

Abstract
Sirtuin 3 (SIRT3) is a potential therapeutic target for cardiovascular, metabolic, and other aging-related diseases. In this study, we investigated the role of SIRT3 in diabetic cardiomyopathy (DCM). Mice were injected with streptozotocin (STZ, 60 mg/kg, ip) to induce diabetes mellitus. Our proteomics analysis revealed that SIRT3 expression in the myocardium of diabetic mice was lower than that of control mice, as subsequently confirmed by real-time PCR and Western blotting. To explore the role of SIRT3 in DCM, SIRT3-knockout mice and 129S1/SvImJ wild-type mice were injected with STZ. We found that diabetic mice with SIRT3 deficiency exhibited aggravated cardiac dysfunction, increased lactate dehydrogenase (LDH) level in the serum, decreased adenosine triphosphate (ATP) level in the myocardium, exacerbated myocardial injury, and promoted myocardial reactive oxygen species (ROS) accumulation. Neonatal rat cardiomyocytes were transfected with SIRT3 siRNA, then exposed to high glucose (HG, 25.5 mM). We found that downregulation of SIRT3 further increased LDH release, decreased ATP level, suppressed the mitochondrial membrane potential, and elevated oxidative stress in HG-treated cardiomyocytes. SIRT3 deficiency further raised expression of necroptosis-related proteins including receptor-interacting protein kinase 1 (RIPK1), RIPK3, and cleaved caspase 3, and upregulated the expression of inflammation-related proteins including NLR family pyrin domain-containing protein 3 (NLRP3), caspase 1 p20, and interleukin-1β both in vitro and in vivo. Collectively, SIRT3 deficiency aggravated hyperglycemia-induced mitochondrial damage, increased ROS accumulation, promoted necroptosis, possibly activated the NLRP3 inflammasome, and ultimately exacerbated DCM in the mice. These results suggest that SIRT3 can be a molecular intervention target for the prevention and treatment of DCM.
Keywords: Sirtuin 3, diabetic cardiomyopathy, necroptosis, mitochondria, ROS, NLRP3, RIPK3
Introduction
Diabetic cardiomyopathy (DCM) is characterized by structural abnormalities and cardiac dysfunction independent of coronary artery disease, hypertension, or valvular heart disease [1, 2]. The potential pathogenesis of DCM may be associated with hyperglycemia, hyperinsulinemia, increases in circulating fatty acids and triacylglycerols, endothelial dysfunction, microangiopathic disorder, autonomic neuropathy, and so on [3]. In addition, evidence suggests that inflammation and reactive oxygen species (ROS) overproduction, mitochondrial dysfunction, long-term autophagy activation, apoptosis, calcium disorders, glycation, and fatty acid metabolism abnormalities may play key roles in DCM [4–8]. Currently, it is believed that high-glucose-induced metabolic imbalances, oxidative stress, and cumulative inflammation are common characteristics of the initial stage of DCM.
Sirtuin 3 (SIRT3) is a member of the class III histone deacetylases dependent on nicotinamide adenine dinucleotide (NAD+), and primarily resides in mitochondria [9–11]. Previous studies confirmed that SIRT3 is involved in cell energy metabolism, oxidative stress, the electron transport chain, the mitochondrial membrane potential, and fatty acid oxidation [12, 13]. SIRT3 deletion is likely to induce cardiovascular diseases, liver diseases, renal diseases, metabolic diseases, tumors, age-related hearing loss, and neurological diseases [14–18]. These outcomes suggest that SIRT3 is a potential therapeutic target for cardiovascular, metabolic and other aging-related diseases. However, the detailed mechanism of SIRT3 in DCM has not been fully clarified.
Necroptosis, as a programmed cell death pathway, is different from classical necrosis and apoptosis. Necroptosis typically causes cell membrane rupture, organelle swelling, and the subsequent massive release of intracellular contents [19, 20]. Necroptosis is mainly regulated by receptor-interacting protein kinase 1 (RIPK1) and RIPK3 [21–23]. Mixed lineage kinase domain-like protein (MLKL) has been identified as a key downstream enzyme of RIPK3 in the necroptosis pathway. Necroptosis has been reported to participate in atherosclerosis, myocardial ischemia-reperfusion injury, myocardial hypertrophy, myocardial infarction, and aortic aneurysms [24–26]. However, whether SIRT3 deficiency affects necroptosis in DCM is unknown.
Necroptosis, considered a cell death mode associated with high inflammation levels, may activate inflammasomes and promote inflammatory cell recruitment [27, 28]. As an important component of the immune response, NLR family pyrin domain-containing protein 3 (NLRP3) is activated to recruit procaspase 1 and then promote procaspase 1 hydrolysis, resulting in the dissociation of large and small active subunits (p20 and p10) and the formation of a tetramer, namely, activated caspase 1. Active caspase 1 induces the maturation and secretion of proinflammatory cytokines, including pro-interleukin (IL)-1β and pro-IL-18 [29]. It is unclear whether SIRT3 deficiency affects the development of DCM via the NLRP3 inflammasome.
Our study aimed to determine whether SIRT3 deficiency exacerbates DCM to further clarify the pharmacological role and possible mechanisms of SIRT3 in DCM and to provide a novel target for the prevention and treatment of DCM.
Materials and methods
Animal treatment
Eight-week-old male C57BL/6 mice, wild-type (WT) 129S1/SvImJ mice, and SIRT3-knockout (KO) mice were randomly allocated into a diabetes mellitus (DM) group and a control group. After fasting for 12 h, fasting blood glucose (FBG) levels were measured. Then, the mice in the DM group and control group were intraperitoneally injected with streptozotocin (STZ, Sigma-Aldrich, St. Louis, MO, USA; 60 mg/kg) in freshly prepared sodium citrate buffer (0.1 mol/L, pH 4.4) or the same amount of sodium citrate buffer once daily. After 5 days of STZ administration, FBG was measured from the tail vein using a One-Touch blood glucose meter (Ultra Vue, Johnson, New Brunswick, NJ, USA). Mice with FBG levels higher than 16.7 mmol/L were considered diabetic and were chosen for further experiments with FBG monitoring every 2 weeks [30, 31].
Animal experiments were performed in accordance with the NIH guidelines for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Committee of Nantong University (approval no. S-20180323-012).
Proteomics analysis
Twelve weeks after the STZ injection, relative quantification of the proteins in the myocardium of the mice was measured using tandem mass tag mass spectrometry, according to a previous description [32]. A heat map analysis was used to verify the differences in protein levels in the myocardium of the diabetic mice and control mice.
