

Abstract
Dysregulation of the immune system is one potential mechanism by which acute stress may contribute to downstream disease etiology and psychopathology. Here, we tested the role of β-adrenergic signaling as a mediator of acute stress-induced changes in immune cell gene expression. In a randomized, double-blind, and placebo-controlled trial, 90 healthy young adults (44% female) received a single 40 mg dose of the β-blocker propranolol (n = 43) or a placebo (n = 47) and then completed the Trier Social Stress Test (TSST). Pre- and post-stress blood samples were assayed for prespecified sets of pro-inflammatory and antiviral/antibody gene transcripts. Analyses revealed increased expression of both inflammatory and antiviral/antibody-related genes in response to the TSST, and these effects were blocked by pre-treatment with propranolol. Bioinformatics identified natural killer cells and dendritic cells as the primary cellular context for transcriptional upregulation, and monocytes as the primary cellular carrier of genes downregulated by the TSST. These effects were in part explained by acute changes in circulating cell types. Results suggest that acute psychosocial stress can induce an “acute defense” molecular phenotype via β-adrenergic signaling that involves mobilization of natural killer cells and dendritic cells at the expense of monocytes. This may represent an adaptive response to the risk of acute injury. These findings offer some of the first evidence in humans that β-blockade attenuates psychosocial stress-induced increases in inflammatory gene expression, offering new insights into the molecular and immunologic pathways by which stress may confer risks to health and well-being.
Subject terms: Gene expression, Cytokines, Human behaviour
Introduction
Stress is a critical risk factor in the etiology of cardiovascular disease, diabetes, and many psychiatric disorders such as depression [1–5]. Recent research implicates immune dysregulation as one biological mechanism that contributes to adverse health outcomes in the face of stress [6–10]. Complementary work in social genomics reveals that different types of chronic stress (e.g., early life adversity, social isolation, and low social status) are associated with a leukocyte genomic profile called the conserved transcriptional response to adversity (CTRA; [11]). The CTRA is characterized by increased expression of inflammatory genes (e.g., IL1B, IL6, and TNF) and decreased expression of genes supporting innate antiviral responses (e.g., IFNB, IFI, MX1/2, and OAS), as well as some antibody-related transcripts (e.g., IGLL1, JCHAIN). This chronic stress-related shift toward a pro-inflammatory phenotype is implicated in the etiology of several adverse physical and mental health outcomes [7, 11–13]. Although both clinical and preclinical studies report associations between chronic stressors and the CTRA [14–22], little experimental work investigates if similar patterns of gene expression are observed when humans experience an acute stressor [23]. Mapping the gene regulatory impact of acute stress and the mechanisms involved could help clarify whether acute effects are similar to the CTRA pattern associated with chronic stress.
Preclinical research suggests that sympathetic nervous system (SNS) signaling through β-adrenergic receptors plays a central role in immune responses to acute stress [24–26]. Several seminal studies in humans further implicate β-adrenergic regulation of immune cell function and prevalence (i.e., via ex vivo cellular function assays and leukocyte subset redistribution analyses [16, 27–32]), with weaker evidence in small samples for β-adrenergic effects on circulating inflammatory proteins such as interleukin-6 or IL-6 [33, 34]. However, little experimental work in humans documents the role of SNS signaling through β-adrenergic receptors on inflammatory and interferon gene expression during acute stress [16, 32, 35]. As such, we conducted the present study to clarify whether β-adrenergic signaling pathways mediate effects of acute psychological stress on gene expression profiles in circulating immune cells.
Specifically, we investigated β-adrenergic regulation of inflammatory and interferon/antibody gene expression in humans in response to an acute psychosocial stressor. We designed this study with two aims in mind: (i) to characterize the impact of acute stress on inflammatory and antiviral/antibody gene expression, and (ii) to examine how pre-treatment with propranolol which blocks SNS signaling via β-adrenergic receptors might impact gene expression and circulating inflammatory proteins. We thus recruited 90 healthy young adults as part of a randomized, double-blind, placebo-controlled mechanistic trial. Individuals received a single dose of either placebo or 40 mg propranolol and then completed the Trier Social Stress Test (TSST). Blood sampled before and after the stressor was assayed for levels of leukocyte pro-inflammatory and antiviral/antibody gene expression and circulating levels of the inflammatory marker IL-6. This approach allowed us to determine whether β-adrenergic signaling causally contributes to stress-induced changes in immune cell gene expression and inflammatory proteins. In addition, we conducted secondary analyses to determine how any change in the prevalence of circulating leukocyte subsets might potentially contribute to observed differences in the overall blood transcriptome profile.
Methods and materials
Participants
Healthy young adults were recruited from the University of North Carolina at Chapel Hill and its surrounding community via flyers and electronic mailing lists. All potential participants were first screened for eligibility via a structured telephone interview. Individuals were excluded if they reported prior or current use of β-blockers, a history of self-reported mental or physical health problems, self-reported weight and height that indicated a BMI over 33, any current prescription medication, or regular nicotine or recreational drug use. During an initial prescreening visit, individuals could not exhibit a resting heart rate or blood pressure below safety recommendations for propranolol use (i.e., <60 bpm, 80 mm/Hg). On the study day, participants arrived well-hydrated, having eaten their normal meals (but refraining from caffeine or excessive sugar), and having avoided aerobic exercise that day. We further checked that all participants felt currently healthy (e.g., no allergies, common cold, headache, etc.), had not recently been ill nor experienced recent sleep disturbances (e.g., changing time zones, working night shift). Drug conditions (0 = placebo, 1 = propranolol) were matched on sex, age, race, and did not significantly differ in terms of depressive or anxiety symptoms, recent perceived life stress, body mass index (BMI), or objective socioeconomic status (all p > 0.05). The final sample included 90 healthy young adults (44% female; 56.7% White; Mage: 20.28 ± 1.42 years) with n = 43 receiving propranolol and n = 47 receiving placebo. See Supplementary Information for sample characteristics. Sample size (N = 90) was determined ahead of time; assuming a small effect size of propranolol on inflammation based on prior studies [33, 34], power estimates in G*power [36] suggested at least 72 participants total (38 within each group) to achieve 95% power for testing interactions of group × time. See Supplementary Information for a CONSORT-consistent flow diagram.
