

Abstract
Protein phosphatase 2A (PP2A) composed of a scaffold, a catalytic and multiple regulatory subunits is a ubiquitously expressed serine/threonine phosphatase. We have previously shown that the PP2A catalytic subunit is increased in T cells from patients with systemic lupus erythematosus (SLE) and promotes IL-17 production by enhancing the activity of Rho-associated kinase (ROCK) in T cells. However, the molecular mechanism whereby PP2A regulates ROCK activity is unknown. Here we show that the PP2A regulatory subunit PPP2R2A is increased in T cells from people with SLE and binds to, dephosphorylates and activates the guanine nucleotide exchange factor GEF-H1 at Ser885 which in turn increases the levels of RhoA-GTP and the activity of ROCK in T cells. Genetic PPP2R2A deficiency in murine T cells reduced Th1 and Th17 but not regulatory T cell differentiation and mice with T cell-specific PPP2R2A deficiency displayed less autoimmunity when immunized with myelin oligodendrocyte glycoprotein peptide. Our studies indicate that PPP2R2A is the regulatory subunit that dictates the PP2A-directed enhanced Th1 and Th17 differentiation and therefore it represents a therapeutic target for pathologies linked to Th1 and Th17 cell expansion.
Keywords: PPP2R2A, Th17, Th1, GEF-H1, Systemic lupus erythematosus, ROCK, T cells
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease with a complex pathogenesis. The expression of autoimmunity and organ inflammation involves altered production of cytokines by T cells (1–3). Specifically, decreased IL-2 production may account for the reported decreased cytotoxic T cell function and the decreased function and numbers of regulatory T cells (2), whereas increased production of IL-17 and IFN-γ may contribute directly to organ inflammation (4).
Small GTPase proteins of the Rho family are critical regulators of T-cell-mediated immune responses, including T-cell receptor (TCR)-mediated signaling, cytoskeletal reorganization, and the acquisition of the appropriate T-cell effector program (5). Rho-GTPase mediates downstream signaling through the activation of serine/threonine Rho-associated kinase (ROCK) (6). ROCK activity is up-regulated in patients with SLE (7) and its inhibition effectively decreases the secretion of proinflammatory cytokines IL-21, IL-17, and IFN-γ in T cells (8, 9), differentiation of Th1 and Th17 cells (10) and the autoimmune disease in animals (8, 11).
PP2A is a ubiquitously expressed and highly conserved serine/threonine phosphatase that plays an essential role in multiple cellular processes including cell division, cytoskeletal dynamics and various signaling pathways (12). The PP2A core enzyme consists of the scaffold subunit A (PP2AA) and the catalytic subunit C (PP2AC). To form a functional holoenzyme, the core enzyme interacts with one of the many regulatory subunits (PP2AB), which define its specificity (13). We have previously shown that the mRNA and protein levels as well as the catalytic activity of PP2Ac are increased in T cells from patients with SLE (14, 15). PP2Ac has been identified as a lupus susceptibility gene (16) with an intronic SNP to control its transcription (17). Transgenic mice which overexpress PP2Ac in T cells develop glomerulonephritis in an IL-17-dependent manner (18) which was linked to increased ROCK activity (19). However, to date the molecular mechanism by which PP2Ac regulates ROCK activity is unknown.
Previously, we showed that the PP2A regulatory subunit Bβ (PPP2R2B) accounts for the decreased IL-2 deprivation-induced T-cell apoptosis (20) and limits CD8+ T cell lifespan (21), and that another regulatory subunit PPP2R2D suppresses IL-2 production and Treg function (22). Since the specificity of PP2A function is governed by its use of a particular regulatory subunit (13) it is logical to propose that other regulatory subunits control additional distinct T cell functions. Based on our previous finding that PP2Ac leads to enhanced ROCK activity (19) and a report that the PP2A regulatory subunit Bα (PPP2R2A) is involved in the ROCK pathway-mediated control of the endothelial contractility and vessel lumen integrity (23), we hypothesized that PPP2R2A is involved in ROCK activation in T cells. Here we demonstrate that PPP2R2A dephosphorylates and activates the guanine nucleotide exchange factor GEF-H1 which in turn increases the levels of RhoA-GTP and the activity of the ROCK which promotes IL-17 and IFN-γ production. Mice lacking PPP2R2A in T cells develop less autoimmune disease.
Materials and methods
Patients enrolled in the study and T cell isolation
A total of 15 patients with systemic lupus erythematosus and 11 healthy donors were enrolled in this study. Primary T cells were purified from peripheral venous blood obtained from healthy volunteers as well as patients. The blood was incubated for 30 min with a rosette T cell purification kit (Stem Cell Technologies) that contained a tetrameric Ab mixture against CD14, CD16, CD19, CD56, and glyA that attaches non-T cells to erythrocytes. Lymphocyte separation medium (17-829E, Lonza) was subsequently used to separate these complexes from T cells. Studies were approved by the Institutional Review Board of Beth Israel Deaconess Medical Center (BIDMC).
Antibodies
Primary antibodies against PPP2R2A (#5689), p-MYPT1 (#5163) and p-SMAD2 (#3108) were purchased from Cell Signaling Technology. Primary antibody against p-SMAD3 (07-1389) was purchased from Millipore. Primary antibody against β-actin (#A5316) was from Sigma. Primary antibody against p-GEF-H1 (S-C380996) was bought from LifeSpan Biosciences Inc. Primary antibody against GEF-H1 (MA5-15027), and secondary antibodies including Goat anti-Mouse IgG (H+L) (#31430) and Goat anti-Rabbit IgG (H+L) (#31460) were purchased from ThermoFisher Scientific. Anti-human CD3 antibody (clone OKT3) was purchased from Biolegend. Anti-human CD28 antibody (RUO, #556620) was from BD Biosciences. Normal mouse IgG (#sc-2025) was from Santa Cruz Biotechnology.
Cell Culture and stimulation
Human T cells were cultured in RPMI 1640 medium (Corning Life Sciences) supplemented with 10% fetal bovine serum (FBS, Corning Life Sciences) and 1% penicillin/streptomycin (Corning Life Sciences). All cells were maintained in a humidified incubator at 37 °C with 5% CO2. T cells were stimulated with plate bound OKT3/anti-CD3 antibody (1 μg/ml) and anti-CD28 antibody (1 μg/ml) for indicated time points. Cells were used for protein extraction and ROCK activity assays.
Western blotting
Cell lysates were prepared using RIPA buffer (Boston BioProducts) containing protease cocktail inhibitor (cOmplete Mini EDTA- free, Roche) and phosphatase cocktail inhibitor (Phostop, Roche). Protein concentration was determined by coomassie protein assay reagent (Sigma-Aldrich). Twenty μg of total protein was resolved by a NuPAGE 4-12% Bis-Tris gel (Life Technologies), and transferred to PVDF membrane (Thermo Fisher Scientific). After blocking with 5% non-fat milk (M-0841, LabScientific), the membrane was incubated with a primary antibody overnight at 4 °C. Subsequently, the membrane was incubated with a secondary antibody for 90 minutes at room temperature. Western ECL substrate (1705061, Bio-Rad) was used to develop the immunoblot. The picture was captured and analyzed by Image Lab (Version 5.2.1) using ChemiDoc™ XRS+ System (Bio-Rad). When probing for multiple targets, stripping and re-probing a single membrane were applied. The membrane was stripped using mild stripping buffer (46430, Thermo Scientific) for 5 minutes at room temperature in order to avoid stripping out the sample protein on the membrane. The results were quantified by plotting the intensity of the band. β-actin was used as the loading control.
Transfections in T cells
Control siRNA (Cat#AM4611) and siRNA targeting PPP2R2A (Cat#AM16708) were purchased from Ambion Inc. PPP2R2A plasmid was obtained from Addgene (Plasmid #13804). Plasmid or siRNA transfections in primary human T cells were carried out using the Nucleofector system (Lonza). Five million freshly isolated T cells were resuspended in 100 ul of Nucleofector solution and the respective amounts of plasmid or siRNA were added. For plasmid transfection, 2 μg of plasmid per million cells were used. In the case of siRNA, 37.5, 75, or 150 nM of siRNA were transfected into T cells. Cells were transfected using the program U-014 and were rescued immediately in pre-warmed RPMI media supplemented with 10% FBS and 1% penicillin-streptomycin. Cells transfected with plasmid or siRNA were harvested at the indicated post-transfection time and subjected to protein analysis.