Echocardiography assessment
Twelve weeks after the STZ injection, all the mice were anesthetized with 1.5% isoflurane. The cardiac configuration was observed by an ultrasonic diagnostic apparatus (Visual Sonic Vevo 2100, Toronto, ON, Canada) with a probe frequency of 30 MHz to obtain the parasternal long-axis view. Cardiac images generated with two-dimensional M-mode echocardiography were recorded. Ejection fraction (EF) and fractional shortening (FS) were calculated to evaluate systolic function. Diastolic function was assessed by the ratio of early diastolic peak (E) to late diastolic peak (A) of mitral valve blood flow as measured by pulse Doppler ultrasound.
Hematoxylin–eosin staining
The left ventricles of the myocardia were collected and fixed in 4% paraformaldehyde for 24 h. Then, paraffin-embedded tissue sections with a thickness of 5 μm were dewaxed by two treatments of dimethyl benzene and then rehydrated by ethanol in a gradient from high to low concentrations. After staining with hematoxylin for 5 min, the sections were immersed in 0.25% ammonia solution for 30 s, gradient ethanol dehydration, eosin staining for 1 min, dimethyl benzene for 5 min, and finally successive periods of neutral gum sealing. The pathological structures of the myocardium were observed under a microscope.
Transmission electron microscope (TEM) examination
Small pieces of myocardium, ~1 mm3, were fixed overnight in 2.5% glutaraldehyde fixative solution at 4 °C followed by 1% osmium tetroxide treatment for 2 h at 4 °C. The samples were dehydrated, infiltrated, embedded in Epon812, and cut into ultrathin sections of 70 nm followed by staining with uranyl acetate and lead citrate. The ultrastructures of the myocardium were observed with a TEM (HT7700, HITACHI, Japan).
Dihydroethidium (DHE) staining
The left ventricular myocardium was embedded with optimal cutting temperature compound (Sakura Finetek, Japan) and frozen in a freezer at −80 °C. After the tissue was cut into 4 μm sections, the slices were incubated with DHE (2 μM, Beyotime, Shanghai, China) for 30 min at 37 °C and stained with DAPI (Beyotime, Shanghai, China) for 5 min. The superoxide anion production in the myocardium was assessed by DHE fluorescence intensity with a laser confocal microscope (Leica, Wetzlar, Germany).
Neonatal cardiomyocyte culture
Sprague-Dawley rats (1–3 days old) were soaked in iodophor and disinfected with alcohol. After decapitation, the heart was quickly excised and immediately washed in precooled phosphate-buffered saline to remove the blood. Subsequently, the heart tissues were minced into fragments and digested in 0.25% trypsin (Beyotime, Shanghai, China) for 10–12 cycles at 37 °C until the tissues were completely digested. All supernatants from the digested tissues, except the first supernatant, were filtered through a 200-mesh sieve and centrifuged at 1200 rpm for 5 min. The cells were resuspended in culture medium with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA) and cultured in an incubator with 5% CO2 at 37 °C for 4 h, and the supernatant was carefully transferred to a new culture container to remove cardiac fibroblasts and continued to be cultured in the incubator.
SIRT3 siRNA interference
After the cardiomyocytes reached 70%–80% confluency, the culture medium was replaced by medium without FBS or antibiotics for 12 h. Specific double-stranded RNA oligonucleotides targeting SIRT3 (SIRT3 siRNA, sense, 5′-CCAUCUUUGAACUAGGCUUTT-3′, and antisense, 5′-AAGCCUAGUUCAAAGAUGGTT-3′; Sangon Biotech, Shanghai, China) and nonspecific control RNA (NC siRNA, sense, 5′-UUCUCCGAACGUGUCACGUTT-3′, and antisense, 5′-ACGUGACACGUUCGGAGAATT-3′) were transfected into the cardiomyocytes with Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA). After 4 h, the culture medium was exchanged for medium with a normal glucose level (5.5 mmol/L, NG) or high-glucose level (25.5 mmol/L, HG) and 10% FBS.
Lactate dehydrogenase (LDH) and adenosine triphosphate (ATP) measurements
LDH levels in the serum and cell culture medium and the ATP levels in the myocardium and cardiomyocytes were determined according to the instructions of the respective commercial kit (Beyotime, Shanghai, China).
TdT-mediated dUTP nick end labeling (TUNEL) staining
The myocardium was fixed overnight with 4% paraformaldehyde, embedded in paraffin, and then cut into sections with a thickness of 5 μm. After staining with TUNEL, the sections were observed and photographed under an optical microscope.
After fixation with polyformaldehyde for 30 min, the cardiomyocytes were stained with fluorescein-dUTP in the presence of terminal deoxynucleotidyl transferase (TdT) according to the instructions of a One-Step TUNEL apoptosis assay kit (Beyotime, Shanghai, China), and the apoptotic cardiomyocytes, which fluoresced green, were observed with a laser confocal microscope.
Mitochondrial membrane potential (Δψm) measurement in the cardiomyocytes
The Δψm of the cardiomyocytes was measured with a JC-1 kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Briefly, the cardiomyocytes were stained for 20 min with JC-1 working solution in the dark at 37 °C and then gently washed with JC-1 staining buffer. Afterward, DAPI was added for nuclear staining. Finally, the cells were observed and photographed by a laser confocal microscope.
Mitochondrial superoxide (MitoSOX) evaluation in the cardiomyocytes
The level of superoxide in mitochondria was measured by a MitoSOX probe that fluoresces red. Briefly, the primary cardiomyocytes were incubated for 20 min with MitoSOX red (5 μM, Yeasen, Shanghai, China) and MitoTracker green or red (100 nM, Beyotime, Shanghai, China) at 37 °C without light. Then, DAPI was added to stain the nuclei for 5 min. Finally, the cells were observed and photographed by a laser confocal microscope.
Immunofluorescence staining
The cardiomyocytes were fixed for 20 min with immunofluorescent fixation solution and incubated for 60 min with blocking solution containing 0.5% Triton X-100. Next, the cells were incubated overnight with the desired primary anti-RIPK1, anti-RIPK3 (1:100, Cell Signaling Technology, Danvers, MA, USA), anti-NLRP3 (1:200, AdipoGen, San Diego, CA, USA), or anti-caspase 1 (1:200, Abcam, Cambridge, UK) antibody at 4 °C, followed by incubation for 2 h with IgG conjugated with Cy3 or Alexa Fluor 488 (1:500, Beyotime, Shanghai, China) at room temperature, and DAPI was added to stain the nuclei. Finally, the fluorescence was observed with a laser confocal microscope.