Procedure
The study was pre-registered with ClinicalTrials.gov (Trial ID: NCT02972554) and approved by the university’s IRB. The study was conducted in accordance with IRB guidelines for human participants, including full consent and debriefing. Between 3 and 7 days after initial enrollment, all participants completed the lab visit from 12 to 5 p.m., to help control for potential diurnal effects. Upon arrival, a nurse blind to condition inserted a catheter with a heparin lock into the nondominant forearm for blood draws. Catheters were inserted no later than 12:30 p.m., with a minimum of 30 min acclimation period before an initial predrug baseline was taken.
After the initial baseline blood sample and 50 min after catheter insertion, each participant received their randomly assigned dose of either propranolol or placebo, self-administered orally under supervision. Tablets were provided by the university’s research pharmacy which also managed the drug random assignment. Participants were randomized in blocks based on gender and race (white vs. non white) to ensure equal representation across groups. All study staff and participants were blind to condition. Given that effects of propranolol peak 1–2 h after oral administration [37], the postdrug baseline blood sample (at 60 min following drug/placebo administration) and acute stress induction (at 75 min postdrug administration) took place when β-blockade was in maximum effect.
Acute psychosocial stress was induced using the TSST [38], a gold-standard method in the human stress literature. Participants had 2 min to prepare a speech about why they would be a good candidate for their dream job, then gave a 10 min speech and completed 5 min of mental arithmetic aloud before an evaluative panel of two condition-blinded, neutral researchers wearing laboratory coats. Blood draws for IL-6 were collected in EDTA tubes at predrug baseline (BL1), postdrug baseline (BL2), and 30, 60, and 90 min following completion of the stressor (T30, T60, and T90). Blood samples for gene expression were collected in PAXgene Blood RNA tubes at BL1, BL2, and at post-TSST T30 (given that gene expression changes occur more rapidly than protein-level changes). The postdrug baseline (BL2) sample was acquired before participants were informed about the details of the TSST procedure and thus does not reflect anticipatory stress.
Upon session completion, participants were debriefed, paid (US$100), and discharged once physiological vitals returned to baseline. Study intake and data collection began November 2016 and ended October 2017. All participants remained in their originally assigned conditions, with no changes in study design, selection and exclusion criteria, or analysis procedures. Propranolol and TSST effects on physiological and emotional reactions were also assessed as a second aim of this project and are reported elsewhere [39].
Measures
Gene expression and transcriptional bioinformatics
At the end of each session, all PAXgene RNA tubes (Qiagen PAXgene Blood RNA) were stored upright at room temperature for 24 h, then frozen for 24 h at −20 °C, before being transferred into storage at −80 °C until study completion. Total RNA was extracted and checked for suitable mass (>200 ng by NanoDrop ND1000 spectrophotometry) and integrity (RNA integrity number >3 by Agilent TapeStation capillary electrophoresis). Genome-wide transcriptional profiles were assayed by RNA sequencing (RNAseq) in the UCLA Neuroscience Genomics Core Laboratory using Lexogen QuantSeq 3’ FWD cDNA library synthesis and multiplex DNA sequencing on an Illumina HiSeq 4000 instrument with single-strand 65-nucleotide sequence reads, all following manufacturers’ standard protocols. Sequencing yielded an average 12.7 million sequence reads per sample, each of which was mapped to the RefSeq human genome sequence using the STAR aligner [40], initially quantified as gene transcript counts per million mapped reads and further normalized to equate the median value of expressed transcripts across all samples.
Inflammatory protein markers
Plasma samples were assayed for IL-6 using Meso Scale Discovery V-Plex Pro-inflammatory Human Panel 1 (Rockville, MA, USA), according to manufacturer’s instructions. Given prior literature focusing on IL-6 reactivity to acute stress [41, 42], our primary a priori inflammatory marker of interest was IL-6. However, as an exploratory addition, assay kits included detection antibodies for analytes IL-1β, IL-4, IL-10, and TNF-α. All plates were read on a Meso Quickplex machine and data analyzed using Discovery Workbench software 4.0. The intra- and inter-assay coefficients for IL-6 were below 9%. We present IL-6 findings in the main text but see Supplementary Information for other inflammatory marker findings.
Analysis strategy
Gene expression and transcriptional bioinformatics
Normalized gene transcript counts per million values were floored at 1 and log2 transformed for standard linear statistical model analyses. Primary analyses focused on two key groups of genes: an a priori-defined composite of 19 pro-inflammatory transcripts and an a priori-defined composite of 34 interferon- and antibody-related transcripts [20, 43]. To characterize overall effects of experimental conditions on inflammatory and antiviral gene expression, data were first analyzed in a 2 (group: placebo vs. propranolol) × 3 (time: BL1, BL2, T30) × 2 (gene set: inflammatory vs. antiviral/antibody) mixed effect linear model analysis treating time and gene set as repeated measures. Analyses were conducted using SAS PROC MIXED, specifying a fully saturated (unstructured) variance-covariance matrix for residuals. Following significant omnibus ANOVA results, we conducted a series of planned contrasts to characterize the effect of experimental group on pre- to post-TSST changes [T30—baseline (pooled BL1, BL2)] in gene expression for each specific gene set.
