

Androgen deprivation therapy (ADT) is an important treatment strategy that can be used to delay the progression of prostate cancer. However, after an initial response, which varies significantly among patients, the disease eventually progresses despite the low levels of testosterone in the systemic circulation.1 The mechanism underlying the progression of prostate cancer to castration-resistant prostate cancer (CRPC) is not yet clear, and further investigation is needed.2 Tumor-associated macrophages (TAMs) are associated with resistance to ADT in prostate cancer.3,4 This study discovered that SEMA3A expression was upregulated in CRPC, which resulted in enhanced recruitment of monocytes and their polarization into the M2 subtype. Activated M2 TAMs could in turn promote cancer cell resistance to ADT. Mechanistically, SEMA3A could bind to the NRP1 receptor of TAMs, boosting the phosphorylation of downstream PI3K and AKT. Moreover, targeting SEMA3A and its receptor NRP1 proved to be a potent approach for CRPC treatment, offering a novel method for ameliorating ADT resistance in prostate cancer.
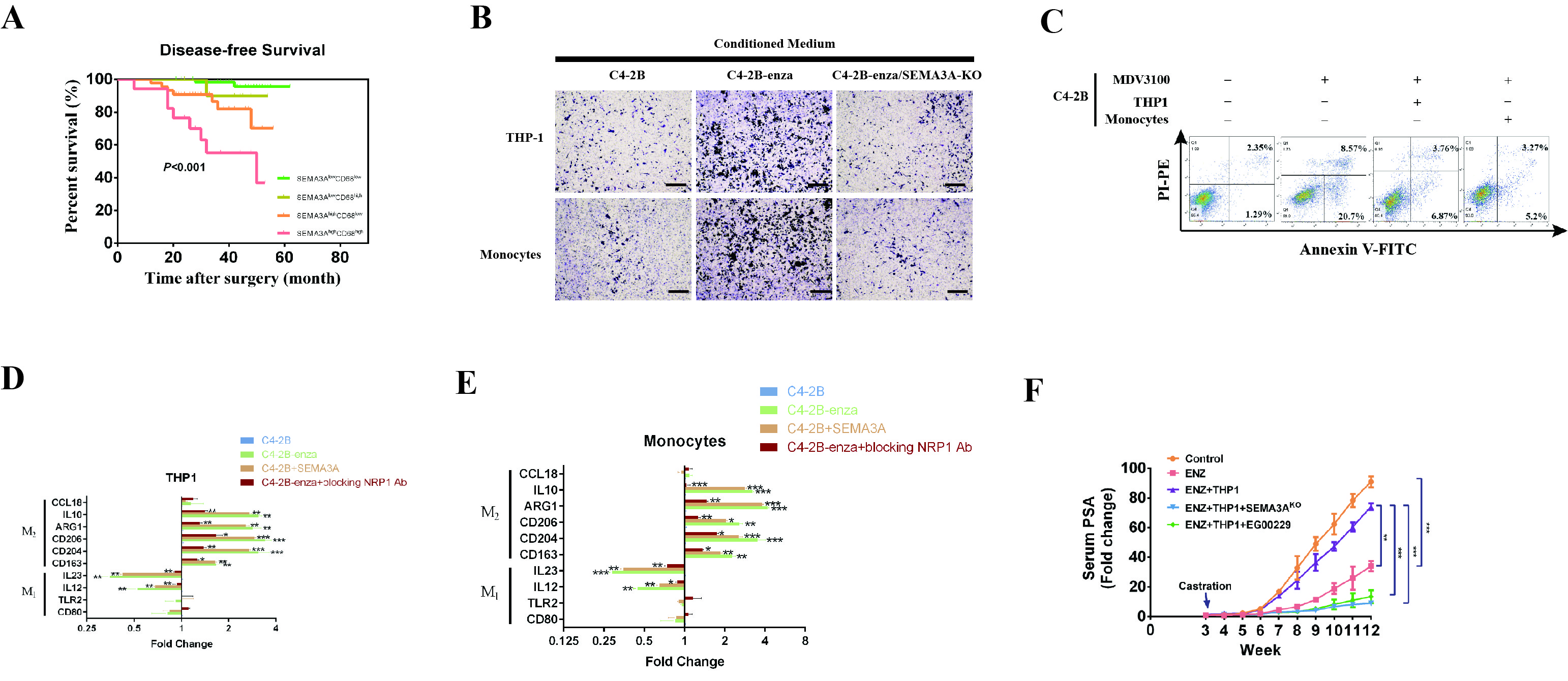
Our previous research found that the semaphorin pathway plays an important role in the progression of prostate cancer.5 RNA collected from prostate cancer patients before and after ADT was extracted to perform quantitative real-time PCR experiments. The results showed that most patients (7/9) had significantly increased SEMA3A transcription levels after ADT (Fig. 1A), which confirmed that SEMA3A was linked with the progression of CRPC. Since TAMs are also associated with CRPC,6–8 we examined the relationship between SEMA3A and TAMs. Tissue samples were collected for 25 cases of HSPC and 25 cases of CRPC to perform an IHC experiment. The results showed that with the development of prostate cancer, the expression of SEMA3A was positively correlated with that of the TAM marker CD68 (Fig. 1B, C). To explore the impact of this positive correlation on patient prognosis, we investigated the expression of these markers in 140 consecutive samples of prostate cancer, which were divided into four groups according to the levels of SEMA3A and CD68 expression. The results revealed that biochemical recurrence and disease-free survival times were significantly lower in the SEMA3AhighCD68high group than in the other three groups (Fig. 1D and Fig. S1A), indicating that coexpression of SEMA3A and CD68 was negatively correlated with patient prognosis.