Quantitative real-time PCR
Total RNA of the myocardium and cardiomyocytes was extracted with TRIzol. After reverse transcription, the cDNA was amplified by SYBR Green qPCR mixture (Takara, Otsu, Shiga, Japan) and by real-time PCR systems (ABI 7500, Carlsbad, CA, USA). The primers for mouse SIRT3 mRNA (F, 5′-GGATTCGGATGGCGCTTGA-3′ and R, 5′-GCTTCCTCTAGTGACACTGTTAG-3′) and rat SIRT3 mRNA (F, 5′-GAGGTTCTTGCTGCATGTGGTTG-3′ and R, 5′-AGTTTCCCGCTGCACAAGGTC-3′) were provided by Sangon Biotech Co., Ltd. (Shanghai, China) with 18S (F, 5′-AGTCCCTGCCCTTTGTACACA-3′ and R, 5′-CACCTGTAACACTCCCGGAC-3′) used as a housekeeping gene. Relative mRNA expression was calculated as the fold based on the expression of the control.
Western blotting
Protein samples from the myocardium and cardiomyocytes were extracted with lysis buffer containing 1% phenylmethylsulfonyl fluoride. Next, the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk for 2 h at room temperature and then incubated overnight with the relevant primary antibody, namely, anti-RIPK1, anti-RIPK3, anti-caspase 3, anti-cleaved caspase 3, anti-MLKL, anti-p-MLKL, and anti-SIRT3 (1:1000, Cell Signaling Technology, Danvers, MA, USA); anti-NLRP3 (1:1000, AdipoGen, San Diego, CA, USA); anti-caspase 1 (1:200) and anti-IL-1β (1:1000) (Abcam, Cambridge, UK); or anti-β-tubulin (1:3000, CMCTAG, Milwaukee, WI, USA) and anti-GAPDH (1:5000, Sigma-Aldrich, St. Louis, MO, USA) on a shaker at 4 °C. Subsequently, the membranes were incubated for 2 h with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000, Zhongshan Jinqiao, Beijing, China) at room temperature. Enhanced chemiluminescence (Thermo Fisher Scientific Inc., Rockford, IL, USA) was applied to the membrane to visualize the protein bands, which were quantitatively analyzed using ImageJ software.
Statistical analysis
All the data are presented as the means ± standard error of the mean (SEM), based on statistical analysis by one-way ANOVA followed by Bonferroni post hoc test. P values less than 0.05 were considered significant.
Results
SIRT3 expression is decreased in the myocardium of the STZ-induced diabetic mice
Twelve weeks after STZ injection, several major differential proteins were identified by comparative proteomic analysis of the myocardium between diabetic mice and control mice. Through heat map analysis, SIRT3 expression in the myocardium of diabetic mice was lower than that of control mice (Fig. 1a). Further real-time PCR and Western blotting measurements also confirmed less SIRT3 expression in the myocardium of diabetic mice (both P < 0.01, Fig. 1b, c). These data suggested that the SIRT3 pathway was impaired in the myocardium with diabetes. Consequently, SIRT3-KO mice were used to investigate the real role of SIRT3 in DCM.
Fig. 1. SIRT3 expression is decreased in the myocardium of the STZ-induced diabetic mice.

Eight-week-old male C57BL/6 mice were injected intraperitoneally daily with STZ (60 mg/kg, DM group) or sodium citrate buffer (control group) for 5 days. a After 12 weeks, quantification of the relative protein in the myocardium in the mice was measured using tandem mass tag (TMT) mass spectrometry, and a heat map was generated. b SIRT3 mRNA expression in the myocardium was measured by real-time PCR. c SIRT3 protein expression in the myocardium was detected by western blotting. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. **P < 0.01 vs control, n = 5.
SIRT3 deficiency aggravates cardiac dysfunction in the diabetic mice
There was no difference in the FBG level of all mice before STZ administration (P > 0.05). Three days after STZ injection, the FBG levels in both WT mice and SIRT3-KO mice were more than 16.7 mmol/L, indicating that DM was successfully induced. All diabetic mice remained hyperglycemic throughout the study (Fig. 2a, b). It is worth noting that there was no difference in blood glucose levels between WT mice and SIRT3-KO mice in the DM group, suggesting that SIRT3 deficiency did not further alter FBG in diabetic mice.
Fig. 2. SIRT3 deficiency aggravates cardiac dysfunction in the diabetic mice.

Eight-week-old male wild-type (WT) 129S1/SvImJ mice and SIRT3-knockout (SIRT3-KO) mice were injected intraperitoneally daily with STZ (60 mg/kg, DM group) or sodium citrate buffer (control group) for 5 days. a, b The level of fasting blood glucose (FBG) was detected at different time points. c After 12 weeks, typical two-dimensional M-mode echocardiography and pulse Doppler ultrasound measurements were recorded. d Ejection fraction (EF) and fractional shortening (FS) were calculated. e The ratio of the early diastolic peak (E) to the late diastolic peak (A) of mitral valve blood flow was measured. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. *P < 0.05, **P < 0.01 vs the control group of the same genotype; ##P < 0.01 vs the DM group of WT mice, n = 8.
Twelve weeks after STZ administration, the EF, FS, and E/A ratio of the mice in the DM group were lower than those in the control group (P < 0.05), indicating impaired diastolic and systolic functions in diabetic mice. This result suggested that the mice after STZ administration suffered from serious DCM in the present study. Compared with the DM of WT mice, the EF, FS, and E/A ratio in the DM of SIRT3-KO mice decreased (P < 0.01, Fig. 2c–e). This result indicated that SIRT3 deficiency aggravated cardiac systolic and diastolic dysfunction in diabetic mice.
SIRT3 deficiency exacerbates myocardium injury in the diabetic mice
Compared with those in the control mice, the cardiomyocytes in the diabetic mice exhibited disordered arrangement with a large number of vacuoles and distortions, indicating that the myocardial structures of the diabetic mice were destroyed. Compared with the WT diabetic mice, the disorder of the myocardial structure in the SIRT3-KO diabetic mice was more profound (Fig. 3a).
Fig. 3. SIRT3 deficiency exacerbates myocardial injury in the diabetic mice.

After 12 weeks, the left ventricle of the myocardium was collected. a The sample was stained with HE and photographed. Bar = 50 μm. b The ultrastructure of the myocardium was examined with transmission electron microscopy. Bar = 2 μm (upper) and 1 μm (lower). c Lactate dehydrogenase (LDH) level in the serum was detected. d Adenosine triphosphate (ATP) level in the myocardium was measured. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. **P < 0.01 vs the control group of the same genotype; #P < 0.05, ##P < 0.01 vs the DM group of WT mice, n = 8.
