

Abstract
Gene replacement for laminin-α2-deficient congenital muscular dystrophy 1A (MDC1A) is currently not possible using a single adeno-associated virus (AAV) vector due to the large size of the LAMA2 gene. LAMA2 encodes laminin-α2, a subunit of the trimeric laminin-211 extracellular matrix (ECM) protein that is the predominant laminin expressed in skeletal muscle. LAMA2 expression stabilizes skeletal muscle, in part by binding membrane receptors via its five globular (G) domains. We created a small, AAV-deliverable, micro-laminin gene therapy that expresses these G1–5 domains, LAMA2(G1–5), to test their therapeutic efficacy in the dyW mouse model for MDC1A. We also fused the heparin-binding (HB) domain from HB epidermal growth factor-like growth factor (HB-EGF) to LAMA2(G1–5) to test whether this would increase muscle ECM expression. dyW mice treated intravenously with rAAV9.CMV.HB-LAMA2(G1–5) showed increased muscle ECM expression of transgenic protein relative to mice treated with rAAV9.CMV.LAMA2(G1–5) and showed improved weight-normalized forelimb grip strength relative to untreated dyW mice. Additionally, dyW muscle fibers expressing either micro-laminin protein showed some measures of reduced pathology, although levels of muscle cell apoptosis and inflammation were not decreased. Although systemic expression of rAAV9.CMV.HB-LAMA2(G1–5) did not inhibit all disease phenotypes, these studies demonstrate the feasibility of using a micro-laminin gene therapy strategy to deliver gene replacement for MDC1A.
Keywords: gene therapy, muscular dystrophy, laminin, AAV, extracellular matrix
Graphical Abstract
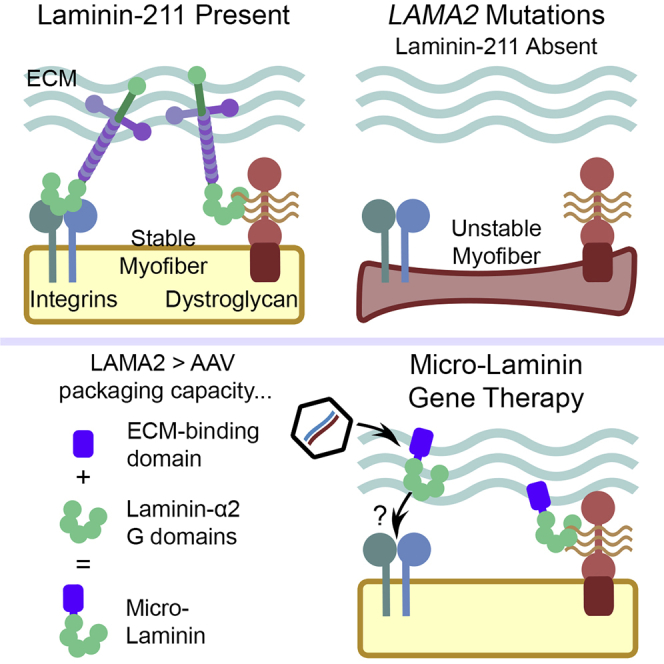
Packer and Martin describe a novel gene therapy for laminin-α2-deficient congenital muscular dystrophy in which the LAMA2 gene, which is too large to fit in current gene therapy vectors, is shortened to a vector-deliverable “micro” gene linked to a heparin-binding motif that facilitates extracellular matrix binding.
Introduction
Mutations in the LAMA2 gene can cause laminin-α2-deficient congenital muscular dystrophy 1A (MDC1A),1,2 a rare but severe disorder characterized by neonatal hypotonia, progressive muscle weakness, loss of ambulation, and premature death, often caused by respiratory muscle failure.3,4 The LAMA2 gene encodes laminin-α2, one subunit in the trimeric laminin-211 protein, the predominant laminin expressed in the skeletal muscle extracellular matrix (ECM).5 Laminin-211 supports muscle structure and force transmission by anchoring myofibers to the ECM.6, 7, 8 The N-terminal regions of the three laminin protein chains (laminin-α2, -β1, and -γ1) enable laminin-211 polymerization in the ECM,9 while additional domains bind other ECM components, such as agrin,10 nidogen, and collagen IV.11 The five globular (G) domains, unique to laminin-α subunits, bind the myofiber membrane receptors integrins12 and dystroglycan,13 both of which are essential to skeletal muscle membrane stability.14,15 Loss of laminin-211, as a consequence of LAMA2 mutations, allows contracting myofibers to detach from the ECM and undergo apoptosis, resulting in progressive muscular dystrophy.6 There are currently no effective FDA-approved treatments known to modify disease outcome in patients, although various strategies are being developed, including apoptosis inhibition,16, 17, 18, 19 non-AAV gene20 or protein21,22 replacement, CRISPR-mediated LAMA2 mutation correction,23 compensatory laminin-α1 gene upregulation,24, 25, 26 and expression of mini-agrin and laminin-α1 “linker” proteins.27,28
At 9.5 kb, the entire LAMA2 gene is too large to package into a single adeno-associated virus (AAV).29,30 Complete gene replacement is therefore impossible using AAV, unless recombination of LAMA2 fragments is induced by expressing gene fragments in separate AAV vectors, a less efficient strategy than using a single AAV vector.31 Gene miniaturization is one possible solution to this problem. The DMD gene encoding dystrophin was previously shortened into micro-dystrophin by Chamberlain and colleagues,32 allowing it to fit into AAV. Overexpression of micro-dystrophin reduced dystrophic skeletal and cardiac muscle pathology in the mdx mouse model of Duchenne muscular dystrophy.32, 33, 34, 35 A number of other mini- or micro-dystrophin forms have been tested, some of which have shown similar therapeutic effects.36, 37, 38, 39 Three different AAV.micro-dystrophins are now in phase 1/2a clinical trials (ClinicalTrials.org: NCT03375164, NCT03368742, and NCT03362502). Ruegg and colleagues28 have shown that expression of miniaturized agrin, termed mini-agrin, can be therapeutic in the dyW mouse model of MDC1A. This linker protein fuses the three G domains in the C terminus of agrin, which bind muscle membrane proteins α-dystroglycan and several integrins,40, 41, 42 to a separate region in the N terminus of agrin that binds laminin in the ECM (the NtA domain). Although this approach showed significant therapeutic effects in transgenic dyW mice,28,43,44 and also in AAV-treated dyW mice,45 neither strategy resulted in full recovery of normal muscle function.28 Ruegg, Yurchenco, and colleagues27 have shown that co-expression of a second linker protein, αLNNd, which is a fusion of the LN domain of laminin-α1 and the laminin-binding domain of nidogen, along with mini-agrin yields additional therapeutic improvements. Expression of full-length laminin-α2 or laminin-α1, in transgenic mice or via CRISPR-mediated mutation correction, also significantly inhibits disease in dyW mice,20,23,24 as does laminin-111 protein therapy.21,22
Our goal here was to build on micro-gene and linker protein strategies to create an AAV-deliverable micro-laminin gene therapy that would allow us to test partial gene replacement in a disease model for MDC1A. In doing so, we also tested a method for increased expression of recombinant laminin-α2 G1–5 protein fragments in the ECM. The skeletal muscle ECM contains abundant heparan sulfate glycosaminoglycans (GAGs), and these GAGs are often linked to ECM proteoglycans.46,47 Recently, we found that overexpression of a secreted form of the heparin-binding (HB) epidermal growth factor-like growth factor (HB-EGF) in mouse skeletal muscle led to high expression of this protein within the muscle ECM.48 We therefore also tested whether adding the HB domain from HB-EGF to micro-laminin would increase ECM expression.
Results
Micro-laminins are expressed in wild-type (WT) muscle after intramuscular (i.m.) injection of AAV
Gene therapy vectors rAAV9.CMV.LAMA2(G1–5) and rAAV9.CMV.HB-LAMA2(G1–5) expressing micro-laminins were made using the human gene segments as described in Figure 1A. HB-LAMA2(G1–5) protein bound α-dystroglycan, a known laminin receptor,13 in gel overlays (Figure 1B), and both proteins displayed calcium-dependent binding in the nanomolar (nM) concentration range to a partially purified muscle membrane preparation containing α-dystroglycan (Figure 1C). Both HB-LAMA2(G1–5) and LAMA2(G1–5) protein were secreted from transfected CHO cells, yielding a 130- to 140-kDa protein, close to the 109–118 kDa weight expected from the amino acid sequence, along with proteolytic fragments on immunoblots using anti-HB-EGF and anti-human laminin-α2 G5 antibodies (Figure 1D). HB-LAMA2(G1–5), however, also demonstrated staining of the muscle ECM in protein overlay staining experiments (Figure 1E), which was confirmed by co-staining with laminin-α2, while LAMA2(G1–5) staining overlays did not display ECM localization when a FLAG epitope tag was on the N terminus of this protein (Figure S1).