Real-time PCR analysis
Total RNA was isolated from T cells with RNAeasy Plus micro kit (QIAGEN). The isolated RNA was transcribed into cDNA using the RNA to cDNA premix (Clontech) according to the manufacturer’s instructions. SYBR green were purchased from Roche and the assays were performed on 96-well reaction plates (Life Technologies). The real time PCR was performed on StepOnePlus system (Life Technologies). In all experiments β-actin was used as a reference gene to normalize gene expression. Mouse β-actin (Forward: CTAAGGCCAACCGTGAAAAG; Reverse: ACCAGAGGCATACAGGGACA), PPP2R2A (Forward: AAGGTGGGAGAGTTGTCATCTT; Reverse: AGCTTTTCAAGTAGTCAAATTCTGG), IFN-γ (Forward: CTCTTCCTCATGGCTGTTTCT; Reverse: TTCTTCCACATCTATGCCACTT), IL-17A (Forward: TCCAGAAGGCCCTCAGACTA; Reverse: AGCATCTTCTCGACCCTGAA), T-bet (Forward: TCAACCAGCACCAGACAGAG; Reverse: AAACATCCTGTAATGGCTTGTG), RORC (Forward: ACCTCTTTTCACGGGAGGA; Reverse: TCCCACATCTCCCACATTG) primers were used.
ROCK activity assay
Activity of Rho-associated Protein Kinase was assessed by Millipore’s ROCK Activity Assay Kit (#CSA001) which is an enzyme immunoassay for detection of the active ROCK and DMPK family kinases. Plates were pre-coated with recombinant MYPT1, which contains a Thr696 residue that may be phosphorylated upon addition of ROCK-I, ROCK-II, or DMPK. A detection antibody that specifically detects only MYPT1 phosphorylated at Thr696 was then applied. Subsequently, an HRP-conjugated secondary detection antibody was added. The amount of phoshorylated substrate was measured by addition of the chromogenic substrate tetra-methylbenzidine (TMB), which when bound to HRP converts from a colorless solution to a blue solution (or yellow after the addition of stop solution). The absorbance signal at 450 nm reflects the relative amount of ROCK activity in the sample. Lysates from T cells exogenously expressing PPP2R2A or having been subjected to siRNA-mediated silencing were used for determining ROCK activity as described above.
RhoA activation assay
RhoA activation was determined using RhoA Activation Assay Kit (STA-403-A, Cell Biolabs Inc.) per manufacturer’s instruction. Briefly, protein lysate was prepared using lysis buffer provided in the kit. Then protein lysate was incubated with Rhotekin RBD Agarose bead slurry which can bind to RhoA-GTP (active form) for 1 hour with rotation at 4 °C. After that, Rhotekin RBD Agarose bead slurry was pelleted by centrifugation and the bound protein was eluted by sample buffer for western blot analysis using a RhoA antibody provided in the kit.
Co-immunoprecipitation (Co-IP)
Co-IP experiments were conducted using Pierce™ Classic Magnetic IP/Co-IP Kit (88804, ThermoFisher) according to manufacturer’s instructions. Briefly, protein lysates were prepared using ice-cold IP lysis buffer containing protease cocktail inhibitor (cOmplete Mini EDTA- free, Roche). Then antigen/antibody mixture was made by overnight incubation of protein lysate with an antibody against PP2Ac (#05-421, Millipore), or normal mouse IgG (sc-2025, Santa Cruz Biotechnology), as indicated. Subsequently, the mixture was added to the tube containing pre-washed magnetic beads and incubated for 1 hour at room temperature under continuous mixing. The beads were then collected on a magnetic stand and washed, and the protein complex was eluted from the beads for western blot analysis.
Generation of PPP2R2A conditional knockout mice
PPP2R2A conditional ready mice containing a neomycin genetic insert in a non-coding region of PPP2R2A that is flanked with FRT recombinase sites were purchased from the Nanjing Biomedical Research Institute (NBRI) of Nanjing University (Strain # T001370). To generate PPP2R2A-flox (R2Afl/fl) mice, PPP2R2A conditional ready mice were crossed with ROSA26::FLPe knock in mice (The Jackson Laboratory, Stock No: 009086) to excise the neomycin insert flanked by FRT sites leaving a wild type gene with loxp sites flanking exon 3 of PPP2R2A. Then R2Afl/fl mice were crossed with B6.Cg-Tg-Lck-iCre (LckCre) mice (distal promoter, The Jackson Laboratory, Stock No: 012837) to generate conditional knockout mice (LckCre R2Afl/fl) in which PPP2R2A is deleted in T cells. Both age- and sex-matched male and female mice at the age of 8-12 weeks were used for experiments. All mice were bred and housed in a specific pathogen-free environment in a barrier facility in accordance with the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee (IACUC).
Apoptosis detection
Two million of thymocytes cultured in 96-well plates with 1 μg/mL of anti-CD3 overnight were stained subsequently with Zombie Aqua (ZA, BioLegend, 423101) for 15 minutes at room temperature, surface antigens (CD4, CD8) at 4 °C for 15 minutes, and 2 μL of annexin V (BD Pharmingen, 556570) in 200 μL of 1× binding buffer at room temperature for 15 minutes. Cells cultured under Th1 or Th17 polarization conditions were stained with ZA and annexin V as mentioned above. The cells were washed by PBS between each stain. Apoptotic cells were detected by FACS. Apoptotic thymocytes were analyzed by gating single cells and exclusion of dead cells, while apoptotic Th1 and Th17 cells were analyzed by gating single cells.
In vitro T cell differentiation
Naive CD4+ T cells were purified by a mouse CD4+CD62L+ T Cell Isolation Kit II (Miltenyi Biotec). Purified naive T cells were stimulated with plate-bound goat anti-hamster antibodies, soluble anti-CD3 (0.25 μg/ml,145-2C11; Biolegend) and anti-CD28 (0.5 μg/ml, 37.51; Biolegend) for Th0-non-polarized condition culture. In addition to Th0-non-polarized condition, the following stimulation was used for each polarizing condition: IL-12 (20 ng/ml; R&D Systems) and anti-IL-4 (10 μg/ml, C17.8; Biolegend) for Th1; IL-6 (3 ng/ml; R&D Systems), TGF-β1 (0.3 ng/ml; R&D Systems), anti-IL-4 (10 μg/ml, C17.8; Biolegend) and anti-IFNγ (10 μg/ml; XMG1.2; Biolegend) for Th17; and IL-2 (20 ng/ml; R&D Systems), TGF-β1 (3 ng/ml), anti-IL-4 (10 μg/ml) and anti-IFNγ (10 μg/ml) for Treg.
Cell proliferation
Naive CD4+ T cells were stained with CellTrace Violet (C34557, ThermoFisher Scientific) according to the manufacturer’s instructions. Then, cells were cultured under Th1- or Th17-polarization conditions for two days before FACS analysis.
Flow cytometry
Cells were stained with fluorescence-tagged antibodies purchased from eBioscience, BD Biosciences, Invitrogen or BioLegend (Supplemental Table 1) and analyzed using a Cytoflex flow cytometer. Flow cytometry data were analyzed using CytExpert version 2.0. For intracellular cytokine staining, cells were stimulated for 4 hours with 50 ng/ml of phorbol myristate acetate (PMA) and 1 μM of ionomycin in the presence of 1 μg/ml of brefeldin A, harvested, fixed and stained with BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit.
Experimental autoimmune encephalomyelitis (EAE)
EAE was conducted as previously described (24). Briefly, on day 0, 8-week-old mice were subcutaneously immunized with 100 μg MOG35-55 peptide emulsified in complete Freund’s adjuvant (Sigma) containing 4 mg/mL Mycobacterium tuberculosis extract (H37Ra, Difco), distributed between the two hind flanks. On days 0 and 2, 100 ng/mouse pertussis toxin (PT) (List Biological Laboratories) was given by intraperitoneal injection. Mice were monitored and weighed daily until day 28 of the experiment. The following clinical scores were used: 1, limp tail; 2, hind-limb paresis; 3, hind-limb paralysis; 4, tetraplegia; and 5, moribund.