The ultrastructural examination revealed that the myocardial mitochondria in the diabetic mice were obviously swollen and arranged in a disorderly manner, and the cristae became increasingly condensed, indicating that the myocardial mitochondria were damaged. Compared with the mitochondria in the WT diabetic mice, the ultrastructural disorder of myocardial mitochondria in SIRT3-KO diabetic mice was more serious (Fig. 3b).
We also found that the LDH level was increased in the serum, while the ATP level decreased in the myocardium of the diabetic mice (P < 0.01). Compared with the WT mice with DM, the LDH level in the SIRT3-KO mice with DM was remarkably increased (P < 0.01, Fig. 3c), but ATP level was further suppressed (P < 0.05, Fig. 3d).
SIRT3 deficiency promotes myocardial necroptosis in the diabetic mice
ROS accumulation is a dominant factor that induces necroptosis, which might attenuate cardiomyocyte survival and aggravate myocardial injury [19, 33]. We found that the intensity of the DHE staining of the myocardium was enhanced in the mice with DM, which was further increased in the diabetic SIRT3-KO mice (Fig. 4a).
Fig. 4. SIRT3 deficiency promotes myocardial necroptosis in the diabetic mice.

After 12 weeks, the left ventricle of the myocardium was collected. a The level of superoxide anion in the myocardium was measured by DHE fluorescence probe. Bar = 75 μm. b The rate of apoptosis was assessed by TUNEL staining. Bar = 50 μm. c–f The protein expression levels of RIPK1, RIPK3, caspase 3, and MLKL in the myocardium were detected by Western blotting. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. **P < 0.01 vs the control group of the same genotype; ##P < 0.01 vs the DM group of WT mice, n = 6.
Next, apoptosis, a critical manifestation of necroptosis, was determined after 12 weeks. There were more brownish yellow nuclei in the TUNEL-stained myocardium of the DM mouse group than in the control mouse group. Compared with the WT diabetic mice, the number of positive cells in the SIRT3-KO diabetic mice was further increased (Fig. 4b).
RIPK1 and RIPK3 play critical roles in the activation of necroptosis. Caspase 3 activation is a typical characteristic of necroptosis, and MLKL is a key downstream substrate of RIPK3 [33]. Therefore, these proteins are regarded as sensitive and robust markers of necroptosis. Our present study revealed that the expression levels of RIPK1, RIPK3, and cleaved caspase 3 increased in the myocardium of both the WT and SIRT3-KO mice with DM (P < 0.01). Compared with the WT diabetic mice, the expression of the above three proteins in the myocardium of the SIRT3-KO diabetic mice was increased (P < 0.01, Fig. 4c–e). However, no significant difference in the levels of MLKL and phosphorylated MLKL was found in the myocardium of mice in either group (P > 0.05, Fig. 4f). These data suggested that SIRT3 deficiency promoted necroptosis in the myocardium of the diabetic mice.
SIRT3 silencing exacerbates cell injury and enhances oxidative stress in the high-glucose-stimulated cardiomyocytes
To verify the role and mechanism of SIRT3 in diabetic myocardial injury, primary cultured cardiomyocytes were studied in vitro. After SIRT3 siRNA transfection, both SIRT3 mRNA and protein expression levels were decreased (P < 0.01, Fig. 5a, b).
Fig. 5. SIRT3 silencing exacerbates cell injury and enhances oxidative stress in the high-glucose-stimulated cardiomyocytes.

SIRT3 siRNA and nonspecific control (NC) siRNA were transfected into cardiomyocytes. a, b After 48 h, SIRT3 mRNA and protein expression levels were measured by real-time PCR and Western blotting, respectively. **P < 0.01 vs the NC group, n = 6. c After 4 h, the cardiomyocytes were stimulated with normal glucose (5.5 mmol/L, NG) or high glucose (25.5 mmol/L, HG) for 48 h. LDH level in the medium was measured. d ATP levels in cardiomyocytes were measured. e The mitochondrial membrane potential (Δψm) of the cardiomyocytes was measured with JC-1 staining. Bar = 50 μm. f Mitochondrial superoxide was detected with MitoSOX. Mitochondrial localization of the MitoSOX signal was confirmed by MitoTracker green. Bar = 75 μm. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. **P < 0.01 vs the NG-treated cardiomyocytes transfected with the same siRNA; ##P < 0.01 vs the NC + HG-treated cardiomyocytes, n = 8.
Then, LDH release and ATP levels were detected to evaluate the degree of cell injury in the high-glucose-stimulated cardiomyocytes after SIRT3 silencing. Compared with the NG group, LDH release was increased, while the ATP levels decreased in the HG group, which was intensified after SIRT3 siRNA transfection (P < 0.01, Fig. 5c, d), suggesting that SIRT3 silencing exacerbated cell injury caused by high-glucose stimulation.
Energy metabolism abnormalities are likely to impair the structure and function of mitochondria, as indicated by a decrease in Δψm [9, 10]. Our study found, the green fluorescence intensity of the JC-1 monomer, indicating a decreased Δψm, increased after HG stimulation. The red fluorescence intensity of the JC-1 aggregates, indicating normal membrane potential, decreased. Moreover, SIRT3 silencing further increases the green fluorescence intensity but diminished the red fluorescence intensity (Fig. 5e). These data demonstrated that SIRT3 silencing decreased Δψm after high-glucose stimulation.
Studies have shown that a decrease in Δψm might lead to ROS accumulation and cell damage [9, 10, 34]. There was stronger fluorescence intensity in MitoSOX after high-glucose stimulation, which was further enhanced after SIRT3 silencing (Fig. 5f).
Taken together, the data indicated that SIRT3 silencing exacerbated cell injury and enhanced the oxidative stress in high-glucose-stimulated cardiomyocytes.
SIRT3 silencing promotes necroptosis in the high-glucose-stimulated cardiomyocytes
Apoptosis was also determined in cardiomyocytes after high-glucose exposure. TUNEL staining showed that high-glucose levels increased the number of positive cells after HG stimulation, which were further increased in the high-glucose-stimulated cardiomyocytes after SIRT3 siRNA transfection (Fig. 6a). This result suggested that the downregulation of SIRT3 increased cardiomyocyte apoptosis upon high-glucose stimulation.
Fig. 6. SIRT3 silencing promotes necroptosis in the high-glucose-stimulated cardiomyocytes.