Figure 1.

Micro-laminin design and in vitro characterization
(A) Using human gene segments, LAMA2(G1–5) was designed by adding the HBEGF gene segment encoding the signal peptide (s) to the 5′ end of the LAMA2 gene segment encoding G domains 1–5. HB-LAMA2(G1–5) was designed by adding the HBEGF gene segment encoding the signal peptide, prepropeptide domain (which is normally cleaved from the protein), and the heparin-binding (HB) domain to the 5′ end of the LAMA2 gene segment encoding G domains 1–5. The HB domain of HB-EGF was expected to enhance laminin-α2 G1–5 binding to the muscle ECM. The G1–G5 domains of LAMA2 were expected to bind muscle membrane receptors, including integrins and dystroglycan. (B) Whole-muscle lysates and Wheat Germ Agglutinin (WGA)-purified dystroglycan were overlaid with HB-LAMA2(G1–5) protein or incubated in dilution buffer alone (None), then immunoblotted with antibody recognizing the HB domain of HB-EGF. IIH6 immunoblot recognized α-dystroglycan and demonstrated similar band location to HB-LAMA2(G1–5) overlay. (C) ELISA binding assay of HB-LAMA2(G1–5) and LAMA2(G1–5) proteins to WGA-purified muscle membrane proteins, including α-dystroglycan. Protein binding in buffer containing 1 mM Ca2+ and 1 mM Mg2+ (open circles and squares) was compared with binding in buffer with 10 mM EGTA (filled circles and squares). (D) Whole conditioned media (Whole), media precipitated by heparin-agarose (Precip), or supernatant following precipitation (Supern) from CHO cells transfected with HB-LAMA2(G1–5) or LAMA2(G1–5) expression vectors was compared with media from mock-transfected controls. Secreted protein was recognized by blotting with anti-HB-EGF or anti-human laminin-α2 G5 (Lmn-α2 G5) antibodies. From the amino acid sequence, the expected weight of HB-LAMA2(G1–5) is 118 kDa, and that of LAMA2(G1–5) is 109 kDa. (E) Muscle sections were overlaid with HB-LAMA2(G1–5) protein, and protein binding was visualized by immunostaining with an anti-HB-EGF antibody, then compared with staining for endogenous laminin-α2 using an antibody to the short-arm domain not present in micro-laminin. In (B), (D), and (E), antibodies used for staining or blotting are shown on the left side of the figures.
To determine whether micro-laminins could be expressed in the muscle ECM, we performed i.m. injections in WT C57BL/6J mice. We injected 1E+11 vector genomes (vg) of rAAV9.CMV.LAMA2(G1–5) or rAAV.CMV.HB-LAMA2(G1–5) into the tibialis anterior (TA) muscles of 2-month-old WT mice and analyzed transduction, gene expression, and protein expression 2 months later. PBS was injected as a negative control, and rAAV9.CMV.HB-EGF (soluble form) was injected as a positive control, as we had already shown that this form of HB-EGF binds the ECM when expressed in skeletal muscle.48 In mice injected with rAAV9.CMV.HB-LAMA2(G1–5), we identified micro-laminin protein using both an anti-HB-EGF polyclonal antibody and an anti-human-specific laminin-α2 G4–5 antibody. HB-LAMA2(G1–5) staining co-localized with endogenous laminin-α2 staining in the ECM, identified with an antibody against the short arm of mouse laminin-α2 that does not recognize the G1–G5 domains (Figure 2A). In mice injected with rAAV9.CMV.LAMA2(G1–5), we identified micro-laminin protein by the human G4–5 domain, while no staining was evident with the HB-EGF antibody, as expected. LAMA2(G1–5) staining co-localized with mouse laminin-α2 protein in the ECM, but staining was less pronounced than for HB-LAMA2(G1–5).
Figure 2.

Micro-laminin protein expression in mouse skeletal muscle following intramuscular injection
TA muscles from adult wild-type mice were injected with 1E+11 vg rAAV9.CMV.HBEGF, rAAV9.CMV.HB-LAMA2(G1–5), rAAV9.CMV.LAMA2(G1–5), or an equivalent volume of PBS. Muscles were analyzed at 2 months post-injection. (A) TA muscle sections stained with antibodies against HB-EGF (red), human laminin-α2 G4–5 (Lmn-α2 G4–5, green), and mouse laminin-α2 short-arm (Lmn-α2 short-arm, far-red). (B) Recombinant human HB-EGF protein (rHB-EGF) and TA muscle lysates were blotted with antibodies against HB-EGF and human laminin-α2 G5 domain (Lmn-α2 G5). Anti-GAPDH blotting was performed as a loading control. Heparin-agarose precipitation of muscle lysates was performed to demonstrate heparin binding. Genes expressed are shown on the top of the figures, with PBS injection shown as a negative control. Antibodies used for staining or blotting are shown on the left side of the figures.
We also assessed protein expression by western blotting of whole-muscle protein lysates before or after heparin-agarose precipitation (Figure 2B). rAAV9.CMV.HB-LAMA2(G1–5)-injected muscle produced a large amount of a 130- to 140-kDa band. This band appeared in blots using both HB-EGF and laminin-α2 G5 antibodies. The anti-laminin-α2 G5 antibody also blotted a second band, similar in size to a known ca. 80-kDa cleavage fragment previously identified in a mouse muscle.49 rAAV9.CMV.LAMA2(G1–5)-injected muscle also produced a 130- to 140-kDa band, reactive to the laminin-α2 G5 antibody, but not to the HB-EGF antibody. Consistent with immunostaining results (Figure 2A), LAMA2(G1–5) band density was reduced compared with HB-LAMA2(G1–5) (Figure 2B). Both micro-laminin proteins bound heparin-agarose in precipitation assays. There was no significant difference in vg muscle transduction between the rAAV9.CMV.HB-LAMA2(G1–5) and rAAV9.CMV.LAMA2(G1–5) groups (data not shown). Immunoblotting for Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control for equivalent protein loading and transfer.
Micro-laminins are expressed in dyW muscle after intravenous (i.v.) injection of AAV
To test micro-laminin therapy in dyW mice, we injected 1E+12 vg of rAAV9.CMV.LAMA2(G1–5) or rAAV9.CMV.HB-LAMA2(G1–5) i.v. into the temporal vein of post-natal day (P) 1 dyW pups. These mice were analyzed for muscle function at 2 and 3 months of age, then were euthanized at 4 months to determine transduction of tissues with AAV vg (Figure 3A), as well as gene (Figure 3B) and protein expression (Figure 4). Although survival time is a standard outcome measurement for dyW mice, it has been found to vary with the genetic background and, to some extent, husbandry practices between laboratories.50 All dyW mice in our sample groups survived up to 4 months of age, longer than expected from previous studies, and we therefore we did not report lifespan as an endpoint. Transduction of hindlimb skeletal muscles (gastrocnemius [Gastroc], TA, and quadriceps [Quad]) was low, on the order of 0.1–0.2 vg/nucleus, as was transduction of kidney and brain (ca. 0.1 vg/nucleus). Transduction was higher in forelimb muscle (0.3–0.4 vg/nucleus, triceps brachii) and diaphragm (0.8 vg/nucleus), while transduction was highest in heart and liver (ca. 5–10 vg/nucleus). Importantly, transduction levels for mice treated with rAAV9.CMV.HB-LAMA2(G1–5) or rAAV9.CMV.LAMA2(G1–5) were roughly equivalent in most tissues. Use of the CMV (Cytomegalovirus) promoter allowed for transgene expression throughout the body, which was compared in each tissue with the level of endogenous mouse Lama2 gene expression in WT mice (Figure 3B).
Figure 3.

Biodistribution of AAV vector genomes and transgene expression in dyW mice following intravenous injection
Neonatal dyW mice were injected intravenously with 1E+12 vg rAAV9 containing the micro-laminin genes under the CMV promoter. DNA was extracted from tissues at 4 months of age. (A) The number of AAV vector genomes per nucleus was quantified using qPCR. Errors bars are standard error of the mean (SEM) for n = 6 mice per condition. (B) Fold change in transgene expression was calculated relative to expression of the endogenous mouse Lama2 gene in corresponding tissues of age-matched WT littermates, with 18S ribosomal RNA gene expression as an internal control. Errors bars are SEM for n = 6 mice per condition.
Figure 4.

Micro-laminin protein expression in dyW mouse skeletal muscle following intravenous AAV injection
Tissues from dyW mice treated with intravenous rAAV9 and from untreated WT and dyW littermates were collected at 4 months of age. (A) Triceps brachii muscle sections stained with antibodies against HB-EGF (red), human laminin-α2 G4–5 (Lmn-α2 G4–5, green), and mouse laminin-α2 short-arm (Lmn-α2 short-arm, far-red). (B) Recombinant human HB-EGF protein and triceps brachii muscle lysates were immunoblotted with antibodies against HB-EGF and human laminin-α2 G5. Anti-GAPDH blotting was performed as a loading control. A heparin-agarose precipitation of triceps brachii muscle lysates was performed to demonstrate binding to heparin. Antibodies used for staining or blotting are shown on the left side of the figures.