Single cell isolation
Spines were perfused with PBS and digested with collagenase type IV (300 U/ml, Worthington Biochemical) and DNAse I (100 μg/ml, Roche) in Hank’s balanced salt solution (HBSS) for 30 min at 37 °C for single cell dissociation as previously described (24).
Histological staining and analysis
Sections from 10% formalin-fixed spinal cords were stained with H&E. Spinal cord sections were scored by an investigator blinded to experimental group as follows (24): 0, no infiltration (<50 cells); 1, mild infiltration of nerve or nerve sheath (50-100 cells); 2, moderate infiltration (100-150 cells); 3, severe infiltration (150-200 cells); and 4, massive infiltration (>200 cells).
Statistics
All statistical analyses were conducted using GraphPad Prism 7 (GraphPad software Inc.). Statistical differences between 2 populations were calculated by unpaired t test (two-tailed). Pearson correlation coefficient was used to measure the strength of the linear relationship between PPP2R2A and p-MYPT expression. For the EAE model, clinical scores of each group were compared using two-way ANOVA. A P value of < 0.05 was considered statistically significant.
Results
PPP2R2A expression is increased in T cells from patients with SLE and correlates with ROCK activity.
We have previously shown that increased PP2A activity in T cells (14) contributes to increased IL-17 production in mice and patients with SLE (18). Because PP2A regulatory subunits appear to account for specific cell functions in a specific manner (13), we asked which regulatory subunit accounts for the increased production of IL-17. Western blot analysis of lysates of T cells from patients with SLE revealed increased expression of PPP2R2A compared to T cells from healthy subjects (Table 1, Figures 1A and 1B). We had previously shown that increased PP2A activity in murine T cells causes increased ROCK activity (19). We determined phosphorylation of myosin phosphatase target subunit 1 (MYPT1), an indicator of ROCK activity, in T cell lysates from patients with SLE and found it increased compared to the ROCK activity in T cell lysates from healthy subjects (Figures 1C and 1D), which corroborated our previous findings in engineered mice overexpressing PP2A in T cells (19). Interestingly the levels of PPP2R2A correlated strongly with the levels of phosphorylated MYPT1 (P= 0.009, Figure 1E). These results suggested that PPP2R2A is probably the PP2A regulatory subunit which is involved in the activation of ROCK.
Table 1.
Demographic and clinical characteristics of the patients with systemic lupus erythematosus (SLE) and healthy control subjects
| SLE patients (15) |
Healthy controls (11) |
|
|---|---|---|
| Age, mean (range) years | 43.1 (26-62) | 45.3 (28-60) |
| Sex, no. (%) | ||
| Female | 15 (100%) | 11 (100%) |
| Male | 0 | 0 |
| Ethnicity, no. (%) | ||
| Caucasian | 7 (57.9%) | 5 (57.1%) |
| African American | 5 (26.3%) | 3 (28.6%) |
| Asian | 1 (15.8%) | 1 (14.3%) |
| Hispanic | 2 (13.3%) | 2 (13.3%) |
| SLEDAI, mean (range) | 3.53 (0-8) | - |
| Lupus nephritis, no. (%) | 4 (26.7%) | - |
| Immunosuppressive medications, no. (%) | 15 (100%) |
- |
| Prednisone | 8 (53.3%) | - |
| Hydroxychloroquine | 11 (73.3%) | - |
| Azathioprine | 3 (20%) | - |
| Mycophenolate Mofetil | 5 (33.3%) | - |
| Belimumab | 1 (6.7%) | - |
| Methotrexate | 2 (13.3%) | - |
Figure 1. PPP2R2A expression is increased in T cells from patients with SLE and correlates with ROCK activity.
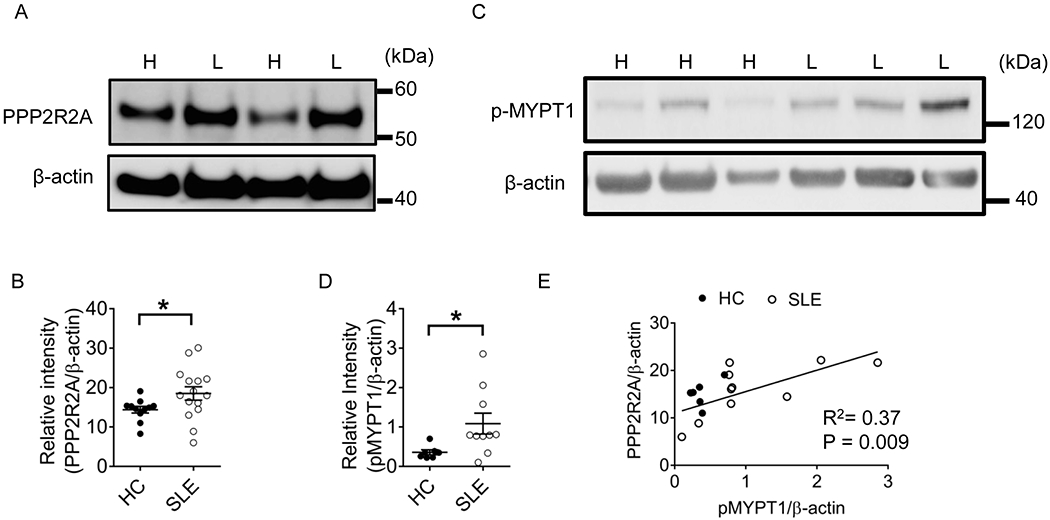
(A) Western blot showing PPP2R2A expression in T cells from healthy donors and SLE patients. H: Healthy donors; L: Lupus patients. (B) Quantification of PPP2R2A expression in T cells from healthy donors and SLE patients. Healthy donors: n=11; SLE patients: n=15. (C) Western blot showing p-MYPT1 expression in T cells from healthy donors and SLE patients. (D) Quantification of p-MYPT in T cells from healthy donors and SLE patients. Healthy donors: n=7; SLE patients: n=10. (E) Correlation between expression levels of PPP2R2A and pMYPT1 from T cells from healthy donors (filled dots, n=7) and patients with SLE (open dots, n=10). HC: Healthy controls. *P<0.05 by unpaired t test with Welch’s correction. The samples were collected from three independent times.
PPP2R2A controls the RhoA/ROCK activity in human T cells
Because PPP2R2A levels correlated with ROCK activity, we investigated whether PPP2R2A regulates the activation of ROCK in human T cells. As shown in Figure 2A, reduction of PPP2R2A levels by siRNA in T cells mitigated and conversely, forced expression of PPP2R2A increased the ROCK activity in T cells (Figure 2B). In agreement with this finding, Western blot analysis showed that silencing PPP2R2A suppressed whereas overexpression of PPP2R2A promoted the levels of phosphorylated MYPT1 (Figures 2C and 2D), the downstream substrate of ROCK. Since ROCK is a downstream effector molecule of the GTPase RhoA we determined the levels of RhoA-GTP (active form) in T cells after silencing PPP2R2A and found them to be decreased (Figure 2E). These results confirmed that PPP2R2A controls the activity of the RhoA/ROCK pathway in T cells.
Figure 2. PPP2R2A controls the RhoA/ROCK activity in T cells.