SIRT3 siRNA and nonspecific control (NC) siRNA were transfected into cardiomyocytes. After 4 h, the cardiomyocytes were stimulated with normal glucose (5.5 mmol/L, NG) or high glucose (25.5 mmol/L, HG) for 48 h. a The rate of apoptosis was assessed by TUNEL staining. Bar = 75 μm. b, c RIPK1 and RIPK3 were stained with immunofluorescent Alexa Fluor 488 (green)-conjugated IgG. The nuclei were stained using DAPI (blue). Bar = 25 μm. d–g The protein expression levels of RIPK1, RIPK3, caspase 3, and MLKL in the cardiomyocytes were measured by Western blotting. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. *P < 0.05, **P < 0.01 vs NG-treated cardiomyocytes transfected with the same siRNA; ##P < 0.01 vs NC + HG-treated cardiomyocytes, n = 6.
Both immunofluorescence staining and Western blotting showed that the expression levels of RIPK1, RIPK3, and cleaved caspase 3 were increased after high-glucose stimulation and were further enhanced after SIRT3 was silenced (P < 0.05 or P < 0.01, Fig. 6b–f). There was no significant difference in the levels of MLKL and phosphorylated MLKL in either group (P > 0.05, Fig. 6g). All these data suggested that SIRT3 silencing promoted necroptosis in the high-glucose-stimulated cardiomyocytes.
SIRT3 deficiency promotes NLRP3 activation in the myocardium of the diabetic mice and high-glucose-stimulated cardiomyocytes
When cardiomyocytes undergo necroptosis, much of the cell contents leaks out, and damage-associated molecular patterns (DAMPs) are released, which directly induce the immune response and NLRP3 activation. The NLRP3 inflammasome is a key participant in the pathological progression of several diseases [35–37]. We found that the expression levels of NLRP3, caspase 1 p20, and IL-1β in the myocardium of the diabetic mice were higher than those in the control group mice (P < 0.05 or P < 0.01), indicating that inflammasome activation was promoted in mice with DCM. Compared with the WT diabetic mice, the levels of these proteins in the myocardium of the SIRT3-KO diabetic mice were further increased (P < 0.05, Fig. 7a–c).
Fig. 7. SIRT3 deficiency promotes NLRP3 activation in the myocardium of the diabetic mice and high-glucose-stimulated cardiomyocytes.

a–c The protein expression levels of NLRP3, caspase 1, and IL-1β in the myocardium were measured by Western blotting. Significance was determined by one-way ANOVA. Data are presented as the means ± SEM. *P < 0.05, **P < 0.01 vs the control group of the same genotype; #P < 0.05 vs the DM group of WT mice, n = 6. d Mitochondrial NLRP3 was stained using immunofluorescent Alexa Fluor 488 (green)-conjugated IgG and colocalized with MitoTracker red . The nuclei were stained using DAPI (blue). Bar = 25 μm. e NLRP3 and caspase 1 were stained with immunofluorescent Alexa Fluor 488 (green)- and Cy3 (red)-conjugated IgG. The nuclei were stained using DAPI (blue). Bar = 25 μm.
Next, MitoTracker and NLRP3 staining was used to measure the recruitment of NLRP3 to mitochondria in vitro. The results showed that HG increased the NLRP3 levels in mitochondria, especially after SIRT3 siRNA transfection (Fig. 7d). The expression of caspase 1 was also verified by immunofluorescence staining (Fig. 7e). The caspase 1 fluorescence intensity in the cardiomyocytes after HG stimulation was 1.54 ± 0.35-fold that of the cardiomyocytes with NG stimulation, and this increase was further exacerbated after SIRT3 siRNA transfection (2.12 ± 0.38-fold). These results suggested that SIRT3 deficiency promoted NLRP3 activation in the myocardium of the diabetic mice and high-glucose-stimulated cardiomyocytes.
Discussion
Long-term diabetes gradually destroys cardiac structures, reduces cardiac function, and eventually leads to heart failure, which is one of the major causes of mortality in diabetic patients [38]. STZ is a highly selective islet β-cytotoxic agent that can disturb the transport of glucose, affect the function of glucokinase, promote DNA methylation and DNA strand breakage in islet cells, destroy islet β cells, and lead to insufficient insulin secretion [39]. In our present study, the dosage of STZ was sufficient to destroy most islet β cells and enhance blood glucose to a large extent. Therefore, no difference was found in the blood glucose levels of the WT mice and SIRT3-KO mice in the DM group. STZ successfully elevated blood glucose in the mice throughout the experiment. As a mitochondrial deacetylase, SIRT3 is a critical regulator of mitochondrial energy metabolism and has an important function in the heart, liver, and kidney [40, 41]. The KO of the SIRT3 gene alone did not affect blood glucose, cardiac function, and LDH or ATP levels without STZ injection. Similarly, the knockdown of SIRT3 did not affect cell injury upon normal glucose induction. SIRT3 deficiency significantly aggravated DCM after STZ injection or high-glucose induction. These findings suggest that SIRT3 has a powerful protective effect in the pathological but not in the physiological state. Growing evidence suggests that SIRT3 protects the heart from the cardiac hypertrophy and cardiac dysfunction associated with heart failure and protects cardiomyocytes from stress-induced cell death [16, 42]. SIRT3 deficiency aggravates cell injury in myocardial ischemia and delays cardiac functional recovery following myocardial ischemia, while SIRT3 overexpression protects cardiac function from myocardial ischemia-reperfusion injury [43, 44]. These results may be related to mitochondrial function, energy homeostasis and oxidative stress, but the exact mechanism remains unclear. In our present study, WT and SIRT3-KO diabetic mice were established by STZ administration, and then, the levels of oxidative stress, necroptosis and inflammation in the myocardium were measured to explore the mechanisms by which DCM was exacerbated in the SIRT3-KO mice.
