

Abstract
Antibodies play essential roles in both diagnostics and therapeutics. Epitope mapping is essential to understand how an antibody works and to protect intellectual property. Given the millions of antibodies for which epitope information is lacking, there is a need for high-throughput epitope mapping. To address this, we developed a strategy, Antibody binding epitope Mapping (AbMap), by combining a phage displayed peptide library with next-generation sequencing. Using AbMap, profiles of the peptides bound by 202 antibodies were determined in a single test, and linear epitopes were identified for >50% of the antibodies. Using spike protein (S1 and S2)-enriched antibodies from the convalescent serum of one COVID-19 patient as the input, both linear and potentially conformational epitopes of spike protein specific antibodies were identified. We defined peptide-binding profile of an antibody as the binding capacity (BiC). Conceptually, the BiC could serve as a systematic and functional descriptor of any antibody. Requiring at least one order of magnitude less time and money to map linear epitopes than traditional technologies, AbMap allows for high-throughput epitope mapping and creates many possibilities.
Keywords: epitope mapping, phage display, random peptide library, next-generation sequencing, COVID-19
Abbreviations: AbMap, antibody binding epitope mapping; BiC, binding capacity; ELISA, enzyme-linked immunosorbent assay; HDX-MS, hydrogen–deuterium exchange mass spectrometry; IgG, immunoglobulin G; NGS, next-generation sequencing; WB, Western blotting
Graphical Abstract
Highlights
-
•
The first technology enables epitope mapping of 200 antibodies in a single run.
-
•
Applications of antibodies, not for original antigens, were further exploited.
-
•
The epitopes of antibodies in one COVID-19 patient were identified.
-
•
The BiC is a new systematic and functional character of antibody.
In Brief
In this study, we developed a strategy, Antibody binding epitope Mapping (AbMap), by combining a phage displayed peptide library with next-generation sequencing. By AbMap, epitopes of 202 antibodies were determined in a single test. Also, the epitopes of spike protein-specific antibodies from convalescent serum of one COVID-19 patient were identified. We defined the profile of the binding peptides of an antibody as binding capacity (BiC), conceptually, BiC could serve as a systematic and functional character for any antibody.
Antibodies are a group of naturally occurring proteins that can specifically bind to antigens, especially proteins. Taking advantage of the high specificity of antibody–antigen binding, antibodies are widely applied as reagents for many different assays, e.g., western blotting, enzyme-linked immunosorbent assay (ELISA), flow cytometry, and immunoprecipitation. According to CiteAb (1), there are 5,726,682 antibodies available on July 13, 2020 from 186 suppliers (https://www.citeab.com/). This number does not include the large number of homemade antibodies by researchers around the world. In addition, the market for antibodies is growing rapidly, with a projected annual rate of 6.1% until 2025 (https://www.grandviewreresearch.com). Since the first therapeutic antibody, i.e., Muromonab OKT3, we have witnessed the dramatic growth in this market. There are 70 FDA- and EMA-approved therapeutic antibodies on the market by 2017 (2, 3) for the treatment of a variety of diseases, including cancer, autoimmune diseases (4), and pathogenic bacteria (5). In addition, there are over 570 therapeutic antibodies in various phases of clinical trials, including 62 in late stages (3).
The binding of an antibody to an antigen occurs via its epitope, which is a small area of a few amino acids on the antigen. Typically, there are two types of epitopes, i.e., linear epitopes and conformational epitopes (6). The epitope is one of the most critical parameters for antibody–antigen binding. Knowing the epitope when using an antibody as a diagnostic reagent makes the experimental conclusions clear and relevant. For a therapeutic antibody, knowing the epitope helps us understand the precise mechanism of action. More importantly, it allows the developer to protect intellectual property.
There are several technologies used for epitope mapping, which can generally be categorized into two types, i.e., structure-based and peptide-based (7). Structure-based technologies, including X-ray crystallography (8), nuclear magnetic resonance (9), and more recently, Cryo-EM (10), are the golden standards. As of May 5, 2020, there were 934 antibody–antigen cocrystal structures in PDB (www.rcsb.org). For any antibody, the comprehensive binding interface can readily be obtained based on the antibody–antigen cocrystal structure. However, it is difficult to acquire high-quality antibody–antigen cocrystals, which is the key step for successful X-ray crystallography. It is even more difficult when the antigen is a membrane protein (11). Peptide-based technologies include peptide microarray (12), SPOT (13), and phage- or E. coli-displayed peptides or protein fragments (14). Recently, next-generation sequencing (NGS) was also introduced into this field in combination with a display platform, such as phage display (15) and E. coli display (16). Typically, peptides covering a single antigen or a set of antigens are synthesized and immobilized on a planar microarray or presented on phage/E. coli. The peptides that an antibody binds to are then enriched and identified. Usually, large numbers of peptides are required, and the construction of the microarray and the library is complex. Compared with conformational epitopes, linear epitopes are much easier to obtain using peptide-based technologies. In addition to the structure-based and peptide-based technologies, other technologies, including site-specific mutagenesis (17), mass spectrometry (18), and hydrogen–deuterium exchange mass spectrometry (HDX-MS) (19), have been developed. All these technologies are time-consuming, labor-intensive, and costly. Using these, epitopes can only be resolved in a case-by-case manner. Success with one antibody–antigen pair is difficult to repeat in another pair. Only one antibody or a set of antibodies can be analyzed at a time, and none of them are suitable for high-throughput analysis.
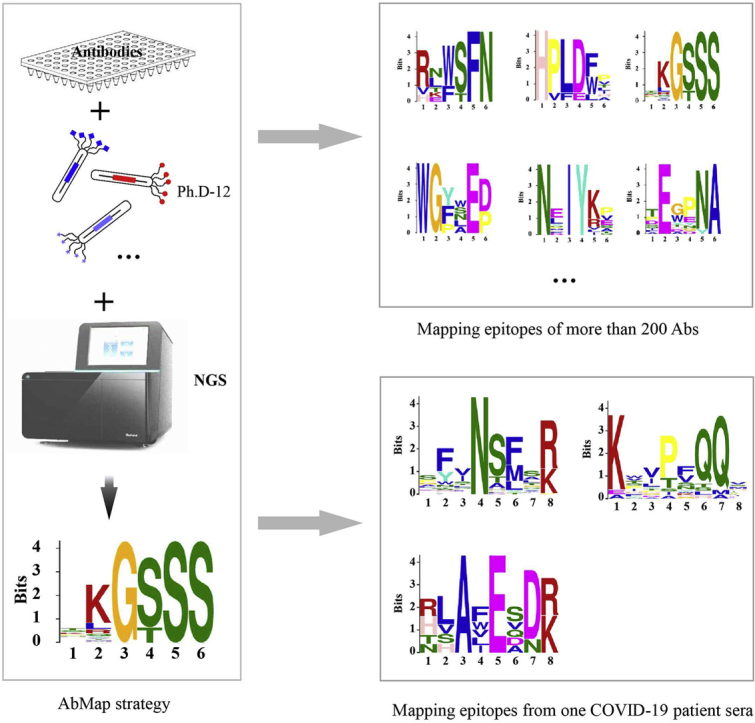
With millions of antibodies on the market and epitope information lacking for most of them, we are aiming to develop an enabling technology to massively map the epitope of antibodies. Once the epitopes are mapped, myriad possibilities can be explored. A schematic of our strategy is depicted in Figure 1, and we named this strategy AbMap. Key components of AbMap are the phage displayed random peptide library and the power of NGS for decoding barcoded and mixed samples in an unprecedented way. Our underlying hypothesis is that the highly diversified phage displaying random peptide library can cover many of the peptides that can be recognized by a given antibody.