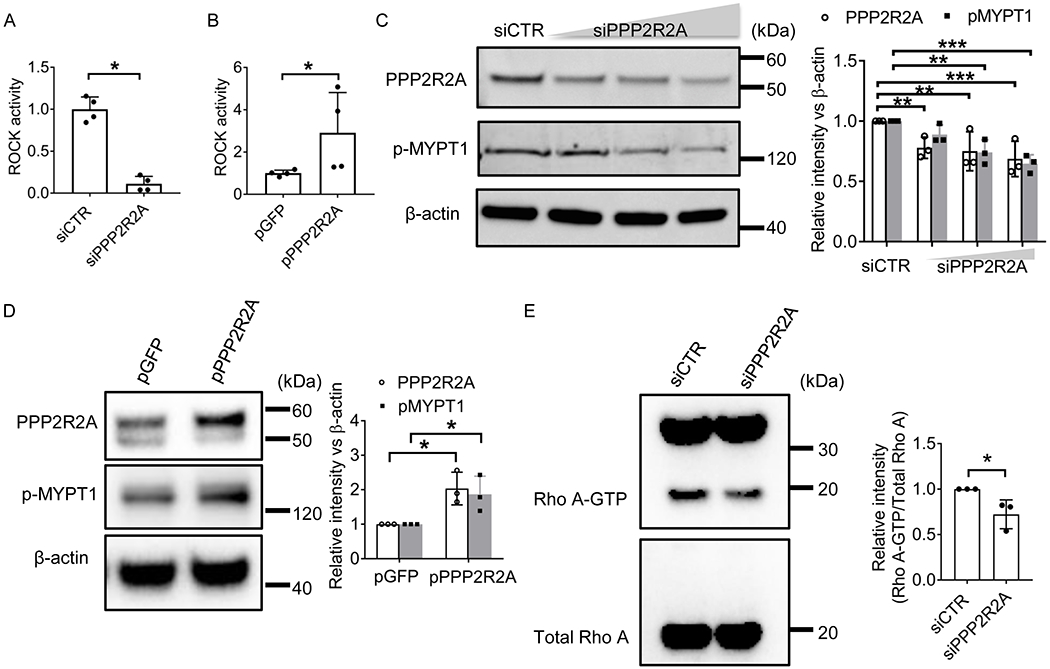
(A-B) T cells were nucleofected with (A) Control siRNA (siCTR) or siRNA targeting PPP2R2A, or (B) GFP or PPP2R2A plasmid by using Amaxa Nucleofector electroporation for 24 hours. Then T cells were stimulated with anti-CD3 and anti-CD28 antibodies for 48 hours before protein lysate was extracted for Rock activity measurement using ROCK Activity Assay Kit. (C-D) T cells were nucleofected with (C) siControl or various amounts of siRNA targeting PPP2R2A, or (D) GFP or PPP2R2A plasmid by using Amaxa Nucleofector electroporation for 24 hours. Then T cells were stimulated with anti-CD3 and anti-CD28 antibodies for 48 hours before protein lysate was extracted for western blot analysis using antibodies as indicated. The concentrations were for siCTR 150 nM, and for siPPP2R2A 37.5, 75, or 150 nM. Left: immunoblot; Right: quantification. (E) T cells were nucleofected with siControl or siRNA targeting PPP2R2A by using Amaxa Nucleofector electroporation for 24 hours. Then T cells were stimulated with CD3 and CD28 antibodies for 48 hours before protein lysate was extracted for RhoA-GTP measurement using RhoA Activation Assay Kit. Left: immunoblot; Right: quantification data. *P<0.05, **P<0.01, and ***P<0.001 by unpaired t test. The data were obtained from four (A-B) or three (C-E) independent experiments and each experiment was duplicate.
PPP2R2A deficiency in T cells does not impair T cell development in the thymus and the spleen
To address the function of PPP2R2A in T cells in a definitive manner, we crossed PPP2R2Afl/fl (R2Afl/fl) mice with LckCre mice (distal promoter) to generate conditional knockout mice (LckCre R2Afl/fl) where PPP2R2A deletion is limited to T cells, as described in Materials and methods. We next determined the role of PPP2R2A in T cell development in R2Afl/fl and LckCreR2Afl/fl mice. We examined the deletion efficiency in thymocytes and found that a relative deletion of PPP2R2A in thymocytes which is probably due to the presence of approximately 18% of single positive (CD4+CD8−or CD4−CD8+) cells (Figures 3B and 3C) which are note deleted when the cre is driven by the distal promoter of lck. In addition, there were no significant differences in the percentage and absolute numbers of T cell subsets in the thymi from R2Afl/fl and LckCreR2Afl/fl mice (Figures 3B–3D). In order to determine the effect of PPP2R2A deficiency in the survival of thymocytes, we used a CD3 antibody to stimulate thymocytes imitating TCR stimulation during double positive (DP) cell development. Thymocytes isolated from R2Afl/fl and LckCreR2Afl/fl mice were stimulated with CD3 antibody overnight and stained for Annexin V expression. As shown in Figures 3E–3G, CD3 antibody stimulation produced a similar percentage of apoptotic double negative (DN) or DP cells in R2Afl/fl and LckCreR2Afl/fl thymocytes. Furthermore, there were no significant differences in the percentage and absolute numbers of T cell subsets in spleens from R2Afl/fl and LckCreR2Afl/fl mice (Figures 3H–3J). These results suggested that PPP2R2A deficiency in T cells does not impair T cell development in the thymus and T cell subset distribution in the spleen.
Figure 3. PPP2R2A deficiency in T cells does not impair T cell development in the thymus and the subset distribution in the spleen.
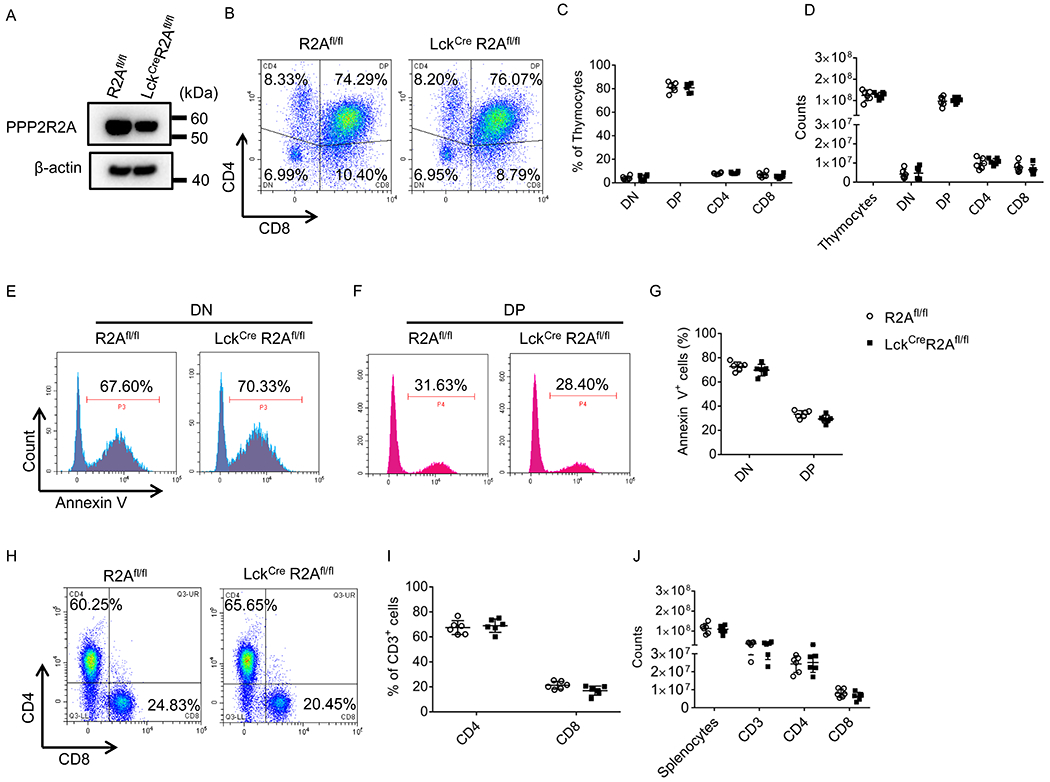
(A) Total protein was extracted from thymocytes isolated from R2Afl/fl or LckcreR2Afl/fl mice and subjected to Western blot analysis. (B-D) Thymocytes isolated from R2Afl/fl or LckcreR2Afl/fl mice were stained with CD4 and CD8 antibodies and analyzed by flow cytometry. (B) Representative flow cytometry plots. Cumulative data (n=6 mice/group) depicting the percentages (C) and absolute numbers (D) of thymocyte cell populations. DN: Double negative; DP: Double positive. (E-G) Thymocytes isolated from R2Afl/fl and LckcreR2Afl/fl mice were stimulated with CD3 antibody (1 μg/ml) overnight, and subsequently stained with Annexin V, CD4 and CD8 antibodies followed by FACS analysis. (E and F) Representative flow cytometry plots. (G) Cumulative data (n=6 mice/group) depicting the percentage of Annexin V-positive DN or DP thymocytes. (H-J) Splenocytes isolated from R2Afl/fl or LckcreR2Afl/fl mice were stained with CD3, CD4 and CD8 antibodies and analyzed by flow cytometry. (H) Representative flow cytometry plots. Cumulative data (n = 6 mice/group) depicting the percentages (I) and absolute numbers (J) of splenic CD3, CD4 or CD8 positive cells. All the data were obtained from two independent experiments.