To cross-validate findings from primary analyses of a priori-defined gene sets, we conducted two sets of secondary analyses. First, we sought to identify empirical differences in gene expression (defined as genes showing a maximum likelihood point estimate of >20% change in response to TSST). Second, we tested the extent to which those genes reflected differential prevalence and/or activity of specific cell types involved in β-adrenergic cell redistribution (e.g., NK cells) and CTRA induction (e.g., classical and nonclassical monocyte subsets and dendritic cells) using Transcript Origin Analyses [44]. These Transcript Origin Analyses were based upon previous transcriptome profiling of physically-isolated samples of ten major leukocyte subsets [45] and classical vs. nonclassical monocytes [46]. Importantly, Transcript Origin Analyses are sensitive to changes in both cell prevalence and changes in cellular activation status within fixed cell prevalence.
To more specifically assess changes in cell prevalence within the circulating leukocyte pool (i.e., as distinct from activation differences), we conducted Transcriptome Representation Analyses [16] using the same reference cell transcriptome profiles as used for Transcript Origin Analyses, and treating all genes showing >6 SD higher abundance in a given cell type relative to all others as diagnostic of that cell subpopulation [16]. In all analyses, statistical testing was based on standard errors derived from bootstrap resampling of linear model residual vectors, which provides a nonparametric assessment of statistical significance while appropriately controlling for correlation among genes.
Interleukin-6
As the IL-6 data had a right-skewed distribution, it was log transformed. Analyses were completed in R (lme4 package) using repeated-measures ANOVA with a random intercept to account for within-subject nesting of the repeated measures. We then conducted follow-up planned paired samples t tests to help illustrate within-condition changes in IL-6 at T30, T60, and T90 from baseline.
Covariates
We conducted ancillary analyses for both the primary gene expression models and the secondary circulating inflammatory protein analyses, controlling for a standard set of covariates typically controlled for in the social genomics research literature including age, sex, race/ethnicity, BMI, and minor illness symptoms (e.g., allergies) in the week prior to study participation.
Results
Effect of acute stress and propranolol on CTRA-related gene sets
Results from a 2 (group: placebo vs. propranolol) × 3 (time: BL1, BL2, and T30) × 2 (gene set: inflammatory vs. antiviral/antibody) mixed effect linear model analysis treating time and gene set as repeated measures indicated a significant group × time interaction, F(2, 87) = 3.77, p = 0.027, but no significant 3-way gene set × group × time interaction, F(2, 87) = 0.84, p = 0.437. Similar findings emerged from ancillary analyses that adjusted for covariates (group × time interaction: F(2, 79) = 3.72, p = 0.029; gene set × group × time interaction: F(2, 79) = 0.80, p = 0.452). Results from follow-up preplanned contrast analyses (Fig. 1) found placebo-treated participants to differ significantly from propranolol-treated participants in the profile of pre- to post-TSST change in expression of both inflammatory genes and antiviral/antibody genes (i.e., a significant group × time interaction for both inflammatory genes: b = −0.42 ± SE 0.18, p = 0.033; and antiviral/antibody-related genes: −0.43 ± 0.19, p = 0.035; resulting in a nonsignificant interaction for the overall CTRA composite score that represents the difference between those two gene sets: +0.11 ± 0.06, p = 0.069).
Fig. 1. Change in gene expression across inflammatory, antiviral, and overall CTRA responses in TSST + Placebo vs. TSST + Propranolol groups.
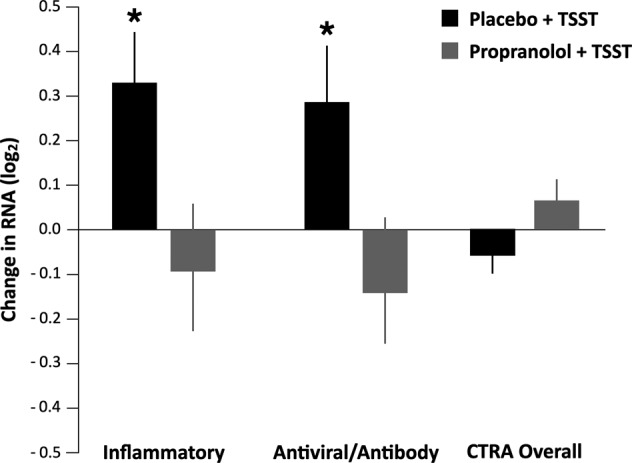
Black stars indicate significance at p < 0.05.
To help interpret the significant group × time interaction patterns for the inflammatory and antiviral/antibody-related gene sets, we conducted follow-up simple effects tests examining the magnitude of pre- to post-TSST change for each group separately. Placebo-treated participants showed significant increases in expression of both pro-inflammatory and antiviral/antibody-related genes at 30 min post-TSST relative to baseline (inflammatory: mean change = +0.33 ± 0.13 log2 mRNA abundance, p = 0.018; antiviral/antibody: +0.29 ± 0.14, p = 0.039). Thus, the acute stress pattern identified here shows upregulation of both pro-inflammatory and antiviral/antibody-related genes, whereas chronic stress is associated with upregulated inflammatory but downregulated expression of antiviral/antibody genes [7, 11, 16]. Consistent with this parallel rather than reciprocal change in the two CTRA-related gene sets, the standard CTRA composite score (i.e., scoring inflammatory positively and antiviral/antibody-related genes negatively) showed no significant change in expression from pre- to post-TSST (−0.06 ± 0.04, p = 0.143).
In contrast to placebo-treated participants, propranolol-treated participants showed no significant change in expression of either gene set from pre- to post-TSST (inflammatory: −0.10 ± 0.15, p = 0.533; antiviral/antibody: −0.13 ± 0.15, p = 0.379). Consistent with the absence of any significant change in pro-inflammatory or antiviral/antibody-related genes in propranolol-treated individuals, the standard CTRA contrast score also showed no significant change in expression from pre- to post-TSST for propranolol-treated individuals (+0.05 ± 0.04, p = 0.265).