Fig. 1.

The schematic and workflow of AbMap. A, antibodies are incubated with a phage displayed random peptide library. B and C, the phages bound by the antibody are enriched by using Protein G-coated magnetic beads. D, the DNA is released and barcoded with a unique barcode pair in each well. E, a unique index is selected for each sample. F, the PCR products are mixed. G, quality control of the barcoded and indexed PCR products. H, NGS analysis of the samples. I, data processing. J, epitope/mimotope calculation.
Taking several affinity-tagged specific antibodies as examples, we first prove the feasibility of AbMap. A high-throughput protocol is then established, and its capability is demonstrated by testing 202 antibodies in a single run. The results show that linear epitopes can be determined for >55% of the antibodies. The identified peptides/epitopes are then explored for other interesting applications. Lastly, we demonstrate the use of AbMap to determine epitopes of SARS-CoV-2-specific antibodies enriched from the serum of a convalescent patient with COVID-19.
Experimental Procedures
Enrichment of Phage Displayed Peptides Through Antibody Binding
Screening the Antibodies’ Binding Peptides
The Ph.D-12 phage display library was purchased from New England Biolabs. Subsequently, this library was amplified on approximately 200 solid plates, each 150 mm in diameter. According to the protocol, the amplified Ph.D-12 phage display library was purified, titered, and stored at −20 °C in 50% glycerin. For screening of antibodies’ binding peptides, each well of a 96-well PCR plate was blocked with 200 μl of 3% bovine serum albumin in TBST (containing 0.5% Tween 20 in TBS) overnight on a rotator at 4 °C. Immunoglobulin G (IgG) antibodies (0.02–0.8 μg) of and 10 μl of the amplified PhD.-12 library (∼100 fold representation, 1011 plaque-forming units for a library of 109 clones) were added to each preblocked well. The antibodies and phages from the PhD.-12 library were incubated overnight on a rotator at 4 °C. Ten microliters of Dynabeads protein G (Thermo Scientific) was added into each well and incubated for 4 h on a rotator at 4 °C to capture the antibody–phage complex. With a 96-well magnetic stand, the Dynabeads protein G was washed three times with 200 μl of TBST, followed by an additional wash with deionized water. The beads in each well were resuspended in 15 μl of deionized water, and the phages were lysed at 95 °C for 10 min. The phage lysates were stored at −80 °C.
Constructing the Library for NGS
The library for NGS was constructed following a previously protocol (20), with slight modifications. Briefly, two rounds of extension PCR were performed by using Q5 Hot Start High-Fidelity DNA Polymerase from New England Biolabs. In the first round of extension PCR, the phage lysate from each well was used as template. The primers used are shown in supplemental Table S1. A unique combination of upstream (X-SXX-23R) and downstream primers (X-NXX-18) was set for each antibody. A total of 25 cycles were performed. After evaluation by electrophoresis, the products of the first round of extension PCR were purified by using a gel extraction kit (TIANGEN Biotech), and 5 μl of each purified sample was used as the template in the second extension PCR. In the second extension PCR, the primer combination S502 and N701 or S520 and N720 was used, and only ten cycles were performed. The PCR products were evaluated by electrophoresis and purified as in the first round of PCR. The concentration of each sample was determined by using a NanoDrop 2000c spectrophotometer (Thermo Scientific). All the PCR products were mixed in equimolar amounts. The quality of the NGS library was checked by TA cloning and Sanger sequencing. After examining the structure of each insert and frequency of the different barcode pairs, the NGS library was subjected to Illumina sequencing using the paired-end 2 × 150 sequencing mode.
NGS Data Processing and Analysis
Quality Control of NGS Data
To filter out the low-quality reads from the raw data (Fig. 2A), the software Trimmomatic (version 0.35) was used with the MINLEN parameter set to 150 and other parameters set to default (21). Since the insert size of our library was exactly 103 bp, the remainder (47 bp) of each read was trimmed as 3’ adaptors (Fig. 2B). To ensure high sequencing fidelity, only paired reads with the exact same insert sequence (i.e., no mismatches in the 103-bp region between the paired reads) were kept as clean data for downstream analysis.
Fig. 2.

Workflow of data processing.A, the structure of the construct for NGS. The solid black lines represent fixed sequences and colored boxes represent varied sequences. B, the filtered NGS data. C, illustration of the reads assigned to antibodies. D, illustration of the peptides assigned to antibodies. E, the algorithm used to obtain binding peptides. For each peptide, both E (red dots) and E−1 (blue dots) are plotted. Only peptides with red dots above the dashed line of the cutoff (E−1max) were kept as binding peptides for a given antibody. F, an example of an epitope identified with MEME based on the binding peptides, the number, i.e., 44, of binding peptides for generating the logo is depicted on the right as “44 Peps.” The epitope was accompanied by a position-specific probability matrix from MEME. G, an example of a similarity calculation between the epitopes and the antigens. The red boxes (each consisting of six amino acids) on the antigens indicate the best-matched position-specific probability matrix (epitope) based on which the epitope–antigen similarity was calculated.
Peptide Count Normalization
For each read, there were two 8-bp barcodes and a 36-bp variable DNA sequence. Each pair of 8-bp barcodes uniquely defined the antibody sample from which the insert sequence originated (Fig. 2C). The 36-bp DNA sequence, which corresponded to the peptide sequence displayed by phage, was translated into a 12-aa peptide and assigned to the corresponding antibody according to the unique barcode pair (Fig. 2D). Subsequently, the number of peptides for each antibody was counted and put into an N x M matrix (defined as a raw count matrix) with rows representing N different 12-aa peptides and columns representing M different antibodies. Thus, each cell of the matrix was the raw count of a peptide. Then the raw count was normalized by the total count of peptides in the column. If a peptide appeared in a sample, and did not appear in the control sample, the count of that peptide was set to 1 in the control sample. The counts of these peptides were not calculated for total count of control sample. Additionally, the arithmetic mean count of each peptide was calculated for all control samples and was further added into the matrix as the control column. The resulting matrix was defined as the normalized count matrix.
Identifying Binding Peptides for Each Antibody
Based on the normalized count matrix, the binding peptides for each antibody were determined by comparing between the antibody and control columns. Since no antibody was added to the control samples, all peptide counts observed in the control samples were considered background noise. For any antibody in the matrix, the enrichment factors (E) of all peptides were defined as the antibody column divided by the control column. Due to the background noise, the values in the control column were often much greater than those in the antibody column. Thus, for the same antibody, the reverse enrichment factors (E−1) were calculated by dividing the control column by the antibody column, and the maximum reverse enrichment factor (E−1max) was used as the 1× cutoff for the enrichment factors. The 1× and 3× cutoff for each antibody was calculated separately, and for different antibodies, the cutoffs might be different. Unless otherwise specified, only peptides with E > 1× cutoff were kept as binding peptides for downstream analysis (Fig. 2E).
Epitope Analysis for Each Antibody
Binding peptides for each antibody were submitted to the standa-lone version of MEME for epitope discovery using the parameters -evt 0.001 -nepitopes 8 -minw 6 -maxw 10 with the rest set to default (22). Epitopes with E-values <0.05 were considered to be reliable epitopes for each antibody (Fig. 2F). All the epitopes were based on the peptides with enrichment factors larger than 3× the cutoff unless otherwise stated.