Fig. 1.

Cancer cells interact with tumor-associated macrophages through SEMA3A. A Quantitative real-time PCR (qRT-PCR) analysis of the expression of SEMA3A in nine prostate cancer tissue samples collected before and after androgen deprivation therapy (ADT). Data are expressed as the fold change relative to SEMA3A expression before ADT. B, C Representative IHC staining (scale bar = 50 μm, n = 5/group, red arrows indicate CD68-positive cells) of hormone-sensitive prostate cancer (HSPC) and castration-resistant prostate cancer (CRPC) and the correlation between IHC scores for SEMA3A and CD68. D Kaplan–Meier curves for biochemical recurrence (BCR) in prostate cancer patients were analyzed according to combined SEMA3A and CD68 expression (n = 140). E Cell counts based on migration assays performed with THP1 cells or monocytes sorted from healthy donors in the presence of conditioned medium (CM) from C4-2B, enzalutamide-treated C4-2B (C4-2B-enza) or SEMA3AKO C4-2B cells. Expression of M1- and M2-related genes in THP1 cells (F) or sorted monocytes (G) in the presence of CM from C4-2B, C4-2B-enza or C4-2B-enza/SEMA3AKO cells. Cell counts based on migration assays performed with THP1 cells (H) or monocytes (I) in the presence of CM from C4-2B cells, CM from C4-2B-enza cells, a recombinant human SEMA3 Fc chimeric protein or an NRP1 inhibitor. Western blotting was performed to investigate the expression of p-PI3K, PI3K, AKT1, p-ERK1/2, ERK1/2, and IL-10 in THP1 cells (J) or monocytes (K) in the presence of CM from C4-2B cells, CM from C4-2B-enza cells, the SEMA3/Fc or the NRP1 inhibitor. Beta-actin served as the internal control. L C4-2B cells cultured with or without THP1 cells in the presence of SEMA3AKO cells or the NRP1 inhibitor EG00229 (500 µg/mL) were injected into the prostate glands of NOD/SCID mice subjected to castration/enzalutamide (ENZ) treatment. The mice were sacrificed at the 12th week after injection. M Photon flux was examined in different groups of mice. The results are presented as the fold increase in tumor growth over time. *p < 0.05; **p < 0.01; ***p < 0.001; and NS not significant
The above results showed that SEMA3A was coexpressed with TAMs and negatively correlated with prognosis. To further clarify the relationship between SEMA3A and TAMs, SEMA3A was knocked out utilizing Cas9 technology in the enzalutamide-resistant cell line C4-2B-enza. Then, THP1 cells and monocytes were stimulated with three (C4-2B, C4-2B-enza and C4-2B-enza/SEMA3A-KO) cell culture supernatants. A cell migration experiment was conducted to examine the ability of SEMA3A to recruit either THP1 cells or monocytes. As shown in Fig. 1E and Fig. S1B, C4-2B-enza cells promoted enhanced migration of THP1 cells and monocytes compared with C4-2B cells. However, when SEMA3A was knocked out, this enhanced migration was weakened. In addition, the results of qPCR experiments showed that the supernatant of the C4-2B-enza group could downregulate the expression of M1 markers in THP1 cells or monocytes and upregulate that of M2 markers in these cells compared to that of the C4-2B group. However, when SEMA3A was knocked out, this regulation was diminished (Fig. 1F, G). These results suggested that tumor cells could affect the migration and differentiation of THP1 cells or monocytes and that SEMA3A might participate in this process. A coculture model was constructed to investigate the effect of THP1 cells or monocytes on cancer cells. The proportion of apoptotic cells in cocultures with THP1 cells or monocytes and enzalutamide was significantly lower than that in control cultures (Fig. S1C). Thus, experimental data showed that SEMA3A secreted by prostate cancer cells could recruit monocytes and differentiate them toward the M2 subtype, which in turn promoted drug resistance in tumor cells.