Cellular origin of differential gene expression
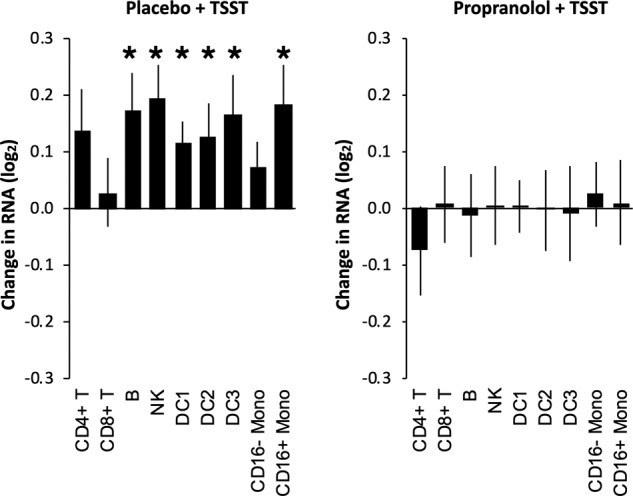
To identify cellular mechanisms that might potentially structure TSST-induced upregulation of pro-inflammatory and antiviral/antibody-related gene sets, we conducted Transcript Origin Analyses of all 9388 gene transcripts that showed >20% difference in expression between baseline to 30 min post-TSST in placebo-treated participants (Fig. 2). Analyses using reference profiles from major leukocyte subsets (Fig. 2A) implicated NK cells as a potential cellular source of genes upregulated in response to the TSST, and CD8+ T cells and classical (CD16-) monocytes as potential cellular sources of genes downregulated in response to the TSST. Results also indicated a nonsignificant trend (p = 0.052) for downregulated genes to derive from DC3 plasmacytoid dendritic cells. Follow-up analyses focusing specifically on classical and nonclassical monocytes again implicated classical (CD16-) monocytes as the primary cellular source of TSST downregulated genes (Fig. 2B).
Fig. 2. Effect of beta-blockade on stress-induced immune transcription profiles.

Transcript origin analyses determined diagnosticity scores for upregulated vs. downregulated immune-related cell gene expressions. Black stars indicate significance at p < 0.05 while gray stars indicate nonsignificant trends at p < 0.10. A shows the effect of TSST in placebo-treated individuals. B shows the effect of TSST in propranolol-treated individuals.
In parallel Transcript Origin Analysis of the 3564 gene transcripts showing >20% change over time in propranolol-treated participants, results showed no indication that differential gene expression derived from either NK cells, CD8+ T cells, or classical (CD16-) monocytes (all p > 0.05). However, these analyses did suggest a potential upregulation of DC2 and DC3 dendritic cell populations to TSST-related upregulation in the propranolol-treated group, and an unanticipated contribution of DC1 to TSST-related downregulation.
Role of changing cell prevalence
The Transcript Origin Analyses reported above are designed to assess effects of both differential cell prevalence and differential cellular activation (i.e., holding constant cell prevalence). To more specifically assess changes in cell prevalence, we conducted Transcriptome Representation Analyses using reference genes that are predominately expressed by a single cell type based on previous gene expression profiling analyses of isolated cell populations. As shown in Fig. 3, analyses of change over time indicated upregulation in the prevalence of multiple leukocyte subsets in placebo-treated participants, including B lymphocytes, NK cells, all three types of dendritic cells (DC1, DC2, and DC3), and nonclassical (CD16+) monocytes (but not classical CD16-monocytes). By contrast, these bioinformatic analyses showed no significant indication of change over time for any cell population analyzed in propranolol-treated participants.
Fig. 3. The effect of acute stress on prevalence of circulating immune cells.
Transcriptome representation analysis quantified the effect of TSST on circulating leukocyte subsets in individuals treated with placebo or propranolol. Black stars indicate significance at p < 0.05.
To determine whether changing cell prevalence was a plausible mechanism of TSST-associated changes in expression of the pro-inflammatory and antiviral/antibody-related gene sets analyzed above, we conducted ANOVAs that also controlled for changes in major leukocyte subset markers (CD3D/E, CD4, CD8A, CD19, CD56/NCAM1, CD16/FCGR3A, and CD14) while assessing stressor effects in the placebo group and the additional effects of propranolol. Control for major leukocyte subset markers rendered nonsignificant both the basic effects of the stressor in the placebo-treated group (+0.05 ± 0.05, p = 0.080; antiviral/antibody: +0.00 ± 0.03, p = 0.936) and the differential change over time in the propranolol vs. placebo-treated groups (+0.00 ± 0.04, p = 0.995; antiviral/antibody: +0.04 ± 0.05, p = 0.364), suggesting that the observed changes in CTRA gene expression in response to the TSST in the placebo-treated group are due almost entirely to stress-induced changes in cell prevalence.
Effect of acute stress and propranolol on IL-6
To examine the humoral inflammatory response in parallel with the cellular inflammatory mechanisms assessed by RNA profiling, we examined whether the TSST increased levels of the circulating inflammatory marker IL-6 and, in turn, whether pre-treatment with propranolol blunted stress-related IL-6 reactivity, adjusted for covariates (Fig. 4). Consistent with prior stress literature [41], there was a significant stress effect of the TSST on IL-6 reactivity across time. Specifically, relative to the baselines, there was a significant increase in IL-6 at T30 (b = 0.24, SE = 0.10, p = 0.014), at T60 (b = 0.23, SE = 0.10, p = 0.021), and in particular by T90 (b = 0.46, SE = 0.10, p < 0.0001). However, this effect was not significantly abrogated by propranolol, neither overall as a main effect of drug (p = 0.41) nor in interaction with any given timepoint (p > 0.25). Furthermore, there were no significant covariate effects of sex, age, BMI, ethnicity, nor recent illness symptoms in relation to IL-6 (p > 0.25). See the Supplementary Information for a full table with IL-6 results as well as exploratory analyses for IL-1β, TNF-α, IL-4, and IL-10. In brief, IL-1β and IL-4 were below the limits of detection and thus not examined. There were no effects of propranolol on TNF-α nor IL-10 (consistent with IL-6 findings).