Reproducibility Analysis
The reproducibility of our data was assayed at the peptide level. The Pearson correlation coefficient was calculated using the raw peptide counts from two experimental replicates of the same antibody at a comparable sequencing depth.
Sequencing Depth for Linear Epitope Discovery
To analyze the impact of sequencing depth on linear epitope discovery, we performed downsampling to randomly draw reads from the raw sequencing data (i.e., FASTQ files). Additionally, in the secondary biological repeat, the samples were sequenced twice. The raw data, approximately 143 M reads, were obtained. To balance the sequence depth and cost, this sample was sequenced again. As a result, the raw data, approximately 75 M reads, were obtained. In other side, we randomly sampled the raw data (143 M paired reads) of the secondary biological repeat into 12 data files consisting of 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, and 130 M reads each, performed the same analysis to obtain the linear epitopes for each antibody, and compared the linear epitopes with those from the raw data. Based on linear epitope reproducibility, the optimal cost-effective sequencing depth for linear epitope discovery was determined.
Statistical Analyses
All statistical analyses were performed using the R language (http://www.r-project.org/). In the case of multiple hypothesis testing, p-values were corrected with the BH method unless otherwise specified (23).
Validation of Antibody–Peptide Binding
Measuring the Antibody–Peptides Binding Affinities
All the peptides were synthesized by GL Biochem (Shanghai) Ltd. According to the manufacturer’s guidelines, the peptides were biotin labeled with by using Sulfo-NHS-LC-Biotin (Thermo Scientific). The unconsumed Sulfo-NHS-LC-Biotin was quenched with a 1 M glycine solution. The biotinylated peptides were diluted to 2 μM and loaded on a prewetted SA sensor from Pall ForteBio. A serial dilution of antibodies was tested against the peptide-loaded sensors. The results were recorded using the Octet Red 96 system and analyzed by Octet software v7.x from Pall ForteBio.
Validation of Peptides Binding by Corresponding Antibodies via Western Blot
A total of 43 antibodies and one binding peptide for each antibody were selected. The peptide sequences were linked to several widely applied affinity tags, and a synthetic protein (SP) was constructed. The sequence of the SP was translated into a DNA sequence, codon optimized, synthesized by GenScript, and integrated into the expression vector pET-28a. The SP was induced via incubation in 0.5 mM IPTG at 16 °C overnight. A blank control, E. coli containing empty pET-28a, was cultured and induced under the same conditions. To prepare the samples, the lysate from E. coli containing the SP was diluted via twofold serial dilutions with the lysate from the blank control. All the samples were mixed with 5× loading buffer and boiled for 5 min. Subsequently, SDS-PAGE and Western blotting (WB) were carried out using a 10% SDS-PAGE gel to investigate binding between the SP and the 43 antibodies. For WB, the dilution factor of the 43 antibodies was set at 5000. Furthermore, the E. coli lysates containing different amounts of the SP were mixed with tissue lysates from a variety of species and a variety of cell lines in order to investigate antibody specificity.
Applications of the Identified Peptides/Linear Epitopes
Matching the Linear Epitopes to Human and E. coli Proteins
To demonstrate the relationship between epitope and protein, we designed a custom, sliding-window strategy to calculate linear epitope–protein similarity as follows. First, we used MEME to generate a position-specific probability matrix that showed the frequency of each amino acid at each position of the identified linear epitope (22). Second, we used this matrix as a sliding window (moving one amino acid at a time) to scan the peptide from N terminus to C terminus and determine which section of the peptide best matched the matrix. Third, based on the best-matched section, we calculated the similarity of the whole protein. Based on the best-matched part, the value for each defined residue in the corresponding positions of the linear epitope was searched. Subsequently, all the values for each residue in the best-matched section were added together and then divided by the length of the linear epitope. The result was defined as the protein–epitope similarity.
The linear epitopes (based on binding peptides) of the 202 antibodies were matched to corresponding immunogens, and their similarities were calculated in the same way. The linear epitopes of the 202 antibodies were matched to all the human and E. coli proteins. A list of human and E. coli proteins with high sequence similarity to the identified linear epitopes were obtained.
Also, to test the results of binding peptide identification for each antibody, anti-6xHis, anti-V5, and three other antibodies with linear epitopes based the binding peptide with enrichment factor larger than 3× the cutoff were taken as example. The three antibodies were selected randomly. The similarity between linear epitope and each peptide in the raw data, or peptides with enrichment factor greater than 1× the cutoff, or peptide with enrichment factor greater than 3× the cutoff, was calculated. All the results were generated and the violin plot was plotted by using the vioplot package (24).
Validating Antibody–Protein Binding by Western Blotting
Approximately 11 human proteins with similarities greatly than 0.6 were selected for validation. In the selection process, a few proteins with two linear epitope matches at different sites were also included. Using the human ORF library from Invitrogen, the plasmids carrying the selected protein genes were transferred to the expression vector pDEST 15 via LR reactions (Thermo Scientific). Subsequently, all the reaction products were transformed into BL21 (DE3) chemically competent cells and confirmed by Sanger sequencing. All correct proteins were induced via incubation in 0.5 mM IPTG at 37 °C for 2 h. All the samples were used for western blotting to validate binding of the target protein by antibodies. The dilution factor used for antibodies in western blotting was 1000.
For proteins from E. coli, approximately 11 proteins were selected for validation. In the selection process, a few proteins with two linear epitope matches at different sites were also included. The subsequent procedures were the same as those for human proteins.
Antibody Pairing for ELISA
Protein Purification
The gene for protein Q6UXI7 was amplified with the primers F30B, with the sequence CGGGAATTCATGAGGACTGTTGTTCTCACTATGAAG, and R30B, with the sequence CCCAAGCTTTCAGTTCCGAGGCTGTGAGTTGAACTCTG, which contained the restriction sites for Eco RI and Hind III, respectively. The amplified fragment and expression vector pET-28a were digested with Eco RI and Hind III. After purification, the gene fragment and the linearized vector were linked by using T4 DNA ligase (New England Biolabs). Subsequently, the plasmid was transformed into BL21 (DE3) chemically competent cells. The final expression vector containing the gene for protein Q6UXI7 was confirmed by Sanger sequencing. Proteins Q6UXI7, P16431, and P23930 were induced via incubation in 0.5 mM IPTG at 37 °C for 2 h. The cells were harvested and lysed by using lysis buffer (TBST containing 2% SDS). The protein Q6UXI7 was purified using GST affinity columns. The proteins P16431 and P23930 were purified using Ni-NTA affinity columns. The purified proteins were stored at −80 °C.
Biotinylation of the Antibodies
Ab019 and Ab161 were purified via incubation with Protein G beads, and their concentrations were determined by using a BCA protein assay kit (Thermo Scientific). According to the manufacturer’s protocol, Ab019 and Ab161 were labeled with Sulfo-NHS-LC-Biotin (Thermo Scientific). The free Sulfo-NHS-LC-Biotin was removed by dialysis. The concentrations of the biotinylated antibodies were determined by BCA assay.
