early endosome fusion in vitro. Cell 64, 915–925. 10.1016/0092-8674(91)90316-q. [DOI] [PubMed] [Google Scholar]
- Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N, 2007. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet 16, 678–690. 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- Hou X, Watzlawik JO, Fiesel FC, Springer W, 2020. Autophagy in Parkinson’s disease. J. Mol. Biol 432, 2651–2672. 10.1016/j.jmb.2020.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Niu J, Xiong J, Nie S, Zeng F, Zhang Z, 2018. LRRK2 G2019S mutation inhibits degradation of α-Synuclein in an in vitro model of Parkinson’s Disease. Curr Medical Sci 38, 1012–1017. 10.1007/s11596-018-1977-z. [DOI] [PubMed] [Google Scholar]
- Huang P, Bertaccini E, Loew GH, 1995. Molecular dynamics simulation of anesthetic-phospholipid bilayer interactions. J. Biomol. Struct. Dyn 12, 725–754. 10.1080/07391102.1995.10508773. [DOI] [PubMed] [Google Scholar]
- Humans I.W.G. on the E. of C.R. to, 2014. Trichloroethylene, TETRACHLOROETHYLENE, and some other chlorinated agents. Iarc Monogr Evaluation Carcinog Risks Humans World Heal Organization Int Agency Res Cancer 106, 1–512. [PMC free article] [PubMed] [Google Scholar]
- Jankovic J, 2008. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Kim J, Pajarillo E, Rizor A, Son D-S, Lee J, Aschner M, Lee E, 2019. LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS One 14, e0210248. 10.1371/journal.pone.0210248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluss JH, Conti MM, Kaganovich A, Beilina A, Melrose HL, Cookson MR, Mamais A, 2018. Detection of endogenous S1292 LRRK2 autophosphorylation in mouse tissue as a readout for kinase activity. Npj Park Dis 4, 13. 10.1038/S41531-018-0049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluss JH, Mamais A, Cookson MR, 2019. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem Soc T 47, 651–661. 10.1042/bst20180462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, Iwatsubo T, 2020. The emerging functions of LRRK2 and Rab GTPases in the Endolysosomal system. Front. Neurosci 14, 227. 10.3389/fnins.2020.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, Inoue K, D’Agati VD, Fujimoto T, Eguchi T, Saha S, Wolozin B, Iwatsubo T, Abeliovich A, 2016. LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci Rep-uk 6, 29945. 10.1038/srep29945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavalley NJ, Slone SR, Ding H, West AB, Yacoubian TA, 2015. 14–3-3 proteins regulate mutant LRRK2 kinase activity and neurite shortening. Hum. Mol. Genet 25, 109–122. 10.1093/hmg/ddv453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang QJ, Pan N, Lee S, Zhao Y, Chait BT, Yue Z, 2011. Phosphorylation-dependent 14–3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS One 6, e17153. 10.1371/journal.pone.0017153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie PPY, Nixon RA, 2018. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis 122, 94–105. 10.1016/j.nbd.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Choi D-Y, Hunter RL, Pandya JD, Cass WA, Sullivan PG, Kim H-C, Gash DM, Bing G, 2010. Trichloroethylene induces dopaminergic neurodegeneration in fisher 344 rats. J. Neurochem 112, 773–783. 10.1111/j.1471-4159.2009.06497.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Shin E-J, Dang D-K, Jin C-H, Lee PH, Jeong JH, Park S-J, Kim Y-S, Xing B, Xin T, Bing G, Kim H-C, 2018. Trichloroethylene and Parkinson’s Disease: risk assessment. Mol. Neurobiol 55, 6201–6214. 10.1007/s12035-017-0830-x. [DOI] [PubMed] [Google Scholar]
- Madureira M, Connor-Robson N, Wade-Martins R, 2020. LRRK2: autophagy and Lysosomal activity. Front. Neurosci 14, 498. 10.3389/fnins.2020.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamais A, Chia R, Beilina A, Hauser DN, Hall C, Lewis PA, Cookson MR, Bandopadhyay R, 2014. Arsenite stress down-regulates phosphorylation and 14–3-3 binding of leucine-rich repeat kinase 2 (LRRK2), promoting self-association and cellular redistribution. J. Biol. Chem 289, 21386–21400. 10.1074/jbc.m113.528463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, Mamais A, Dihanich S, Abeti R, Soutar MPM, Plun-Favreau H, Giunti P, Tooze SA, Bandopadhyay R, Lewis PA, 2013a. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochimica Et Biophysica Acta Bba - Mol Cell Res 1833, 2900–2910. 10.1016/j.bbamcr.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, Mamais A, Dihanich S, McGoldrick P, Devine MJ, Zerle J, Kara E, Taanman J-W, Healy DG, Marti-Masso J-F, Schapira AH, Plun-Favreau H, Tooze S, Hardy J, Bandopadhyay R, Lewis PA, 2013b. Pathogenic Parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Bioph Res Co 441, 862–866. 10.1016/j.bbrc.2013.10.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras C, Canning CG, Goldman SM, 2019. Environment, lifestyle, and Parkinson’s disease: implications for prevention in the next decade. Movement Disord 34, 801–811. 10.1002/mds.27720. [DOI] [PubMed] [Google Scholar]
- Martinez TN, Greenamyre JT, 2012. Toxin models of mitochondrial dysfunction in Parkinson’s Disease. Antioxid. Redox Signal. 16, 920–934. 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matysik S, Ramadan AB, Schlink U, 2010. Spatial and temporal variation of outdoor and indoor exposure of volatile organic compounds in greater Cairo. Atmos Pollut Res 1, 94–101. 10.5094/apr.2010.012. [DOI] [Google Scholar]
- Morrison RD, Murphy BL, Doherty RE, 2005. Environmental Forensics 259–277. 10.1016/b978-012507751-4/50034-3. [DOI] [Google Scholar]
- Nagano M, Toshima JY, Siekhaus DE, Toshima J, 2019. Rab5-mediated endosome formation is regulated at the trans-Golgi network. Cornmun Biology 2, 419. 10.1038/S42003-019-0670-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, Dzarnko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR, Alessi DR, 2010. 14–3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 430, 393–404. 10.1042/bj20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, 2005. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 26, 373–382. 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- OSHA, 2019. OSHA Trichloroethylene Tetrachloroethylene [WWW Document]. URL. https://www.osha.gov/dts/sltc/methods/mdt/mdt1001/1001.html.