Fig. 4. Mean reactivity from Baseline for IL-6 comparing placebo vs. propranolol across time.
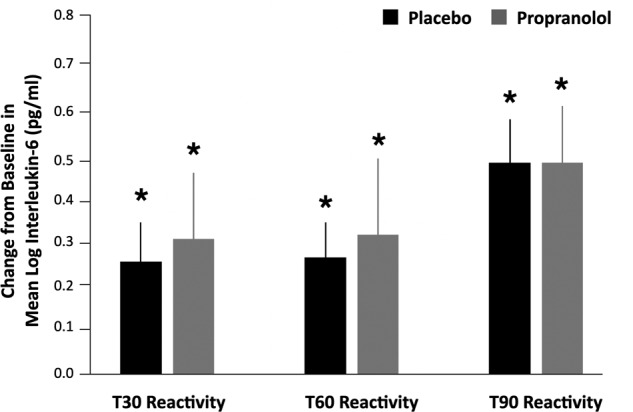
Timepoints cover 30 min post-stressor (T30), 60 min post-stressor (T60), and 90 min post-stressor (T90). Blood for circulating cytokine analysis was collected at these timepoints but additional blood for genomic analyses was only collected during baseline and T30. There were significant increases in IL-6 post stressor within groups (relative to baseline) for all timepoints but no significant differences between the placebo vs. propranolol groups.
Discussion
This study investigated the effect of acute psychosocial stress on inflammatory and antiviral/antibody gene expression in humans. We found that acute stress exposure increased expression of both pro-inflammatory and antiviral/antibody-related genes in the whole-blood transcriptome, and that these effects could potentially be accounted for by changing prevalence of specific leukocyte subsets within the total blood cell pool. Stress-induced transcriptomic changes were blocked by pre-treatment with the β-adrenergic antagonist propranolol, demonstrating β-adrenergic signaling as a causal mediator of these effects. The pattern of transcriptome alteration observed here in response to acute stress (i.e., simultaneous upregulation of both pro-inflammatory and antiviral/antibody-related genes) differs from the classic CTRA profile previously linked to chronic stress, in which there is reciprocal upregulation of pro-inflammatory genes and downregulation of antiviral/antibody genes [11, 14, 18, 23]. As such, it appears that the impact of acute stress on pro-inflammatory and antiviral/antibody-related gene expression in blood is qualitatively different from that observed following chronic stress. Both profiles involve upregulation of pro-inflammatory genes, but acute stress also elicits an upregulation of antiviral/antibody genes, while chronic stress is associated with downregulation of these genes.
Consistent with this distinction for CTRA gene expression induced by acute vs. chronic stress, these results also indicate distinct cellular sources underlying the observed blood transcriptome changes. Whereas the established CTRA profile of chronic stress is mediated in part by upregulation of classical (CD16-) monocytes, the acute stress effects observed herein involved a relative downregulation of genes that are expressed predominately by classical monocytes and a simultaneous upregulation of genes that are predominately expressed in NK cells (Fig. 2). Moreover, bioinformatics suggested that much of the stressor-induced increases in pro-inflammatory and antiviral/antibody-related gene expression could potentially be accounted for by acute changes in the prevalence of circulating leukocyte subsets. Specifically, the bioinformatically-inferred prevalence of NK cells, B cells, dendritic cells, and nonclassical monocytes all showed stress-induced mobilization whereas T cells and classical monocytes showed little evidence of mobilization, presenting a decline in relative prevalence within the blood cell transcriptome as a whole. Consistent with this observation, statistical control for alterations in the abundance of mRNAs encoding major leukocyte subset marker proteins abrogated the TSST-induced alterations in inflammatory and antiviral/antibody gene expression in the circulating blood pool. Moreover, those apparent redistribution effects appear driven in large part by SNS signaling, given that propranolol abrogated bioinformatic indications of multiple leukocyte subset mobilization and given that control for such effects blunted stress-induced changes in pro-inflammatory and antiviral/antibody-related transcript representation within the blood cell pool.
Taken together, these data suggest that acute stress may induce an “acute defense” molecular phenotype that is distinct from the classical CTRA profile associated with chronic stress, which is mediated by β-adrenergic signaling and involves acute mobilization of NK cells, dendritic cells, and nonclassical monocytes at the expense of classical monocytes. One plausible interpretation is that this SNS-mediated acute stress response may help coordinate circulating immune cell changes to preemptively detect and prepare for diverse types of assaults, be it bacterial infection from a potential wound (e.g., via the attack of a predator or threatening social other) or viral infection (e.g., after close contact with social others during acute stress coping). However, this multifront acute defense is likely also metabolically costly; when a stressor becomes chronic, the body may then no longer be able to “afford” maintaining a dual defense. Studies assessing the metabolic costs of acute vs. chronic immune defense would be an important next step in testing this hypothesis; for example, future research should examine how mitochondrial DNA may regulate acute vs. chronic stress-induced immune transcriptional responses [47, 48]. Furthermore, the finding that the transcriptional response to acute stress differs from the chronic stress response raises the possibility that after extended stress exposure, there may be a switch to the classical CTRA upregulation of inflammatory genes and downregulation of antiviral/antibody genes. Future research should test this possibility.
