

Abstract
Sepsis initiates simultaneous pro- and anti-inflammatory processes, the pattern and intensity of which vary over time. The inability to evaluate the immune status of patients with sepsis in a rapid and quantifiable manner has undoubtedly been a major reason for the failure of many therapeutic trials. Although there has been considerable effort to immunophenotype septic patients, these methods have often not accurately assessed the functional state of host immunity, lack dynamic range, and are more reflective of molecular processes rather than host immunity. In contrast, ELISpot assay measures the number and intensity of cytokine-secreting cells and has excellent dynamic range with rapid turnaround. We investigated the ability of a (to our knowledge) novel whole blood ELISpot assay and compared it with a more traditional ELISpot assay using PBMCs in sepsis. IFN-γ and TNF-α ELISpot assays on whole blood and PBMCs were undertaken in control, critically ill nonseptic, and septic patients. Whole blood ELISpot was easy to perform, and results were generally comparable to PBMC-based ELISpot. However, the whole blood ELISpot assay revealed that nonmonocyte, myeloid populations are a significant source of ex vivo TNF-α production. Septic patients who died had early, profound, and sustained suppression of innate and adaptive immunity. A cohort of septic patients had increased cytokine production compared with controls consistent with either an appropriate or excessive immune response. IL-7 restored ex vivo IFN-γ production in septic patients. The whole blood ELISpot assay offers a significant advance in the ability to immunophenotype patients with sepsis and to guide potential new immunotherapies.
Sepsis is life-threatening organ dysfunction caused by a dysregulated host response to infection (1). Sepsis initiates a complex immunologic response that varies depending upon numerous factors, including patient age, the number and severity of comorbidities, nutritional state, genetics, site of infection, and the particular type of pathogen (2–7). Furthermore, the host immunoinflammatory response will vary in the individual patient over time as the infection persists or resolves. Typically, there is an initial or early proinflammatory phase of sepsis that is accompanied by a more prolonged immunosuppressive phase, often termed immunoparalysis (8–10) or the compensatory anti-inflammatory response syndrome.
The historic large number of unsuccessful clinical trials in sepsis therapeutics have garnered considerable pessimism on the development of potentially new immunomodulatory therapies. Nevertheless, there continue to be several trials underway testing multiple new therapeutics (11–13). Although precision biologic therapy for cancer patients can map each patient’s unique tumor mutation profile and therapies for autoimmune diseases can identify and target individual cell type and/or cytokine dysregulation, there remains a void in patient phenotyping for sepsis that would allow for similar application of precise individualized therapies. This void has been compounded by the fact that patients with sepsis often exhibit and transit through several immunological states during the course of their disease, supporting the critical need to functionally endotype individual patients prior to intervention with immunomodulatory drugs. To underscore this need, we have recently seen in the ongoing COVID-19 viral pandemic the failure of several targeted biological response modifiers that further highlights the desperate need for diagnostic tests that can immunophenotype patients (14, 15). Whereas many COVID-19 patients were being treated with drug therapies that block cytokine signaling or suppress immune effector cell function, other COVID-19 patients were being treated with drugs that enhance or restore the immune response. Thus, diametrically opposing therapies were being used in identical COVID-19 cohorts without any approach that could reveal their immunologic phenotype. For the application of new immunomodulatory therapies in sepsis to succeed, there is a critical need for a diagnostic modality that can both determine the functional state of the patient’s immune system in a quantifiable manner as well as evaluate the effectiveness of potential immune restorative therapies.
There have been many efforts to develop predictive indices and to identify specific immune phenotypes for patients with sepsis using genomic or proteomic biomarkers of immunity. Although these methods have been helpful in predicting outcomes in sepsis, in general, they have not been able to provide an accurate assessment of the functional state of host immunity and are generally more reflective of past cellular or molecular responses rather than the present state of the subject’s immune response (16). The ELISpot is a highly sensitive immunoassay that measures the ex vivo frequency of cytokine-secreting cells at the single cell level (17–20). A key advantage of ELISpot is that the assay has an excellent dynamic five-log range, enabling it to accurately define the immune dysfunction. In addition to detecting the number of cytokine-secreting cells, the relative amount of cytokine that is produced by each cell can be determined by measuring the total well intensity (TWI) as a function of the total area of counted spots and the pixel density of each spot.
An additional advantage of the ELISpot assay is its ability to independently assess the function of the two major arms of the immune system, namely the innate and adaptive response (21–24). This ability to selectively assess the function of both innate and adaptive immunity is particularly important because sepsis is widely considered to cause an initial potent activation of innate immunity and an early suppression of adaptive immunity. Precise knowledge of the functional state of innate and adaptive immunity will permit the identification of individual sepsis patients who may benefit from new immunomodulatory drug therapies that selectively target key innate and adaptive immune effector cells (25).
Current ELISpot protocols require the isolation of PBMCs prior to ex vivo stimulation. The purpose of this investigation was to establish a novel whole blood ELISpot method to determine the functional immune status (i.e., proinflammatory versus immunosuppressive) in critically ill patients with sepsis. Successful development of an ELISpot assay using patient whole blood can greatly simplify assay performance and would generate findings that are much more likely to reflect the actual immunologic state of the patient’s immune response because the assay is performed in the presence of the patient’s own plasma and includes all leukocyte populations. IFN-γ production was used to assess adaptive immune function and TNF-α as an indicator of the innate response. IFN-γ was selected as the T cell cytokine of interest because of its central role in host defense, and loss of T cell IFN-γ production is a hallmark of “exhausted” T cells in patients with sepsis (26). TNF-α was selected as an indicator of the state of TLR4-mediated innate immune function because TNF-α is one of the major cytokines produced by activated myeloid cells (27). The results of the ELISpot assays for IFN-γ and TNF-α were obtained serially throughout the hospital course in septic patients to determine the differential effects on innate and adaptive immune function over time and to relate changes with clinical metrics. Finally, we tested the ability of potential immune therapies ex vivo to restore the immune effector cell function using the whole blood ELISpot assay. We therefore hypothesized that use of the whole blood ELISpot assay could both uncover key functional immune endotypes of patients with sepsis and serve as a viable platform for evaluating the efficacy of different immunotherapies.
Materials and Methods
Study design
This prospective, observational, ex vivo study was performed on adult patients with sepsis, adult patients with critical illness without sepsis, and healthy volunteers acquired at Barnes Jewish Hospital (Washington University School of Medicine, St. Louis, MO). The study was approved by the Human Research Protection Office (Institutional Review Board approval no.: 201603006 and 201808049). Informed consent for participation was provided by all patients or their legally authorized representatives.
Inclusion criteria
Patients hospitalized in the intensive care unit who were 18 y of age or greater were eligible for enrollment. Sepsis was defined based on the 2016 Third International Consensus Conference definition for sepsis and septic shock (Sepsis-3) (1). Patients with a change of two points or greater using the sequential organ failure assessment (SOFA) scoring system were included. In addition, enrolled patients had a clinically suspected or microbiologically proven infection. Control subjects consisted of 1) a cohort of critically ill nonseptic (CINS) patients who were admitted to the intensive care unit for noninfectious causes and 2) a cohort of healthy nonhospitalized subjects.
Exclusion criteria
To minimize the potential confounding effects by immune altering conditions, subjects having any one of the following criteria were excluded: 1) immune-altering chronic infectious diseases such as HIV or chronic hepatitis, 2) immunosuppressive medications including chemotherapy or radiation treatment within the previous 6 wk, 3) current use of high-dose corticosteroid regimens defined as exceeding greater than a dose of 300 mg of hydrocortisone or its equivalent, 4) immune-modifying biological agents or other immunosuppression transplant-associated medications, and 5) patients with systemic autoimmune diseases.
Blood sampling and processing
Patients consented for up to three blood samples obtained serially in sodium heparin tubes. The initial blood sample from septic patients was drawn within the first 24–48 h of sepsis diagnosis. Subsequent blood draws occurred on days 3–5 and 6–10 for up to three samples if the patient remained in the hospital.
Fractionation of PBMCs
Fresh whole blood samples were processed within 90 min of collection as previously described (28). Briefly, blood was diluted in an equal volume of PBS and layered carefully on Ficoll Paque PLUS (GE Healthcare). The PBMC fraction was isolated following centrifugation at 500 × g for 30 min at room temperature. The number of total PBMCs was determined with a Vi-CELL Viability Analyzer (Beckman Coulter, Brea, CA). Flow cytometry was performed on PBMC fraction for cell typing.
Preparation of ELISpot assay for assessment of adaptive and innate immune function
Innate and adaptive immune function was assessed using ELISpot analysis by measurement of the production of IFN-γ and TNF-α in ex vivo–stimulated cells following overnight culture. Capture Ab precoated 96-well polyvinylidene difluoride–backed strip plates were used for single color enzymatic assays (ImmunoSpot; Cellular Technology [CTL], Cleveland, OH) for detection of human IFN-γ and TNF-α. ELISpot culture procedure was followed as directed using instructions from the ELISpot kit. Samples were run in duplicate for each test condition. Plates were prepared with stimulant and were incubated at 37°C and 5% CO2 for 30 min prior to plating cells. Identical conditions were prepared for comparison of whole blood assay to PBMC assay, and culture media alone was used as a negative control. Combination of 500 ng/ml of anti-CD3 (clone HIT3a; BioLegend) with 2.5 μg/ml of anti-CD28 (clone CD28.2; BioLegend) Abs were used to induce IFN-γ, and 2.5 ng/ml LPS (from Salmonella abortus equi S-form, ALX-581-009; Enzo Life Sciences, Farmingdale, NY) was used to induce TNF-α wells. Total well volume for all samples was 200 μl. PBMCs were plated into wells in quantities of 2.5 × 104 cells per well for IFN-γ and 2.5 × 103 cells per well for TNF-α. The relevant volume for 5 × 104 leukocytes of diluted whole blood (in culture media) was plated in each well based on complete blood counts performed in the clinical research core laboratory at Washington University. PBMCs and diluted whole blood were costimulated with and without recombinant human IL-7 (Escherichia coli–derived protein, product no. 207-IL; R&D Systems, Minneapolis, MN). ELISpot assays were incubated overnight for 18–22 h at 37°C and 5% CO2 as previously described (29). Following overnight incubation, a biotinylated secondary detection Ab, streptavidin-bound alkaline phosphatase, and developer solution were applied to samples as per manufacturer instructions prior to image capture and analysis.
ELISpot analysis
Samples were scanned, analyzed, and quality controlled for spot count, spot area, and TWI using a Cellular Technology series 6 ImmunoSpot Universal Analyzer with ImmunoSpot 7.0 professional software (Cellular Technology Analyzers, Shaker Heights, OH). ELISpot analysis parameters were optimized to obtain appropriate spot numbers (cytokine-secreting cells) and were maintained constant throughout each sample.
Evaluation of cytokine production based upon number of spot-forming units
The number of cytokine-secreting cells present in each ELISpot well is referred to as spot-forming units (SFU) and is reported in two distinct ways. SFUs are reported as spots per microliter of diluted whole blood and as spots per 1000 lymphocytes (for IFN-γ ELISpot) or as spots per 1000 myeloid (monocytes and neutrophils) cells (for TNF-α ELISpot). The number of lymphocytes in each well was determined based upon the absolute lymphocyte count as measured by the patients’ complete blood count. For ELISpot studies involving PBMCs, flow cytometry was performed on the PBMC fraction, and the number of lymphocytes, monocytes, and residual neutrophils were determined. Cells were stained for CD14 (clone M5E2; BioLegend, San Diego, CA). Samples were acquired on FACScan (Becton Dickenson, Franklin Lakes, NJ) with a five-color modification (CyTek Biosciences, Fremont, CA) and analyzed using FlowJo 10.2 (FlowJo, Ashland, OR) as previously described (28). Gating strategy (shown in Supplemental Fig. 1) consisted of drawing a preliminary size gate on a forward × side scatter plot. This gate was interrogated for CD14 positivity (monocytes). A subsequent gate that excluded the monocytes (Boolean “not” gate) was used to determine lymphocyte and granulocyte population percentages on a forward × side scatter plot.
Evaluation of cytokine production based upon spot intensity
In addition to the number of cytokine-producing cells, data are also reported using an automated analytical method (Cellular Technology ImmunoSpot 7.0 software) based upon the pixel density/intensity of each ELISpot well with adjustment for background well intensity (23, 30). The intensity of each well was calculated based upon the total area of the well encompassed by spots with a correction for the background intensity of each well. This analytical method allows easy interassay variability of wells from different experimental settings to be accurately assessed. The mean intensity is then multiplied by the proportion of the well that is covered in spots (total foreground area [× 103 mm2/total well area]) to establish the TWI. This metric, presented as a percentage of the maximum intensity, is a comparable measurement to the results obtained by ELISA for ex vivo–stimulated cytokine production. The total intensity is reported in this article multiplied by 102 for ease of expression. TWI is normalized and reported as TWI per microliter of whole blood as well as per 1000 lymphocytes (IFN-γ) or 1000 myeloid cells (TNF-α).