Staining for micro-laminin proteins was present in the ECM surrounding the membranes of skeletal myofibers 4 months after i.v. delivery in P1 dyW mice (Figure 4A). Staining patterns in the triceps brachii muscle were similar to those observed in WT muscle following i.m. injection (Figure 2A). Again, HB-LAMA2(G1–5) protein was stained by both HB-EGF and laminin-α2 G4–5 antibodies, while LAMA2(G1–5) was stained by only the laminin-α2 G4-5 antibody. As expected, residual mouse laminin-α2 protein could be detected with an anti-laminin-α2 short-arm antibody in dyW muscle.51 As seen in the WT i.m. injections, HB-LAMA2(G1–5) staining was more intense and present in more myofibers than was LAMA(G1–5) staining (Figure 4A).
Similar to WT i.m. injections (Figure 2B), immunoblotting with a laminin-α2 G5 antibody after i.v. injection produced bands around 130–140 kDa in triceps brachii whole-muscle lysate from mice receiving either vector (Figure 4B), while immunoblotting with an HB-EGF antibody produced a band only for HB-LAMA2(G1–5). Both micro-laminin proteins could be precipitated by heparin-agarose. As before, the anti-laminin-α2 G5 antibody also recognized a ca. 65-kDa protein fragment. As with WT i.m. injection (Figure 2), HB-LAMA2(G1–5) protein showed higher overall expression than LAMA2(G1–5) protein, despite no significant difference in vg transduction and transgene expression (Figure 3). This was the case in multiple different comparisons of transduced triceps brachii muscles from rAAV9-treated dyW mice (Figure S2). The average of six such comparisons, all normalized to internal GAPDH blots, showed that overall expression of LAMA2(G1–5) protein was reduced in the triceps brachii by 35% ± 11% relative to HB-LAMA2(G1–5) (n = 5–6 muscles per group; p < 0.05).
On western blot, micro-laminin protein levels varied in muscles throughout the body (Figure S3A). Recombinant protein was evident in the TA, Quad, triceps brachii (triceps), diaphragm, and heart, while expression was lower in Gastroc muscle. Micro-laminin protein expression was not evident by western blot in liver, despite a high vg biodistribution, nor was it evident in kidney or brain (Figure S3A). Immunostaining for HB-LAMA2(G1–5) was prominent in the diaphragm (Figure S3B), although coincidence of HB-EGF and LAMA2 G4–5 staining was less apparent than in the triceps brachii. Staining for HB-LAMA2(G1–5) and LAMA2(G1–5) was also high in cardiomyocytes in the heart (Figure S3C). Human laminin-α2 G4–5 staining in the liver was difficult to evaluate because of high background staining, but staining was elevated in HB-LAMA2(G1–5) relative to untreated dyW, while LAMA2(G1–5) staining appeared to be predominantly intracellular (Figure S3D). Although endogenous laminin-α2 staining was present in the glomeruli of the WT mouse kidney, it was reduced or absent in treated and untreated dyW mice (Figure S3E). There was only a small degree of micro-laminin staining present in the kidney for HB-LAMA2(G1–5). Micro-laminin staining was also present in the choroid plexus of treated dyW brain (Figure S3F).
Analysis of histopathology in i.v.-micro-laminin-treated and control dyW muscles
We assessed the percentage of micro-laminin-expressing myofibers and pathology within expressing triceps brachii muscles at 4 months of age after i.v. delivery of micro-laminin gene therapy in P1 dyW mice: 36.3% ± 3.8% (n = 9 muscles) of myofibers stained for HB-LAMA2(G1–5) protein, and 10.6% ± 3.2% (n = 9 muscles) of myofibers stained for LAMA2(G1–5) (Figure 5A). Within triceps brachii muscles of treated mice, myofibers expressing either micro-laminin had significantly increased diameter (Figure 5B), reduced coefficient of variance in diameter (Figure 5C), and reduced percentage of cells with central nuclei (a marker for a cycle of muscle degeneration and regeneration) (Figure 5D). Increased myofiber size variability and central nucleation are indicators of muscle pathology. Thus, myofibers expressing either micro-laminin protein has less histopathology relative to non-expressing myofibers. There was, however, no significant improvement in muscle pathology between treated and untreated dyW mice when myofibers were not broken down between expressing and non-expressing groups (Figure S4). This may be because of the low percentage of micro-laminin-expressing myofibers or the presence of additional regenerating fibers in treated muscles, which would be consistent with elevated overall central nuclei in HB-LAMA2(G1–5)-treated muscles.
Figure 5.

Triceps brachii muscle histopathology after intravenous treatment of dyW mice with AAV9 expressing micro-laminins
(A) Percentage of myofibers that stained positive for micro-laminin proteins. (B) Minimum (mini-)Feret’s diameter of stained myofibers compared with unstained myofibers. (C and D) Quantification of myofiber pathological measures (coefficient of variance in myofiber diameter and percentage of myofibers with central nuclei) in stained and unstained myofibers. Error bars are SEM for n = 9 muscles per condition. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
There was no difference in muscle wasting, measured by percent muscle area occupied by myofibers, between untreated and HB-LAMA2(G1–5)-expressing dyW mice, but, surprisingly, LAMA2(G1–5)-expressing dyW muscle had increased muscle wasting relative to untreated muscle (Figure S5C). To further assess fibrosis, we measured collagen-1 expression. Triceps brachii muscle in dyW mice had regional variation in collagen-1 protein expression (Figure S5A). In HB-LAMA2(G1–5)-expressing muscle, regions with greater micro-laminin protein frequently coincided with lowered collagen-1 staining, while regions with little or no HB-LAMA2(G1–5) had higher collagen-1 staining. Col1a1 mRNA expression in the whole muscle, however, was still elevated in micro-laminin-treated dyW muscle relative to untreated dyW muscle and was elevated higher still in LAMA2(G1–5)-treated dyW mice (Figure S5B).
To assess muscle inflammation, we stained muscle for CD4, a marker for helper T cells, CD8, a marker for cytotoxic T cells, and CD68, a marker for macrophages (Figure S6A). We then quantified the numbers of positively stained cells for each condition (Figure S6B). There was no significant difference in the number of CD4+, CD8+, or CD68+ cells between micro-laminin-treated and untreated dyW mice. There were, however, increases in mRNA expression for inflammatory cytokines, including interleukin-β1 (Ilb1), C-C chemokine ligand 2 (Ccl2, also called MCP1), and C-C chemokine ligand 3 (Ccl3, also called MIP1α) in LAMA2(G1–5)-treated dyW muscles relative to HB-LAMA2(G1–5) treatment and to untreated dyW muscles (Figure S6C).
We next quantified the number of i.m. apoptotic cells using TUNEL staining with a DAPI co-stain to mark all nuclei. There was an increase in TUNEL+ cells in dyW muscle relative to WT, as previously shown,52 and this was not decreased by either micro-laminin treatment. If anything, LAMA2(G1–5) treatment dyW muscle trended toward higher numbers of apoptotic cells relative to HB-LAMA2(G1–5)-treated and untreated dyW muscles (Figure S7).
Improvement in forelimb grip strength in rAAV9.CMV.HB-LAMA2(G1–5)-treated dyW mice but lack of improvement in hindlimb grip strength or ambulation
We found a statistically significant improvement in body weight-normalized forelimb grip strength in dyW mice treated with rAAV9.CMV.HB-LAMA2(G1–5) (3.1 ± 0.2 g/g, n = 12 mice) compared with untreated dyW littermates (2.5 ± 0.1 g/g, n = 24 mice, p = 0.021) at 3 months of age (Figure 6). Due to dystrophy and peripheral nerve demyelination,51 dyW hindlimbs are stiffened by paralysis and contractures. Likely because of this and lowered transduction of hindlimb muscles relative to forelimb muscles, we found that ambulation speed and duration were not significantly changed by micro-laminin treatment (Figures S8A and S8B). Perhaps for similar reasons, hindlimb grip strength was not significantly increased in the HB-LAMA2(G1–5)-treated group (Figure S8C). Untreated dyW mice had reduced body and muscle weight compared with WT littermates. Neither micro-laminin significantly improved these measures (Figure S9).
Figure 6.