ELISA
The first antibody was diluted to 4 μg/ml with PBST (PBS containing 1% Triton X-100). The 96-well plates were sensitized with 50 μl of antibody per well. After centrifugation for 2 min at 550g in order to distribute the proteins evenly, the 96-well plates were incubated at 4 °C overnight. Subsequently, the plates were emptied and blocked with 300 μl of blocking buffer (Cat. 37572, Thermo Scientific) at RT for 1 h. The plates were washed three times with PBST and incubated with 50 μl of target protein at different concentrations at RT for 1 h. The plates were washed with PBST three more times and incubated with 50 μl of biotinylated antibody at a concentration of 2 μg/ml. After incubation for 1 h at RT, the plates were washed with PBST three more times and incubated with 50 μl of 1000X diluted SA-HRP (Sigma-Aldrich). After three washes with PBST, 50 μl of TMB solution (Sigma-Aldrich) was added to each well, and the plates were incubated for 20 min at 37 °C. The plates were incubated with 50 μl of 1 M H2SO4 to stop the reaction. The results were recorded using a multichannel spectrophotometer (BioTek Instruments) at 450 nm. Three replicates were performed. A typical third-order polynomial (cubic) nonlinear regression model was used for standard curve fitting for ELISAs, which was later used to calculate the concentration of target proteins in samples.
Enriching the S1 and S2 Domain Binding Antibodies From the Convalescent Serum of a COVID-19 Patient
The 20 μg biotinylated S1 (Kactus Biosystems) and S2 were bound to 100 μl of streptavidin magnetic beads (Invitrogen) in PBS buffer at room temperature for 1 h. Meanwhile, the convalescent serum from a COVID-19 patient was diluted twice in PBS and then preincubated with streptavidin beads to avoid nonspecific binding. The protein-coated streptavidin beads were washed four times with PBS containing 0.1% BSA and incubated with aforementioned serum at 4 °C for 4 h. The streptavidin beads were then washed three times with PBS containing 0.1% BSA and eluted with 0.2 M glycine, 1 mM EGTA, pH 2.2. Finally, the antibodies were neutralized with 1M Tris-HCl, pH8.0, and stored at −80 °C until use.
Serum Collection
The Institutional Ethics Review Committee of Foshan Fourth Hospital, Foshan, China, approved this study, and written informed consent was obtained from the patient. The COVID-19 patient was hospitalized and received treatment at Foshan Forth Hospital. The serum sample was collected when the patient was discharged from the hospital. The serum was collected according to a standard protocol (25) and stored at −80 °C until use. Before used, the serum was incubated at 56 °C for 1 h to inactive the virus. The patient was female and 70 years old.
Results
Schematic Diagram of AbMap
The schematic and workflow of AbMap are shown in Figure 1. Antibodies (monoclonal or polyclonal) are aliquoted in 96-well plates (Fig. 1A), and a phage displayed random peptide library, in our case Ph.D-12, is incubated with the antibody in each well (Fig. 1B). The phages bound by the antibody are enriched by using Protein G-coated magnetic beads (Fig. 1, B and C). The DNA is released from the enriched phages, the PCR product is barcoded with a unique barcode pair in each well (Fig. 1D), and a unique index is chosen for each batch of antibodies (Fig. 1E). The barcodes and indexes used in this study are shown in supplemental Table S1. To avoid possible amplification bias, the index is incorporated for each individual antibody in each well and then mixed (Fig. 1F). To control the sample quality, e.g., the purity and the concentration of the DNA samples, the integrity of the PCR products is measured (Fig. 1G). The sample is then subjected to NGS (Fig. 1H). The sequencing data is processed through filtering, normalization, etc (Fig. 2). The peptides that are strongly bound by each antibody are determined (Fig. 1I). The linear epitope or mimotope (the peptide that mimics the structure of an epitope) (26) of a given antibody is revealed by a variety of computational tools (Fig. 1J).
Binding Peptides/Linear Epitopes Resolved for Widely Applied mAbs and pAbs
To assess the suitability of AbMap for monoclonal antibodies, we selected two widely applied affinity tag antibodies, i.e., anti-6xHis and anti-V5, and went through the procedure depicted in Figures 1 and 2. Figure 3A shows four representative peptides recognized by the anti-6xHis antibody. In total, 43 peptides were detected, and 32 of them contained >3 histidines. These peptides are rich in histidines, as expected, and the anti-6xHis antibody was therefore used as a positive control for the following analyses. Clearly, the linear epitope matched well with 6xHis; however, these six histidines did not contribute equally (Fig. 3B). A four-amino-acid linear epitope was determined for the anti-V5 antibody, which perfectly matches the V5 sequence (Fig. 3B). These results indicate that AbMap is reliable for identifying the linear epitopes of monoclonal antibodies, and even the linear epitopes of widely applied tag antibodies could be refined.
Fig. 3.

High-throughput profiling of antibody–peptide binding.A, four binding peptides of an anti-6xHis antibody. B, the binding epitopes for the anti-6xHis antibody and an anti-V5 antibody. C, the reproducibility of AbMap at the peptide level. The Pearson correlation of two independent replicates of two antibodies (left panel). The summary of the Pearson correlation of the two replicates for all the antibodies tested (right panel). D, the reproducibility of AbMap at the epitope level. Two representative antibodies are shown. E, the binding kinetics between representative antibody-binding peptide pairs. BLI was applied for this analysis. F, validation with a de novo synthesized protein, i.e., synthetic protein (SP) (see supplemental Fig. S5 for more details). SP was overexpressed in E. coli, and the cell lysate was prepared. Western blotting was performed on serially diluted SP from E. coli cell lysates. “16” represented 16× dilution. To further assess the specificity of the antibodies, the E. coli cell lysate with overexpressed SP was diluted with cell lysates from other species and cells.
In addition to monoclonal antibodies, polyclonal antibodies are also widely used. To test the possibility of identifying linear epitopes of polyclonal antibodies by using AbMap, three antibodies recognizing three well-studied proteins/tags, i.e., GST, histone H3, and GFP, were chosen. Through the same procedure as shown in Figures 1 and 2, statistically significant linear epitopes were identified for all three of these antibodies. Most of the linear epitopes matched the protein sequences well. In comparison with the two monoclonal antibodies used above, each of the polyclonal antibody tested recovered 2 to 3 different linear epitopes (supplemental Fig. S1). This is consistent with the fact that polyclonal antibodies usually comprise of antibodies from different clones, and the linear epitope for each antibody in the polyclonal antibody sample can be different (27). Therefore, the identification of different motifs matching different locations of the targets is consistent with the polyclonal nature of these antibodies (supplemental Fig. S1).
High-Throughput Antibody–Peptide Binding Profiling
After success with several widely used antibodies, we then set out to apply AbMap for high-throughput analysis. A total of 202 mouse monoclonal antibodies (supplemental Table S2), produced in 2012 via immunization with selected peptide antigens, were included (28). The anti-6xHis antibody was used as a positive control. A blank control without any antibody was also included. Following the procedure, as depicted in Figures 1 and 2, we performed the analysis (supplemental Tables S3 and S4). To assess reproducibility, the binding peptide profiling of these 202 antibodies was independently repeated twice. Pearson analysis showed that between the two repeats, most of the antibodies had a Pearson correlation coefficient of more than 0.8 at the peptide level (Fig. 3C and supplemental Table S5). At epitope level, it is noteworthy that the majority (69/83) of the linear epitopes are determined for both of the two replicates (Fig. 3D). The epitopes that were determined in only one experiment were usually associated with larger E values and less binding peptides.