Determining contribution of monocytes versus neutrophils to ELISpot TNF-α via cell depletion
In additional whole blood measurements, RBCs were eliminated from the blood sample using EasySep RBC Depletion Reagent (STEMCELL Technologies, Vancouver, BC, Canada), according to the manufacturer’s directions. After completing RBC depletion, monocytes were selectively removed using the Human Monocyte Isolation Kit (STEMCELL Technologies). The final product was a whole blood solution without RBCs or monocytes. Samples were washed and reconstituted in culture media with native patient plasma. Purity of the sample was confirmed using flow cytometry.
Assay of cytokines and chemokines
Cytokine quantitation was performed on previously frozen plasma using a human MagPix multiplex cytokine panel (Invitrogen) and analyzed on a Luminex FLEXMAP 3D instrument, according to the manufacturer’s instructions. Cytokines in the 35-plex panel included EGF, Eotaxin, FGF-basic, G-CSF, GM-CSF, HGF, IFN-α, IFN-γ, IL-1 β, IL-1 α, IL-1RA, IL-2, IL-2R, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p40/p70) IL-13, IL-15, IL-17A, IL-17F, IL-22, IP-10, MCP-1, MIG, MIP-1 α, MIP-1 β, RANTES, TNF-α, and VEGF.
Statistical analysis
ELISpot samples were performed in duplicate, and results from the two wells were averaged. ELISpot data were analyzed using GraphPad Prism version 8.4 (GraphPad, San Diego, CA). Analysis of differences between groups was performed using a nonparametric Kruskal–Wallis test with multiple comparisons corrected by controlling for the desired false discovery rate (FDR) of 5% using the Benjamini, Krieger, and Yekutieli method (31). The FDR corrected p values are reported with p = 0.05 indicating statistical significance. Statistical analysis of the change in cytokine production with and without ex vivo IL-7 was analyzed using the Wilcoxon signed-rank test.
Results
Demographic characteristics/clinical parameters
The relevant clinical and laboratory data for the 19 septic, six CINS, and 20 healthy control subjects are presented in Table I. The average Acute Physiology and Chronic Health Evaluation-II and SOFA scores for the septic patients were 17 ± 1 and 6 ± 1, respectively. Of note, there was no significant difference between sepsis survivors and nonsurvivors in terms of severity of illness or comorbidity scores (Supplemental Fig. 2). The in-hospital mortality was 37% (7/19) and 33% (2/6) for septic and CINS patients, respectively. A list of primary diagnoses for individual patients with sepsis and for CINS patients can be found in Supplemental Table I.
Table I.
Patient demographics
| Demographics | Septic Patients (n = 19) | CINS Patients (n = 6) | Healthy Controls (n = 20) | p Value |
|---|---|---|---|---|
| Age, mean (range) | 59 (28–87) | 48 (23–64) | 51 (25–69) | 0.06 |
| Sex | ||||
| Female | 12 (63%) | 2 (33%) | 13 (65%) | 0.36 |
| Male | 7 (37%) | 4 (67%) | 7 (35%) | |
| Race | ||||
| African American | 8 (42%) | 2 (33%) | 3 (15%) | 0.49 |
| White | 11 (58%) | 4 (67%) | 15 (75%) | |
| Asian | 0 | 0 | 1 (5%) | |
| Hispanic | 0 | 0 | 1 (5%) | |
| Comorbidities | ||||
| Cancer | 2 (11%) | 1 (17%) | ||
| Cardiovascular disease | 9 (47%) | 1 (17%) | ||
| Diabetes | 8 (42%) | 0 | ||
| Gastrointestinal disease | 1 (5%) | 0 | ||
| Hepatic disease | 1 (5%) | 0 | ||
| Hyperlipidemia | 2 (11%) | 1 (17%) | ||
| Hypertension | 2 (11%) | 1 (17%) | ||
| Kidney disease | 5 (26%) | 1 (17%) | ||
| Neurologic disease | 2 (11%) | 0 | ||
| Obesity | 4 (21%) | 0 | ||
| Respiratory disease | 6 (32%) | 2 (33%) | ||
| Substance abuse | 1 (5%) | 1 (17%) | ||
| Thyroid disease | 1 (5%) | 0 | ||
| APACHE II score, mean (range) | 17 (9–26) | 11 (3–21) | 0.04 | |
| SOFA score, mean (range) | 6 (0–15) | 4.3 (1–9) | 0.46 | |
| Charlson comorbidity score | 4 (0–8) | 2 (0–7) | 0.1 | |
| Subjects with secondary infections | 4 (11%) | N/A | ||
| In-hospital mortality | 7 (37%) | 2 (33%) | ||
| WBC count × 103 per μl (initial blood draw) | 11.5 (3.4–35.2) | 12.6 (6.7–20.3) | 6 (4.2–13.7) | 0.001 |
| Absolute lymphocyte count × 103 per μ1 (initial blood draw) | 1.04 (0.3–2.5) | 1.82 (1.4–2.5) | 1.89 (1.1–4.1) | 0.0008 |
| Absolute monocyte count × 103 per μ1 (initial blood draw) | 0.77 (0.2–1.9) | 1.25 (0.6–1.9) | 0.49 (0.3–1) | 0.003 |
APACHE II, Acute Physiology and Chronic Health Evaluation-II.
The WBC counts (cells × 1000/μl) were higher in both CINS (12.6 ± 2.4; p < 0.005), septic survivors (12.8 ± 2.7; p < 0.005), and septic nonsurvivors (9.2 ± 1.3; p < 0.05) compared with healthy control subjects (6.1 ± 0.5). Conversely, the absolute lymphocyte count (cells × 1000/μl) in septic patients who died (0.6 ± 0.1) was significantly decreased compared with healthy controls (1.9 ± 0.2; p < 0.0005) and CINS (1.8 ± 0.2; p < 0.01) but not septic survivors (1.3 ± 0.2). The number of monocytes (cells × 1000/μl) was increased in septic survivors (1.0 ± 0.1; p < 0.05) and CINS (1.3 ± 0.2; p < 0.01) compared with both sepsis nonsurvivors (0.4 ± 0.1) and healthy control (0.5 ± 0.04) subjects. Sepsis nonsurvivors had a similar monocyte count to healthy controls (Supplemental Fig. 2).
Unstimulated whole blood production of IFN-γ and TNF-α in patients with sepsis
Data from ex vivo production of IFN-γ and TNF-α in whole blood unstimulated with either anti-CD3/CD28 or LPS are shown in Fig. 1 for the first 15 of the 19 septic patients in the cohort. There was essentially no production of IFN-γ without stimulation by CD3/CD28-activating Abs. This lack of cytokine production was true not only for whole blood ELISpot but also for ex vivo IFN-γ production in PBMCs (data not shown). In contrast to IFN-γ, septic patient samples produced spontaneous, unstimulated TNF-α (i.e., without the addition of LPS). Among septic patients, two distinct groups can be characterized based on low (<100 SFU/μl) or high (>100 SFU/μl) spontaneous TNF-α production (nine versus five patients, respectively). In both groups, there was also a subset of patients who had at least a 20% increase in the number of TNF-α–producing cells following LPS stimulation compared with spontaneous production, as well as a subset of patients who did not respond to LPS stimulation above their baseline. Intriguingly, three of the four septic patients who failed to demonstrate an increase in TNF-α production with LPS stimulation died versus only one of nine septic patients who did have a response to LPS.
FIGURE 1.
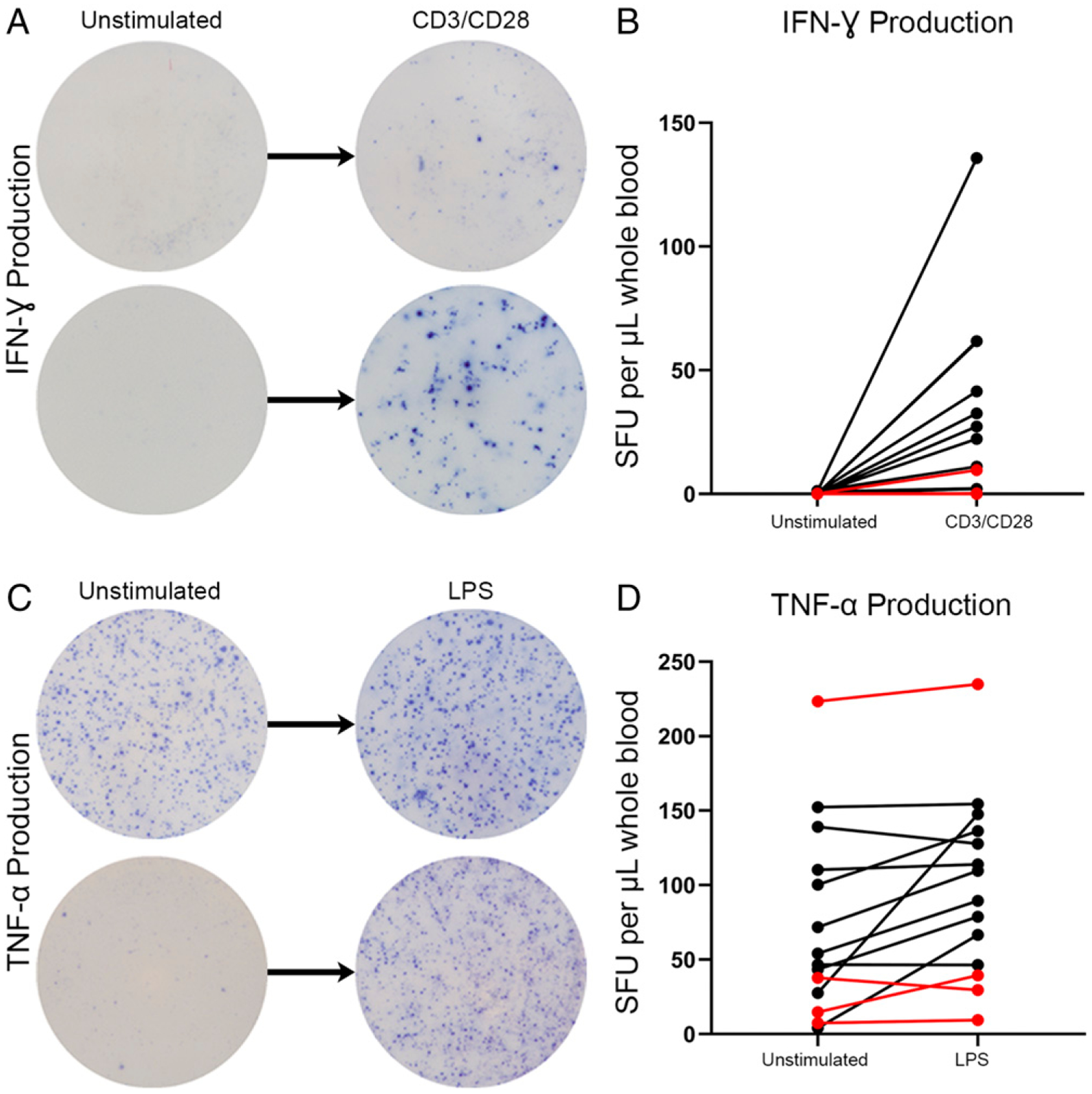
Unstimulated ex vivo production of IFN-γ and TNF-α in whole blood from septic patients using ELISpot assay. (A) Representative ELISpot images depicting IFN-γ production in media alone versus with CD3/CD28 Ab. (B) Graphic representation of n = 15 septic patient responses between unstimulated and stimulated ex vivo cytokine production of IFN-γ. (C) Representative ELISpot images depicting TNF-α production in media alone versus with LPS. (D) Graphic representation of n = 15 septic patient responses between unstimulated and stimulated ex vivo cytokine production of TNF-α. Red lines represent mortalities.
Suppressed ELISpot IFN-γ production is associated with sepsis mortality
Whole blood IFN-γ production after CD3/CD28 stimulation is shown in Fig. 2. Healthy volunteers and CINS patient responses are compared with the immune function of sepsis within 48 h after sepsis diagnosis. Number of cytokine-producing cells (SFU) and overall cytokine production as TWI was measured. Mean number of IFN-γ–producing cells per microliter of blood was 25 ± 4 for healthy volunteers and 44 ± 9 for CINS versus 50 ± 12 and 7 ± 4 for septic survivors and septic nonsurvivors, respectively (Fig. 2A). Per microliter of blood, sepsis nonsurvivors had significantly lower IFN-γ–producing cells and TWI compared with healthy controls (p < 0.05), CINS (p < 0.01), and sepsis survivors (p < 0.01) (Fig. 2A, 2C). Patients who died of sepsis had 3-fold lower numbers of activated T cells per lymphocyte compared with patients who survived sepsis (10 ± 4 for nonsurvivors, 37 ± 8 for survivors; p < 0.01) (Fig. 2B). Intensity per 1000 lymphocytes was also significantly lower in the sepsis nonsurvivors compared with CINS (p < 0.05) and sepsis survivors (p < 0.01) (Fig. 2D). These findings are consistent with an early and severe adaptive immune suppression in patients with sepsis who ultimately succumb. Septic patient survivors and CINS had a non–statistically significant trend toward more IFN-γ SFU/μl compared with healthy volunteers, and septic survivors had a 3-fold increase in activated T cells per lymphocyte compared with healthy controls (37 ± 8 versus 12 ± 2; p < 0.01), as well as a doubling of the total intensity per microliter from 16 ± 3 to 32 ± 13, indicating that a subset of patients with sepsis had an appropriately activated adaptive immune response to infection.