Weight-normalized forelimb grip strength after intravenous treatment of dyW mice with AAV9 expressing micro-laminins
The forelimb grip strength of mice treated with intravenous rAAV9 and that of untreated wild-type and dyW littermates was assessed at 2 and 3 months of age. Grip strength was normalized to body weight for each mouse. Error bars are SEM for n = 7–24 mice per condition. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Changes in AAV biodistribution and gene and protein expression with age in micro-laminin-treated dyW muscles
After transduction of single-stranded rAAV vectors into skeletal muscle, maximum gene expression usually takes 3 or sometimes 4 weeks.53,54 Therefore, we compared micro-laminin gene expression at 1 month post-injection, when gene expression first reaches maximum, with expression at 4 months post-injection, the experimental endpoint, after i.v. injection of dyW mice with 1E+12 vg rAAV9.CMV.HB-LAMA2(G1–5) at P1. vg counts were significantly higher at 1 month of age than at 4 months of age in the TA, Gastroc, and triceps brachii muscles (Figure 7A). Although this reduction in vg counts over time was evident in several limb muscles, it was not observed in the diaphragm, heart, liver, or Quad. Interestingly, gene expression did not necessarily follow the trend set by vg counts (Figure 7B). Notably, the percentage of HB-LAMA2(G1–5)-stained myofibers in the triceps brachii muscle was greater at the 4-month time point (36.3% ± 3.8%, n = 9 muscles) than at the 1-month time point (11.4% ± 2.6%, n = 8 muscles, p < 0.0001) (Figures 7C and 7D). Further, there was more ECM staining at 4 months than at 1 month, when staining was primarily intracellular (Figure 7D). This result could reflect secretion of HB-LAMA2(G1–5) from transduced myofibers onto adjacent non-transduced myofibers over time (bystander effect), slow kinetics of protein expression, or increased promoter activity at the later time point. Although gene expression was increased in the triceps brachii and diaphragm muscles, it was decreased in hindlimb muscles. This suggests that loss of gene expression may also occur because of ongoing muscle damage to expressing myofibers.
Figure 7.

Comparison of muscle biodistribution and gene and protein expression at 1 and 4 months after i.v. treatment of dyW mice with rAAV9.CMV.HB-LAMA2(G1–5)
Neonatal (P1) dyW mice were injected intravenously with 1E+12 vg rAAV9.CMV.HB-LAMA2(G1–5). Tissues were analyzed at 1 (grey circles) or 4 months (dark circles) post-injection. (A) The number of AAV genomes per nucleus was quantified in various tissues. (B) Expression of HB-LAMA2(G1–5) transgene was determined. Tissues were analyzed at 1 (grey bars) or 4 months (dark bars) post-injection. Fold change in gene expression was calculated relative to expression of endogenous mouse Lama2 (mLama2) gene in the corresponding tissues of 4-month-old wild-type littermates. (C) Percentage of myofibers that stained positively for HB-LAMA2(G1–5) protein. (D) Example of HB-LAMA2(G1–5) staining using HB-EGF antibody (green) at 1 and 4 months post-treatment, with dystrophin (red) used as a counterstain to identify myofibers. Composite image includes DAPI (blue) to stain nuclei. Error bars are SEM for n = 4 mice for the 1-month time point and n = 6 mice for the 4-month time point. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Increased expression of micro-laminin protein after i.m. injection in juvenile dyW mice
We next sought to increase the percentage of myofibers expressing micro-laminin by performing i.m. injections in juvenile dyW mice. To do this, we injected 1E+11 vg rAAV9.CMV.HB-LAMA2(G1–5) or rAAV9.CMV.LAMA2(G1–5) bilaterally into dyW TA muscles at 2 weeks of age and analyzed muscles 2 months after injection. Because i.m. injection into a small muscle often leads to variable levels of transduction, we found vg from rAAV9.CMV.LAMA2(G1–5) injection to be 2.7 ± 1-fold higher than for rAAV9.CMV.HB-LAMA2(G1–5). This led to expression of micro-laminin in an equivalent percentage of myofibers for both HB-LAMA2(G1–5) and LAMA2(G1–5), even though LAMA2(G1–5) had lower expression when transduction levels were equivalent (Figures 2, 3, and 4). In both cases, the percentage of myofibers transduced exceeded levels found with i.v. treatment; 45.5% ± 6.1% of muscles expressed HB-LAMA2(G1–5) protein, while 42.6% ± 6.3% of myofibers expressed LAMA2(G1–5) protein (Figures 8A and 8B).
Figure 8.

Tibialis anterior muscle histopathology after intramuscular treatment of dyW mice with rAAV9 expressing micro-laminin
The TA muscles of 2-week-old dyW mice were injected with 1E+11 vg rAAV9 containing either LAMA2(G1–5) or HB-LAMA2(G1–5) genes, or an equal volume of PBS, and analyzed 2 months post-injection. (A) Muscle sections stained with antibodies against human laminin-α2 G4–5 to detect micro-laminins (green) and against dystroglycan (red) to outline myofibers. (B) Percentage of myofibers that stained positively for micro-laminins. (C) Myofiber mini-Feret’s diameter. (D) Myofiber mini-Feret’s diameter coefficient of variance. (E) Percentage of myofibers with central nuclei. Error bars are SEM for n = 9–12 muscles per condition. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Because about half of all myofibers in the TA muscle were transduced, we assessed pathology for the entire muscle to determine the effects of each transgene on muscle pathology. Surprisingly, overall, TA myofiber diameters were, on average, smaller in rAAV9.CMV.HB-LAMA2(G1–5) (20.8 ± 0.7 μm, n = 9 muscles, p = 0.0006) and rAAV9.CMV.LAMA2(G1–5) (20.6 ± 0.7, n = 10 muscles, p = 0.0003) conditions compared with PBS-injected dyW littermates (25.8 ± 0.8 μm, n = 12 muscles) (Figure 8C). Within treated muscle, however, myofibers expressing HB-LAMA2(G1–5) or LAMA2(G1–5) were 54% ± 2% or 50% ± 2% larger, respectively, than non-expressing myofibers (p < 0.001 for both comparisons). Pathological measures of muscular dystrophy, including myofiber diameter variance (52.9 ± 1.0 μm/μm, n = 10 muscles, p < 0.0001) and the percentage of myofibers with central nuclei (21.5% ± 2.4%, n = 10 muscles, p = 0.0002), were reduced in muscles expressing LAMA2(G1–5) compared with PBS-injected dyW littermates (63.8 ± 1.3 μm/μm, n = 12 muscles; 25.7% ± 1.6%, n = 12 muscles) (Figures 8D and 8E). Myofiber diameter variance was also reduced in muscles expressing HB-LAMA2(G1–5) (59.8 ± 1.2 μm/μm, n = 9 muscles). Thus, injection of muscles with AAV.micro-laminin at P14 decreased pathology measures at the whole-muscle level but may have also changed post-natal muscle growth.
There are compensatory increases in other laminins, such as laminin-α4,27 and laminin-associated proteins in dyW muscles following loss of laminin-α2 expression, and upregulation of these proteins may inhibit the extent of disease.27,55 We therefore compared gene and protein expression for potential therapeutic molecules using TA muscles injected i.m. with AAV.micro-laminin (Figure S10). Laminin-α4 (Lama4) gene expression was elevated about 4-fold in treated and untreated dyW muscles relative to WT mice, as was gene expression of Lamb1, which encodes laminin-β1, while Lama2 expression was reduced (Figure S10B). Expression of laminin receptors, including integrin-α7β1 (encoded by Itga7 and Itgb1) and dystroglycan (encoded by Dag1), was not changed more than 2-fold in either direction in micro-laminin-treated muscles relative to untreated dyW muscles. Immunostaining found differences between dyW TA muscles and WT TA muscle in laminin-α2, laminin-α4, integrin-α7, integrin-β1, α-dystroglycan (by IIH6 staining), and total α/β-dystroglycan expression. Comparison of western blots for these proteins between treated and untreated TA muscles, using GAPDH as an internal control for protein loading and transfer (which was lower in dyW than in WT, but which did not significantly vary between treated and untreated dyW muscles), showed that laminin-α4 expression was elevated, although not to a statistically significant level, in HB-LAMA2(G1–5)- and LAMA2(G1–5)-treated dyW muscle relative to untreated dyW muscle, as was expression of α-dystroglycan (by IIH6 blotting) and β-dystroglycan (Figures S10C–S10F). Expression of integrin-α7 and -β1 were more variable. We used a polyclonal antibody to the C terminus of integrin-α7 for immunoblotting that recognizes multiple cleaved protein forms in addition to the native 120-kDa protein, resulting in multiple bands, and so we quantified each protein fragment separately.