To further study the specific mechanism of the action of tumor cells on TAMs, we addressed the role of the SEMA3A receptor NRP1.9 In a stimulation experiment with cell culture supernatant, it was found that C4-2B-enza cells could promote a higher rate of THP1 cell migration than C4-2B cells. Interestingly, when a SEMA3A/Fc fragment was added to the C4-2B group, this group could also promote the migration of THP1 cells or monocytes similar to the C4-2B-enza group, demonstrating the effect of SEMA3A. However, the role of C4-2B-enza cells in promoting cell migration was attenuated when an NRP1 inhibitor was added to the supernatant (Fig. 1H, I). To further investigate the role of NRP1, M1 and M2 markers were examined using qPCR. The results showed that the C4-2B-enza and C4-2B plus SEMA3A groups were able to downregulate the transcriptional level of the M1 marker and increase the level of the M2 marker, while this regulatory effect was reduced when the NRP1 inhibitor was added to the supernatant (Fig. S1D, E). In addition, in a western blot experiment, we also found that the phosphorylation levels of PI3K, AKT1, and ERK1/2 in THP1 cells or monocytes cocultured with the C4-2B-enza group were significantly increased compared with those in corresponding cells cocultured with the C4-2B group. Moreover, the addition of the SEMA3A/Fc could also promote SEMA3A phosphorylation. In contrast, the phosphorylation level was downregulated when the NRP1 inhibitor was added (Fig. 1J, K).
To further verify the earlier experimental results, in vivo animal treatment experiments were conducted using the SEMA3A-knockout cell line and NRP1 inhibitor EG00229. It was found that compared with the control group, the enzalutamide group exhibited a significantly reduced tumor growth rate and blood PSA level. After coculture with THP1 cells, the tumor growth rate and blood PSA level were significantly increased. However, regardless of whether SEMA3A was knocked out or NRP1 was inhibited by EG00229, the growth rate and blood PSA level were significantly decreased (Fig. 1L, M; Fig. S1F). Animal treatment experiments showed that in CRPC models, the interaction between tumor cells and TAMs was closely related to SEMA3A and NRP1. Therapy targeting SEMA3A or NRP1 might inhibit tumor progression. Thus, we discovered that blocking SEMA3A/NRP1 can reduce the tumor growth rate and reverse castration resistance.
Supplementary information
{kind=link}
Acknowledgements
The National Natural Science Foundation of China (81472397 to S.R. and 81773154 to C.W.). The Top-level Clinical Discipline Project of Shanghai Pudong (PWYgf2018-03, C.W.). The Shanghai Natural Science Foundation (20ZR1449600, C.W.). The Leadership Program in Science and Technology of Beijing Municipal Science and Technology Commission (181100006318007, N.X.).
Competing interests
The authors declare no competing interests.
Contributor Information
Fei Liu, Email: liufei_2359@163.com.
Shancheng Ren, Email: renshancheng@gmail.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-021-00637-4.
References
- 1.Velez EM, et al. Comparative prognostic implication of treatment response assessments in mCRPC: PERCIST 1.0, RECIST 1.1, and PSA response criteria. Theranostics. 2020;10:3254–3262. doi: 10.7150/thno.39838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillessen S, et al. Management of patients with advanced prostate cancer: report of the advanced prostate cancer consensus conference 2019. Eur. Urol. 2020;77:508–547. doi: 10.1016/j.eururo.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Di Mitri D, et al. Re-education of tumor-associated macrophages by CXCR2 blockade drives senescence and tumor inhibition in advanced prostate cancer. Cell Rep. 2019;28:2156–2168.e5. doi: 10.1016/j.celrep.2019.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casazza A, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Ren S, et al. Whole-genome and transcriptome sequencing of prostate cancer identify new genetic alterations driving disease progression. Eur. Urol. 2018;73:322–339. doi: 10.1016/j.eururo.2017.08.027. [DOI] [PubMed] [Google Scholar]
- 6.Wang C, et al. Blocking the feedback loop between neuroendocrine differentiation and macrophages improves the therapeutic effects of enzalutamide (MDV3100) on prostate cancer. Clin. Cancer Res. 2018;24:708–723. doi: 10.1158/1078-0432.CCR-17-2446. [DOI] [PubMed] [Google Scholar]
- 7.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tse BWC, et al. Neuropilin-1 is upregulated in the adaptive response of prostate tumors to androgen-targeted therapies and is prognostic of metastatic progression and patient mortality. Oncogene. 2017;36:3417. doi: 10.1038/onc.2016.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.