FIGURE 2.
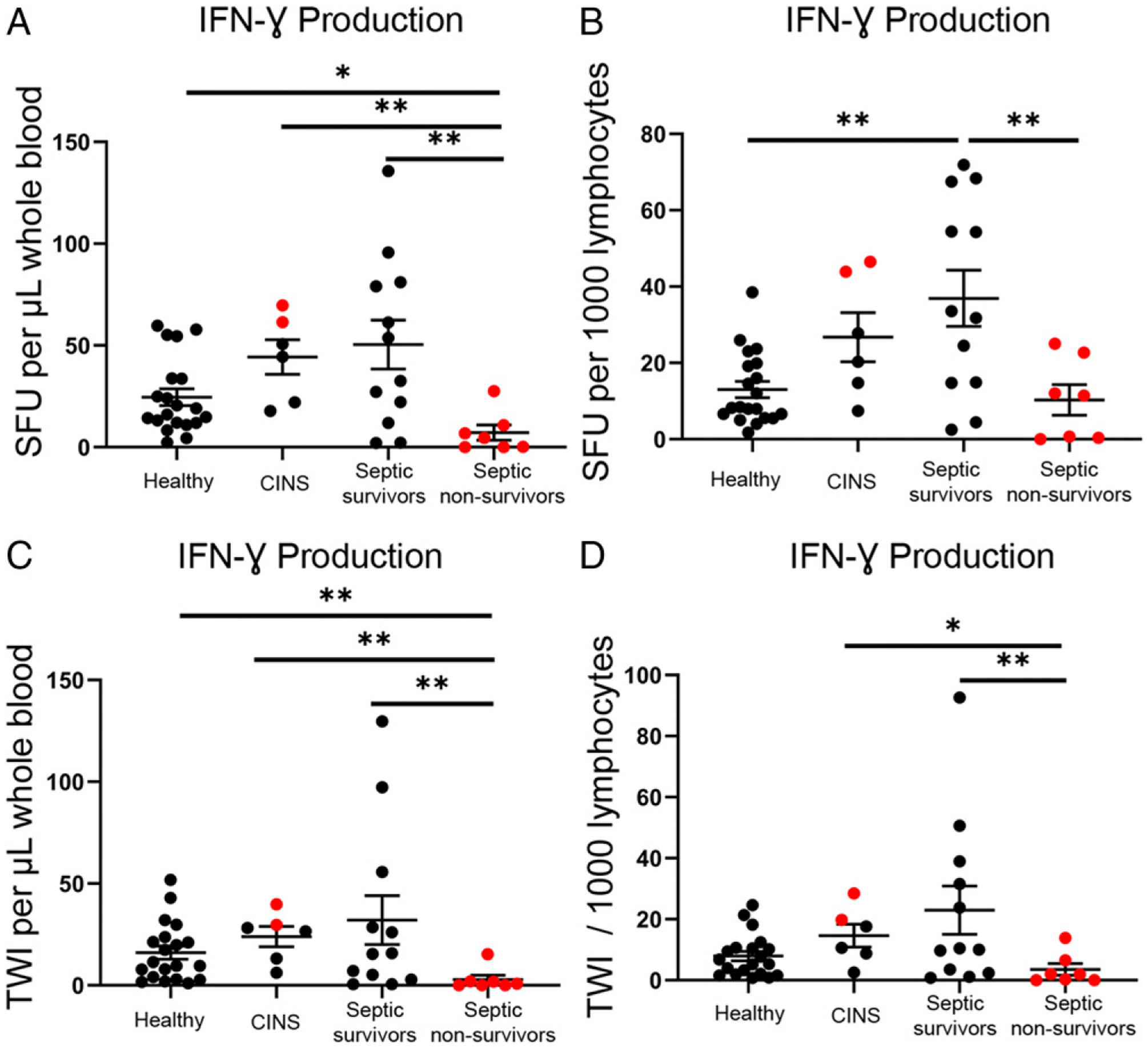
Spot number and TWI of CD3/CD28-stimulated IFN-γ–producing cells using whole blood ELISpot assay. Graphic representation comparing the differential cytokine production, in terms of the number of activated cells, in healthy control (n = 20), CINS (n = 6), sepsis survivors (n = 12), and sepsis nonsurvivors (n = 7). The number of cells is represented as SFU, and the quantity of cytokine production is represented as TWI. (A) Whole blood IFN-γ production as SFU per microliter of blood. (B) IFN-γ production as SFU per 1000 lymphocytes. (C) TWI per microliter of whole blood. (D) TWI per 1000 lymphocytes. Each ELISpot assay was performed in duplicates. Bars represent mean ± SEM. Group comparison using Kruskal–Wallis test with multiple comparisons corrected for FDR. Red dots represent mortalities. *p < 0.05, **p < 0.01.
Suppressed ELISpot TNF-α production is associated with sepsis mortality
Fig. 3 represents whole blood LPS-stimulated TNF-α production using the ELISpot assay. Healthy volunteers and CINS patient responses were compared with the initial sepsis time point (24–48 h postdiagnosis). The mean number of TNF-α–producing cells per microliter of whole blood was 88 ± 10 for healthy controls, 290 ± 60 for CINS, 143 ± 27 for septic patients who survived, and 70 ± 35 for septic patients who did not survive (Fig. 3A). CINS patients had the highest mean TNF-α production per microliter and per myeloid cell in terms of the number of cytokine-producing cells as well as total intensity, with a 3-fold increase compared with healthy volunteers (p < 0.001, p < 0.01, respectively) (Fig. 3A, 3C). Sepsis nonsurvivors had the lowest mean TNF-α production per microliter and per myeloid cell in terms of both the number of cytokine-producing cells as well as total intensity (Fig. 3). Sepsis nonsurvivors had significantly fewer TNF-α–producing cells per microliter compared with sepsis survivors (70 ± 35 versus 143 ± 27; p < 0.05) (Fig. 3A). Although there was no significant difference in terms of spots per 1000 myeloid cells plated, there was a strong trend toward decreased numbers of TNF-α–producing myeloid cells in septic nonsurvivors versus septic survivors (7 ± 3 versus 14 ± 2) (Fig. 3B). Additionally, although TNF-α production per microliter was not statistically different between sepsis nonsurvivors and healthy volunteers, they had significantly suppressed the number of cells and intensity when compared on a per 1000 myeloid cell basis (p < 0.01) (Fig. 3D).
FIGURE 3.
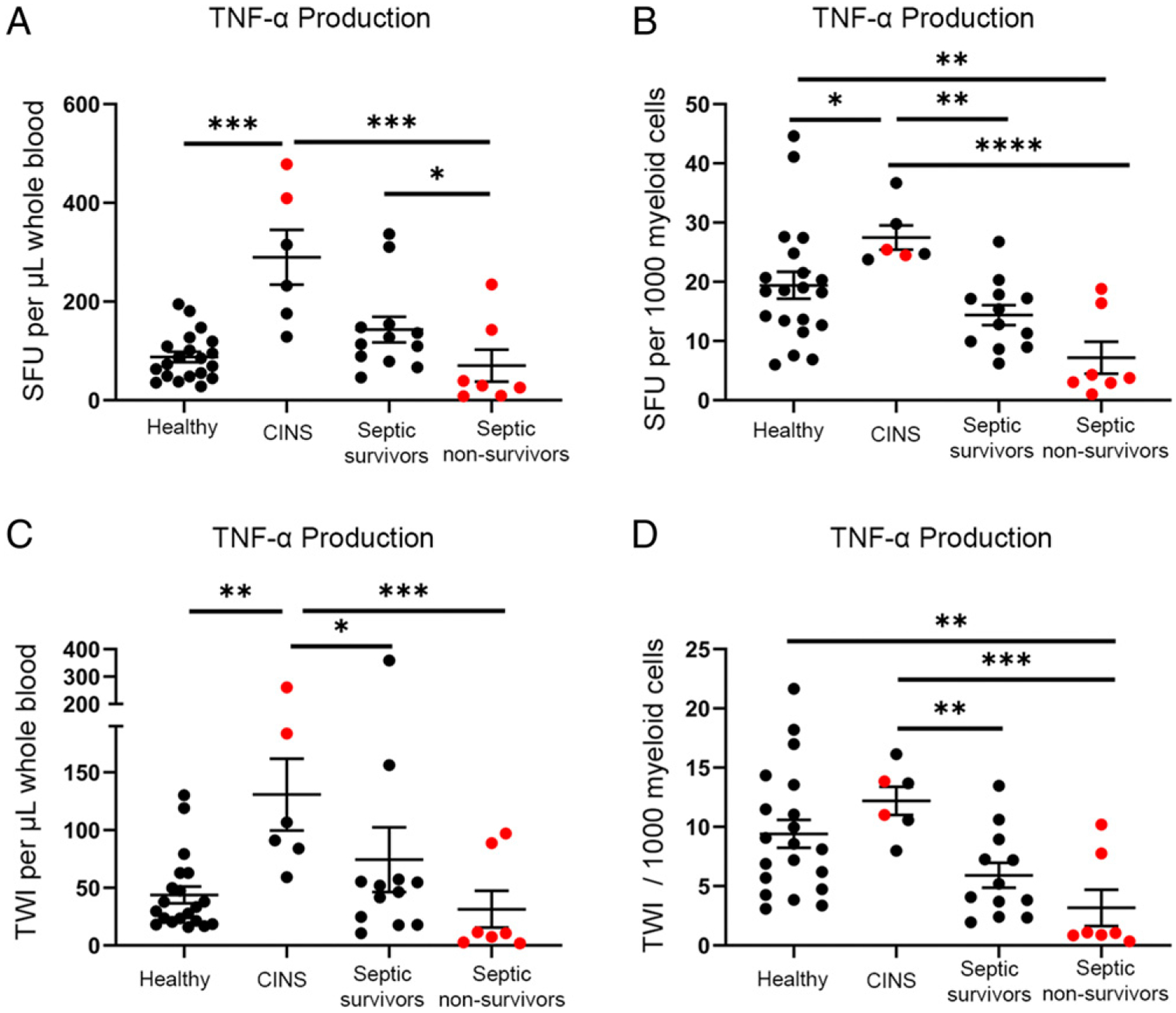
Spot number and TWI of LPS-stimulated TNF-α–producing cells using whole blood ELISpot assay. Graphic representation comparing the differential cytokine production, in terms of the number of activated cells, in healthy control (n = 20), CINS (n = 6), sepsis survivors (n = 12), and sepsis nonsurvivors (n = 7). The number of cells is represented as SFU, and the quantity of cytokine production is shown as TWI. (A) Whole blood TNF-α production as SFU per microliter of blood. (B) TNF-α production as SFU per 1000 lymphocytes plated. (C) TWI per microliter of blood. (D) TWI per 1000 lymphocytes. Each ELISpot assay is performed in duplicates. Bars represent mean ± SEM. Group comparison using Kruskal–Wallis test with multiple comparisons corrected for FDR. Red dots represent mortalities. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
IFN-γ ELISpot responses are comparable in whole blood and PBMCs preparations
A key goal of the study was to compare ELISpot results in diluted whole blood versus PBMCs obtained after Ficoll gradient separation. Note that the ELISpot results for the septic patient using PBMCs have previously been reported (15) for 15 of the 19 patients and are used for a comparison with the whole blood assay.
Representative color photomicrographs of IFN-γ ELISpot wells for three septic patients are presented in Fig. 4A, displaying images of single wells for both whole blood and PBMC ELISpot assay methods. Graphs comparing individual whole blood and PBMC IFN-γ production per 1000 lymphocytes for healthy controls, sepsis survivors, and nonsurvivors are depicted in Fig. 4B–D. Means and ranges for IFN-γ production (SFU/1000 lymphocytes) between whole blood and PBMC stimulations were similar in mean values (healthy: 12 ± 2 lymphocytes versus 20 ± 3; septic survivors: 37 ± 8 versus 18 ± 6; sepsis nonsurvivors: 10 ± 4 versus 5 ± 3) and range of variation among each cohort, with PBMCs having a slightly lower number of IFN-γ–producing cells in each cohort except in the healthy group.
FIGURE 4.