Necroptosis has been implicated in the pathogenesis of nervous system degeneration, tumors, immune system disorders, viral infections, and cardio-cerebrovascular disease [19, 45, 46]. Necroptosis inhibition can reduce arterial plaque formation, attenuate ischemia-reperfusion injury, and improve ventricular remodeling [47]. RIPK1 and RIPK3 are considered two important factors in necroptosis. RIPK3 binds to RIPK1 through the RIPK-homotypic interaction motif domain to form necrosomes, which promote necroptosis [48–50]. Previous studies have confirmed that the inhibition of RIPK1 activity can reduce the infarct area after myocardial ischemia in mice [51]. In a mouse model of cardiac hypertrophy induced by abdominal aortic constriction, the expression levels of RIPK1 and RIPK3 were increased. Downregulation of RIPK1 and RIPK3 significantly inhibited necroptosis and alleviated cardiac hypertrophy [52]. MLKL is a key downstream enzyme of RIPK3. After RIPK1 binds with RIPK3 to form necrosomes, the complex recruits MLKL and then promotes MLKL phosphorylation to aggravate necroptosis [53]. Knocking out MLKL in HT-29 cells significantly prevented necroptosis [54]. Although MLKL is regarded as a key downstream enzyme of RIPK3 in the pathway of necroptosis, the changes in MLKL were found to dissimilar in different pathophysiological processes. For example, MLKL is involved in cerebral ischemia-reperfusion injury [55] and human ovarian cancer cell proliferation [56] but remains stable in myocardial ischemia-reperfusion injury [33, 57]. In our present study, the expression of RIPK1, RIPK3, and cleaved caspase 3 proteins was increased, and necroptosis was exacerbated in the myocardium of mice with DCM. More importantly, these disorders were further exacerbated in the SIRT3-KO diabetic mice, indicating that the deletion of SIRT3 aggravated necroptosis in the myocardium. However, neither diabetes induction nor SIRT3 KO had an effect on MLKL, suggesting that MLKL may not be involved in the occurrence or progression of DCM. Some other RIPK3 substrates, such as CaMKII [57], may be involved in necroptosis induced by DCM. This outcome may be attributable to differences in the course of diabetes progression, animal models and mouse strains. Similar results were also obtained by the downregulation of SIRT3 expression with SIRT3 siRNA in high-glucose-stimulated cardiomyocytes.
Necroptosis can lead to the cell rupture and extensive content leakage, including DAMPs [27]. Many studies have reported that RIPK3 was able to promote cytokine release and inflammasome formation, thus activating procaspase 1 through RIPK3-dependent pathways by caspase 8 or NLPR3 activation [58, 59]. After NLRP3 recognizes DAMPs, the pyridine domain in NLRP3 binds to the apoptosis-associated speck-like proteins containing a caspase-recruitment domain (CARD), which then binds to procaspase 1 through CARD–CARD interactions. Subsequently, procaspase 1 is converted into active caspase 1, and pro-IL-1β is cleaved by the inflammasome to become mature IL-1β, which leads to an inflammatory response [60]. Previous studies found that SIRT3 regulates inflammation in several pathophysiological processes, including diabetic wounds, mycobacterial infection, and acute lung injury [61–63]. Consistent with these results, we confirmed that SIRT3 KO can also increase the level of inflammation in the mice with DCM. SIRT3 KO indeed increased the expression level of these proteins in the myocardium of mice with DCM, and similar results were found in the cell experiments.
ROS accumulation and oxidative stress are considered to be the key factors in the occurrence and development of diabetic complications and may also be the most prominent initiation signals in the pathological cascade of the diabetic vascular system [64]. There is evidence that ROS production during myocardial hypoxia-reoxygenation injury leads to necroptosis. Furthermore, necroptosis can also aggravate ROS production [65], suggesting that necroptosis and ROS levels may mutually form a vicious cycle. Our study found that SIRT3 KO significantly increased the levels of ROS in the myocardium of the mice with DCM, and the downregulation of SIRT3 increased the intracellular ROS of cardiomyocytes stimulated by high glucose. Intracellular ROS could induce the opening of the mitochondrial permeability transition pore, which results in decreased Δψm, blocked electron transport chain and further a burst of ROS production [66]. The mitochondria of mice with DCM were profoundly swollen and disordered in the SIRT3-KO mice, indicating that the mitochondria were damaged, which is also the possible mechanism of excessive ROS production. The Δψm decreased under the stimulation of high glucose, which was significantly aggravated after SIRT3 was knocked down. These results suggested that SIRT3 deletion aggravated myocardial mitochondrial damage and increased oxidative stress, which may have promoted myocardial necroptosis in the mice with DCM.
Several drugs or compounds, including resveratrol [67], dihydromyricetin [68], and hydrogen sulfide [9], have the potential to increase SIRT3 expression, which may serve as a new therapeutic strategy to attenuate DCM in the future.
In summary, SIRT3 deficiency aggravated hyperglycemic mitochondrial damage, increased ROS accumulation, promoted necroptosis, possibly activated the NLRP3 inflammasome, and finally exacerbated DCM in mice. These results suggest that SIRT3 can be used as a molecular intervention target to provide new insights for the prevention and treatment of DCM. An illustration of the mechanism by which SIRT3 deficiency exacerbates DCM is outlined in Fig. 8.
Fig. 8. Illustration of the mechanism by which SIRT3 deficiency exacerbates diabetic cardiomyopathy.
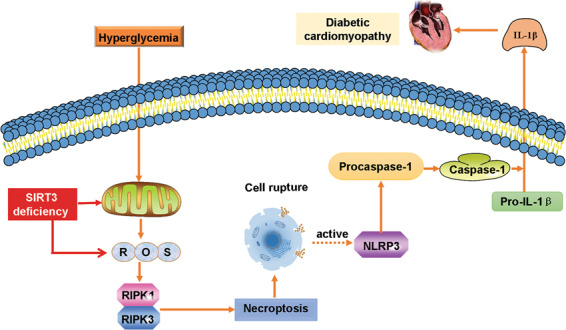
SIRT3 deficiency aggravated hyperglycemia-induced mitochondrial damage, increased ROS accumulation, promoted necroptosis and cell rupture, possibly activated the NLRP3 inflammasome, and finally exacerbated diabetic cardiomyopathy in the mice.
Acknowledgements
The work was funded by a grant from the National Natural Science Foundation of China (81770279), a major project of Natural Science Research in Jiangsu Higher Education Institutions (18KJA310005), the Six Talent Peaks Project in Jiangsu Province (2018-WSN-062), the China Postdoctoral Science Foundation (2019T120449 and 2017M610342), a Jiangsu Planned Project for Postdoctoral Research Funds (1701050A), a Science and Technology Project of Taicang City (TC2019KJFZ02), a Science and Technology Project of Nantong City (JC2018009), and the Nantong University Cooperative Innovation Program of Small Molecular Compound R&D (NTU2016-1).