PPP2R2A deficiency in T cells reduces Th1 and Th17 cell differentiation
To investigate whether PPP2R2A is crucial for in vitro T cell differentiation, we isolated naïve CD4+ T cells from the spleens of R2Afl/fl or LckCre R2Afl/fl mice and cultured them under Th1-, Th17-, and Treg-polarizing conditions in vitro. As presented in Figures 4A–4C, loss of PPP2R2A expression (Figure 4A) reduced the percentage of IFN-γ (Th1)- and IL-17 (Th17)-producing cells but not of Foxp3-expressing cells (Treg cells) (Figures 4B and 4C). Moreover, PPP2R2A deficiency significantly downregulated the mRNA expression level of IFNG and TBX21 (T-bet) in Th1 cells, and IL17A and RORC in Th17 cells (Figure 4D). However, lack of PPP2R2A expression did not affect the proliferation (Figures 4E and 4F) and apoptosis (Figures 4G and 4F) of Th1 or Th17 cells, indicating that the reduced numbers of Th1 and Th17 cells are not due to reduced proliferation and increased apoptosis. To further confirm these findings, splenocytes isolated from R2Afl/fl or LckCre R2Afl/fl mice were ex vivo stimulated with phorbol myristate acetate (PMA) and ionomycin for 4 hours. As shown in Supplemental Figures 1A and 1B, PPP2R2A deficiency in T cells decreased the percentage of IFN-γ- and IL-17-positive T cells among splenocytes. These results suggested that PPP2R2A deficiency in T cells inhibits Th1 and Th17 cell differentiation.
Figure 4. PPP2R2A deficiency in T cells reduces Th1 and Th17 cell differentiation in vitro.
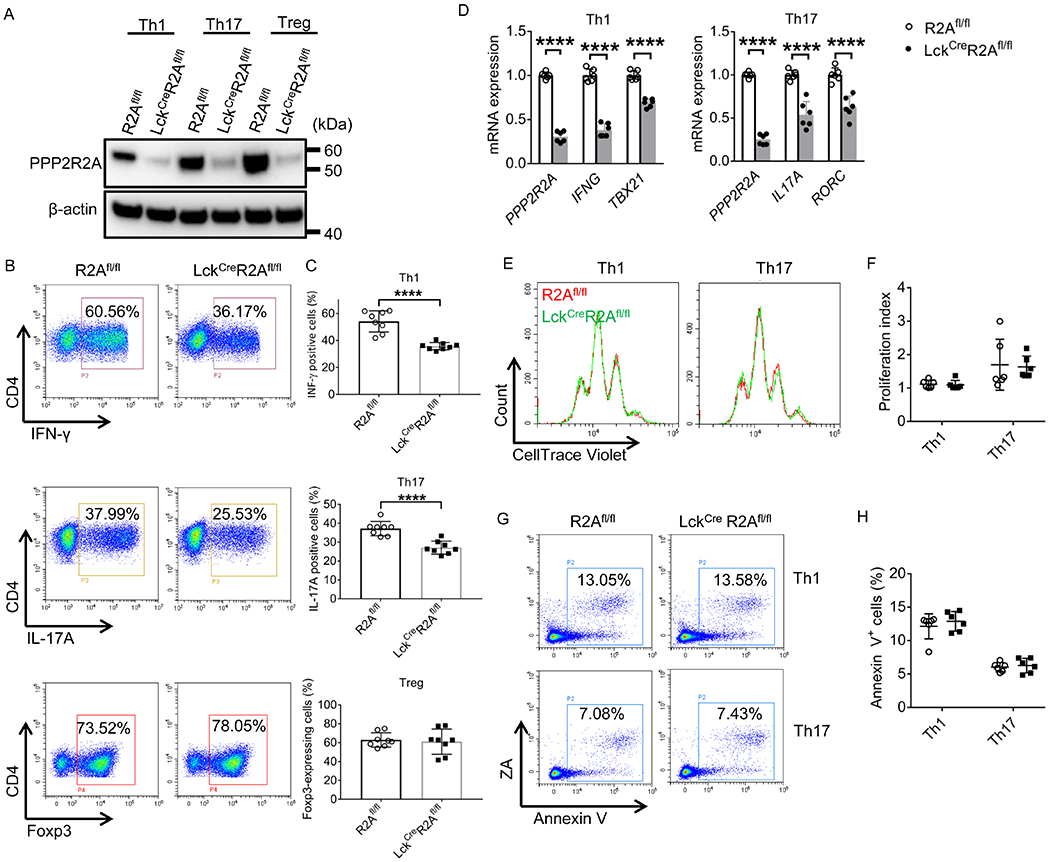
(A-C) After R2Afl/fl and LckCreR2Afl/fl naive CD4+ T cells were cultured under Th1 -, Th17 - or Treg-polarizing conditions for 3 days, total protein was isolated for Western blot analysis (A), or cells were stained with the indicated antibodies before FACS analysis (B-C). Representative flow cytometry plots are shown in (B). Cumulative data (n = 8 mice/group) depicting the percentages of IFN-γ (Th1)- and IL-17A (Th17)-producing cells and Foxp3 (Treg)-expressing cells are shown in (C). (D-H) After R2Afl/fl and LckcreR2Afl/fl naive CD4+ T cells were cultured under Th1-, or Th17- polarizing conditions for 2 days (E and F), or 3 days (D, G and H) in vitro, total RNA (D) was isolated for qPCR analysis (n = 6 mice/group), or cells were collected for cell proliferation (E and F) or apoptosis (G and H) analysis using flow cytometry. Representative flow cytometry plots are shown in (E and G). Cumulative data (n = 6 mice/group) depicting proliferation index (F) or the percentages of apoptotic cells (H). ****P<0.0001 by unpaired T test. The data were obtained from four (A-C) or three (D-H) independent experiments.
PPP2R2A deficiency impairs GEF-H1/RhoA/ROCK pathway in Th1 and Th17 cells
RhoA can be activated through the exchange of GDP for GTP, which is promoted by the Rho guanine nucleotide exchange factor 2 (ARHGEF2), also known as GEF-H1 (25). Phosphorylation of GEF-H1 at Ser 885 renders it inactive (25). Since PP2A is a serine threonine phosphatase, we determined whether PPP2R2A affects the phosphorylation status of GEF-H1 and subsequently the RhoA/ROCK activity comparing GEF-H1 and MYPT1 phosphorylation levels between mice with genetic deletion of PPP2R2A (LckCreR2Afl/fl) in T cells and wild type (R2Afl/fl) mice. As depicted in Figures 5A and 5B, lack of PPP2R2A expression (LckCreR2Afl/fl) increased the phosphorylation levels of GEF-H1 while it decreased the levels of phosphorylated MYPT1, the indicator of ROCK activity in both Th1 and Th17 cells. In addition, RhoA-GTP levels were decreased in PPP2R2A-deficient (LckCreR2Afl/fl) as compared to PPP2R2A-sufficient (R2Afl/fl) Th1 and Th17 cells (Figure 5C). Moreover, co-immunoprecipitation experiments disclosed decreased binding of PP2A catalytic subunit C (PP2Ac) to GEF-H1 in the absence of the regulatory subunit PPP2R2A in Th1 and Th17 cells (Figure 5D). Taken together, these results suggest that PP2Ac binds and activates GEF-H1 through PPP2R2A, leading to increased RhoA/ROCK activity (Figure 5E). In addition, a previous study showed that PP2Ac deficiency decreased Th17 differentiation through decreased phosphorylation of SMAD2 but increased phosphorylation of SMAD3 in T cells upon TGF-β stimulation (26). In order to examine whether this mechanism was involved in the decreased Th17 differentiation caused by PPP2R2A deficiency, we checked the phosphorylation status of SMAD2 and SMAD3 in Th17 cells. As shown in Supplemental Figures 2A and 2B, PPP2R2A deficiency did not significantly affect the phosphorylation status of SMAD2 and SMAD3 in Th17 cells, indicating that another regulatory subunit of PP2A rather than PPP2R2A is probably involved in the phosphorylation of SMAD2 and SMAD3.
Figure 5. PPP2R2A deficiency impairs the GEF-H1/RhoA/ROCK pathway in Th1 and Th17 cells.