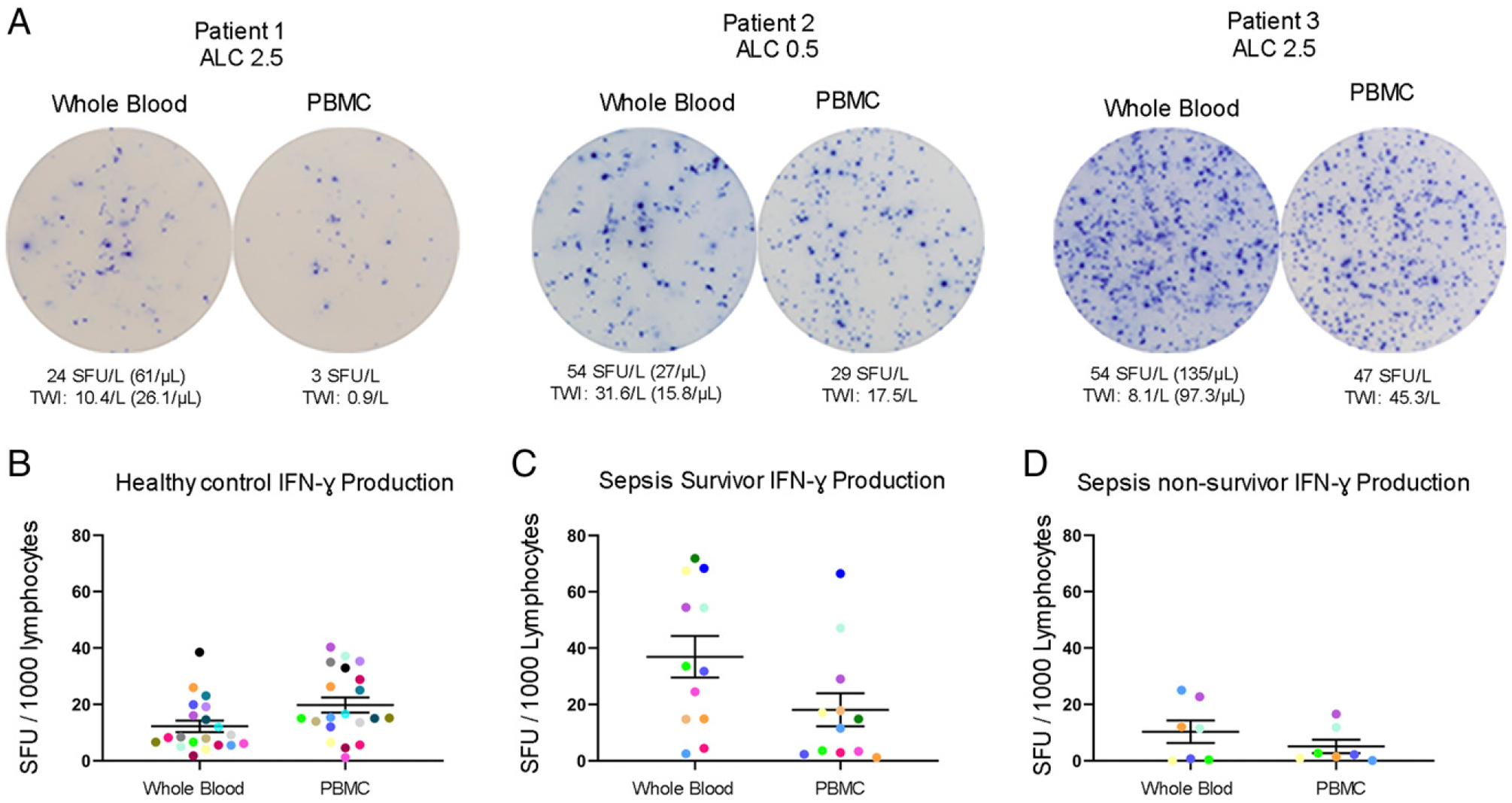
Comparison of T cell IFN-γ production in whole blood versus PBMCs ELISpot assay. (A) Representative figures depicting IFN-γ production of three individual patients using both whole blood and PBMC assays. (B–D) Dot plot graphs comparing data between whole blood and PBMC assays in healthy controls (n = 20), sepsis survivors (n = 12), and sepsis nonsurvivors (n = 7). Colored dots represent individual patients for comparison between assays. Each ELISpot assay was performed in duplicates. Bars represent mean ± SEM.
Increased TNF-α ELISpot response in PBMCs compared with whole blood preparation
Representative color photomicrographs comparing whole blood versus PMBC ELISpot TNF-α for three septic patients are presented in Fig. 5A. Graphs comparing individual whole blood versus PBMC TNF-α production per 1000 myeloid cells for healthy controls, sepsis survivors, and nonsurvivors are depicted in Fig. 5B–D. To account for neutrophil production of TNF-α, results were therefore normalized to the number of total myeloid cells plated in each experiment. Because whole blood and PBMC factions have vastly different cell type proportions, the corrected results are reported as nearly 10-fold higher for the PBMC assay compared with whole blood. Flow cytometry was performed on the PBMC fraction to determine the contaminating proportion of neutrophils in each sample. The percentage of monocytes in the PBMCs varied from ~20–30% of total cells and were not statistically different in four cohorts of subjects (i.e., healthy controls, CINS, septic survivors, and septic nonsurvivors) (Supplemental Fig. 1). In contrast, the percentage of lymphocytes in the PBMCs ranged from ~50–80% of total cells and was statistically higher in healthy controls versus septic survivors (53 ± 6%) (p < 0.01) and sepsis nonsurvivors (50 ± 12%) (p < 0.01). Whereas the percentage of neutrophils in the PBMC fraction was 7% or less in healthy controls and CINS, the percentage of neutrophils was ~15 and 35% in septic survivors and septic nonsurvivors, respectively (p < 0.01 for both groups). The results indicate that standard Ficoll gradient separation fails to deplete a significant percentage of neutrophils of the PBMC blood fraction in patients with sepsis. These contaminating low-density neutrophils produce considerable TNF-α (vide infra) and make the comparison between whole blood and PBMC assays more complex.
FIGURE 5.
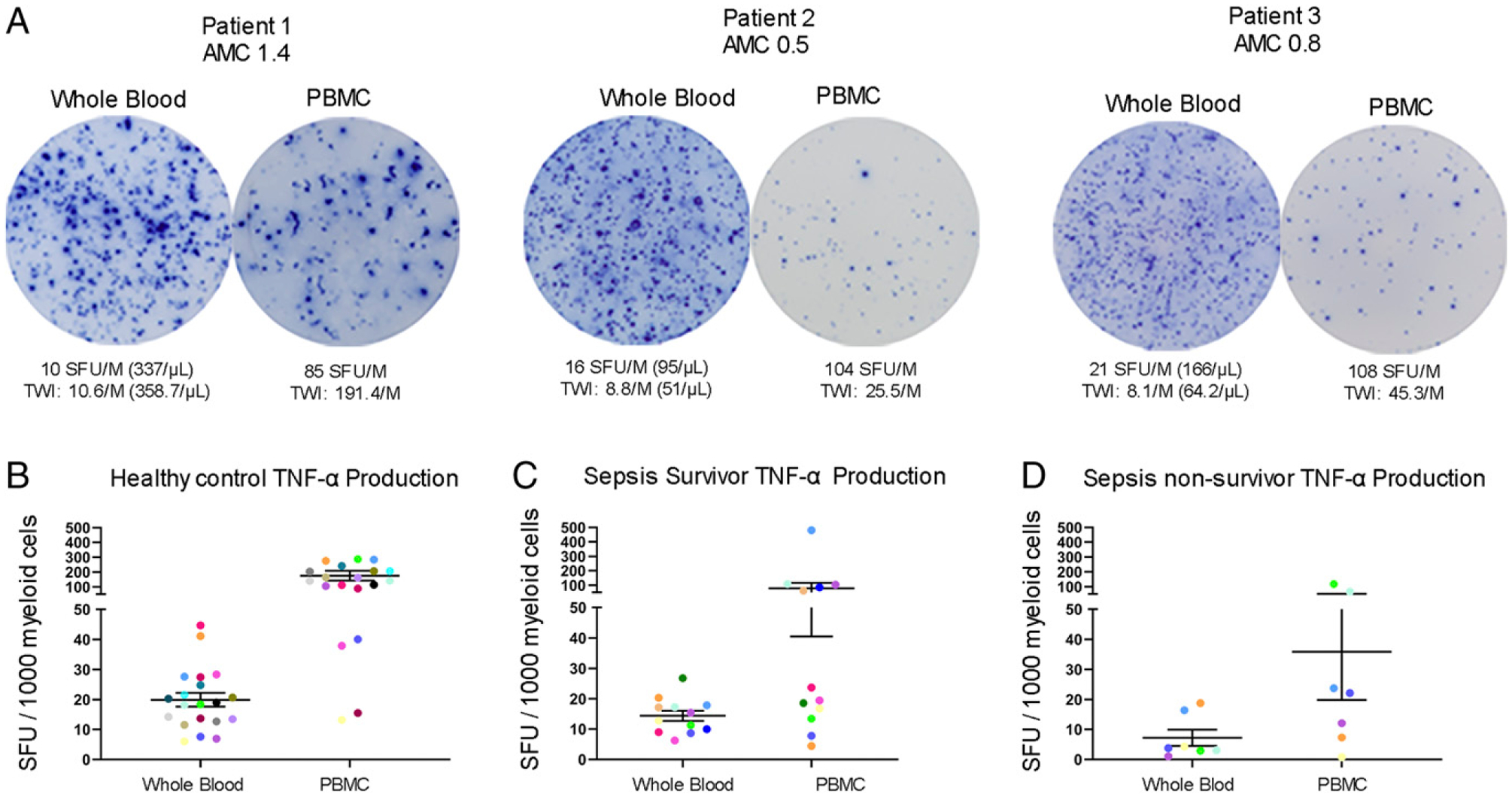
Comparison of myeloid cell TNF-α production in whole blood versus PBMCs ELISpot assay. (A) Representative figures depicting TNF-α production for three individual patients using both whole blood and PBMC assays. (B–D) Dot plot graphs compares data between whole blood and PBMC assays in healthy controls (n = 20), sepsis survivors (n = 12), and sepsis nonsurvivors (n = 7). Colored dots represent individual patients used for comparison between assays. Each ELISpot assay was performed in duplicates. Bars represent mean ± SEM.
Whole blood ELISpot TNF-α production is due to both neutrophils and monocytes
To determine the relative contributions of neutrophils and monocytes to the TNF-α ELISpot production, blood samples from healthy volunteers underwent serial RBC depletion using a magnetic bead erythrocyte depletion kit followed by monocyte magnetic bead depletion using kits from STEMCELL Technologies. Purity of the monocyte depletion was analyzed using flow cytometry for the detection of CD14+ cells. Whole blood contained 6.3% monocytes (±0.5%, n = 8), RBC-depleted blood contained 4.5% (±0.7%) monocytes, and monocyte-depleted blood contained 0.7% (±0.2%) monocytes. The number of cells positive for TNF-α after overnight LPS stimulation was compared in whole blood versus RBC- and monocyte-depleted blood (Fig. 6). On average, 81% ± 4.5% (n = 8) of the spots in RBC-depleted whole blood were produced by monocytes (range 57–96%) and 27% ± 2% (n = 8) of monocytes plated in each well were activated and secreting TNF-α during the assay (range 20–34%). Put another way, almost 20% of the TNF-α–secreting cells in whole blood ELISpot assays from septic patients are coming from nonmonocyte sources, presumably granulocytes.
FIGURE 6.