Author contributions
SS and GLM designed the study; SS, YD, GLD, YZ, MTX, JRS, and TTC performed the research; GLD, MTX, YC, and GLM contributed new reagents and analytical tools; SS, GLD, YC, and GLM analyzed the data; and SS and G-lM wrote the paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Shu Song, Yue Ding, Guo-liang Dai
References
- 1.Filardi T, Ghinassi B, Di Baldassarre A, Tanzilli G, Morano S, Lenzi A, et al. Cardiomyopathy associated with diabetes: the central role of the cardiomyocyte. Int J Mol Sci. 2019;20:E3299. doi: 10.3390/ijms20133299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Hroob AM, Abukhalil MH, Hussein OE, Mahmoud AM. Pathophysiological mechanisms of diabetic cardiomyopathy and the therapeutic potential of epigallocatechin-3-gallate. Biomed Pharmacother. 2019;109:2155–72. doi: 10.1016/j.biopha.2018.11.086. [DOI] [PubMed] [Google Scholar]
- 3.Alonso N, Moliner P, Mauricio D. Pathogenesis, clinical features and treatment of diabetic cardiomyopathy. Adv Exp Med Biol. 2018;1067:197–217. doi: 10.1007/5584_2017_105. [DOI] [PubMed] [Google Scholar]
- 4.Wei X, Tao J, Shen Y, Xiao S, Jiang S, Shang E, et al. Sanhuang Xiexin Tang ameliorates type 2 diabetic rats via modulation of the metabolic profiles and NF-kappaB/PI3K/Akt signaling pathways. Front Pharmacol. 2018;9:955. doi: 10.3389/fphar.2018.00955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao H, Chen T, Shi Y. Glycation of human serum albumin in diabetes: impacts on the structure and function. Curr Med Chem. 2015;22:4–13. doi: 10.2174/0929867321666140912155738. [DOI] [PubMed] [Google Scholar]
- 6.Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018;61:21–8. doi: 10.1007/s00125-017-4390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Athithan L, Gulsin GS, McCann GP, Levelt E. Diabetic cardiomyopathy: Pathophysiology, theories and evidence to date. World J Diabetes. 2019;10:490–510. doi: 10.4239/wjd.v10.i10.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Fail Clin. 2019;15:341–7. doi: 10.1016/j.hfc.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Meng G, Liu J, Liu S, Song Q, Liu L, Xie L, et al. Hydrogen sulfide pretreatment improves mitochondrial function in myocardial hypertrophy via a SIRT3-dependent manner. Br J Pharmacol. 2018;175:1126–45. doi: 10.1111/bph.13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Yu J, Chen Y, Liu L, Xu M, Sun L, et al. Exogenous hydrogen sulfide supplement attenuates isoproterenol-induced myocardial hypertrophy in a sirtuin 3-dependent manner. Oxid Med Cell Longev. 2018;2018:9396089. doi: 10.1155/2018/9396089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng G, Zhao S, Xie L, Han Y, Ji Y. Protein S-sulfhydration by hydrogen sulfide in cardiovascular system. Br J Pharmacol. 2018;175:1146–56. doi: 10.1111/bph.13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun W, Liu C, Chen Q, Liu N, Yan Y, Liu B. SIRT3: a new regulator of cardiovascular diseases. Oxid Med Cell Longev. 2018;2018:7293861. doi: 10.1155/2018/7293861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller FJ., Jr Hypertension and mitochondrial oxidative stress revisited: sirtuin 3, the improved “antioxidant”. Circ Res. 2020;126:453–5. doi: 10.1161/CIRCRESAHA.120.316567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Song S, Xu M, Hua Y, Ding Y, Shan X, et al. Sirtuin3 deficiency exacerbates carbon tetrachloride-induced hepatic injury in mice. J Biochem Mol Toxicol. 2019;33:e22249. doi: 10.1002/jbt.22249. [DOI] [PubMed] [Google Scholar]
- 15.Zhu C, Liu Y, Guan Z, Zhou Y, Liu F, Zhang T. Hypoxia-reoxygenation induced necroptosis in cultured rat renal tubular epithelial cell line. Iran J Basic Med Sci. 2018;21:863–8. doi: 10.22038/IJBMS.2018.26276.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He X, Zeng H, Chen JX. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J Cell Physiol. 2019;234:2252–65. doi: 10.1002/jcp.27200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Zeng Z, Zhang W, Deng Z, Wan Y, Zhang Y, et al. Emerging role of SIRT3 in mitochondrial dysfunction and cardiovascular diseases. Free Radic Res. 2019;53:139–49. doi: 10.1080/10715762.2018.1549732. [DOI] [PubMed] [Google Scholar]
- 18.Storder J, Renard P, Arnould T. Update on the role of sirtuin 3 in cell differentiation: a major metabolic target that can be pharmacologically controlled. Biochem Pharmacol. 2019;169:113621. doi: 10.1016/j.bcp.2019.08.023. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. 2020;11:42. doi: 10.3389/fphar.2020.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the pathophysiology of disease. Am J Pathol. 2020;190:272–85. doi: 10.1016/j.ajpath.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15:199. 10.1186/s12974-018-1235-0. [DOI] [PMC free article] [PubMed]
- 22.Jensen S, Seidelin JB, LaCasse EC, Nielsen OH. SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci Signal. 2020;13:eaax8295. doi: 10.1126/scisignal.aax8295. [DOI] [PubMed] [Google Scholar]
- 23.Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci. 2020;41:209–24. doi: 10.1016/j.tips.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Gupta K, Phan N, Wang Q, Liu B. Necroptosis in cardiovascular disease—a new therapeutic target. J Mol Cell Cardiol. 2018;118:26–35. doi: 10.1016/j.yjmcc.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224. doi: 10.1126/sciadv.1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4:128834. doi: 10.1172/jci.insight.128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du L, Shen K, Bai Y, Chao J, Hu G, Zhang Y, et al. Involvement of NLRP3 inflammasome in methamphetamine-induced microglial activation through miR-143/PUMA axis. Toxicol Lett. 2019;301:53–63. doi: 10.1016/j.toxlet.2018.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Kearney CJ, Martin SJ. An inflammatory perspective on necroptosis. Mol Cell. 2017;65:965–73. doi: 10.1016/j.molcel.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Wang Y, Mu N, Lou X, Li W, Chen Y, et al. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab Investig. 2017;97:922–34. doi: 10.1038/labinvest.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang S, Xu M, Meng G, Lu Y. SIRT3 deficiency delays diabetic skin wound healing via oxidative stress and necroptosis enhancement. J Cell Mol Med. 2020;24:4415–27. doi: 10.1111/jcmm.15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y, et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. 2016;65:3171–84. doi: 10.2337/db16-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L, Elias JE. Relative protein quantification using tandem mass tag mass spectrometry. Methods Mol Biol. 2017;1550:185–98. doi: 10.1007/978-1-4939-6747-6_14. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Chen Y, Luo H, Xu M, Meng G, Zhang W. Ca2+/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem Pharmacol. 