There was no contribution for some of the peptides for calculating the final sequence logo by MEME. For example, only 324 out of the 344 input peptides contributed to the generation of epitope Ab012-M1 (supplemental Table S3, Fig. 3D). To investigate the possible contribution of a peptide for the determination of an epitope, the similarity between the peptide and the epitope was calculated. It is reasonable to hypothesize that if a peptide has contribution to an epitope, it should have a relative higher similarity to the final epitope as compared with peptides that do not have contribution. To test this hypothesis, Anti-6xHis, anti-V5, and three antibodies randomly selected antibodies, i.e., Ab012, Ab019, and Ab096 (supplemental Table S4B), were taken as the examples. As shown in supplemental Fig. S3, before filtered by the 1× or 3× cutoff, the similarity of most peptides to the corresponding epitope was ∼0.2, while after being filtered, the similarities of peptides were increased to 0.5 for anti-6xHis, Ab012, and Ab096 and to 0.6 for anti-V5 and Ab019. These results demonstrate that most of the filtered peptides do contribute to the generation of the final epitopes. And this further confirms the effectiveness of filtering the peptides by the cutoffs.
It is possible that binding peptides of the antibody are biased to reflect the abundance of the peptide-corresponding phages in the original phage display library. To rule out this possibility, we randomly selected three antibodies and chose three binding peptides that represented high, medium, and low enrichment factors for each antibody. The abundances of all these peptides in the original phage library were then quantified by using qPCR. The results clearly showed no statistical difference in abundance between the three peptide-corresponding phages for each antibody (supplemental Fig. S2).
It is critical to strike a balance between the number of NGS reads and cost. We independently analyzed the 202 mAbs twice, and NGS reads for these two analyses were obtained, i.e., 143 M and 75 M. Twelve subsets of reads (ranging from 20 M to 130 M) were randomly selected from the data set of 143 M reads. Linear epitopes for the same antibody were compared among all the 14 data sets. The results showed that more than 60 M, i.e., more than 0.3 M reads per antibody was enough to assure the successful calculation of the linear epitopes (supplemental Fig. S3).
To confirm the interaction between antibodies and the binding peptides, we chose several binding peptides with varied enrichment factors. The binding peptides (supplemental Table S6) were synthesized, biotinylated, and immobilized on a streptavidin-coated sensor. Bio-layer interferometry (BLI) assays were performed. The KD could be measured to the nM level or even greater (Fig. 3E and supplemental Table S6). Also, the mismatched peptide–antibody pairs could not interact with each other.
To further validate more antibody–peptide binding, 43 antibodies were randomly selected and one peptide for each of these antibodies was also selected. The criteria for selecting these peptides were arbitrary set as enrichment factor around 500. (supplemental Table S7). We linked the sequences of all these peptides together and constructed a synthetic protein (SP), which we overexpressed in E. coli (Fig. 3F and supplemental Fig. S5A). To test antibody sensitivity, SP was serially diluted (supplemental Fig. S5B) and western blotted using all 43 antibodies individually. The results showed that SP could be clearly and specifically recognized by all 43 antibodies. Even after 128× dilution, specific bands were still clearly visible for some of the antibodies (Fig. 3F and supplemental Fig. S5C). To test the specificity, complex samples were prepared by diluting the E. coli lysate containing the overexpressed SP with lysates from a variety of species and cell lines. We performed western blotting of these complex samples, using several antibodies randomly selected from the 43 antibodies. The results showed that the sensitivity and specificity of most of the antibodies were consistent even in complex background (Fig. 3F). Also, unmatched antibodies could not recognize the SP protein (supplemental Fig. S5D).
For the high-throughput experiment, as shown in supplemental Fig. S6, 174 antibodies exceeded the cutoff, i.e., had at least one enriched binding peptide. The failure of the other 28 antibodies may due to deleterious effects from the prolonged storage since 2012, the high enrichment cutoff required here, or an insufficient sequencing depth. For example, the cutoff for Ab045 was 50.96, which was greater than that of most antibodies.
If the cutoff was decreased, there would be more peptides remaining, and the significant linear epitope for Ab045 might be calculated. However, the ideal cutoff threshold was not known at current stage. Only after many validation tests could a more precise cutoff be obtained. Additionally, sequencing depth is an important factor. For example, the sequencing depth for Ab019 was 204,464, which was less than the average value of 250,000.
In this study, 66 antibodies each recovered 1∼20 binding peptides that passed the 3× cutoff. Among these antibodies, sequence logos with E value smaller than 0.05 were obtained for 13 of them. For example, Ab030 has 17 binding peptides, and a sequence logo with E value smaller than 0.05 was successfully obtained. More importantly, this logo was validated by WB (supplemental Fig. S7A). Three other motifs (Ab182-M1 calculated from ten binding peptides, Ab185-M2 calculated from five binding peptides, Ab197-M2 calculated from 13 binding peptides) were also validated by WB (supplemental Fig. S7, A and C). Logos generated for antibodies recovering small numbers of peptides were associated with larger E values and lower intensities detected in immunoblotting.
Statistically significant linear epitopes were obtained for 112/174 antibodies based on the binding peptides with enrichment factor greater than 1× the cutoff. We found that the linear epitopes of 49 out of the 112 antibodies were consistent with the peptides against which they were raised (supplemental Fig. S6, A and B and supplemental Table S4A). Why a high fraction of antibodies yielded epitopes discordant with the immunogens remains to be explored.
In summary, we have established a reliable procedure for revealing the linear epitope recognized by an antibody in a high-throughput manner.
The Antibodies Were Applied to Recognize Proteins Other Than the Antigens Against Which They Were Raised
Based on a 3× cutoff value, the linear epitopes of higher stringency were also calculated (supplemental Table S4B). Given the multispecificity of antibodies (Fig. 4A), the linear epitopes in supplemental Table S4B were used to perform matching/homologous searches in human and E. coli, representing eukaryotes and prokaryotes, respectively. For this experiment, we used an arbitrary similarity cutoff 0.5 (Note that a better strategy for cutoff selection would involve a larger validation set). As shown in Figure 4B and supplemental Table S8A, many proteins were matched.
Fig. 4.

The antibodies could be applied to recognize proteins other than the antigens against which they were raised.A, the multispecific nature of antibodies. Usually, an antibody is generated through animal immunization with an antigen of interest. The antibody can recognize a specific epitope or mimotope, and this epitope/mimotope exists in a variety of proteins in nature. B, the epitopes revealed through AbMap from the 202 mAbs match to many proteins. C and D, western blotting of human (C) and E. coli (D) proteins using the matched mAbs. E, ELISA using a pair of Abs matched to different positions of the same protein.
According to the selection procedures described in the Experimental Procedures section, 11 human and 11 E. coli proteins were identified for further validation (supplemental Table S8B).
For the human proteins, if the target proteins were successfully expressed, the interactions between the matched proteins and the corresponding antibodies were validated by western blotting (Fig. 4C, supplemental Fig. S7, A and B). E. coli proteins were selected randomly from supplemental Table 8A. Most of the predicted interactions (14/17) were validated (Fig. 4C and supplemental Fig. S7C).
Furthermore, the linear epitopes of two or more antibodies could match to different positions of a protein (supplemental Table S8A), i.e., linear epitopes Ab019-M1 and Ab156-M1, both of which are well matched with human protein Q6UXI7 (Fig. 4E). In another two examples, linear epitopes Ab161-M1 and Ab185-M2 with protein P16431, and Ab019-M1 and Ab182-M1 with P23930, were well matched and validated by western blotting (Fig. 4E, supplemental Fig. S7, B and C). These antibodies could easily be paired for developing ELISAs. Relatively high dynamic ranges and sensitivities were achieved for detecting Q6UXI7 and P16431 via ELISAs (Fig. 4E).