This study has several strengths: (i) the experimental induction of acute stress in a controlled laboratory setting, (ii) the randomized, placebo-controlled administration of propranolol, (iii) a sample that was well-powered for the proposed questions, and (iv) consideration of leukocyte redistribution as a potential mediating mechanism. Some limitations should also be noted. The study was a first step and not sufficiently powered to identify significant effects for specific individual gene transcripts or for other exploratory analyses seeking to identify new sets of genes that are empirically responsive to acute stress. Similarly, although we did examine potential effects of sex, age, and other such covariates, this study was not designed nor powered to fully identify how individual characteristics may interact with the observed stress and drug-related patterns of immune gene expression. Future research with larger, more diverse samples should examine how sex, age, BMI, socioeconomic status, and recent life stress might moderate findings. Another caveat is that participants did not complete a full fast prior to the session which may have influenced propranolol bioavailability and inflammation [49–53]. Another limitation is a lack of a non-stressor control group; we thus cannot distinguish stress effects from the secular trend. Finally, although our bioinformatic analyses sought to identify the specific cellular origins and cell population structures that might contribute to observed whole-blood transcriptome changes, future research using flow cytometric enumeration and cell sorting is needed to confirm the present bioinformatic inferences and to clarify any per-cell changes in gene expression within specific leukocyte subpopulations.
Beyond the above caveats, although the TSST successfully evoked increases in circulating IL-6, propranolol did not alter stressor-induced IL-6 reactivity. β-adrenergic receptor signaling has been implicated in IL-6 regulation previously (e.g., 29); one possible reason for this null effect is that the post-stress assessment time was insufficiently long to detect effects of propranolol on IL-6. Indeed, IL-6 levels in plasma up to 90 min post stressor may reflect the release of existing cytokines into circulation (e.g., from adipose tissue or lymphoid organs) rather than de novo production. Although stress-induced β-adrenergic signaling may be expected to facilitate greater release of extant cytokines like IL-6 (as we found), it is possible that propranolol (via β-adrenergic blockade) specifically blunts de novo production that is not captured within 90 min. Future work should assay levels of inflammatory cytokines over longer windows of time (e.g., up to 24 h). Importantly, we would not expect changes in circulating inflammatory cells (e.g., as assessed here by RNA profiling) to correlate directly with changes in circulating inflammatory cytokines (e.g., IL-6) because circulating leukocytes are not generally the source of circulating cytokines (e.g., circulating IL-6 derives predominately from adipose tissue and cells in lymphoid organs). Rather, RNA/cellular and protein/humoral blood parameters represent distinct inflammatory mechanisms that jointly impact systemic inflammatory biology.
Given that inflammatory biology is implicated in vulnerability to disease etiology, premature cellular aging, and psychopathology [8, 10, 54, 55], the present findings may have implications for medicine and public health. By upregulating inflammation in addition to antiviral transcriptional profiles, the short-term observed response to acute stress may come at a cost to physical and mental health if sustained over prolonged durations, ultimately transforming from an acute-type “dual activation” profile to the classic reciprocal CTRA profile with its associated chronic disease risks. It is thus critical to identify how acute stress effects on immune-related gene expression transform into chronic stress effects [56]. Furthermore, longitudinal studies are needed to examine acute stress-related gene expression reactivity and future risk for psychopathology. More generally, future work in larger samples should investigate the role of subjective stress (e.g., stress perceptions and appraisals, affective experience during stressors) in modulating immune-related gene expression responses to acute stressors.
In conclusion, findings from the present study suggest that acute stress induces an “acute defense” molecular phenotype that is distinct from the classical CTRA profile associated with chronic stress. This distinctive transcriptional response to acute stress is mediated by β-adrenergic signaling and involves acute mobilization of NK cells and dendritic cells at the expense of classical monocytes. These findings offer some of the first evidence that β-blockade buffers against increases in inflammatory gene expression during acute psychosocial stress in humans. As such, β-adrenergic signaling is a crucial pathway linking acute stress to inflammation and may be one avenue by which stress confers greater risk for disease and psychopathology.
Funding and disclosure
JKM received support from a Ruth L. Kirschstein National Research Service Award predoctoral fellowship from the National Institute on Aging (1F31AG055265-01A1) as well as a T32 postdoctoral fellowship from the National Heart, Lung, and Blood Institute (5T32HL007560-37) via the University of Pittsburgh Department of Psychiatry’s Cardiovascular Behavioral Medicine research training program. SWC received funding support from the National Institute on Aging (P30AG017265), awarded to the USC/UCLA Center on Biodemography and Population Health. The North Carolina Translational and Clinical Sciences (NC TraCS) Institute provided further assistance, via support from the National Center for Advancing Translational Sciences (NCATS; UL1TR002489). EKS is supported by Australian National Health and Medical Research Council APP1147498. The authors declare no competing interests.
Supplementary information
Author contributions
KAM developed the initial study concept with JKM, ELAC, and SMB refining ideas and contributing to the study design. SMB served as physician overseeing participant safety and propranolol administration. Testing and data collection were performed by ELAC, MMGD, and JKM, with additional assistance from undergraduate research assistants in Muscatell’s laboratory. SWC and JMGA performed gene expression processing and data analyses with interpretation input from KAM, EKS, and JKM. MMGD preformed inflammatory protein assays and JKM conducted assay analyses. JKM and SWC drafted the paper, with primary revisions from KAM and critical revisions from EKS. Additional revisions were made by ELAC, MMGD, and SMB. All authors approved the final version of the paper for submission.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41386-020-00897-0).