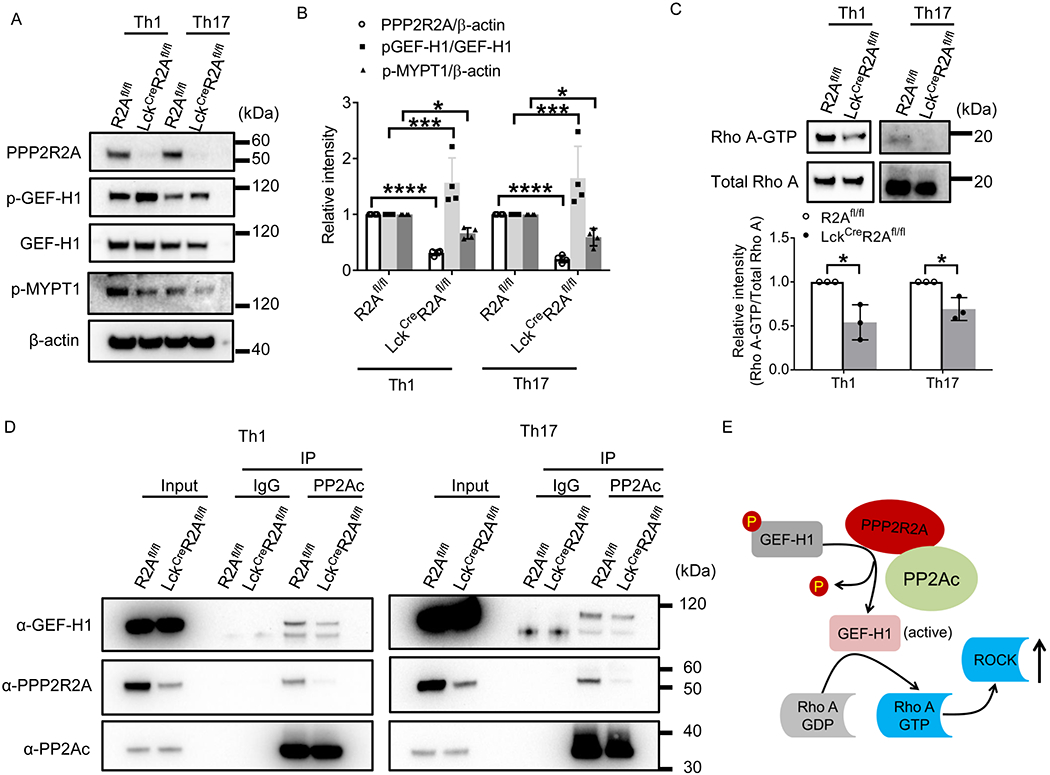
R2Afl/fl and LckcreR2Afl/fl naive CD4+ T cells were cultured under Th1- or Th17-polarizing conditions for 3 days in vitro before analysis. (A-B) Protein lysates were prepared for western blot analysis using the indicated antibodies. (A) immunoblot; (B) cumulative data (n=4 mice/group). (C) Protein lysates were prepared for RhoA-GTP measurement using RhoA Activation Assay Kit. Upper: immunoblot; lower: quantification data (n=3 mice/ group). (D) Co-immunoprecipitation analysis of PP2Ac, GEF-H1, and PPP2R2A. The blot is representative of three independent experiments. (E) Schematic representation of the signaling pathway which is regulated by PPP2R2A in T cells. *P<0.05, ***P<0.001, and ****P<0.0001 by unpaired t test. The data were obtained from two (A-B) or three (C) independent experiments.
PPP2R2A deficiency in T cells mitigates experimental autoimmune encephalomyelitis (EAE)
To determine the role of PPP2R2A-deficient T cells in the regulation of Th1- and Th17-driven autoimmune disease, we immunized R2Afl/fl and LckCreR2Afl/fl mice with myelin oligodendrocyte glycoprotein peptide of amino acids 35-55 (MOG 35-55) to induce EAE. As shown in Figure 6A, LckCreR2Afl/fl mice showed significantly reduced clinical score as compared to R2Afl/fl mice (P = 0.0187). Histology scores of spinal cords from LckCreR2Afl/fl EAE mice were significantly decreased as compared to those from R2Afl/fl EAE mice (Figures 6B and 6C). This observation was further confirmed by assessing the absolute numbers of spinal cord-infiltrating cells by flow cytometry. The number of spinal cord-infiltrating CD4 (Thy1.2+CD4+), CD8 (Thy1.2+CD8+), and double negative (DN, Thy1.2+CD4−CD8−) cells was significantly reduced in the spinal cords from LckCreR2Afl/fl mice when compared to those from R2Afl/fl EAE mice (Figures 6D–6F). Moreover, the number of IFN-γ- (Thy1.2+CD4+IFN-γ+,Thy1.2+CD8+IFN-γ+; and Thy1.2+CD4−CD8−IFN-γ+; Figures 6G–6I) and IL-17A- (Thy1.2+CD4+IL-17A+, Thy1.2+CD4−CD8−IL-17A+ and Thy1.2+CD8+IL-17A+; Figures 6J–6L) producing cells were significantly reduced in LckCreR2Afl/fl mice when compared to R2Afl/fl EAE mice. Collectively, PPP2R2A deficiency in T cells protects against Th1- and Th17-driven autoimmune disease in mice.
Figure 6. PPP2R2A deficiency in T cells mitigates experimental autoimmune encephalomyelitis (EAE).
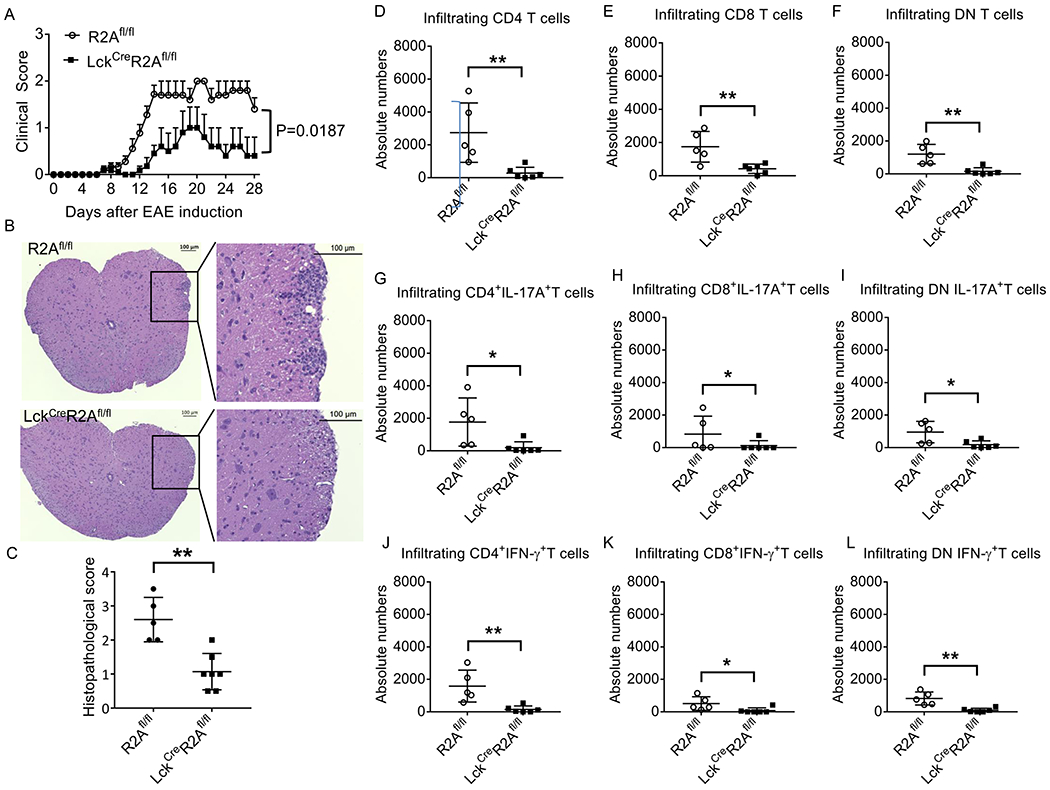
EAE was induced in R2Afl/fl and LckcreR2Afl/fl mice by immunization with MOG35-55 emulsified in complete Freund’s adjuvant. (A) Clinical scores. Cumulative results of three independent experiments with total 10 mice per group are shown. Data are shown as Mean ± SEM. P = 0.0187 by two-way ANOVA analysis. (B) Representative H&E staining of spinal cords. Scale bar: 100 μm. (C) Cumulative data elucidating the histopathologic scores for inflammation (n = 5-7 mice/group). FACS analysis of the number of spinal cord-infiltrating (D) CD4 (Thy1.2+CD4+), (E) CD8 (Thy1.2+CD8+), (F) Double Negative (DN, Thy1.2+CD4−CD8−) cells, (G-I) IFN-γ- (Thy1.2+CD4+IFN-γ+,Thy1.2+CD8+IFN-γ+; and Thy1.2+CD4−CD8−IFN-γ+) and (J-L) IL-17A- (Thy1.2+CD4+IL-17A+, Thy1.2+CD4−CD8−IL-17A+ and Thy1.2+CD8+IL-17A+) producing cells. n = 5-6 mice/group. *P<0.05 and **P<0.01 by unpaired T test. The data in (C-I) were obtained from two independent experiments.