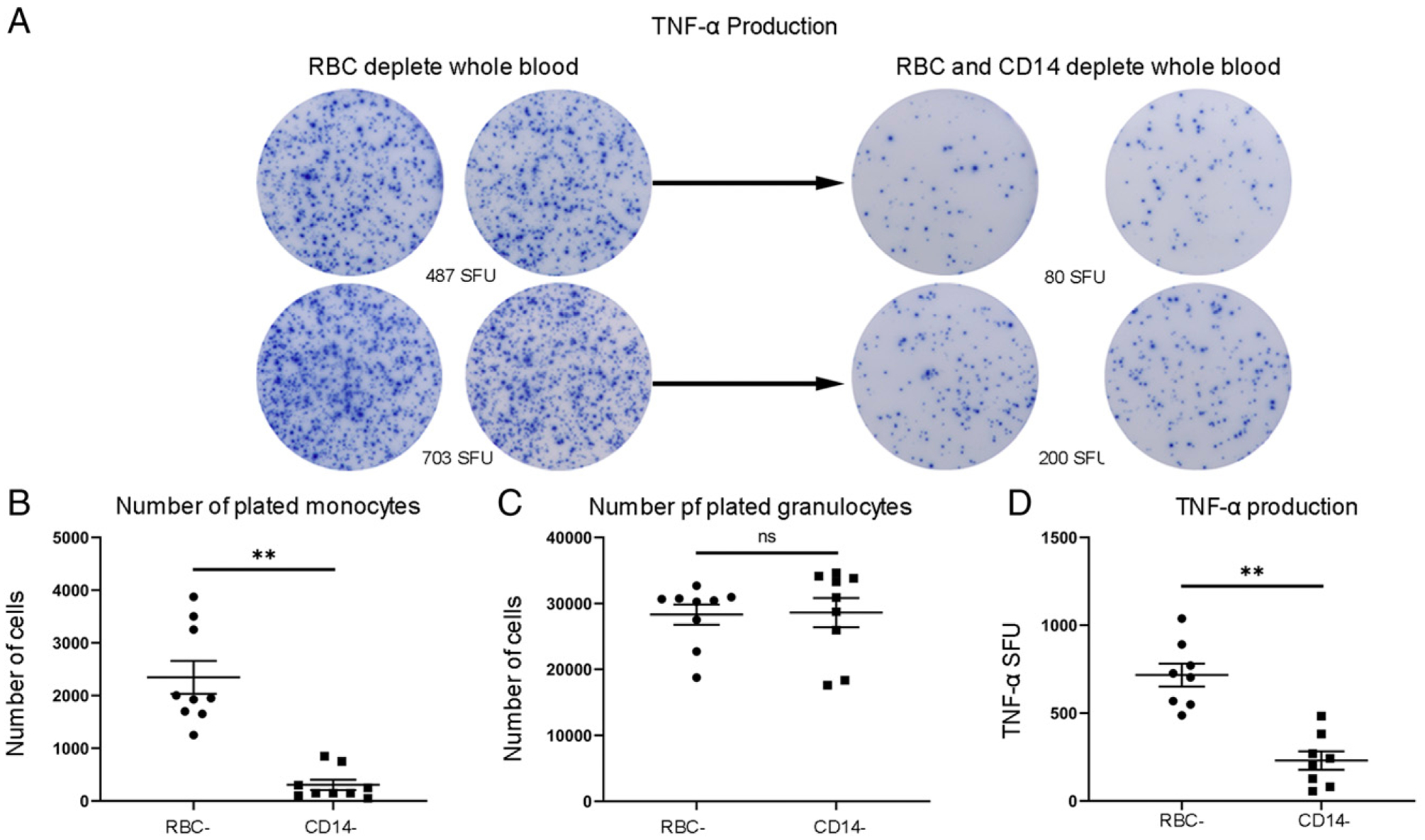
TNF-α production in whole blood ELISpot assay following depletion of monocytes in healthy volunteers. (A) Graphic depiction representing TNF-α production in RBC-depleted blood versus RBC- and monocyte-depleted blood. (B) The number of monocytes plated per individual experiment (n = 9) for RBC-depleted blood and RBC- plus monocyte-depleted blood. (C) The number of granulocytes/neutrophils plated per individual experiment (n = 9) for RBC-depleted blood and RBC- plus monocyte-depleted blood. (D) The number of SFU for TNF-α production for RBC-depleted blood and RBC- plus monocyte-depleted blood. Statistical analysis using Wilcoxon ranked sum test. **p < 0.01.
The early and profound suppression of adaptive immunity is sustained throughout sepsis in nonsurvivors
Serial time course examination of septic patient IFN-γ production via ELISpot is presented for patients who survived sepsis versus those who died of sepsis. Representative color micrographs are presented for two sepsis survivors and two nonsurvivors for three consecutive time points (Fig. 7A, 7B). Data were analyzed by both the number of IFN-γ–producing lymphocytes per 1000 lymphocytes and number of IFN-γ–producing lymphocytes/μl of diluted whole blood. In both cases, septic survivors had increased IFN-γ production compared with nonsurvivors, which was sustained over the course of the 6–10 d after sepsis diagnosis (Fig. 7C, 7D). This impairment in IFN-γ production that was present on their initial presentation was sustained throughout their entire period of study. There was a non–statistically significant trend toward worsening IFN-γ production as sepsis persisted.
FIGURE 7.
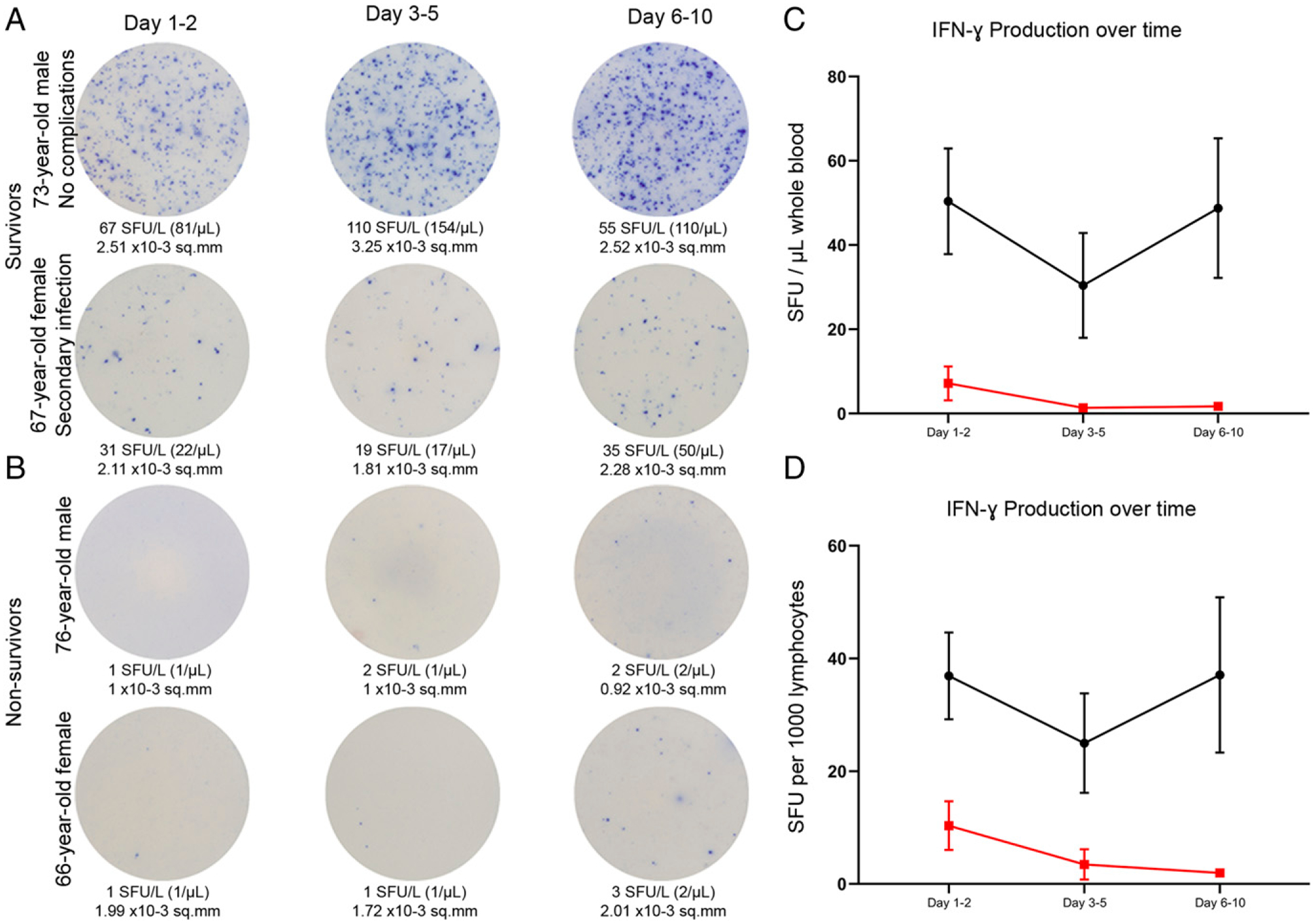
Adaptive immune function timeline for the first 7–10 d following diagnosis of sepsis. Representative images show the change in whole blood production of CD3/CD28-stimulated IFN-γ over time in patients with sepsis who survived (A) and those who did not survive (B). (C) IFN-γ production as SFU/μl comparing sepsis survivors (black line) to sepsis nonsurvivors (red line) on days 1–2, 3–5, and 6–10. (D) IFN-γ production as SFU per 1000 lymphocytes. Dots represent mean value, and bars represent ±SEM.
The early and profound suppression in innate immunity is sustained throughout sepsis in nonsurvivors
Serial time course examination of septic patient TNF-α production via ELISpot is presented for patients who survived sepsis versus patients who died. Representative color micrographs are presented for two sepsis survivors and two nonsurvivors for three consecutive time points (Fig. 8A, 8B). The number of TNF-α–producing myeloid cells was increased in septic survivors compared with septic patients who expired and was sustained throughout the study period of up to 10 d following diagnosis of sepsis. Sepsis nonsurvivors demonstrated sustained innate immune dysfunction with a sustained low number of LPS-stimulated cells producing TNF-α (Fig. 8C, 8D).
FIGURE 8.
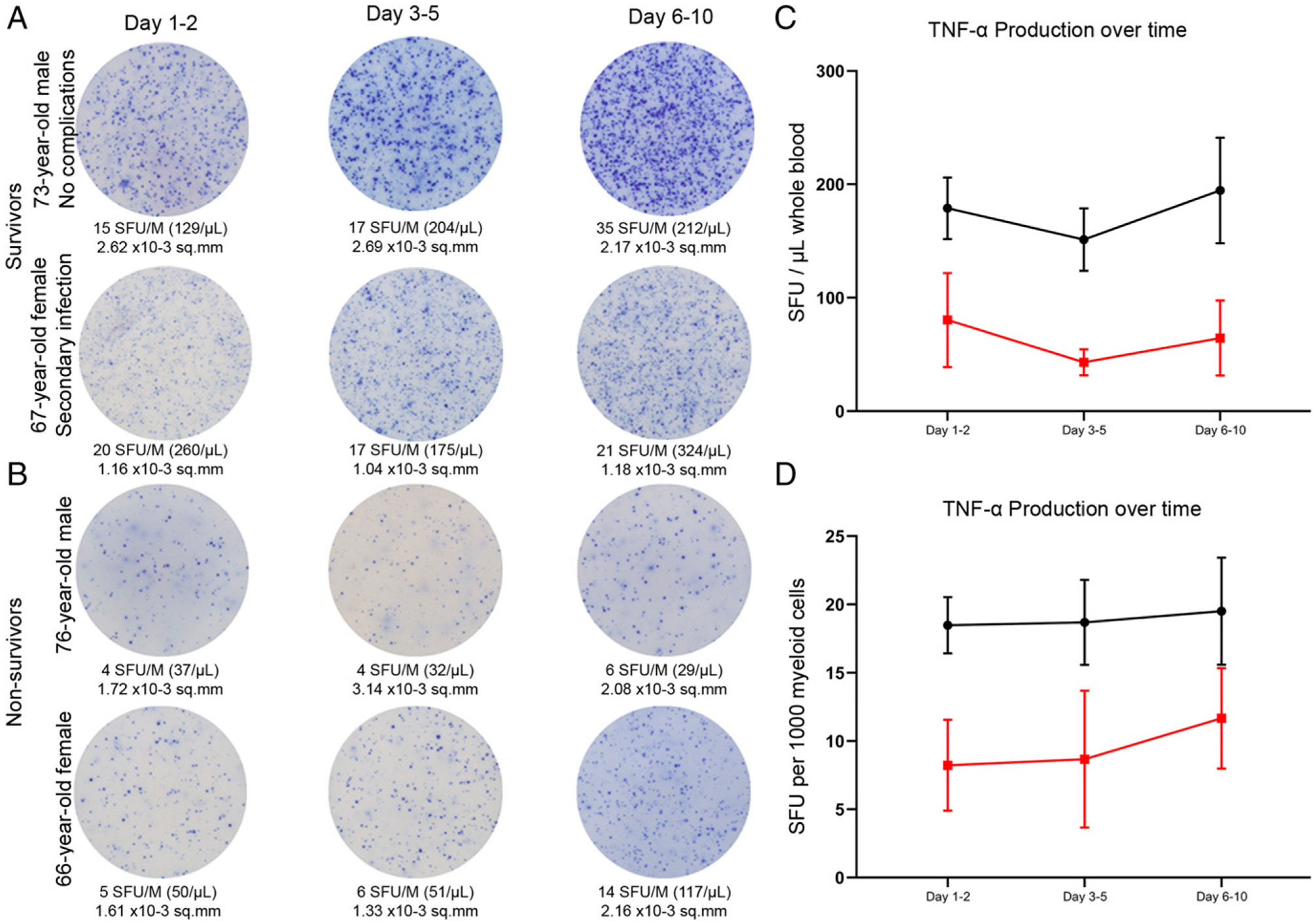
Innate immune function timeline for the first 7–10 d following diagnosis of sepsis. Images show the change in whole blood production of LPS-stimulated TNF-α over time in patients with sepsis who survived (A) and those who did not survive (B). (C) TNF-α production as SFU/μl comparing sepsis survivors (black line) to sepsis nonsurvivors (red line) on days 1–2, 3–5, and 6–10. (D) TNF-α production as SFU per 1000 myeloid cells. Dots represent mean value, and bars represent ±SEM.