We must point out that even if a linear epitope matched many proteins in different species, it might not mean that the antibody was useless. In the real-world applications, there is almost no need to test mixed-species samples, so relative specificity could still be assured when testing samples from a given species. More definitive results could be obtained by using phage peptidomes tiling the amino acids sequences of target proteomes (20, 29, 30). However, the peptidome/peptide library would need to be specifically designed and synthesized, which is costly and time-consuming. Furthermore, it could subsequently only be applied in the corresponding species and would not be as generally applicable as a random peptide library.
In addition, our understanding of antibody multispecificity could be enhanced. According to the conventional concept, the capacity of an antibody to bind several “unrelated” targets is regarded as the multispecificity of the antibody (13, 28, 31). When the antibody-binding linear epitope is not available, the relationship among these “unrelated” targets could not be understood beyond their being recognized by the same antibody. Our results demonstrated that the relationships among the “unrelated” targets might be explained by shared antibody-binding linear epitopes. For example, Ab156 was generated by immunizing mouse with peptide from the human protein SEC62, a translocation protein (Uniprot ID: Q99442). In this study, Ab156 also interacted with the human protein vitrin (Fig. 4E and supplemental Fig. S7B), which plays a role in spinal cord formation (Uniprot ID: Q6UXI7). In another example, Ab019 interacted with protein P177174 (transcription regulatory region sequence-specific DNA binding) (Fig. 4D), Q96MT8 (required for normal spindle assembly) (supplemental Fig. S7A), Q6UXI7 (plays a role in spinal cord formation) (supplemntal Fig. S7B), and P23930 (catalyzes the phospholipid-dependent N-acylation of the N-terminal cysteine of apolipoprotein) (supplemental Fig. S7C).
These results demonstrate that, guided by the defined linear epitope, the multispecificity of an antibody can be explored for potential applications, by targeting matched proteins other than the antigen against which it was raised.
Mapping the Epitopes for SARS-CoV-2 spike Protein-Specific Antibodies
COVID-19 is a global pandemic that we are currently facing (32). To accelerate the development of effective therapeutic antibodies and vaccines, it is crucial to understand the relevant humoral immunity (33), especially the key epitopes recognized by SARS-CoV-2-specific antibodies. Given the critical role that spike protein plays in the viral infection process (34) and high S protein-specific antibody titers in convalescent serum (35), the S1 and S2 subunits were used to enrich spike protein-specific antibodies from convalescent serum from a COVID-19 patient. We chose the serum from of this patient because it showed strong binding to both S1 and S2 in our recent study (35). Following the established AbMap procedure, binding peptides were obtained for both S1- and S2-specific antibodies, and statistically significant motifs were successfully determined (Fig. 5A, supplemental Table S9).
Fig. 5.

Mapping epitopes for spike protein-specific antibodies enriched from convalescent serum from a COVID-19 patient. A, the resolved epitopes for S1- and S2-specific antibodies. B, the locations and similarities of motifs matched to the sequences of S1 and S2. C, the distribution of three epitopes in the 3D structure of the spike protein, in both monomeric (upper) and trimeric (lower) formats.
Two motifs for S1 specific antibodies match the S1 sequence with high similarity. As shown in Figure 5B, one motif, “YXNSFXR,” matches the region 27-AYTNSFTRFV-36 of S1, and the results indicate that this is a linear epitope. This linear epitope is consistent with that in other studies (36, 37, 38). Another motif, “KXLPFQQ,” matches the region 558-KFLPFQQ-564 of S1, which is close to the receptor-binding domain (RBD) region. This linear epitope partially overlaps with a predicted linear epitope 558-KFLPF-562 (39). The linear epitope, “AVEQDK,” matches the region 101-TGIAVEQDKNT-111 of S2 with a similarity of 0.46. This result is also consistent with the finding of another study (40). For better visualization, all the identified linear epitopes were marked on the crystal structure of the spike protein (PDB ID: 6VSB) (41) in both monomeric and trimeric formats (Fig. 5C). One of these linear epitopes, “KXLPFQQ,” located near the RBD of the spike protein, might play an important role in the interaction between the spike protein and receptor ACE2.
While we have used our approach to define the linear epitopes recognized by antibodies, it is possible that conformational epitopes are detected. While we have not performed any validation, it is interesting that of the recovered motifs “DXTLLN” could not be linearly matched to S1, PepSurf could identify clusters that would be consistent with conformational epitopes (supplemental Fig. S8). These remain to be experimentally validated.
Thus, by testing a single serum sample, AbMap successfully revealed both linear and potentially conformational epitopes of the Spike protein, and these epitopes are physiologically relevant.
Discussion
We have established the AbMap strategy for high-throughput antibody epitope mapping by combining a phage displayed random peptide library and NGS. We are not the first to do so, as there are several similar studies (16, 42). The differences in AbMap are the dual-barcoded strategy for NGS library preparation and the algorithm used to remove the false-positive binding peptides in AbMap. With AbMap, one technician is able to map the linear epitopes of more than 200 antibodies in 1 month at an affordable cost. Approximately 50% of the determined linear epitopes matched the original antigens. The peptides/linear epitopes and their corresponding antibodies were explored for a variety of applications, from WB and ELISA to immune precipitation. Also convalescent serum collected from a COVID-19 patient was used to explore the feasibility of using AbMap for complex sample. Both linear and potentially conformational epitopes were revealed. Effective therapeutic antibodies and vaccines are desperately needed for the ever-growing pandemic we are facing. Applying the same protocol, we are analyzing more sera. Once we compile all the epitope information, we may be able to provide precise instructions for the development of therapeutic antibodies and vaccines.
Using AbMap, the profile of the peptides bound by an inputted antibody could be readily resolved. We argue that the peptide-binding profile of an antibody could represent, to a certain extent, all the peptides that it can bind in nature. Thus, conceptually, we define an antibody’s peptide-binding profile as its binding capacity (BiC). The BiC could serve as a systematic and functional descriptor for antibodies and is the first such descriptor, to the best of our knowledge. Based on the BiC, we could easily predict which proteins an antibody could bind and at precisely on which sites. We could also predict where the so-called antibody nonspecificity comes from and compare the similarity of antibodies.
There are several advantages of AbMap compared with traditional epitope mapping technologies. First, AbMap enables the linear epitope mapping of hundreds of antibodies simultaneously. Using traditional technologies, such as X-ray crystallography (8) and microarray (12), only one antibody can usually be analyzed at a time. Using these technologies, it is a common practice to perform epitope mapping only for the antibodies that have the highest potential. In contrast, it is feasible to map 10,000 to even 100,000 antibodies by using AbMap. Second, the cost of AbMap per antibody is 1 to 2 orders of magnitude lower than that of traditional technologies. The most expensive part of AbMap is NGS. In our case, we usually test 200 antibodies in one experiment, and 70 M of NGS reads can ensure the reliability. Thus, the NGS cost per antibody is affordable. This low cost enables us, for the first time, to perform on-demand antibody epitope mapping. Third, the accessibility of AbMap is very high. Except for NGS, neither complicated techniques nor sophisticated instruments are required. The key reagent is the phage library, which is commercially available.