Discussion
In this communication we present first evidence that PPP2R2A, a regulatory subunit of PP2A, controls the production of IL-17 and IFN-γ through a distinct pathway which involves the dephosphorylation and activation of GEF-H1, the production of RhoA-GTP and the activation of ROCK. At the translational level we show that the levels of PPP2R2A are increased in T cells from patients with SLE and correlate with the levels of phosphorylated MYPT1, an indicator of ROCK activity. It has been accepted that regulatory subunits of PP2A define PP2A functional specificity (13). Previously, we had shown that PPP2R2B (Bβ) controls the IL-2 deprivation-induced T cell apoptosis (20) and CD8+ T cell lifespan (21) and that PPP2R2D suppresses IL-2 production and Treg cell function (22). Our current report builds on the premise that each regulatory subunit controls a distinct function of T cells.
SLE is an autoimmune disorder which is characterized by diverse T cell effector dysfunction involving both CD4+ and CD8+ cells (1–3). We have established previously that the catalytic subunit PP2Ac is increased in T cells isolated from SLE patients and causes decreased production of IL-2 by dephosphorylating the transcriptional enhancer pCREB (14). In subsequent studies we showed that transgenic mice overexpressing PP2Ac in T cells develop an IL-17-dependent glomerulonephritis (18), which was attributed to epigenetic modification of proinflammatory genes and increased ROCK activity (19), which in turn increased the activity of the IL-17 nominal transcription factor IRF4 (8). IL-17, a proinflammatory cytokine, is produced rapidly and in large amounts by CD4+ T cells, (TCR)γδ+ and CD4−CD8− (double-negative) TCRαβ+ T cells (27–29) which are present in the periphery and in the kidney tissue of patients with lupus nephritis (29).
The abundance of PP2A in all cells and the large number of its regulatory subunits point to a complicated landscape whereby PP2A controls cell function (13). In T cells two subunits PPP2R2B and PPP2R2A control distinct functions. With regard to the study of T cells from patients with SLE, the involvement of PP2A in the immunopathogenesis of the disease appears to be even more complicated in view of the fact that the expression of PPP2R2B is decreased (20) whereas that of PPP2R2A is increased, as shown in this study.
We provide evidence that PPP2R2A directly interacts with GEF-H1 leading to its dephosphorylation and activation in T cells. GEF-H1 exhibits a variety of cellular functions (25) including the regulation of innate immune defences through various signalling pathways (30, 31). In addition, GEF-H1 is required to maintain neutrophil motility and migration in response to shear stress (32), and contributes to pulmonary endothelial cell barrier dysfunction and exacerbation of lipopolysaccharide-induced inflammation (33). GEF-H1 activates RhoA GTPase through the exchange of GDP for GTP (34) (35). Phosphorylation of GEF-H1 at Ser 885 creates a high-affinity binding site at the 14-3-3 scaffold that inhibits the exchange activity of GEF-H1 (36). A previous report showed that GEF-H1 activation in response to the G-protein-coupled receptor (GPCR) ligands requires dephosphorylation by PP2A (37). Here we demonstrate that PPP2R2A is requisite for GEF-H1 dephosphorylation and IL-17 and IFN-γ production in T cells.
The current set of experiments addresses a previously unanswered question that is how can a phosphatase increase the activity of the kinase ROCK which is active when phosphorylated (19). The ROCK family is composed of two ubiquitously expressed serine/threonine kinases, ROCK1 and ROCK2 (38). These proteins are involved in several important pathways and have important functions in different types of cells, including endothelial cells and smooth muscle cells. Specifically, ROCK has been associated to the pathogenesis of SLE (7) through its capacity to regulate cytoskeletal proteins and enhance the ability of T cells to enter inflamed tissues (39) and its ability to activate the transcription factor IRF4 that enhances IL-17 production (8). More importantly, ROCK inhibitors have been shown to limit skin inflammation in patients with psoriasis by limiting the entry of T cells in the skin (40), the secretion of proinflammatory cytokines IL-21, IL-17, and IFN-γ in T cells (8, 9) and the differentiation of Th1 and Th17 cells (10), and inhibit autoimmune disease in animal models (8, 11). Our experiments provide the missing link: when PP2Ac is associated with the regulatory subunit PPP2R2A it dephosphorylates and activates GEF-H1 which in turn enhances the production of RhoA-GTP and activates ROCK. Data presented in this study demonstrate clearly that PPP2R2A deficiency decreases Th1 and Th17 differentiation and thus making PPP2R2A a promising target to treat autoimmune and inflammatory disorders. Indeed, we present evidence that PPP2R2A deficiency in T cells alleviates Th1- and Th17-driven EAE in mice.
The complexity of the biochemical abnormalities in patients with SLE requires an exact understanding of the malfunctioning signaling pathways. The fact that the regulatory subunit PPP2R2B is decreased whereas PPP2R2D and PPP2R2A are increased in T cells from patients with SLE and each contributes to the pathogenesis of the disease through distinct mechanisms dictates the need to develop drug modulators of each subunit. Such modulators may be used to correct a specific immune cell abnormality. Such an approach has been used successfully with modulators of the regulatory subunits of PP1, another abundant phosphatase (41, 42). In conclusion, PP2A comprising the PPP2R2A regulatory subunit dephosphorylates GEF-H1 which leads to ROCK activation. Targeting PPP2R2A represents an exploitable therapeutic target for SLE and other autoimmune diseases.
Supplementary Material
Key points:
PPP2R2A promotes Th1 and Th17 but not Treg cell differentiation
PPP2R2A deficiency impairs GEF-H1/RhoA/ROCK pathway in Th1 and Th17 cells
PPP2R2A deficiency in T cells mitigates experimental autoimmune encephalomyelitis
Acknowledgments
Work was supported by NIH grant RO1 AI136924.