IL-7 restores adaptive but not innate immune response
Another key goal of this study was to investigate if the ELISpot assay could be used to determine the potential ex vivo efficacy of immune-adjuvant drug therapies using whole blood ELISpot. In this manner, the potential value of specific immune therapies on cell preparations from individual septic patients could be tested. As proof of principle, we evaluated the ability of IL-7, a potent T cell activator that has undergone phase I/II trials in sepsis, to improve the T cell response in patient samples. Previous work by our group has demonstrated a significant increase in septic patient IFN-γ production using IL-7, and these findings are compared in this study with whole blood ELISpot assay (28). This is a critical comparison as whole blood ELISpot has the potential to be used in ex vivo determination of immunotherapy candidacy in several disease states. Overall, septic patients had a 172% ± 77 increase in the number of spots when stimulated with IL-7 (p < 0.005) (Fig. 9A). This effect was greater in the severely immunosuppressed cohort of patients who did not survive. Survivors had a 102% ± 32 increase in spot numbers with IL-7, whereas the nonsurvivors had a 312% ± 240 increase. A similar effect of IL-7 to restore T cell IFN-γ production in septic patients was observed in PBMCs (p < 0.0001) (Supplemental Fig. 3A). In contrast, for whole blood ELISpot, IL-7 does not increase the number of TNF-α–producing cells (Fig. 9C). Surprisingly, for PBMC ELISpot, IL-7 did increase TNF-α production in 13/19 septic patients, resulting in an overall 97% increase in TNF-α SFUs (p < 0.01) (Supplemental Fig. 3B). Similarly, IL-7 significantly increased the SFUs in both healthy subjects and CINS patients in both whole blood and PBMC ELISpot assays (Supplemental Fig. 3C, 3E). For TNF-α, there was no change in SFUs for CINS in the whole blood assay, but there was a significant increase using the PBMC assay. For healthy subjects, there was a significant decrease in TNF-α SFUs for whole blood (p < 0.01) and no change in PBMCs (Supplemental Fig. 3D, 3F).
FIGURE 9.
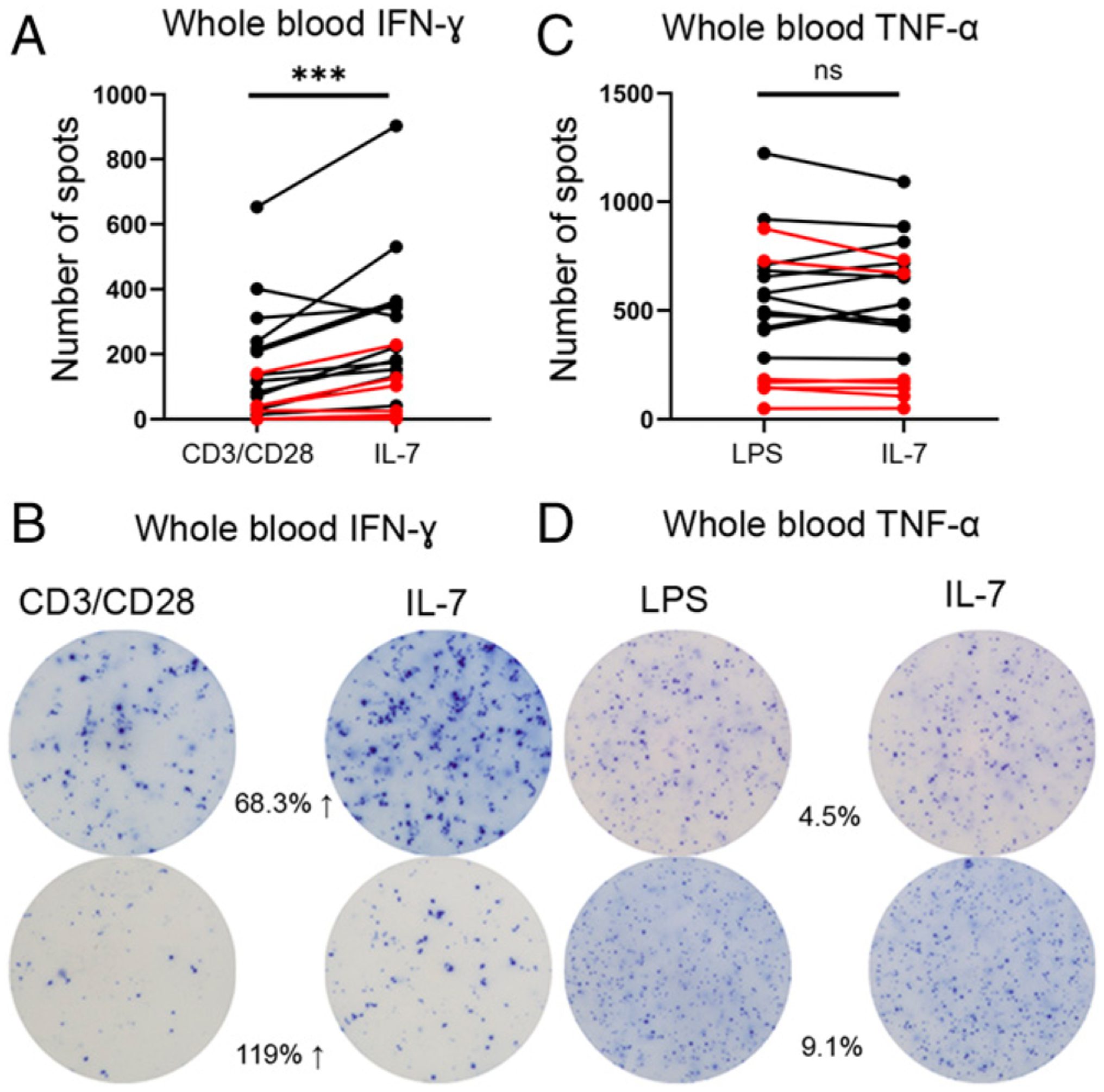
IFN-γ and TNF-α production in response to ex vivo IL-7 administration based on whole blood ELISpot assay in patients with sepsis. (A) Change in number of IFN-γ spots (SFU) in CD3/CD28 versus CD3/CD28 + IL-7–stimulated cultures (n = 19). (B) Representative ELISpot images showing the change in the number of IFN-γ spots in CD3/CD28 versus CD3/CD28 + IL-7–stimulated cultures. (C) Change in the number of TNF-α spots (SFU) in LPS versus LPS + IL-7–stimulated cultures (n = 19). (D) Representative ELISpot images showing the change in number of TNF-α spots in LPS versus LPS + IL-7–stimulated cultures. (A and C) The number of spots are the total number of spots per well and are not corrected for volume or cell number. Lines in red depict mortality. Statistical analysis performed using paired Wilcoxon ranked sum test. ***p < 0.001.
Circulating pro- and anti-inflammatory cytokines in sepsis
Analysis of prototypical pro- and anti-inflammatory cytokines were quantitated using a human cytokine Luminex panel in patients with sepsis (n = 15) during their hospital course and compared with values for healthy controls (n = 9) and CINS (n = 4) patients (Table II) Note that the cytokine data for the healthy control and CINS cohorts, but not the septic patients, have been previously reported (15) and are used in this study to compare with septic patient cytokine response. There was no significant difference in any cytokine concentration between sepsis survivors and non-survivors. Circulating plasma TNF-α levels were significantly higher in septic (p < 0.0001) and CINS (p < 0.005) patients compared with healthy controls. Elevated IL-6 levels were not associated with any IFN-γ or TNF-α phenotype. Interestingly, plasma TNF-α levels that were above 5 pg/ml were associated with higher unstimulated ex vivo TNF-α production. Surprisingly, IL-8 is inversely related with ex vivo TNF-α production, with the higher levels (>80 pg/ml) being associated with low TNF-α production. Circulating IL-10 levels ranged from 1.5 to 524 pg/ml in septic patients. Although the individual patient with the highest (524 pg/ml) IL-10 level died, the remainder of patients with IL-10 levels >15 pg/ml were associated with a favorable outcome and higher ex vivo IFN-γ production. Of the eight patients with elevated IL-6 levels (>70 pg/ml), there were two mortalities.
Table II.
Serum cytokine profile over time in patients with sepsis
| Cytokine | Septic 1–2 d (n = 15), Mean ± SEM (range) | Septic 3–6 d (n = 13), Mean ± SEM (range) | Septic 7–11 d (n = 7), Mean ± SEM (range) | Healthy Control (n = 9). Mean ± SEM (range) | CINSa (n = 4), Mean ± SEM (range) |
|---|---|---|---|---|---|
| IL-1β | 1 ± 0.32 (<0.08–3.4) | 1.5 ± 0.7 (0.1–9.2) | 2.2 ± 1.6 (0.08–10.7) | 1.3 ± 0.8 (1–2) | 0.8 ± 0.3 (0.6–1) (n = 2) |
| IL-6 | 503.4 ± 96.4 (7.3–4849) | 536.8 ± 77.9 (3.9–4849) | 362.7 ± 328.3 (4.7–2179.1) | 2 ± 0.3 (1–3.7) | 137.1 ± 110.3 (21.7–422.3) |
| IL-7 | 7 ± 1.2 (2.5–18.5) | 8.7 ± 1.5 (2.8–20.9) | 7.3 ± 1.8 (1.3–11.6) | 35.8 ± 7.8 (5.1–75.6) | 5.5 ± 0.6 (4.1–6.3) |
| IL-8 | 83.5 ± 27.4 (3.9–377.7) | 92.7 ± 38.7 (1.9–455.3) | 71.9 ± 27 (3–198.7) | 15 ± 1 (11.5–17) | 73 ± 25.8 (27.4–113.8) |
| IL-10 | 62.8 ± 36.7 (1.5–524.6) | 57 ± 30.3 (1.5–359.72) | 57.2 ± 51.9 (1.3–345.2) | 108.1 ± 78.7 (1–667.3) | 25.5 ± 6 (12.5–29.5) |
| IL-12 | 92.7 ± 36.1 (6.9–548.6) | 82.1 ± 31.3 (7.2–417.5) | 107.4 ± 63.2 (24.2–450.4) | 50.4 ± 6.8 (31.4–77) | 44.4 ± 15.3 (6.1–65.3) |
| MCP-1 | 502.4 ± 103.8 (71.7–1385.4) | 596.5 ± 190.8 (127.1–2362.3) | 438.7 ± 124.7 (112.8–1000) | 486.1 ± 50.2 (182.1–687.6) | 414.8 ± 83 (258.6–595.7) |
| IL-1RA | 383.8 ± 205.3 (50.2–2831.9) | 313.8 ± 234.9 (13–3034.2) | 1082.1 ± 1077.7 (26.3–7067.5) | 32 ± 4.3 (18.8–49.9) | 108.5 ± 38.5 (40.5–200.4) |
| IFN-γ | 3.5 ± 2.6 (0.4–38.3) | 1.6 ± 0.6 (0.4–8.5) | 0.7 ± 0.3 (0.4–2.2) | 4.8 ± 0.6 (1.5–7) | 9 (n = 1) |
| TNF-α | 4.3 ± 0.7 (1.9–12.4) | 5.2 ± 0.7 (2.5–10.1) | 3.9 ± 1.5 (1.1–11.6) | 1.34 ± 0.2 (0.7–1.88) | 6.6 ± 3.8 (2.8–16.5) |
| FGF-basic | 15.6 ± 8.8 (2.5–134.5) | 18.2 ± 11.6 (4.1–151.4) | 35.5 ± 31.3 (4.5–209.1) | 23.3 ± 9.1 (0–78.5) | 6.5 ± 2.4 (2.3–12.3) |
| G-CSF/CSF-3 | 224.5 ± 193.2 (6.5–2821.2) | 29.4 ± 9.2 (7.5–103.2) | 19.1 ± 7.3 (6.1–54.5) | 32.5 ± 7.6 (8–80) | 8 ± 2.1 (2.7–10.4) |
| IL-13 | 5.1 ± 1.3 (0.3–19.7) | 5.8 ± 1.7 (1.7–24.4) | 3.7 ± 0.9 (0.9–5.5) | 3.2 ± 0.9 (0.8–9.2) | 2.7 ± 0.5 (1.9–3.7) |
| RANTES | 1310.2 ± 303.3 (457.7–2058.7) | 886.1 ± 158.2 (282.4–2058.7) | 1034.7 ± 312.4 (350.2–2362.4) | 697.9 ± 64 (360.7–888.1) | N/A |
| EOTAXIN | 26.5 ± 4.9 (9.2–69.5) | 28.2 ± 5.5 (4.2–67) | 22.9 ± 2.9 (10.4–30.2) | 92.4 ± 28.1 (8.1–255.7) | 57.5 ± 26.4 (15.8–101.4) |
| IL-17A | 3.1 ± 0.6 (0.1–6.4) | 3.1 ± 0.5 (0.1–5.5) | 2.9 ± 1.4 (0.1–9.4) | 5.2 ± 1.9 (0.7–18.9) | 3.2 ± 1 (1.3–5.4) |
| MIP-1a | 14.9 ± 10.3 (1.4–152.7) | 19.5 ± 16 (1.4–203.2) | 36.9 ± 38.3 (1.4–249.9) | 5.1 ± 2.6 (0.3–18.5) | 11.97 (n = 1) |
| GM-CSF | 2.9 ± 1.3 (0.4–18.2) | 3.2 ± 1.3 (0.8–14.7) | 4.5 ± 1.9 (0.5–11.7) | 9.6 ± 8.4 (0.3–72.6) | 1.6 ± 1 (0.9–4.3) |
| MIP-1b | 137.5 ± 58.2 (19.7–871) | 158.2 ± 79.8 (24.8–1019.9) | 237 ± 195.7 (20.6–1323.3) | 88.4 ± 11 (65.1–152.7) | 70.7 ± 18.4 (33.2–109.4) |
| IL-15 | 228.5 ± 169.1 (1.8–2490.4) | 261.5 ± 204.6 (1.8–2602.7) | 601 ± 592.7 (9.5–3892.4) | 42 ± 9.7 (13.3–88.3) | 122.8 ± 66.9 (12.7–274.5) |
| EGF | 89.7 ± 35.7 (3.2–546.1) | 102.1 ± 50.4 (3.2–660.4) | 193.1 ± 155.9 (3.2–1056.4) | 69.1 ± 10.7 (41.1–128.9) | 95.2 ± 27.2 (57.9–159.5) |
| IL-5 | 4 ± 1.5 (0.4–22.7) | 4.2 ± 1.3 (1.3–17.1) | 7.7 ± 4.5 (0.8–31.3) | 2.8 ± 0.6 (1.1–7) | 2.2 ± 0.4 (1.3–3.1) |
| HGF | 7710.6 ± 4960.9 (27–54,374.5) | 2265.6 ± 824.6 (198.6–8177.8) | 3435 ± 2641.6 (59.6–17.727.6) | 45 ± 9.4 (22.1–89.2) | 556.9 ± 338.8 (49.4–1325.8) |
| VEGF | 2.9 ± 0.7 (0.03–11.2) | 1.8 ± 0.7 (0.03–9.23) | 3.1 ± 1.4 (0.8–9.9) | 2.1 ± 0.7 (0.2–5.7) | 3.6 ± 1.7 (0.7–7.2) |
| IL-1a | 3.3 ± 0.9 (0.7–12.7) | 4.3 ± 1.7 (0.3–22.3) | 7 ± 4.3 (0.5–30) | 3.2 ± 1.4 (0.5–12.6) | 9.6 ± 7.7 (1.3–29.5) |
| IL-17F | 170.4 ± 43.7 (53.8–710.9) | 173.3 ± 50 (71.2–685.8) | 278.6 ± 94.2 (58.7–641.1) | 53.5 ± 6.8 (37.6–101.6) | 111.3 ± 51.6 (42.1–242.5) |
| IFN-α | 31.4 ± 18.7 (3.5–281.1) | 36.7 ± 24.9 (2.5–322.1) | 69.3 ± 64.2 (6.2–425.8) | 26.7 ± 11.9 (7.2–113.6) | 13.6 ± 5.5 (6.4–27.7) |
| IL-9 | 7.4 ± 5.4 (0.6–79.7) | 7.6 ± 5.8 (0.09–74.3) | 11.7 ± 10 (0.4–66.6) | 1.3 ± 0.3 (0.2–3.5) | 3 ± 0.5 (2.1–3.7) |
| IL-3 | 33.7 ± 18.4 (0.3–263.9) | 34 ± 17.3 (1–183.1) | 38.6 ± 26.2 (0.5–180) | 26.4 ± 7.9 (2.8–73) | 37.7 ± 23.9 (12.3–99.7) |
| IL-2 | 62.6 ± 48.6 (3.3–714.3) | 91.5 ± 77.8 (3.9–984.1) | 227.1 ± 231.9 (3–1515.2) | 14.8 ± 3.6 (5.6–33.3) | 23.5 ± 16.5 (5–65.4) |