There are also some limitations of AbMap. First, AbMap is intrinsically difficult to use to map conformational epitopes. The length of the peptides that we used was 12-aa, making it hard to preserve any effective conformational information. To address this limitation, one solution would be to prepare a phage display library with peptides of much larger size, for example, 56-aa (20). Another solution would be to predict conformational epitope through computational tools, such as PepSurf (43), as we attempted to do here for antibodies to the spike protein of SARS-CoV-2. Second, the theoretical space of the random 12-mer library is much larger than the real one. This is a general problem for the preparation of phage displayed random library, not just for the library (Ph.D-12) that we used. In fact, Ph.D-12 phage display library is one of the best libraries that have been widely applied. Due to technical limitations, practically speaking, it is impossible to reach the theoretical space. As long as the displayed peptides are randomly/evenly distributed in the theoretical space, the library can somewhat represent the space. Then, by calculating based on the set of binding peptides, we can obtain epitopes close to the “real one.” This is why AbMap works. Additionally, if a high-quality library of higher diversity is available, better results are possible. Third, current bioinformatic tools are not optimized for AbMap. It is critical to calculate/predict the linear or conformational epitopes after we have obtained the BiC of an antibody. MEME (22) is used for the calculation of linear epitopes, and PepSurf is used for conformational epitopes. In these two tools, information on peptide abundance is not considered. To obtain more accurate epitope information from the binding peptides, specialized bioinformatic tools optimized for AbMap are needed in future studies.
The utility of AbMap for affordable, high-throughput, and reliable mapping of antibody linear epitopes makes it an enabling technology. Using AbMap, a variety of applications for the development of both therapeutic antibodies and diagnostic reagents are made possible. For therapeutic antibodies, we can apply AbMap early in the development pipeline, and by comparing the resolved binding epitopes among the antibodies, we can discard the redundant antibodies with similar epitopes and focus limited resources on the unique ones. There is the possibility of producing an antibody that recognizes an epitope similar to that of an existing therapeutic antibody, thus leading to intellectual property infringement by the developer (44). To avoid this, we can compare the epitopes among the candidates to those of approved therapeutic antibodies as early as possible. For diagnostic reagent development, AbMap could be applied in two directions: 1. quality control of antibodies; 2. repurposing of the existing antibodies. It is possible to resolve the binding peptides/epitopes of any antibody by using AbMap; thus, we can add another layer of quality control to antibody development. This additional information can guide researchers to select the appropriate antibody with a high degree of confidence. By searching the high-confident epitope, an antibody could be rediscovered for the purpose of testing other proteins. When a large number of antibodies are analyzed by AbMap, it is possible to rediscover antibodies with much better performance than what we showed in Figure 4.
In summary, facilitated by the high-throughput power of a phage displayed peptide library and NGS, we have developed the AbMap strategy based on existing technologies. AbMap enables, for the first time, linear epitope mapping of hundreds of antibodies simultaneously, at a much lower cost than that of traditional technologies, while maintaining high accuracy. The identified peptides/linear epitopes could be explored for a wide range of applications. AbMap is highly accessible and generally applicable. We believe that this enabling technology will be applied widely wherever epitope mapping is necessary and provide unprecedented acceleration in both clinical and basic studies.
Data Availability
The sequencing data have been deposited in the NCBI SRA (https://www.ncbi.nlm.nih.gov/sra). The BioProject accession number is PRJNA660904. The index and barcode information of the NGS results is included in supplemental Table S10.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgments
This work was partially supported by the National Key Research and Development Program of China (No. 2018YFC1003501), National Key Research and Development Program of China Grant (No. 2016YFA0500600), National Natural Science Foundation of China (Nos. 31970130, 31600672, 31670831, 31370813, and 31501054), and Open Foundation of Key Laboratory of Systems Biomedicine (No. KLSB2017QN-01). We thank Abmart (Shanghai) for providing the 202 monoclonal antibodies. We thank PTM Biolabs for providing antibodies. We thank Dr Lan Wang and Dr Xiaojuan Yu for providing key reagents. We thank Mr Wei Wang and Mr Jie Zhou for collecting the COVID-19 serum.
Author contributions
S.-c. T., X. Z., H. L., and H. Q. designed the study; H. Q. performed the experiments related to the 202 mouse monoclonal antibodies and the serum of COVID-19 patient; C. H. and H. L. performed the NGS and the data analysis; All the authors analyzed the data; S.-c. T, H. Q., M. M., C. H., X. Z., and H. L. wrote the article.
Contributor Information
Xiaodong Zhao, Email: xiaodongzhao@sjtu.edu.cn.
Hua Li, Email: kaikaixinxin@sjtu.edu.cn.
Sheng-ce Tao, Email: taosc@sjtu.edu.cn.
Supplemental Data
References
- 1.Helsby M.A., Leader P.M., Fenn J.R., Gulsen T., Bryant C., Doughton G., Sharpe B., Whitley P., Caunt C.J., James K., Pope A.D., Kelly D.H., Chalmers A.D. CiteAb: A searchable antibody database that ranks antibodies by the number of times they have been cited. BMC Mol. Biol. 2014;15:6. doi: 10.1186/1471-2121-15-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grilo A.L., Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9–16. doi: 10.1016/j.tibtech.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Kaplon H., Reichert J.M. Antibodies to watch in 2019. mAbs. 2019;11:219–238. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machado A., Torres T. Guselkumab for the treatment of psoriasis. Biodrugs. 2018;32:119–128. doi: 10.1007/s40259-018-0265-6. [DOI] [PubMed] [Google Scholar]
- 5.Greig S.L. Obiltoxaximab: First global approval. Drugs. 2016;76:823–830. doi: 10.1007/s40265-016-0577-0. [DOI] [PubMed] [Google Scholar]
- 6.Van Regenmortel M.H.V. What is a B-cell epitope? In: Schutkowski M., Reineke U., editors. Epitope Mapping Protocols. 2nd Ed. Humana Press; Totowa, NJ: 2009. pp. 3–20. [Google Scholar]