References
- 1.McEwen BS. Neurobiological and systemic effects of chronic stress. Chronic Stress. 2017;1:247054701769232. doi: 10.1177/2470547017692328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheth C, McGlade E, Yurgelun-Todd D. Chronic stress in adolescents and its neurobiological and psychopathological consequences: an RDoC perspective. Chronic Stress. 2017;1:247054701771564. doi: 10.1177/2470547017715645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammen C. Stress sensitivity in psychopathology: mechanisms and consequences. J Abnorm Psychol. 2015;124:152–4. doi: 10.1037/abn0000040. [DOI] [PubMed] [Google Scholar]
- 4.Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA. 2007;298:1685. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- 5.Brotman DJ, Golden SH, Wittstein IS. The cardiovascular toll of stress. Lancet. 2007;370:1089–1100. doi: 10.1016/S0140-6736(07)61305-1. [DOI] [PubMed] [Google Scholar]
- 6.Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. 2016;21:1696–709. doi: 10.1038/mp.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol Bull. 2014;140:774–815. doi: 10.1037/a0035302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ménard C, Pfau ML, Hodes GE, Russo SJ. Immune and neuroendocrine mechanisms of stress vulnerability and resilience. Neuropsychopharmacology. 2017;42:62–80. doi: 10.1038/npp.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25:1822–32. doi: 10.1038/s41591-019-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole SW. The conserved transcriptional response to adversity. Curr Opin Behav Sci. 2019;28:31–7. doi: 10.1016/j.cobeha.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisenberger NI, Moieni M, Inagaki TK, Muscatell KA, Irwin MR. In sickness and in health: The co-regulation of inflammation and social behavior. Neuropsychopharmacology. 2017;42:242–53. doi: 10.1038/npp.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Donovan A, Sun B, Cole S, Rempel H, Lenoci M, Pulliam L, et al. Transcriptional control of monocyte gene expression in post-traumatic stress disorder. Dis Markers. 2011;30:123–32. doi: 10.3233/DMA-2011-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connor M-F, Schultze-Florey CR, Irwin MR, Arevalo JMG, Cole SW. Divergent gene expression responses to complicated grief and non-complicated grief. Brain Behav Immun. 2014;37:78–83. doi: 10.1016/j.bbi.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller GE, Murphy MLM, Cashman R, Ma R, Ma J, Arevalo JMG, et al. Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun. 2014;41:191–9. doi: 10.1016/j.bbi.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA. 2013;110:16574–9. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole SW, Capitanio JP, Chun K, Arevalo JMG, Ma J, Cacioppo JT. Myeloid differentiation architecture of leukocyte transcriptome dynamics in perceived social isolation. Proc Natl Acad Sci USA. 2015;112:15142–7. doi: 10.1073/pnas.1514249112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole SW, Conti G, Arevalo JM, Ruggiero AM, Heckman JJ, Suomi SJ. Transcriptional modulation of the developing immune system by early life social adversity. Proc Natl Acad Sci USA. 2012;109:20578–83. doi: 10.1073/pnas.1218253109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole SW, Levine ME, Arevalo JMG, Ma J, Weir DR, Crimmins EM. Loneliness, eudaimonia, and the human conserved transcriptional response to adversity. Psychoneuroendocrinology. 2015;62(Complete):11–17. doi: 10.1016/j.psyneuen.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fredrickson BL, Grewen KM, Algoe SB, Firestine AM, Arevalo JMG, Ma J, et al. Psychological Well-Being and the Human Conserved Transcriptional Response to Adversity. PLoS ONE. 2015;10:e0121839. doi: 10.1371/journal.pone.0121839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohrt BA, Worthman CM, Adhikari RP, Luitel NP, Arevalo JM, et al. Psychological resilience and the gene regulatory impact of posttraumatic stress in Nepali child soldiers. Proc Natl Acad Sci USA. 2016;113:8156–61. doi: 10.1073/pnas.1601301113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller GE, Chen E, Sze J, Marin T, Arevalo JMG, Doll R, et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-κB signaling. Biol. Psychiatry. 2008;64:266–72. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwaiger M, Grinberg M, Moser D, Zang JCS, Heinrichs M, Hengstler JG, et al. Altered stress-induced regulation of genes in monocytes in adults with a history of childhood adversity. Neuropsychopharmacology. 2016;41:2530–40. doi: 10.1038/npp.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engler H, Dawils L, Hoves S, Kurth S, Stevenson JR, Schauenstein K, et al. Effects of social stress on blood leukocyte distribution: the role of α and β-adrenergic mechanisms. J Neuroimmunol. 2004;156:153–62. doi: 10.1016/j.jneuroim.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Exton MS, Gierse C, Meier B, Mosen M, Xie Y, Frede S, et al. Behaviorally conditioned immunosuppression in the rat is regulated via noradrenaline and beta-adrenoceptors. J Neuroimmunol. 2002;131:21–30. doi: 10.1016/s0165-5728(02)00249-7. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz D, Wilsenack K, Lendemanns S, Schedlowski M, Oberbeck R. β-Adrenergic blockade during systemic inflammation: Impact on cellular immune functions and survival in a murine model of sepsis. Resuscitation. 2007;72:286–94. doi: 10.1016/j.resuscitation.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Benschop RJ, Nieuwenhuis EE, Tromp EA, Godaert GL, Ballieux RE, van Doornen LJ. Effects of beta-adrenergic blockade on immunologic and cardiovascular changes induced by mental stress. Circulation. 1994;89:762–9. doi: 10.1161/01.cir.89.2.762. [DOI] [PubMed] [Google Scholar]