Discussion
Although LAMA2 whole gene replacement is known to be therapeutically efficacious in mouse models of MDC1A using transgenic or gene-editing approaches,20,23 the LAMA2 gene is too large to fit into a single AAV vector, the best current clinical method for gene therapy. In this work, we have designed a micro-laminin gene therapy and demonstrated that it can prevent several aspects of muscle histopathology and even, in one instance, improve muscle function in the dyW mouse model for MDC1A. This strategy is akin to the linker protein therapies used by Ruegg, Yurchenco and colleagues27 to create a LAMA2 surrogate out of forms of agrin28 or laminin-α1. In both cases, functional protein-binding regions were linked to an ECM-binding region to create structural scaffoldings akin to those present in the native laminin-211 protein. We have used a similar linker protein strategy with the G1–5 domains of LAMA2 protein itself, which allows for a partial gene replacement, by adding the HB motif from HB-EGF, a protein that can be highly expressed in the muscle ECM.48 Interaction of the HB domain with muscle ECM likely results from binding to GAGs that are copiously present in the muscle ECM.47 In doing so, this very small HB domain may replace the much larger ECM-binding domains in the N termini of the α2, β1, and γ1 chains of laminin-211 that normally dictate ECM localization.9,11,56 The G1–5 region of LAMA2 also has the ability to bind heparin,57,58 but this HB region likely overlaps with its dystroglycan binding domain,57,58 which can be blocked by heparin. Thus, addition of the HB domain may free the G1–5 domains of laminin-α2 to properly bind dystroglycan at the muscle membrane. The HB domain may also serve to facilitate the secretion of the large G1–5 protein fragment, which is less well expressed, less effective, and in some instances, even deleterious in muscle in the absence of the HB domain.
Although our gene therapy approach is the first to show that the micro-laminin can have clinical benefits in a mouse model for MDC1A, it clearly is only one step toward maximizing this approach for patients. For example, although micro-laminin expression could prevent muscle damage, increase myofiber size, and even increase weight-normalized forelimb grip strength, dyW mice treated with micro-laminin displayed no improvement in ambulation or hindlimb grip strength. This may be because of limited treatment effect on the peripheral nervous system, because dyW mice display demyelination of peripheral nerves and gradual hindlimb paralysis,20 or it may be because of the limited transduction in hindlimb muscles. Additionally, the AAV vectors we have used may need to be optimized further to allow more efficient protein expression and secretion. The expression of such a large ECM protein clearly requires time. Analysis of HB-LAMA2(G1–5) staining at 1 month post-treatment, when gene expression has become saturating in dyW mice,54 showed a protein expression pattern far less coincident with the ECM, while staining at 4 months post-treatment showed higher ECM localization. dyW mice have reduced ability to regenerate muscle and display increased muscle cell apoptosis, both of which contribute to increased muscle histopathology.18,19,59, 60, 61 The kinetic lag in achieving maximum gene expression54 and full protein expression using AAV may have worked against our testing of this therapy in the dyW model. Immune responses to the human laminin protein, or HB-LAMA2 fusion domain epitopes, may also have worked against achieving maximal protein expression.
In its current form, micro-laminin gene therapy does not compare favorably with other treatment strategies tested in dyW mice, including rAAV9-delivered mini-agrin,45 apoptosis inhibitor omigapil,17,62 and transgenic expression of the linker protein αLNNd. Each of these treatments achieved improvements in body weight, grip strength, and/or ambulation and reduced fibrosis and/or muscle cell apoptosis. By contrast, HB-LAMA2(G1–5) micro-laminin gene therapy demonstrated limited therapeutic effect in dyW mice, having no effect on muscle cell apoptosis or muscle inflammation, but it was successfully expressed, localized to muscle ECM, and reduced some aspects of muscle histopathology. It also was generally more effective than LAMA2(G1–5) without the HB domain, suggesting that additional modifications to improve protein secretion, expression, and functionality may further improve treatment outcomes for MDC1A.
Materials and methods
Production of AAV9 vectors containing micro-laminin genes
The LAMA2(G1–5) and HB-LAMA2(G1–5) micro-laminin genes were designed using the human LAMA2 G domain coding sequence from RefSeq GenBank: NM_000426.3 (bp 6,538–9,436), the human HBEGF signal peptide from GenBank: NM_001945.2 (bp 276–348) for LAMA2(G1–5), and the human HBEGF signal peptide through HB domain from GenBank: NM_001945.2 (bp 276–597) for HB-LAMA2(G1–5). The recombinant genes were synthesized by GeneArt Gene Synthesis (Regensburg, Germany) in a pMA-T vector backbone. The genes were subcloned into a pcDNA3.3-TOPO backbone for use in cell culture transfections and into a recombinant Adeno Associated Virus (rAAV) plasmid with CMV promoter, SV40 enhancer, and SV40 poly-adenylation signal for vector production. From 5′ Inverted Terminal Repeat (ITR) to 3′ ITR, the genome length of rAAV.CMV.HB-LAMA2(G1–5) was 4.649 kb, and the length of rAAV.CMV.LAMA2(G1–5) was 4.377 kb. Plasmids were packaged in recombinant Adeno Associated Virus Serotype 9 (rAAV9) vectors by the Nationwide Children’s Hospital Viral Vector Core (Columbus, OH, USA) using the triple-transfection technique in HEK293 cells53 and were highly purified using sucrose density centrifugation and anion exchange chromatography, as previously described.63
Cell transfection
CHO-K1 cells (ATCC, Manassas, VA, USA) were grown in 10-cm dishes in DMEM (Thermo Fisher Scientific) with 10% fetal bovine serum (Millipore-Sigma). When at 80% confluence, cells were transfected with pcDNA3.3-TOPO.CMV.HB-LAMA2(G1–5) or pcDNA3.3-TOPO.CMV.FLAG-LAMA2(G1–5) using TransIT-X2 Dynamic Delivery System (Mirus, Madison, WI, USA), according to the manufacturer’s instructions for CHO-K1 cells in 10-cm dishes.
Mice
All mouse experiments were performed using protocols approved by the Institutional Animal Care and Use Committee at The Abigail Wexner Research Institute at Nationwide Children’s Hospital (Columbus, OH, USA). C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). dyW mice bred on a pure C57BL/6 background were a generous gift from Dr. Dean Burkin at University of Nevada, Reno, Medical School.
i.m. AAV injections
Adult mice were anesthetized with isofluorane (Baxter, Deerfield, IL, USA), and hindlimbs were shaved over the TA muscles. 1E+11 vg rAAV9 in 25 μL PBS was injected into each TA muscle using a 0.33-cc Thinpro Insulin Syringe (Terumo Medical, Somerset, NJ, USA). Two-week-old mice were restrained, and 1E+11 vg rAAV9 in 5 μL PBS was injected into each TA muscle using a 0.3-cc Thinpro Insulin Syringe.
i.v. AAV injections
Mouse pups up to 48 h old were anesthetized on ice and injected with 1E+12 vg rAAV9 into the temporal vein using a 0.3-cc Thinpro Insulin Syringe. The minimum injection volume allowable by virus titer was used.
Grip strength
On 3 consecutive days, 1 week prior to grip strength assessment, mice were trained on the Grip Strength Meter (Columbus Instruments, Columbus, OH, USA). Each training day consisted of five forelimb and five hindlimb grip strength trials, which were not recorded. The week of assessment, grip strength was assessed on 5 consecutive days. Each assessment consisted of five forelimb trials using the “T Peak” setting and five hindlimb trials using the “C Peak” setting on the Grip Strength Meter and measured in grams, followed by body weight measurement. For each mouse, on each day of the assessment week, the five trials were averaged and normalized to body weight obtained on that day. For each mouse, the final grip strength value was the average of the body weight-normalized grip strength values obtained over the 5 days.
Ambulation
On 3 consecutive days, 1 week before ambulation assessment, mice were trained on the Treadmill Meter (Columbus Instruments, Columbus, OH, USA). The week of assessment, mice were placed on the stationary Treadmill Meter, one or two mice per enclosed lane. The treadmill was started at 5 m/min, increasing 1 m/min every 30 s until 10 m/min was reached. The treadmill was run at 10 m/min for 10 min. For each mouse, the trial ended when the mouse fell off the end of the treadmill three times in a span of 20 s. At the end of the trial, the maximum speed reached and time of ambulation at 10 m/min were recorded. If a mouse failed to reach 10 m/min, 0 s was recorded for ambulation time. For each mouse, the final values for speed and ambulation time were the averages of those obtained over the 5 days.
Tissue processing
Skeletal muscles were removed, weighed, mounted with O.C.T. (Thermo Fisher Scientific) on cork, and snap frozen in liquid-nitrogen-cooled 2-methylbutane. The internal organs and heart were removed, placed in Peel-A-Way Disposable Embedding Molds (Thermo Fisher Scientific), covered in Tissue Plus O.C.T. (Thermo Fisher Scientific), and frozen in a 2-methylbutane (Thermo Fisher Scientific)/dry ice bath. One hemisphere of the brain was snap frozen for molecular analysis. The other hemisphere was fixed overnight in 4% paraformaldehyde (EMS, Hatfield, PA, USA) in PBS at 4°C, washed overnight in 0.1 M glycine (Thermo Fisher Scientific) in PBS at 4°C, then incubated sequentially in 10%, 20%, and 30% sucrose each overnight at 4°C, before freezing in an embedding mold covered with O.C.T.