- Park S, Han S, Choi I, Kim B, Park SP, Joe E-H, Suh YH, 2016. Interplay between Leucine-rich repeat kinase 2 (LRRK2) and p62/SQSTM-1 in selective autophagy. PLoS One 11, e0163029. 10.1371/journal.pone.0163029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer SR, 2018. LRRK2 and Rab GTPases. Biochem Soc T 46, 1707–1712. 10.1042/bst20180470. [DOI] [PubMed] [Google Scholar]
- Ramirez-Moreno MJ, Duarte-Jurado AP, Gopar-Cuevas Y, Gonzalez-Alcocer A, Loera-Arias MJ, Saucedo-Cardenas O, de Oca-Luna RM, Rodriguez-Rocha H, Garcia-Garcia A, 2019. Autophagy stimulation decreases dopaminergic neuronal death mediated by oxidative stress. Mol. Neurobiol 56, 8136–8156. 10.1007/s12035-019-01654-1. [DOI] [PubMed] [Google Scholar]
- Reeve A, Simcox E, Turnbull D, 2014. Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res. Rev 14, 19–30. 10.1016/j.arr.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, De Miranda BR, Sanders LH, 2017. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257. 10.1016/j.nbd.2017.04.004. [DOI] [PubMed] [Google Scholar]
- Rocha EM, De Miranda BR, Castro S, Drolet R, Hatcher NG, Yao L, Smith SM, Keeney MT, Maio RD, Kofler J, Hastings TG, Greenamyre JT, 2019. LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol. Dis. 134, 104626. 10.1016/j.nbd.2019.104626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko IN, Cookson MR, 2014. Heterogeneity of Leucine-rich repeat kinase 2 mutations: genetics, mechanisms and therapeutic implications. Neurotherapeutics 11, 738–750. 10.1007/s13311-014-0284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudyk C, Dwyer Z, Hayley S, Schurr E, Brown E, Gibbings D, Hayley S, Park D, Philpott DC, Rioux JD, Schlossmacher M, 2019. Leucine-rich repeat kinase-2 (LRRK2) modulates paraquat-induced inflammatory sickness and stress phenotype. J Neuroinflamm 16, 120. 10.1186/s12974-019-1483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Ash PEA, Gowda V, Liu L, Shirihai O, Wolozin B, 2015. Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol. Neurodegener 10, 26. 10.1186/s13024-015-0022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage EP, 1989. Termiticide use and indoor air quality in the United States. Rev Environ Contam T 110, 117–130. [DOI] [PubMed] [Google Scholar]
- Schapansky J, LaVoie MJ, 2013. Endogenous LRRK2 dimerizes and translocates to novel membrane compartments during monocyte activation. FASEB J. 27, 873.17. 10.1096/fasebj.27.1_supplement.873.17. [DOI] [Google Scholar]
- Simonetti G, Filippo PD, Riccardi C, Pomata D, Sonego E, Buiarelli F, 2020. Occurrence of halogenated pollutants in domestic and occupational indoor dust. Int J Environ Res Pu 17, 3813. 10.3390/ijerph17113813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TS, Trimmer PA, Khan SM, Tinklepaugh DL, Bennett JP, 1997. Mitochondrial toxins in models of neurodegenerative diseases. II: elevated zif268 transcription and independent temporal regulation of striatal D1 and D2 receptor mRNAs and D1 and D2 receptor-binding sites in C57BL/6 mice during MPTP treatment. Brain Res. 765, 189–197. 10.1016/s0006-8993(97)00430-7. [DOI] [PubMed] [Google Scholar]
- Stevers L, de Vries R, Doveston R, Milroy L-G, Brunsveld L, Ottmann C, 2017. Structural interface between LRRK2 and 14–3-3 protein. Biochem. J. 474, 1273–1287. 10.1042/bcj20161078. [DOI] [PubMed] [Google Scholar]
- Stoiber T, Naidenko O, 2020. Environmental working group tap water database [WWW document]. URL. https://www.ewg.org/tapwater/contaminant.php?contamcode=2984 (accessed 9.30.20).
- Tapias V, Greenamyre JT, Watkins SC, 2013. Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiol. Dis. 54, 158–168. 10.1016/j.nbd.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JPL, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, Fraser K, Kennedy RE, West AB, 2016. G2019S-LRRK2 expression augments α-Synuclein sequestration into inclusions in neurons. J. Neurosci 36, 7415–7427. 10.1523/jneurosci.3642-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y-J, Liu P-WG, Hsu Y-S, Whang L-M, Lin T-F, Hung W-N, Cho K-C, 2019. Application of molecular biological tools for monitoring efficiency of trichloroethylene remediation. Chemosphere 233, 697–704. 10.1016/j.chemosphere.2019.05.203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.