- 28.Benschop RJ, Jacobs R, Sommer B, Schürmeyer TH, Raab HR, Schmidt RE, et al. Modulation of the immunologic response to acute stress in humans by β-blockade and benzodiazepines. FASEB J. 1996;10:517–24. doi: 10.1096/fasebj.10.4.8647351. [DOI] [PubMed] [Google Scholar]
- 29.Sanders VM, Straub RH. Norepinephrine, the β-adrenergic receptor, and immunity. Brain Behav Immun. 2002;16:290–332. doi: 10.1006/brbi.2001.0639. [DOI] [PubMed] [Google Scholar]
- 30.Jetschmann JU, Benschop RJ, Jacobs R, Kemper A, Oberbeck R, Schmidt RE, et al. Expression and in vivo modulation of alpha- and beta-adrenoceptors on human natural killer (CD16+) cells. J Neuroimmunol. 1997;74:159–64. doi: 10.1016/s0165-5728(96)00221-4. [DOI] [PubMed] [Google Scholar]
- 31.Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007;19:251–60. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Nater UM, Whistler T, Lonergan W, Mletzko T, Vernon SD, Heim C. Impact of acute psychosocial stress on peripheral blood gene expression pathways in healthy men☆. Biol Psychol. 2009;82:125–32. doi: 10.1016/j.biopsycho.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steptoe A, Ronaldson A, Kostich K, Lazzarino AI, Urbanova L, Carvalho LA. The effect of beta-adrenergic blockade on inflammatory and cardiovascular responses to acute mental stress. Brain Behav Immun. 2018;70:369–75. doi: 10.1016/j.bbi.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Känel R, Kudielka BM, Metzenthin P, Helfricht S, Preckel D, Haeberli A, et al. Aspirin, but not propranolol, attenuates the acute stress-induced increase in circulating levels of interleukin-6: A randomized, double-blind, placebo-controlled study. Brain Behav Immun. 2008;22:150–7. doi: 10.1016/j.bbi.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 35.Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci USA. 2003;100:1920–5. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faul F, Erdfelder E, Lang A-G, Buchner A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 37.Paterson JW, Conolly ME, Dollery CT, Hayes A, Cooper RG. The pharmacodynamics and metabolism of propranolol in man. Pharm Clin. 1970;2:127–33. doi: 10.1042/cs038010p. [DOI] [PubMed] [Google Scholar]
- 38.Kirschbaum C, Pirke KM, Hellhammer DH. The’Trier Social Stress Test’-a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology. 1993;28:76–81. doi: 10.1159/000119004. [DOI] [PubMed] [Google Scholar]
- 39.MacCormack JK, Armstrong-Carter EL, Gaudier-Diaz MM, Meltzer-Brody S, Sloan EK, Lindquist KA, et al. Beta-adrenergic contributions to emotion and physiology during acute psychosocial stress. 2020 under Rev. [DOI] [PMC free article] [PubMed]
- 40.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marsland AL, Walsh C, Lockwood K, John-Henderson NA. The effects of acute psychological stress on circulating and stimulated inflammatory markers: a systematic review and meta-analysis. Brain Behav Immun. 2017;64:208–19. doi: 10.1016/j.bbi.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. 2007;21:901–12.. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 43.Fredrickson BL, Grewen KM, Coffey KA, Algoe SB, Firestine AM, Arevalo JM, et al. A functional genomic perspective on human well-being. Proc Natl Acad Sci USA. 2013;110:13684–9. doi: 10.1073/pnas.1305419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole SW, Hawkley LC, Arevalo JMG, Cacioppo JT. Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci USA. 2011;108:3080–5. doi: 10.1073/pnas.1014218108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Black DS, Cole SW, Christodoulou G, Figueiredo JC. Genomic mechanisms of fatigue in survivors of colorectal cancer. Cancer. 2018;124:2637–44. doi: 10.1002/cncr.31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J Immunol. 2006;177:7303–11. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 47.Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, et al. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci USA. 2015;112:E6614–23. doi: 10.1073/pnas.1515733112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA. 2014;111:E4033–42. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olanoff LS, Walle T, Cowart TD, Walle KU, Oexmann MJ, Conradi EC. Food effects on propranolol systemic and oral clearance: Support for a blood flow hypothesis. Clin Pharmacol Ther. 1986;40:408–14. doi: 10.1038/clpt.1986.198. [DOI] [PubMed] [Google Scholar]
- 50.Melander A, Danielson K, Scherstén B, Wåhlin E. Enhancement of the bioavailability of propranolol and metoprolol by food. Clin Pharmacol Ther. 1977;22:108–12. doi: 10.1002/cpt1977221108. [DOI] [PubMed] [Google Scholar]
- 51.Manning PJ, Sutherland WHF, McGrath MM, de Jong SA, Walker RJ, Williams MJA. Postprandial cytokine concentrations and meal composition in obese and lean women. Obesity. 2008;16:2046–52. doi: 10.1038/oby.2008.334. [DOI] [PubMed] [Google Scholar]
- 52.Hansen K, Sickelmann F, Pietrowsky R, Fehm HL, Born J. Systemic immune changes following meal intake in humans. Am J Physiol Integr Comp Physiol. 1997;273:R548–53. doi: 10.1152/ajpregu.1997.273.2.R548. [DOI] [PubMed] [Google Scholar]
- 53.Orban Z, Remaley AT, Sampson M, Trajanoski Z, Chrousos GP. The differential effect of food intake and β-adrenergic stimulation on adipose-derived hormones and cytokines in man. J Clin Endocrinol Metab. 1999;84:2126–33. doi: 10.1210/jcem.84.6.5747. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y-Z, Wang Y-X, Jiang C-L. Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci. 2017;11:316. doi: 10.3389/fnhum.2017.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Squassina A, Pisanu C, Vanni R. Mood disorders, accelerated aging, and inflammation: is the link hidden in telomeres? Cells. 2019;8:52. doi: 10.3390/cells8010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rohleder N. Stress and inflammation—the need to address the gap in the transition between acute and chronic stress effects. Psychoneuroendocrinology. 2019;105:164–71. doi: 10.1016/j.psyneuen.2019.02.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.