| IP-10 | 98.1 ± 75.1 (4.1–1110) | 33.1 ± 12.3 (7.4–162.6) | 16.9 ± 3 (6.5–25.1) | 20.1 ± 3.4 (5.7–33.9) | 24.1 ± 10.8 (5.7–48.6) |
| IL-2R | 197.5 ± 60.1 (19.7–829.2) | 147.7 ± 43.2 (21.1–462.1) | 178.6 ± 49.5 (80.8–429.9) | 24.4 ± 6.2 (2.5–51.7) | 275.9 ± 236.6 (39.5–889.5) |
| IL-22 | 74.6 ± 23.7 (14.8–350.2) | 76.4 ± 24.1 (20.3–321.7) | 71.5 ± 23 (17.3–167) | 52.2 ± 14 (20.4–136.1) | 38.8 ± 18.4 (14.5–84) |
| MIG | 65.3 ± 18.6 (7.8–231.4) | 63.6 ± 12.4 (7.8–141.2) | 69.4 ± 24.9 (8.8–147.5) | 15.5 ± 2.9 (5.4–27.1) | 24.3 ± 3.3 (10–45.5) |
| IL-4 | 4.2 ± 0.7 (0.8–9) | 3.9 ± 0.6 (0.8–7.5) | 2.7 ± 0.8 (0.5–5.7) | 3.5 ± 0.6 (1.5–6.6) | 3.3 ± 2.9 (0.3–8) (n = 3) |
Discussion
Sepsis remains a major cause of death and has been remarkably resistant to any new therapies (1–5). Undoubtedly, a key problem in developing immunomodulatory therapies for sepsis is the difficulty in evaluating the immunologic status of the individual patient. The functional state of patients’ immune systems during sepsis is complex (6–9). Currently, there is an enormous effort underway to develop methods to immune phenotype patients with sepsis. Knowledge of the status of patients’ immunity could guide the administration of effective new immune-based therapies that can either dampen damaging cytokine-mediated inflammation or restore immune function in patients who are profoundly immune suppressed.
The present study demonstrates a major advance in the ability to immune phenotype patients. The whole blood ELISpot assay is an effective method to quantify the functional state of patient adaptive and innate cellular function with excellent dynamic range. Circulating peripheral blood, which combines RBCs, WBCs, platelets, cytokines, and chemokines, is considered in combination to be a vital functional organ. In this sense, it is highly informative to measure ex vivo cytokine production as a response to external stimuli in patient samples. Additionally, circulating chemokines and cytokines in the blood plasma fraction from patients with sepsis has potent immunologic effects on the function of the circulating WBCs (32), and removal through PBMC fractionation can dramatically change cellular response to stimuli or therapeutic molecules. Thus, studies testing diluted whole blood are more likely to reflect the in vivo state. Reporting this response per volume of blood is fundamentally comparable between individual patients and offers more practical applicability than absolute cell counts. Finally, use of diluted whole blood has significant technical advantages of reduced preparation time and effort as well as avoiding potential biologic changes to fragile cells from patients with sepsis because of Ficoll gradient separation or any other processing and handling of the sample.
Findings from the current study demonstrate variability and heterogeneity in the innate and adaptive immune response to sepsis. Many septic patients had increased immune activation, indicated by increased IFN-γ and TNF-α production compared with healthy controls, whereas other septic patients were near completely incapable of cell cytokine production (Figs. 2, 3). Importantly, the whole blood ELISpot results provide insight into a major driving force for mortality in sepsis. Septic patients who died had early, severe, and sustained suppression of adaptive immunity, as measured by ex vivo IFN-γ production. A significant percentage of the nonsurvivors had marked suppression of innate immunity as well. There was a reduction not only in the number of immune effector cells producing key cytokines but also in the amount of cytokines produced by each cell as measured by spot intensity (Figs. 2, 3). These results are consistent with the contention that immunosuppression is a key pathophysiologic process in sepsis, and therapies that may restore immunity in vivo could be beneficial on patient outcomes (2–4).
Findings in our study using the whole blood ELISpot also revealed a subgroup of septic patients who had an increase in IFN-γ and TNF-α production compared with healthy control subjects (Fig. 2). This subset of septic patients likely consists of patients who are either mounting an appropriate robust immunologic response to the invading pathogens or, if excessively elevated, a damaging exaggerated proinflammatory response. Findings of an elevated adaptive ex vivo IFN-γ response could be perceived as surprising given the numerous previous observations that sepsis induces significant impairment of adaptive immunity. We speculate that there are several reasons for this observation. First, it is likely that many of the lymphocytes from septic and CINS patients had been primed (because of the infection or injury) prior to being plated in the ELISpot wells. Thus, these lymphocytes were ready to produce IFN-γ upon costimulation during the 18–22-h incubation (33). Conversely, the lymphocytes from healthy control subjects were not primed prior to being plated in the ELISpot wells and therefore did not respond as rapidly to stimulation. Second, it is likely, given the differential propensity of lymphocyte subsets to undergo sepsis-induced apoptosis, that the types of circulating lymphocytes (i.e., naive, effector memory, central memory, etc.) are different in patients with sepsis versus healthy control subjects (34). Naive lymphocytes are slower than effector memory cells to respond to stimulation as they first require differentiation before producing cytokines (35). Finally, it is also important to note that patients with sepsis-induced immunosuppression have significant depletion of CD4+ and CD8+ T cells in spleen, gastrointestinal lymphoid-associated tissues, and secondary lymphoid organs, which is a major cause of sepsis-induced impaired immunity (34).
Another key finding in our study is the high level of spontaneous TNF-α production in the unstimulated patient samples (Fig. 1D). This result could be useful in identifying septic patients who exhibit a proinflammatory phenotype. Differential TNF-α responses to LPS could also serve as an important indicator of immune system exhaustion (36). Our team is currently performing ELISpot assays on additional patients to define the level of boundaries of an appropriate versus an excessive proinflammatory response and to build prognostic models.
The role played by neutrophils in the global immune response and their specific response in the setting of sepsis further highlights the importance of performing the ELISpot assay using whole blood. Neutrophils make large amounts of both pro- and anti-inflammatory cytokines, including TNF-α and IL-10 (37). Results from healthy control subjects showed that ~20% of the TNF-α produced in the diluted whole blood ELISpot assay is derived from neutrophils. A significant amount of TNF-α that is present in blood from septic patients is likely to have been derived from neutrophils because of the neutrophilia that occurs in sepsis. Neutrophils also express multiple negative costimulatory molecules including programmed cell death 1 (PD-1) and PD-1 ligand (PD-L1) that can suppress T cell function (38). Thus, the results from ELISpot studies using diluted whole blood are much more likely to reflect the actual state of the patient’s immune status compared with neutrophil-depleted PBMCs.
Another significant benefit of the ELISpot assay is that not only can it identify patients with sepsis who are at high risk of dying because of immunosuppression, it can also reveal potential immune adjuvant therapies that might effectively reverse the immunosuppression. Importantly, the ELISpot assay can independently assess the functional status of the two major arms of immunity (i.e., adaptive and innate immunity). This ability to discriminate between the effects of sepsis on the two key components of immunity is particularly important given the availability of new immune adjuvants that selectively target key immune effector cell types. There are several immune adjuvants that are undergoing clinical trials in sepsis (e.g., anti–PD-1, anti–PD-L1, GM-CSF, and IL-7) (11–13). In the current study, IL-7 added ex vivo to septic patient samples effectively restored T cell IFN-γ production in the majority of septic patients. By restoring host immunity, IL-7 could potentially accelerate eradication of the primary infection and decrease secondary hospital-acquired infections. Previously, our group reported that anti–PD-1, anti–PD-L1, and OX-40 agonistic Abs are also effective in restoring T cell IFN-γ production in a variable percentage of septic patients using a PBMC ELISpot assay (28). Thus, the ELISpot assay could be used to identify the optimal immune therapy for use in individual septic patients. This method undoubtedly holds translatable potential to many other fields within critical care, oncology, and autoimmune disease.