AAV biodistribution
DNA was extracted from tissue shavings using a DNeasy Blood & Tissue Kit (QIAGEN, Germantown, MD, USA). vg quantity in 100 ng sample DNA was determined using 2X TaqMan Gene Expression Master Mix, a standard curve of linearized rAAV plasmid from 5 vg to 5E+6 vg, and the following primer/probe set amplifying a region spanning the SV40 enhancer and the transgene: 5′-ACTTCTAGGCCTGTACGGAAGTG-3′ (forward), 5′-56-FAM/AAAGCTGCG/ZEN/GAATTGTACCCGCGGC/3IABkFQ/-3′ (probe), and 5′- CTGCAGCCAGAAAGAGCTTCAG-3′ (reverse). vg per mouse nucleus was then calculated using an estimate of 1.7E+5 mouse nuclei per 1 μg genomic DNA.
Gene expression measurement
RNA was extracted from tissue shavings using a Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA, USA), and cDNA was prepared using a High-Capacity cDNA Reverse Transcriptase kit (Applied Biosystems, Bedford, MA, USA). HB-LAMA2(G1–5) and LAMA2(G1–5) transgene expression were measured in cDNA samples using TaqMan Gene Expression Master Mix and LAMA2 (human) Assay Hs01124072_m1 (Applied Biosystems). Additional assays included mouse Lama4, mouse Itga7, and mouse Itgb1 (IDT, Coralville, IA, USA), mouse Lamb1 Assay Mm008801853_m1, and mouse Dag1 Assay Mm00802400 (Applied Biosystems). 18S rRNA was used as an internal control (Applied Biosystems). The 2−ΔΔCT method was used to calculate fold change in expression.64
WGA-agarose precipitation and protein isolation
Immediately following dissection, Gastroc and Quad muscles from WT mice were minced with a sterile scalpel, placed in 5 mL Nonidet P-40 (NP-40) lysis buffer, 1% (w/v) NP-40 (Millipore-Sigma, St. Louis, MO, USA) in 50 mM Tris buffer with 150 mM NaCl, 1 mM EDTA (Invitrogen, Grand Island, NY, USA), and cOmplete EDTA-free Protease Inhibitor Cocktail (Millipore-Sigma) and rocked overnight at 4°C. Supernatant was transferred to 200 μL Wheat Germ Agglutinin (WGA)-agarose (EY Laboratories, San Mateo, CA, USA) and rocked overnight at 4°C. Precipitated sample was washed thoroughly in 0.1% (w/v) NP-40 in 50 mM Tris buffer with 150 mM NaCl, then eluted in 500 μL 0.3 M N-acetyl-D-glucosamine (GlcNAc; Millipore-Sigma, USA) in 0.1% NP-40 buffer for 20 min with rocking at 4°C. To reduce GlcNAc concentration, we dialyzed eluted samples in 1,000-fold volume 0.1% NP-40 buffer overnight at 4°C using 10K MWCO Slide-A-Lyzer Dialysis Cassettes (Thermo Fisher Scientific).
Heparin-agarose precipitation and protein isolation
200 μL Heparin-Agarose Type I saline suspension (Millipore-Sigma) was added per milliliter of conditioned media, cell lysate, or muscle lysate to be precipitated and mixed overnight at 4°C. Precipitated samples were washed thoroughly with PBS. For western blotting, precipitated samples were resuspended in 50 μL 1X NuPage LDS Sample Buffer with 100 mM β-mercaptoethanol. 20 μL of the resuspension was boiled and used for immunoblotting. For isolation of micro-laminin proteins, precipitated samples were eluted with a high-salt solution, 200 μL 1.5 M NaCl in 0.01 M Tris-HCl (pH 7.5). To reduce salt concentration, we dialyzed eluted samples in 1,000-fold volume 0.01 M Tris-HCl overnight at 4°C using 10K MWCO Slide-A-Lyzer Dialysis Cassettes (Thermo Fisher Scientific).
Anti-FLAG-agarose precipitation and protein isolation
We performed anti-FLAG-agarose precipitation and protein isolation identically to heparin-agarose precipitation and protein isolation, replacing Heparin-Agarose Type I with Anti-FLAG M2 Affinity Gel (Millipore-Sigma). High-salt solution was used for elution, as in heparin-agarose precipitation and isolation.
Immunoblotting
Protein was extracted from tissue shavings using NP-40 lysis buffer. 40 μg of each protein lysate was added to NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) with 100 mM β-mercaptoethanol (Thermo Fisher Scientific) and boiled for 10 min prior to gel electrophoresis and transfer to nitrocellulose membranes for immunoblotting. The antibodies against HB-EGF (AF-259), laminin-α4 (AF3837), and α/β-dystroglycan (AF6868) were obtained from R&D Systems (Minneapolis, MN, USA). The antibody against laminin-α2 G5 (2D4) was obtained from Abnova (Taipei, Taiwan). The antibodies against GAPDH (G9545) and α-dystroglycan, clone IIH6C4 (05-593) were obtained from Millipore-Sigma. The antibody against laminin-β1 (PA5-27271) was obtained from Invitrogen. The antibodies against integrin-α7B and -β1 were obtained as described by Martin et al.65 The horseradish peroxidase (HRP)-conjugated secondary antibodies against goat and rabbit IgG were obtained from Jackson ImmunoResearch (West Grove, PA, USA), and the secondary antibody against mouse IgG1 was obtained from Bethyl Laboratories (Montgomery, TX, USA). Protein band densities were measured using ImageJ 1.46r.
For protein overlays, protein gels were transferred to Immobilon-PSQ polyvinylidene fluoride (PVDF) membranes (Millipore-Sigma) and washed briefly in laminin-binding buffer (LBB), 140 mM NaCl, 1 mM CaCl2, and 1 mM MgCl2 in 10 mM Tris-HCl, before blocking in 5% milk in LBB. Membranes were overlaid with micro-laminin protein samples in LBB overnight at 4°C. Primary antibody in 3% BSA in LBB was added for 2 h at room temperature, followed by secondary antibody in 3% BSA in LBB for 1 h.
Enzyme-linked immunosorbent assays (ELISAs)
200 ng WGA-agarose-isolated sample was diluted in 50 mM bicarbonate buffer (pH 9.5) and added per well to NUNC-Immuno MicroWell plates, then incubated overnight at 4°C. Plates were blocked in 3% BSA in 50 mM Tris-HCl with 100 mM NaCl, 1 mM CaCl2, and 1 mM MgCl2, then incubated with micro-laminin ligands in blocking buffer. Ligands were detected using anti-laminin-α2 G5 (2D4) primary antibody and HRP-conjugated secondary antibody. Substrate Reagent Pack colorimetric kit (R&D Systems) was used to measure the signal at 450 nm.
Immunofluorescent staining
10-μm tissue sections, cut on a Cryostat, were placed on Fisher Superfrost Plus glass slides (Thermo Fisher Scientific). A hydrophobic border was drawn around each section using an ImmEdge Hydrophobic Barrier PAP pen (Vector Labs, Burlingame, CA, USA). Sections were fixed in 2% paraformaldehyde (EMS) in PBS for 10 min, permeabilized in 0.1% Triton X-100 (Millipore-Sigma) in PBS for 10 min, incubated in Mouse-on-Mouse Blocking Reagent (Vector Labs) for 1 h, and blocked with 5% donkey serum in PBS for 30 min prior to incubation overnight at 4°C in primary antibodies. Sections were then incubated with secondary antibodies for 1 h at room temperature and then treated with ProLong Gold with DAPI (Invitrogen) before adding a coverslip. The antibodies against HB-EGF (AF-259), laminin-α4 (AF3837), integrin-α7 (MAB3518), and α/β-dystroglycan (AF6868) were obtained from R&D Systems. The antibodies against human laminin-α2 G4-5 (5H2), laminin-α2 short-arm (4H8-2), and α-dystroglycan, clone IIH6C4 (05-593) were obtained from Millipore-Sigma. The antibody against dystrophin (ab15277) was obtained from Abcam (Cambridge, UK). The antibody against integrin-β1 clone 9EG7 (553715) was obtained from BD Biosciences (San Jose, CA, USA). The antibody against laminin-α5 (bs-1086R) was obtained from Bioss (Boston, MA, USA). Rabbit anti-mouse IgG1 secondary antibody (fluorescein isothiocyanate [FITC]; NBP1-73636) was obtained from Novus Biologicals (Centennial, CO, USA). All other secondary antibodies were obtained from Jackson ImmunoResearch.