- 7.Abbott W.M., Damschroder M.M., Lowe D.C. Current approaches to fine mapping of antigen-antibody interactions. Immunology. 2014;142:526–535. doi: 10.1111/imm.12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grover R.K., Zhu X., Nieusma T., Jones T., Boreo I., MacLeod A.S., Mark A., Niessen S., Kim H.J., Kong L., Assad-Garcia N., Kwon K., Chesi M., Smider V.V., Salomon D.R. A structurally distinct human mycoplasma protein that generically blocks antigen-antibody union. Science. 2014;343:656–661. doi: 10.1126/science.1246135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blech M., Peter D., Fischer P., Bauer M.M., Hafner M., Zeeb M., Nar H. One target-two different binding modes: Structural insights into gevokizumab and canakinumab interactions to interleukin-1beta. J. Mol. Biol. 2013;425:94–111. doi: 10.1016/j.jmb.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 10.Fibriansah G., Ibarra K.D., Ng T.S., Smith S.A., Tan J.L., Lim X.N., Ooi J.S., Kostyuchenko V.A., Wang J., de Silva A.M., Harris E., Crowe J.E., Jr., Lok S.M. DENGUE VIRUS. Cryo-EM structure of an antibody that neutralizes dengue virus type 2 by locking E protein dimers. Science. 2015;349:88–91. doi: 10.1126/science.aaa8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parker J.L., Newstead S. Membrane protein crystallisation: Current trends and future perspectives. Adv. Exp. Med. Biol. 2016;922:61–72. doi: 10.1007/978-3-319-35072-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buus S., Rockberg J., Forsstrom B., Nilsson P., Uhlen M., Schafer-Nielsen C. High-resolution mapping of linear antibody epitopes using ultrahigh-density peptide microarrays. Mol. Cell. Proteomics. 2012;11:1790–1800. doi: 10.1074/mcp.M112.020800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kramer A., Keitel T., Winkler K., Stocklein W., Hohne W., Schneider-Mergener J. Molecular basis for the binding promiscuity of an anti-p24 (HIV-1) monoclonal antibody. Cell. 1997;91:799–809. doi: 10.1016/s0092-8674(00)80468-7. [DOI] [PubMed] [Google Scholar]
- 14.Petersen G., Song D., Hugle-Dorr B., Oldenburg I., Bautz E.K. Mapping of linear epitopes recognized by monoclonal antibodies with gene-fragment phage display libraries. Mol. Genet. Genomics. 1995;249:425–431. doi: 10.1007/BF00287104. [DOI] [PubMed] [Google Scholar]
- 15.Rentero Rebollo I., Sabisz M., Baeriswyl V., Heinis C. Identification of target-binding peptide motifs by high-throughput sequencing of phage-selected peptides. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paull M.L., Johnston T., Ibsen K.N., Bozekowski J.D., Daugherty P.S. A general approach for predicting protein epitopes targeted by antibody repertoires using whole proteomes. PLoS One. 2019;14 doi: 10.1371/journal.pone.0217668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson E., Doranz B.J. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology. 2014;143:13–20. doi: 10.1111/imm.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Opuni K.F.M., Al-Majdoub M., Yefremova Y., El-Kased R.F., Koy C., Glocker M.O. Mass spectrometric epitope mapping. Mass Spectrom. Rev. 2018;37:229–241. doi: 10.1002/mas.21516. [DOI] [PubMed] [Google Scholar]
- 19.Wei H., Mo J., Tao L., Russell R.J., Tymiak A.A., Chen G., Iacob R.E., Engen J.R. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: Methodology and applications. Drug Discov. Today. 2014;19:95–102. doi: 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu G.J., Kula T., Xu Q., Li M.Z., Vernon S.D., Ndung'u T., Ruxrungtham K., Sanchez J., Brander C., Chung R.T., O'Connor K.C., Walker B., Larman H.B., Elledge S.J. Comprehensive serological profiling of human populations using a synthetic human virome. Science. 2015;348 doi: 10.1126/science.aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolger A.M., Lohse M., Usadel B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. Meme SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995;57:289–300. [Google Scholar]
- 24.Hintze J.L., Nelson R.D. Violin plots: A box plot-density trace synergism. Am. Stat. 1998;52:181–184. [Google Scholar]
- 25.Qi H., Zhou H., Czajkowsky D.M., Guo S., Li Y., Wang N., Shi Y., Lin L., Wang J., Wu D., Tao S.-C. Rapid production of virus protein microarray using protein microarray fabrication through gene synthesis (PAGES) Mol. Cell. Proteomics. 2017;16:288–299. doi: 10.1074/mcp.M116.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geysen H.M., Rodda S.J., Mason T.J. A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol. Immunol. 1986;23:709–715. doi: 10.1016/0161-5890(86)90081-7. [DOI] [PubMed] [Google Scholar]
- 27.Ditto N.T., Brooks B.D. The emerging role of biosensor-based epitope binning and mapping in antibody-based drug discovery. Expert Opin. Drug Discov. 2016;11:925–937. doi: 10.1080/17460441.2016.1229295. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z., Li Y., Hou B., Pronobis M.I., Wang Y., Wang M., Cheng G., Zhang Z., Weng W., Wang Y., Tang Y., Xu X., Pan R., Lin F., Wang N. An array of 60,000 antibodies for proteome-scale antibody generation and target discovery. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aax2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larman H.B., Zhao Z., Laserson U., Li M.Z., Ciccia A., Gakidis M.A., Church G.M., Kesari S., Leproust E.M., Solimini N.L., Elledge S.J. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Donovan B., Mandel-Brehm C., Vazquez S.E., Liu J., Parent A.V., Anderson M.S., Kassimatis T., Zekeridou A., Hauser S.L., Pittock S.J., Chow E., Wilson M.R., DeRisi J.L. High-resolution epitope mapping of anti-Hu and anti-Yo autoimmunity by programmable phage display. Brain Commun. 2020;2 doi: 10.1093/braincomms/fcaa059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrader J.W., McLean G.R. Multispecificity of a recombinant anti-Ras monoclonal antibody. J. Mol. Recognit. 2018;31 doi: 10.1002/jmr.2683. [DOI] [PubMed] [Google Scholar]
- 32.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni L., Ye F., Cheng M.-L., Feng Y., Deng Y.-Q., Zhao H., Wei P., Ge J., Gou M., Li X., Sun L., Cao T., Wang P., Zhou C., Zhang R. Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52:971–977.e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang H.-W., Li Y., Zhang H.-N., Wang W., Men D., Yang X., Qi H., Zhou J., Tao S.-C. Global profiling of SARS-CoV-2 specific IgG/IgM responses of convalescents using a proteome microarray. Nat. Commun. 2020;11:3581. doi: 10.1038/s41467-020-17488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prachar M., Justesen S., Steen-Jensen D.B., Thorgrimsen S., Jurgons E., Winther O., Bagger F.O. COVID-19 vaccine candidates: Prediction and validation of 174 SARS-CoV-2 epitopes. bioRxiv. 2020 doi: 10.1101/2020.03.20.000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parvez S., Preeti S. Prediction of T and B cell epitopes in the proteome of SARS-CoV-2 for potential use in diagnostics and vaccine design. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12116943.v12116941. [DOI] [Google Scholar]
- 38.Bhattacharya M., Sharma A.R., Patra P., Ghosh P., Sharma G., Patra B.C., Lee S.-S., Chakraborty C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J. Med. Virol. 2020;92:618–631. doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grifoni A., Sidney J., Zhang Y., Scheuermann R.H., Peters B., Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27:671–680.e672. doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng M., Song L. Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS-CoV-2 compared to SARS-CoV. Cell. Mol. Immunol. 2020;17:536–538. doi: 10.1038/s41423-020-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pantazes R.J., Reifert J., Bozekowski J., Ibsen K.N., Murray J.A., Daugherty P.S. Identification of disease-specific motifs in the antibody specificity repertoire via next-generation sequencing. Sci. Rep. 2016;6:30312. doi: 10.1038/srep30312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayrose I., Penn O., Erez E., Rubinstein N.D., Shlomi T., Freund N.T., Bublil E.M., Ruppin E., Sharan R., Gershoni J.M., Martz E., Pupko T. Pepitope: Epitope mapping from affinity-selected peptides. Bioinformatics. 2007;23:3244–3246. doi: 10.1093/bioinformatics/btm493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng X., Storz U., Doranz B.J. Enhancing antibody patent protection using epitope mapping information. mAbs. 2018;10:204–209. doi: 10.1080/19420862.2017.1402998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data have been deposited in the NCBI SRA (https://www.ncbi.nlm.nih.gov/sra). The BioProject accession number is PRJNA660904. The index and barcode information of the NGS results is included in supplemental Table S10.