2019;163:194–205. doi: 10.1016/j.bcp.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 34.Luo H, Song S, Chen Y, Xu M, Sun L, Meng G, et al. Inhibitor 1 of protein phosphatase 1 regulates Ca2+/Calmodulin-dependent protein kinase II to alleviate oxidative stress in hypoxia-reoxygenation injury of cardiomyocytes. Oxid Med Cell Longev. 2019;2019:2193019. doi: 10.1155/2019/2193019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pirzada RH, Javaid N, Choi S. The roles of the NLRP3 inflammasome in neurodegenerative and metabolic diseases and in relevant advanced therapeutic interventions. Genes. 2020;11:E131. doi: 10.3390/genes11020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maroni L, Ninfole E, Pinto C, Benedetti A, Marzioni M. Gut-liver axis and inflammasome activation in cholangiocyte pathophysiology. Cells. 2020;9:E736. doi: 10.3390/cells9030736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Zhang S, Xiao Y, Zhang W, Wu S, Qin T, et al. NLRP3 inflammasome and inflammatory diseases. Oxid Med Cell Longev. 2020;2020:4063562. doi: 10.1155/2020/4063562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–38. doi: 10.1161/CIRCRESAHA.117.311586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baig MA, Panchal SS. Streptozotocin-induced diabetes mellitus in neonatal rats: an insight into its applications to induce diabetic complications. Curr Diabetes Rev. 2019;16:26–39. doi: 10.2174/1573399815666190411115829. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Chiang YL, Lyssiotis CA, Teater MR, Hong JY, Shen H, et al. Non-oncogene addiction to SIRT3 plays a critical role in lymphomagenesis. Cancer Cell. 2019;35:916–31. doi: 10.1016/j.ccell.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebaa R, Johnson J, Pileggi C, Norgren M, Xuan J, Sai Y, et al. SIRT3 controls brown fat thermogenesis by deacetylation regulation of pathways upstream of UCP1. Mol Metab. 2019;25:35–49. doi: 10.1016/j.molmet.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benigni A, Cassis P, Conti S, Perico L, Corna D, Cerullo D, et al. Sirt3 deficiency shortens life span and impairs cardiac mitochondrial function rescued by Opa1 gene transfer. Antioxid Redox Signal. 2019;31:1255–71. doi: 10.1089/ars.2018.7703. [DOI] [PubMed] [Google Scholar]
- 43.He X, Zeng H, Chen JX. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int J Cardiol. 2016;215:349–57. doi: 10.1016/j.ijcard.2016.04.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Yan W, Zhao X, Jia Q, Wang J, Zhang H, et al. Sirt3 attenuates post-infarction cardiac injury via inhibiting mitochondrial fission and normalization of AMPK-Drp1 pathways. Cell Signal. 2019;53:1–13. doi: 10.1016/j.cellsig.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 45.Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review) Int J Mol Med. 2019;44:771–86. doi: 10.3892/ijmm.2019.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhe-Wei S, Li-Sha G, Yue-Chun L. The role of necroptosis in cardiovascular disease. Front Pharmacol. 2018;9:721. doi: 10.3389/fphar.2018.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4:eaaw2004. doi: 10.1126/sciimmunol.aaw2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:391–5. doi: 10.1038/s41586-020-2129-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan H, Qi J, Fan BY, Zhang J, Su FF, Wang HT. MicroRNA-24-3p attenuates myocardial ischemia/reperfusion injury by suppressing RIPK1 expression in mice. Cell Physiol Biochem. 2018;51:46–62. doi: 10.1159/000495161. [DOI] [PubMed] [Google Scholar]
- 52.Zhao M, Lu L, Lei S, Chai H, Wu S, Tang X, et al. Inhibition of receptor interacting protein kinases attenuates cardiomyocyte hypertrophy induced by palmitic acid. Oxid Med Cell Longev. 2016;2016:1451676. doi: 10.1155/2016/1451676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrie EJ, Czabotar PE, Murphy JM. The structural basis of necroptotic cell death signaling. Trends Biochem Sci. 2019;44:53–63. doi: 10.1016/j.tibs.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 54.Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 2018;9:816. doi: 10.1038/s41419-018-0868-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian J, Guo S, Chen H, Peng JJ, Jia MM, Li NS, et al. Combination of emricasan with ponatinib synergistically reduces ischemia/reperfusion injury in rat brain through simultaneous prevention of apoptosis and necroptosis. Transl Stroke Res. 2018;9:382–92. doi: 10.1007/s12975-017-0581-z. [DOI] [PubMed] [Google Scholar]
- 56.Zheng XL, Yang JJ, Wang YY, Li Q, Song YP, Su M, et al. RIP1 promotes proliferation through G2/M checkpoint progression and mediates cisplatin-induced apoptosis and necroptosis in human ovarian cancer cells. Acta Pharmacol Sin. 2020. 10.1038/s41401-019-0340-7. Online ahead of print. [DOI] [PMC free article] [PubMed]
- 57.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–82. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 58.Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem. 2016;85:743–63. doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- 59.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 60.Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol. 2019;10:276. doi: 10.3389/fimmu.2019.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boniakowski AM, denDekker AD, Davis FM, Joshi A, Kimball AS, Schaller M, et al. SIRT3 regulates macrophage-mediated inflammation in diabetic wound repair. J Investig Dermatol. 2019;139:2528–37. doi: 10.1016/j.jid.2019.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim TS, Jin YB, Kim YS, Kim S, Kim JK, Lee HM, et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy. 2019;15:1356–75. doi: 10.1080/15548627.2019.1582743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kurundkar D, Kurundkar AR, Bone NB, Becker EJ, Jr, Liu W, Chacko B, et al. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight. 2019;4:e120722. doi: 10.1172/jci.insight.120722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med. 2020;7:12. doi: 10.3389/fcvm.2020.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46:1650–67. doi: 10.1159/000489241. [DOI] [PubMed] [Google Scholar]
- 66.Zha ZM, Wang JH, Li SL, Guo Y. Pitavastatin attenuates AGEs-induced mitophagy via inhibition of ROS generation in the mitochondria of cardiomyocytes. J Biomed Res. 2018;32:281–7. doi: 10.7555/JBR.31.20160116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chelladurai P, Boucherat O, Stenmark K, Kracht M, Seeger W, Bauer UM, et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. 2019. 10.1111/bph.14932. Online ahead of print. [DOI] [PubMed]
- 68.Zhang J, Chen Y, Luo H, Sun L, Xu M, Yu J, et al. Recent update on the pharmacological effects and mechanisms of dihydromyricetin. Front Pharm. 2018;9:1204. doi: 10.3389/fphar.2018.01204. [DOI] [PMC free article] [PubMed] [Google Scholar]