References
- 1.Liu Z, and Davidson A. 2012. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med 18: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moulton VR, and Tsokos GC. 2015. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest 125: 2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsokos GC 2011. Systemic lupus erythematosus. N Engl J Med 365: 2110–2121. [DOI] [PubMed] [Google Scholar]
- 4.Beringer A, Noack M, and Miossec P. 2016. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol Med 22: 230–241. [DOI] [PubMed] [Google Scholar]
- 5.Tybulewicz VL, and Henderson RB. 2009. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol 9: 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riento K, and Ridley AJ. 2003. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456. [DOI] [PubMed] [Google Scholar]
- 7.Isgro J, Gupta S, Jacek E, Pavri T, Duculan R, Kim M, Kirou KA, Salmon JE, and Pernis AB. 2013. Enhanced rho-associated protein kinase activation in patients with systemic lupus erythematosus. Arthritis Rheum 65: 1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, Bhagat G, and Pernis AB. 2010. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest 120: 3280–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, Mo R, Depoil D, Rao N, Liu B, Wei J, Lucas S, Koslow M, Roche M, Schueller O, Weiss S, Poyurovsky MV, Tonra J, Hippen KL, Dustin ML, Blazar BR, Liu CJ, and Waksal SD. 2014. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A 111: 16814–16819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J, Xi JY, Yu WB, Yan C, Luo SS, Zhou L, Zhu WH, Lu JH, Dong Q, Xiao BG, and Zhao CB. 2019. Inhibition of ROCK activity regulates the balance of Th1, Th17 and Treg cells in myasthenia gravis. Clin Immunol 203: 142–153. [DOI] [PubMed] [Google Scholar]
- 11.Sun X, Minohara M, Kikuchi H, Ishizu T, Tanaka M, Piao H, Osoegawa M, Ohyagi Y, Shimokawa H, and Kira J. 2006. The selective Rho-kinase inhibitor Fasudil is protective and therapeutic in experimental autoimmune encephalomyelitis. J Neuroimmunol 180: 126–134. [DOI] [PubMed] [Google Scholar]
- 12.Ruvolo PP 2016. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin 6: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139: 468–484. [DOI] [PubMed] [Google Scholar]
- 14.Katsiari CG, Kyttaris VC, Juang YT, and Tsokos GC. 2005. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J Clin Invest. 115: 3193–3204. Epub 2005 Oct 3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunahori K, Juang YT, Kyttaris VC, and Tsokos GC. 2011. Promoter hypomethylation results in increased expression of protein phosphatase 2A in T cells from patients with systemic lupus erythematosus. J Immunol. 186: 4508–4517. doi: 4510.4049/jimmunol.1000340. Epub 1002011 Feb 1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan W, Sunahori K, Zhao J, Deng Y, Kaufman KM, Kelly JA, Langefeld CD, Williams AH, Comeau ME, Ziegler JT, Marion MC, Bae SC, Lee JH, Lee JS, Chang DM, Song YW, Yu CY, Kimberly RP, Edberg JC, Brown EE, Petri MA, Ramsey-Goldman R, Vila LM, Reveille JD, Alarcon-Riquelme ME, Harley JB, Boackle SA, Stevens AM, Scofield RH, Merrill JT, Freedman BI, Anaya JM, Criswell LA, Jacob CO, Vyse TJ, Niewold TB, Gaffney PM, Moser KL, Gilkeson GS, Kamen DL, James JA, Grossman JM, Hahn BH, Tsokos GC, Tsao BP, and Alarcon GS. 2011. Association of PPP2CA polymorphisms with systemic lupus erythematosus susceptibility in multiple ethnic groups. Arthritis Rheum 63: 2755–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagpal K, Watanabe KS, Tsao BP, and Tsokos GC. 2014. Transcription factor Ikaros represses protein phosphatase 2A (PP2A) expression through an intronic binding site. J Biol Chem 289: 13751–13757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crispin JC, Apostolidis SA, Rosetti F, Keszei M, Wang N, Terhorst C, Mayadas TN, and Tsokos GC. 2012. Cutting edge: protein phosphatase 2A confers susceptibility to autoimmune disease through an IL-17-dependent mechanism. J Immunol 188: 3567–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apostolidis SA, Rauen T, Hedrich CM, Tsokos GC, and Crispin JC. 2013. Protein phosphatase 2A enables expression of interleukin 17 (IL-17) through chromatin remodeling. J Biol Chem 288: 26775–26784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crispin JC, Apostolidis SA, Finnell MI, and Tsokos GC. 2011. Induction of PP2A Bbeta, a regulator of IL-2 deprivation-induced T-cell apoptosis, is deficient in systemic lupus erythematosus. Proc Natl Acad Sci U S A. 108: 12443–12448. doi: 12410.11073/pnas.1103915108. Epub 1103912011 Jul 1103915111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Rodriguez N, Madera-Salcedo IK, Cisneros-Segura JA, Garcia-Gonzalez HB, Apostolidis SA, Saint-Martin A, Esquivel-Velazquez M, Nguyen T, Romero-Rodriguez DP, Tsokos GC, Alcocer-Varela J, Rosetti F, and Crispin JC. 2020. Protein phosphatase 2A B55beta limits CD8+ T cell lifespan following cytokine withdrawal. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan W, Sharabi A, Ferretti A, Zhang Y, Burbano C, Yoshida N, Tsokos MG, and Tsokos GC. 2020. PPP2R2D suppresses IL-2 production and Treg function. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin M, Geudens I, Bruyr J, Potente M, Bleuart A, Lebrun M, Simonis N, Deroanne C, Twizere JC, Soubeyran P, Peixoto P, Mottet D, Janssens V, Hofmann WK, Claes F, Carmeliet P, Kettmann R, Gerhardt H, and Dequiedt F. 2013. PP2A regulatory subunit Balpha controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO J 32: 2491–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kono M, Yoshida N, Maeda K, and Tsokos GC. 2018. Transcriptional factor ICER promotes glutaminolysis and the generation of Th17 cells. Proc Natl Acad Sci U S A 115: 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birkenfeld J, Nalbant P, Yoon SH, and Bokoch GM. 2008. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol 18: 210–219. [DOI] [PubMed] [Google Scholar]
- 26.Xu Q, Jin X, Zheng M, Rohila D, Fu G, Wen Z, Lou J, Wu S, Sloan R, Wang L, Hu H, Gao X, and Lu L. 2019. Phosphatase PP2A is essential for TH17 differentiation. Proc Natl Acad Sci U S A 116: 982–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crispin JC, and Tsokos GC. 2010. Interleukin-17-producing T cells in lupus. Curr Opin Rheumatol 22: 499–503. [DOI] [PubMed] [Google Scholar]
- 28.Crispin JC, and Tsokos GC. 2010. IL-17 in systemic lupus erythematosus. J Biomed Biotechnol 2010: 943254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crispin JC, and Tsokos GC. 2009. Human TCR-alpha beta+ CD4− CD8− T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J Immunol 183: 4675–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Alonso C, Ballester I, Song JH, Chang SY, Guleng B, Arihiro S, Murray PJ, Xavier R, Kobayashi KS, and Reinecker HC. 2012. Control of NOD2 and Rip2-dependent innate immune activation by GEF-H1. Inflamm Bowel Dis 18: 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang HS, Zhao Y, Song JH, Liu S, Wang N, Terhorst C, Sharpe AH, Basavappa M, Jeffrey KL, and Reinecker HC. 2014. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat Immunol 15: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fine N, Dimitriou ID, Rullo J, Sandi MJ, Petri B, Haitsma J, Ibrahim H, La Rose J, Glogauer M, Kubes P, Cybulsky M, and Rottapel R. 2016. GEF-H1 is necessary for neutrophil shear stress-induced migration during inflammation. J Cell Biol 215: 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kratzer E, Tian Y, Sarich N, Wu T, Meliton A, Leff A, and Birukova AA. 2012. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am J Respir Cell Mol Biol 47: 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt A, and Hall A. 2002. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev 16: 1587–1609. [DOI] [PubMed] [Google Scholar]
- 35.Fukazawa A, Alonso C, Kurachi K, Gupta S, Lesser CF, McCormick BA, and Reinecker HC. 2008. GEF-H1 mediated control of NOD1 dependent NF-kappaB activation by Shigella effectors. PLoS Pathog 4: e1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meiri D, Greeve MA, Brunet A, Finan D, Wells CD, LaRose J, and Rottapel R. 2009. Modulation of Rho guanine exchange factor Lfc activity by protein kinase A-mediated phosphorylation. Mol Cell Biol 29: 5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meiri D, Marshall CB, Mokady D, LaRose J, Mullin M, Gingras AC, Ikura M, and Rottapel R. 2014. Mechanistic insight into GPCR-mediated activation of the microtubule-associated RhoA exchange factor GEF-H1. Nat Commun 5: 4857. [DOI] [PubMed] [Google Scholar]
- 38.Amano M, Nakayama M, and Kaibuchi K. 2010. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 67: 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, Tung K, and Tsokos GC. 2007. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol 178: 1938–1947. [DOI] [PubMed] [Google Scholar]
- 40.Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, Arencibia C, Polimera S, Schueller O, Fuentes-Duculan J, Bonifacio KM, Kunjravia N, Cueto I, Soung J, Fleischmann RM, Kivitz A, Lebwohl M, Nunez M, Woodson J, Smith SL, West RF, Berger M, Krueger JG, Ryan JL, and Waksal SD. 2017. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J Immunol 198: 3809–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsaytler P, Harding HP, Ron D, and Bertolotti A. 2011. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332: 91–94. [DOI] [PubMed] [Google Scholar]
- 42.Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, Sigurdardottir A, and Bertolotti A. 2015. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348: 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.