Although the ELISpot assay can quantitate numerous cytokines, we elected to examine IFN-γ in the current study for several reasons. T cell exhaustion is a key pathophysiologic mechanism of sepsis-induced immunosuppression and decreased T cell production of IFN-γ is the hallmark of exhausted T cells (39, 40). Furthermore, IFN-γ plays a critical role in host defense against invading pathogens by activating monocytes and macrophages to eliminate invading microbes. Decreased IFN-γ production correlates with worsened survival in animal models of sepsis (41), and administration of IFN-γ showed clinically beneficial effects on infectious outcomes in patients with sepsis and trauma (26, 42). Because of the overabundance of IFN-γ receptors that are present on virtually all nucleated cells, circulating levels of IFN-γ are typically either minimally elevated or not above baseline detection in patients with sepsis. Thus, the ELISpot assay for IFN-γ production is the ideal method to evaluate adaptive immune function and assess immune-adjuvant therapies that impact IFN-γ production because of its exquisite sensitivity and the inability to follow IFN-γ blood levels. Although IFN-γ plays a central role in host antimicrobial defenses, it will be important to define the impact of sepsis on T cell–stimulated production of other cytokines (e.g., IL-2 and TNF-α) that are also critical for a coordinated response to invading pathogens. Similarly, ELISpot assay of monocyte production of additional cytokines such as IL-6 and IL-12 will provide important mechanistic insights into sepsis-induced immunosuppression.
In this regard, the LPS-stimulated whole blood TNF-α release assay developed by Hall et al. (43) has been useful in identifying pediatric patients with sepsis or influenza who have impaired immunity and are more likely to have an increased prevalence of secondary infections and death (44). Although this LPS-stimulated whole blood method has been useful in pediatric patients, it has not yet been shown to have similar utility in adult patients with sepsis and lacks a readout of the adaptive immune response. Additionally, the ELISpot assay has the ability to isolate more discrete immune-suppressive phenotypes by determining the number of cells producing the desired cytokine and can differentiate between low and high cytokine-producing cells using the total area and intensity of each spot (45).
There are a number of limitations to the current study. A key limitation is the relatively small numbers of patients included in this trial. Sepsis is a heterogeneous disorder, and it will be important to perform whole blood ELISpot on a larger cohort of patients to prospectively predict outcome and potential responsiveness to therapeutic interventions. These studies currently are underway. The present study related the ELISpot immunologic findings to the key end point of hospital survival. Future studies are needed to correlate the ELISpot assays to additional clinical metrics that reflect the integrity of the patients’ immunity. IFN-γ and TNF-α whole blood ELISpot results should be correlated, for example, with the prevalence of secondary hospital-acquired infections, duration of sepsis, and hospital readmissions. Finally, it will be important to evaluate whole blood ELISpot data in patients with sepsis because of a variety of diverse bacterial and fungal pathogens that may have unique effects on host immunity.
In conclusion, there is significant heterogeneity in the immune response in patients with sepsis. Whereas some septic patients have increased IFN-γ and TNF-α production compared with healthy volunteers, many septic patients have severe suppression of immunity. Septic patients who died had early, severe, and sustained immune suppression, as indicated both by a decrease in the number of cytokine-producing immune effector cells and a decrease in the amount of cytokine produced on a per cell basis. Performing the ELISpot assay in patient-diluted whole blood is feasible, easy to perform, and likely to reflect the actual clinical state of the patient’s immunity. The whole blood ELISpot assay offers a significant advance in the ability to immune phenotype patients with sepsis and to guide therapy of new potential immune adjuvants that are currently being tested in the treatment of sepsis. For example, administration of corticosteroids in patients with septic shock might be guided by ELISpot analysis of T cell function. Patients in septic shock who have severe depression of T cell function on ELISpot assay might not be good candidates for corticosteroids, which could further exacerbate the T cell depression. The whole blood ELISpot assay may have broad clinical applicability in guiding immune therapies in many disorders, including patients with autoimmunity and cancer and patients who have undergone organ transplantation.
Supplementary Material
Acknowledgments
We thank our research coordinators Jane Blood and Catherine Dalton for coordinating the study, enrolling patients, and collecting samples. Additionally, we thank Zoltan Megyesi from Cellular Technology ImmunoSpot, who provided consultative assistance in our ability to use spot intensity from the ImmunoSpot software output as a measure in this study.
This work was supported by grants from the National Institutes of Health/National Institute of General Medical Sciences (GM126928-01 and K23GM129660-03).
Abbreviations used in this article:
- CINS
critically ill nonseptic
- FDR
false discovery rate
- PD-1
programmed cell death 1
- PD-L1
PD-1 ligand
- SFU
spot-forming unit
- SOFA
sequential organ failure assessment
- TWI
total well intensity
Footnotes
The online version of this article contains supplemental material.
Disclosures
Patent pending for intellectual property for whole blood ELISpot immune phenotyping for R.S.H., M.B.M., C.C.C., I.R.T., K.E.R. The other authors have no financial conflicts of interest.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. 2016. The Third International Consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venet F, and Monneret G. 2018. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol 14: 121–137. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, and van der Poll T. 2013. Severe sepsis and septic shock. N. Engl. J. Med 369: 2063. [DOI] [PubMed] [Google Scholar]
- 4.Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, Efron PA, Bihorac A, Segal M, Moore FA, and Moldawer LL. 2018. Chronic critical illness and the persistent inflammation, immunosuppression, and catabolism syndrome. Front. Immunol 9: 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin MD, Badovinac VP, and Griffith TS. 2020. CD4 T cell responses and the sepsis-induced immunoparalysis state. Front. Immunol 11: 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danahy DB, Kurup SP, Winborn CS, Jensen IJ, Harty JT, Griffith TS, and Badovinac VP. 2019. Sepsis-induced state of immunoparalysis is defined by diminished CD8 T cell-mediated antitumor immunity. J. Immunol 203: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wakeley ME, Gray CC, Monaghan SF, Heffernan DS, and Ayala A. 2020. Check point inhibitors and their role in immunosuppression in sepsis. Crit. Care Clin 36: 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotchkiss RS, and Sherwood ER. 2015. Immunology. Getting sepsis therapy right. Science 347: 1201–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotchkiss RS, Monneret G, and Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hutchins NA, Unsinger J, Hotchkiss RS, and Ayala A. 2014. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol. Med 20: 224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, Monneret G, Rimmelé T, Blood T, Morre M, et al. 2018. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight 3: e98960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, Martin GS, Coopersmith CM, Brakenridge S, Mayr FB, et al. 2019. Immune checkpoint inhibition in sepsis: a phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell deathligand 1 antibody (BMS-936559). Crit. Care Med 47: 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, Bartz RR, Brakenridge SC, Delano MJ, Park PK, et al. 2019. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med 45: 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Remy KE, Brakenridge SC, Francois B, Daix T, Deutschman CS, Monneret G, Jeannet R, Laterre PF, Hotchkiss RS, and Moldawer LL. 2020. Immunotherapies for COVID-19: lessons learned from sepsis. Lancet Respir. Med 8: 946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Remy KE, Mazer M, Striker DA, Ellebedy AH, Walton AH, Unsinger J, Blood TM, Mudd PA, Yi DJ, Mannion DA, et al. 2020. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight 5: e140329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierrakos C, Velissaris D, Bisdorff M, Marshall JC, and Vincent JL. 2020. Biomarkers of sepsis: time for a reappraisal. Crit. Care 24: 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Czerkinsky CC, Nilsson LA, Nygren H, Ouchterlony O, and Tarkowski A. 1983. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods 65: 109–121. [DOI] [PubMed] [Google Scholar]
- 18.Sedgwick JD, and Holt PG. 1983. A solid-phase immunoenzymatic technique for the enumeration of specific antibody-secreting cells. J. Immunol. Methods 57: 301–309. [DOI] [PubMed] [Google Scholar]
- 19.Helms T, Boehm BO, Asaad RJ, Trezza RP, Lehmann PV, and Tary-Lehmann M. 2000. Direct visualization of cytokine-producing recall antigen-specific CD4 memory T cells in healthy individuals and HIV patients. J. Immunol 164: 3723–3732. [DOI] [PubMed] [Google Scholar]
- 20.Augustine JJ, and Hricik DE. 2012. T-cell immune monitoring by the ELISPOT assay for interferon gamma. Clin. Chim. Acta 413: 1359–1363. [DOI] [PubMed] [Google Scholar]
- 21.Möbs C, and Schmidt T. 2016. Research techniques made simple: monitoring of T-cell subsets using the ELISPOT assay. J. Invest. Dermatol 136: e55–e59. [DOI] [PubMed] [Google Scholar]
- 22.Cox JH, Ferrari G, and Janetzki S. 2006. Measurement of cytokine release at the single cell level using the ELISPOT assay. Methods 38: 274–282. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Chen W, Herberman RB, Plotnikoff NP, Youkilis G, Griffin N, Wang E, Lu C, and Shan F. 2014. Immunotherapy of cancer via mediation of cytotoxic T lymphocytes by methionine enkephalin (MENK). Cancer Lett 344: 212–222. [DOI] [PubMed] [Google Scholar]
- 24.Patil NK, Bohannon JK, and Sherwood ER. 2016. Immunotherapy: a promising approach to reverse sepsis-induced immunosuppression. Pharmacol. Res 111: 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotchkiss RS, and Moldawer LL. 2014. Parallels between cancer and infectious disease. N. Engl. J. Med 371: 380–383. [DOI] [PubMed] [Google Scholar]
- 26.Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, and Kox W. 1997. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat. Med 3: 678–681. [DOI] [PubMed] [Google Scholar]
- 27.Munoz C, Carlet J, Fitting C, Misset B, Blériot JP, and Cavaillon JM. 1991. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Invest 88: 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thampy LK, Remy KE, Walton AH, Hong Z, Liu K, Liu R, Yi V, Burnham CD, and Hotchkiss RS. 2018. Restoration of T Cell function in multi-drug resistant bacterial sepsis after interleukin-7, anti-PD-L1, and OX-40 administration. PLoS One 13: e0199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazer M, Unsinger J, Drewry A, Walton A, Osborne D, Blood T, Hotchkiss R, and Remy KE. 2019. IL-10 has differential effects on the innate and adaptive immune systems of septic patients. J. Immunol 203: 2088–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrando-Martinez S, Huang K, Bennett AS, Sterba P, Yu L, Suzich JA, Janssen HLA, and Robbins SH. 2019. HBeAg seroconversion is associated with a more effective PD-L1 blockade during chronic hepatitis B infection. JHEP Rep 1: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benjamini YKA, and Yekutieli D. 2006. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93: 491–507. [Google Scholar]
- 32.Gentile LF, Cuenca AG, Vanzant EL, Efron PA, McKinley B, Moore F, and Moldawer LL. 2013. Is there value in plasma cytokine measurements in patients with severe trauma and sepsis? Methods 61: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlingmann TR, Shive CL, Targoni OS, Tary-Lehmann M, and Lehmann PV. 2009. Increased per cell IFN-gamma productivity indicates recent in vivo activation of T cells. Cell. Immunol 258: 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, and Karl IE. 1999. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med 27: 1230–1251. [DOI] [PubMed] [Google Scholar]
- 35.Schroder K, Hertzog PJ, Ravasi T, and Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75: 163–189. [DOI] [PubMed] [Google Scholar]
- 36.Heagy W, Hansen C, Nieman K, Cohen M, Richardson C, Rodriguez JL, and West MA. 2000. Impaired ex vivo lipopolysaccharide-stimulated whole blood tumor necrosis factor production may identify “septic” intensive care unit patients. Shock 14: 271–276, discussion 276–277. [DOI] [PubMed] [Google Scholar]
- 37.Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, and Pillay J. 2016. The role of neutrophils in immune dysfunction during severe inflammation. Crit. Care 20: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, and Hotchkiss RS. 2016. Frontline science: defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J. Leukoc. Biol 100: 1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wherry EJ 2011. T cell exhaustion. Nat. Immunol 12: 492–499. [DOI] [PubMed] [Google Scholar]
- 40.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD II, Kreisel D, Krupnick AS, et al. 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306: 2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hotchkiss RS, Chang KC, Grayson MH, Tinsley KW, Dunne BS, Davis CG, Osborne DF, and Karl IE. 2003. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc. Natl. Acad. Sci. USA 100: 6724–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dries DJ, Jurkovich GJ, Maier RV, Clemmer TP, Struve SN, Weigelt JA, Stanford GG, Herr DL, Champion HR, Lewis FR, et al. 1994. Effect of interferon gamma on infection-related death in patients with severe injuries. A randomized, double-blind, placebo-controlled trial. Arch. Surg 129: 1031–1041, discussion 1042. [DOI] [PubMed] [Google Scholar]
- 43.Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, and Carcillo JA. 2011. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med 37: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muszynski JA, Nofziger R, Greathouse K, Nateri J, Hanson-Huber L, Steele L, Nicol K, Groner JI, Besner GE, Raffel C, et al. 2014. Innate immune function predicts the development of nosocomial infection in critically injured children. Shock 42: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beckmann N, Huber F, Hanschen M, St Pierre Schneider B, Nomellini V, and Caldwell CC. 2020. Scald injury-induced T cell dysfunction can Be mitigated by Gr1 + cell depletion and blockage of CD47/CD172a signaling. Front. Immunol 11: 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.