For protein overlay assays, following blocking, sections were incubated in heparin-agarose-isolated micro-laminin protein from transfected cell lysate in 3% BSA in LBB overnight at 4°C. Primary antibody in 3% BSA in LBB was added for 2 h at room temperature, followed by secondary antibody in 3% BSA in LBB for 1 h.
Microscopy and imaging
For all immunofluorescent images, a Zeiss Imager.Z1 with an AxioCam MRm camera, Plan-Apochromat 10×/0.45 M27 and Plan-Apochromat 20×/0.8 M27 objectives, and AxioVision40 v4.7.1.0 software were used to take single-plane multi-channel epifluorescence images. Equal exposure times were used to image corresponding channels across samples within each experiment. In all staining, DAPI and Alexa 488, CY2, or FITC, CY3, and CY5 fluorophores were used and visualized using fluorophore-appropriate filters.
Histological analysis
Myofiber diameter, central nucleation, and muscle wasting analyses were performed using an ImageJ macro on 10× images of muscle sections stained with an antibody against dystrophin to outline myofibers, much as previously described.66 TUNEL+ nuclei were counted using an ImageJ macro on 10× images of muscle sections. Immune cells were counted manually on 10× images of muscle sections.
Statistical analysis
The statistical significance of differences between two groups was tested using an unpaired, two-tailed Student’s t test. The significance of differences between more than two groups was tested using a one-way ANOVA, with post hoc Tukey’s test for multiple comparisons.
Acknowledgments
We thank Deborah Zygmunt and Anna Ashbrook for technical assistance in the performance of certain experiments. This work was funded by National Institutes of Health (NIH) grant P50 AR070604 to P.T.M. D.P. was funded, in part, by a Research Trainee Award from Nationwide Children's Hospital and a Distinguished University Fellowship from The Ohio State University.
Author contributions
Conceptualization, P.T.M.; methodology, P.T.M. and D.P.; investigation, D.P.; resources, P.T.M.; writing – original draft, D.P.; writing – reviewing & editing, P.T.M. and D.P.; visualization, P.T.M. and D.P.; supervision, P.T.M.; funding acquisition, P.T.M. and D.P.
Declaration of interests
P.T.M. discloses a potential financial conflict of interest regarding licensing fees he and Nationwide Children’s Hospital receive from Sarepta Therapeutics for the clinical development of the rAAVrh74.MCK.GALGT2 gene therapy. P.T.M. is also President and CSO of Genosera, Inc. This study represents part of D.P.’s doctoral thesis work at The Ohio State University.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2021.02.004.
Supplemental information
References
- 1.Tomé F.M., Evangelista T., Leclerc A., Sunada Y., Manole E., Estournet B., Barois A., Campbell K.P., Fardeau M. Congenital muscular dystrophy with merosin deficiency. C. R. Acad. Sci. III. 1994;317:351–357. [PubMed] [Google Scholar]
- 2.Hillaire D., Leclerc A., Fauré S., Topaloglu H., Chiannilkulchaï N., Guicheney P., Grinas L., Legos P., Philpot J., Evangelista T. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum. Mol. Genet. 1994;3:1657–1661. doi: 10.1093/hmg/3.9.1657. [DOI] [PubMed] [Google Scholar]
- 3.Geranmayeh F., Clement E., Feng L.H., Sewry C., Pagan J., Mein R., Abbs S., Brueton L., Childs A.-M.M., Jungbluth H. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010;20:241–250. doi: 10.1016/j.nmd.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Oliveira J., Santos R., Soares-Silva I., Jorge P., Vieira E., Oliveira M.E., Moreira A., Coelho T., Ferreira J.C., Fonseca M.J. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin. Genet. 2008;74:502–512. doi: 10.1111/j.1399-0004.2008.01068.x. [DOI] [PubMed] [Google Scholar]
- 5.Patton B.L., Connoll A.M., Martin P.T., Cunningham J.M., Mehta S., Pestronk A., Miner J.H., Sanes J.R. Distribution of ten laminin chains in dystrophic and regenerating muscles. Neuromuscul. Disord. 1999;9:423–433. doi: 10.1016/s0960-8966(99)00033-4. [DOI] [PubMed] [Google Scholar]
- 6.Hall T.E., Bryson-Richardson R.J., Berger S., Jacoby A.S., Cole N.J., Hollway G.E., Berger J., Currie P.D. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. USA. 2007;104:7092–7097. doi: 10.1073/pnas.0700942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purslow P.P., Trotter J.A. The morphology and mechanical properties of endomysium in series-fibred muscles: variations with muscle length. J. Muscle Res. Cell Motil. 1994;15:299–308. doi: 10.1007/BF00123482. [DOI] [PubMed] [Google Scholar]
- 8.Huijing P.A., Baan G.C., Rebel G.T. Non-myotendinous force transmission in rat extensor digitorum longus muscle. J. Exp. Biol. 1998;201:683–691. [PubMed] [Google Scholar]
- 9.Schittny J.C., Yurchenco P.D. Terminal short arm domains of basement membrane laminin are critical for its self-assembly. J. Cell Biol. 1990;110:825–832. doi: 10.1083/jcb.110.3.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denzer A.J., Brandenberger R., Gesemann M., Chiquet M., Ruegg M.A. Agrin binds to the nerve-muscle basal lamina via laminin. J. Cell Biol. 1997;137:671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aumailley M., Wiedemann H., Mann K., Timpl R. Binding of nidogen and the laminin-nidogen complex to basement membrane collagen type IV. Eur. J. Biochem. 1989;184:241–248. doi: 10.1111/j.1432-1033.1989.tb15013.x. [DOI] [PubMed] [Google Scholar]
- 12.von der Mark H., Dürr J., Sonnenberg A., von der Mark K., Deutzmann R., Goodman S.L. Skeletal myoblasts utilize a novel beta 1-series integrin and not alpha 6 beta 1 for binding to the E8 and T8 fragments of laminin. J. Biol. Chem. 1991;266:23593–23601. [PubMed] [Google Scholar]
- 13.Gee S.H., Blacher R.W., Douville P.J., Provost P.R., Yurchenco P.D., Carbonetto S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J. Biol. Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- 14.Hayashi Y.K., Chou F.L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- 15.Cohn R.D., Henry M.D., Michele D.E., Barresi R., Saito F., Moore S.A., Flanagan J.D., Skwarchuk M.W., Robbins M.E., Mendell J.R. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- 16.Girgenrath M., Beermann M.L., Vishnudas V.K., Homma S., Miller J.B. Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann. Neurol. 2009;65:47–56. doi: 10.1002/ana.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erb M., Meinen S., Barzaghi P., Sumanovski L.T., Courdier-Früh I., Rüegg M.A., Meier T. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J. Pharmacol. Exp. Ther. 2009;331:787–795. doi: 10.1124/jpet.109.160754. [DOI] [PubMed] [Google Scholar]
- 18.Girgenrath M., Dominov J.A., Kostek C.A., Miller J.B. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J. Clin. Invest. 2004;114:1635–1639. doi: 10.1172/JCI22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dominov J.A., Kravetz A.J., Ardelt M., Kostek C.A., Beermann M.L., Miller J.B. Muscle-specific BCL2 expression ameliorates muscle disease in laminin alpha2-deficient, but not in dystrophin-deficient, mice. Hum. Mol. Genet. 2005;14:1029–1040. doi: 10.1093/hmg/ddi095. [DOI] [PubMed] [Google Scholar]
- 20.Kuang W., Xu H., Vachon P.H., Liu L., Loechel F., Wewer U.M., Engvall E. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J. Clin. Invest. 1998;102:844–852. doi: 10.1172/JCI3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rooney J.E., Knapp J.R., Hodges B.L., Wuebbles R.D., Burkin D.J. Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am. J. Pathol. 2012;180:1593–1602. doi: 10.1016/j.ajpath.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Ry P.M., Minogue P., Hodges B.L., Burkin D.J. Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum. Mol. Genet. 2014;23:383–396. doi: 10.1093/hmg/ddt428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kemaladewi D.U., Maino E., Hyatt E., Hou H., Ding M., Place K.M., Zhu X., Bassi P., Baghestani Z., Deshwar A.G. Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat. Med. 2017;23:984–989. doi: 10.1038/nm.4367. [DOI] [PubMed] [Google Scholar]
- 24.Gawlik K., Miyagoe-Suzuki Y., Ekblom P., Takeda S., Durbeej M. Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum. Mol. Genet. 2004;13:1775–1784. doi: 10.1093/hmg/ddh190. [DOI] [PubMed] [Google Scholar]
- 25.Gawlik K.I., Mayer U., Blomberg K., Sonnenberg A., Ekblom P., Durbeej M. Laminin alpha1 chain mediated reduction of laminin alpha2 chain deficient muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 2006;580:1759–1765. doi: 10.1016/j.febslet.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 26.Gawlik K.I., Li J.Y., Petersén A., Durbeej M. Laminin alpha1 chain improves laminin alpha2 chain deficient peripheral neuropathy. Hum. Mol. Genet. 2006;15:2690–2700. doi: 10.1093/hmg/ddl201. [DOI] [PubMed] [Google Scholar]
- 27.Reinhard J.R., Lin S., McKee K.K., Meinen S., Crosson S.C., Sury M., Hobbs S., Maier G., Yurchenco P.D., Rüegg M.A. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 2017;9:eaal4649. doi: 10.1126/scitranslmed.aal4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moll J., Barzaghi P., Lin S., Bezakova G., Lochmüller H., Engvall E., Müller U., Ruegg M.A. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- 29.Dong J.Y., Fan P.D., Frizzell R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996;7:2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- 30.Vuolteenaho R., Nissinen M., Sainio K., Byers M., Eddy R., Hirvonen H., Shows T.B., Sariola H., Engvall E., Tryggvason K. Human laminin M chain (merosin): complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. J. Cell Biol. 1994;124:381–394. doi: 10.1083/jcb.124.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan D., Yue Y., Yan Z., Engelhardt J.F. A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat. Med. 2000;6:595–598. doi: 10.1038/75080. [DOI] [PubMed] [Google Scholar]
- 32.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., Chamberlain J.S. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 33.Liu M., Yue Y., Harper S.Q., Grange R.W., Chamberlain J.S., Duan D. Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salva M.Z., Himeda C.L., Tai P.W., Nishiuchi E., Gregorevic P., Allen J.M., Finn E.E., Nguyen Q.G., Blankinship M.J., Meuse L. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 35.Townsend D., Blankinship M.J., Allen J.M., Gregorevic P., Chamberlain J.S., Metzger J.M. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol. Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- 36.Watchko J., O’Day T., Wang B., Zhou L., Tang Y., Li J., Xiao X. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- 37.Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L., Duan D. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakamoto M., Yuasa K., Yoshimura M., Yokota T., Ikemoto T., Suzuki M., Dickson G., Miyagoe-Suzuki Y., Takeda S. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 2002;293:1265–1272. doi: 10.1016/S0006-291X(02)00362-5. [DOI] [PubMed] [Google Scholar]
- 39.McGreevy J.W., Hakim C.H., McIntosh M.A., Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 2015;8:195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gesemann M., Cavalli V., Denzer A.J., Brancaccio A., Schumacher B., Ruegg M.A. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- 41.Burgess R.W., Dickman D.K., Nunez L., Glass D.J., Sanes J.R. Mapping sites responsible for interactions of agrin with neurons. J. Neurochem. 2002;83:271–284. doi: 10.1046/j.1471-4159.2002.01102.x. [DOI] [PubMed] [Google Scholar]
- 42.Martin P.T., Sanes J.R. Integrins mediate adhesion to agrin and modulate agrin signaling. Development. 1997;124:3909–3917. doi: 10.1242/dev.124.19.3909. [DOI] [PubMed] [Google Scholar]
- 43.Meinen S., Barzaghi P., Lin S., Lochmüller H., Ruegg M.A. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J. Cell Biol. 2007;176:979–993. doi: 10.1083/jcb.200611152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bentzinger C.F., Barzaghi P., Lin S., Ruegg M.A. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J. 2005;19:934–942. doi: 10.1096/fj.04-3376com. [DOI] [PubMed] [Google Scholar]
- 45.Qiao C., Dai Y., Nikolova V.D., Jin Q., Li J., Xiao B., Li J., Moy S.S., Xiao X. Amelioration of Muscle and Nerve Pathology in LAMA2 Muscular Dystrophy by AAV9-Mini-Agrin. Mol. Ther. Methods Clin. Dev. 2018;9:47–56. doi: 10.1016/j.omtm.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singhal N., Martin P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 2011;71:982–1005. doi: 10.1002/dneu.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jenniskens G.J., Oosterhof A., Brandwijk R., Veerkamp J.H., van Kuppevelt T.H. Heparan sulfate heterogeneity in skeletal muscle basal lamina: demonstration by phage display-derived antibodies. J. Neurosci. 2000;20:4099–4111. doi: 10.1523/JNEUROSCI.20-11-04099.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cramer M.L., Xu R., Martin P.T. Soluble Heparin Binding Epidermal Growth Factor-Like Growth Factor Is a Regulator of GALGT2 Expression and GALGT2-Dependent Muscle and Neuromuscular Phenotypes. Mol. Cell. Biol. 2019;39:e00140-19. doi: 10.1128/MCB.00140-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrig K., Leivo I., Argraves W.S., Ruoslahti E., Engvall E. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc. Natl. Acad. Sci. USA. 1990;87:3264–3268. doi: 10.1073/pnas.87.9.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willmann R., Gordish-Dressman H., Meinen S., Rüegg M.A., Yu Q., Nagaraju K., Kumar A., Girgenrath M., Coffey C.B.M., Cruz V. Improving Reproducibility of Phenotypic Assessments in the DyW Mouse Model of Laminin-α2 Related Congenital Muscular Dystrophy. J. Neuromuscul. Dis. 2017;4:115–126. doi: 10.3233/JND-170217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo L.T., Zhang X.U., Kuang W., Xu H., Liu L.A., Vilquin J.T., Miyagoe-Suzuki Y., Takeda S., Ruegg M.A., Wewer U.M., Engvall E. Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul. Disord. 2003;13:207–215. doi: 10.1016/s0960-8966(02)00266-3. [DOI] [PubMed] [Google Scholar]
- 52.Kumar A., Yamauchi J., Girgenrath T., Girgenrath M. Muscle-specific expression of insulin-like growth factor 1 improves outcome in Lama2Dy-w mice, a model for congenital muscular dystrophy type 1A. Hum. Mol. Genet. 2011;20:2333–2343. doi: 10.1093/hmg/ddr126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu R., Chandrasekharan K., Yoon J.H., Camboni M., Martin P.T. Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am. J. Pathol. 2007;171:181–199. doi: 10.2353/ajpath.2007.060927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doe J.A., Wuebbles R.D., Allred E.T., Rooney J.E., Elorza M., Burkin D.J. Transgenic overexpression of the α7 integrin reduces muscle pathology and improves viability in the dy(W) mouse model of merosin-deficient congenital muscular dystrophy type 1A. J. Cell Sci. 2011;124:2287–2297. doi: 10.1242/jcs.083311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denzer A.J., Schulthess T., Fauser C., Schumacher B., Kammerer R.A., Engel J., Ruegg M.A. Electron microscopic structure of agrin and mapping of its binding site in laminin-1. EMBO J. 1998;17:335–343. doi: 10.1093/emboj/17.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hohenester E., Tisi D., Talts J.F., Timpl R. The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol. Cell. 1999;4:783–792. doi: 10.1016/s1097-2765(00)80388-3. [DOI] [PubMed] [Google Scholar]
- 58.Yurchenco P.D., Cheng Y.S., Schittny J.C. Heparin modulation of laminin polymerization. J. Biol. Chem. 1990;265:3981–3991. [PubMed] [Google Scholar]
- 59.Kuang W., Xu H., Vilquin J.T., Engvall E. Activation of the lama2 gene in muscle regeneration: abortive regeneration in laminin alpha2-deficiency. Lab. Invest. 1999;79:1601–1613. [PubMed] [Google Scholar]
- 60.Mukasa T., Momoi T., Momoi M.Y. Activation of caspase-3 apoptotic pathways in skeletal muscle fibers in laminin alpha2-deficient mice. Biochem. Biophys. Res. Commun. 1999;260:139–142. doi: 10.1006/bbrc.1999.0829. [DOI] [PubMed] [Google Scholar]
- 61.Miller J.B., Girgenrath M. The role of apoptosis in neuromuscular diseases and prospects for anti-apoptosis therapy. Trends Mol. Med. 2006;12:279–286. doi: 10.1016/j.molmed.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 62.Meinen S., Lin S., Thurnherr R., Erb M., Meier T., Rüegg M.A. Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice. EMBO Mol. Med. 2011;3:465–479. doi: 10.1002/emmm.201100151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 64.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 65.Martin P.T., Kaufman S.J., Kramer R.H., Sanes J.R. Synaptic integrins in developing, adult, and mutant muscle: selective association of alpha1, alpha7A, and alpha7B integrins with the neuromuscular junction. Dev. Biol. 1996;174:125–139. doi: 10.1006/dbio.1996.0057. [DOI] [PubMed] [Google Scholar]
- 66.Thomas P.J., Xu R., Martin P.T. B4GALNT2 (GALGT2) Gene Therapy Reduces Skeletal Muscle Pathology in the FKRP P448L Mouse Model of Limb Girdle Muscular Dystrophy 2I. Am. J. Pathol. 2016;186:2429–2448. doi: 10.1016/j.ajpath.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.