

Abstract
Premise
Aquilegia is an ideal taxon for studying the evolution of adaptive radiation. Current phylogenies of Aquilegia based on different molecular markers are inconsistent, and therefore a clear and accurate phylogeny remains uncertain. Analyzing the chloroplast genome, with its simple structure and low recombination rate, may help solve this problem.
Methods
Next‐generation sequencing data were generated or downloaded for Aquilegia species, enabling their chloroplast genomes to be assembled. The assemblies were used to estimate the genome characteristics and infer the phylogeny of Aquilegia.
Results
In this study, chloroplast genome sequences were assembled for Aquilegia species distributed across Asia, North America, and Europe. Three of the genes analyzed (petG, rpl36, and atpB) were shown to be under positive selection and may be related to adaptation. The phylogenetic tree of Aquilegia showed that its member species formed two clades with high support, North American and European species, with the Asian species being paraphyletic; A. parviflora and A. amurensis clustered with the North American species, while the remaining Asian species were found in the European clade. In addition, A. oxysepala var. kansuensis should be considered as a separate species rather than a variety.
Discussion
The complete chloroplast genomes of these Aquilegia species provide new insights into the reconstruction of the phylogeny of related species and contribute to the further study of this genus.
Keywords: adaptive evolution, chloroplast genomes, columbine, phylogeny
The genus Aquilegia L. (columbine), comprising approximately 70 perennial herb species, belongs to the family Ranunculaceae and is widely distributed in North America and Eurasia (Munz, 1946). Recently, several new species were reported, bringing the number of columbine taxa to about 110 species (Erst et al., 2017, 2020; Luo et al., 2018). Although the morphologies and habitats of columbine species differ, the phylogenetic resolution of this genus at the molecular level is very low, and therefore the genus is considered to be a widespread population complex. The morphological differences of the floral spurs between species of Aquilegia are easily observed and attract different pollinators, which has led to the rapid divergence of the columbines to form a large number of species (Hodges and Derieg, 2009). Moreover, natural hybrids among columbine species have also been frequently reported (Taylor, 1967). As a result, Aquilegia species have become a model for evolution studies; however, the phylogenetic trees presented in previous studies contain multifurcations, which may be caused by a lack of informative sites (Hodges and Arnold, 1994; Bastida et al., 2010; Fior et al., 2013), complicating subsequent research on the speciation of this genus. It is therefore very important to construct a relatively clear phylogenetic relationship of these species for future evolutionary studies.
Genomic sequencing could compensate for the lack of informative sites in shorter sequences. Notably, the decline in sequencing costs in recent years has made this approach possible for all parts of the plant genome (nuclear, mitochondrial, and chloroplast). Because of the easy interspecific hybridization among Aquilegia species, the nuclear genome structure is complex, with a high recombination rate (Filiault et al., 2018). The mitochondrial genomes of the angiosperms are relatively complex; the order of genes differs among species, and only some regions of the genome are conserved (Kubo et al., 2000). In contrast, the monophyletic inheritance of the chloroplast genome sequence is more suitable for the phylogenetic analysis of Aquilegia due to its low recombination rate and high level of conservation (Dong et al., 2012; Curci et al., 2015; Downie and Jansen, 2015; Nadachowska‐Brzyska et al., 2015). Fior et al. (2013) selected 21 chloroplast genes with rapid evolutionary rates to establish the phylogenetic relationships among Aquilegia species. Although the topology of this phylogeny had a lower resolution and support for some branches (Fior et al., 2013) than previously constructed trees based on fewer chloroplast sequences (Hodges and Arnold, 1994; Bastida et al., 2010), the resolution and support rate were improved. Hence, the complete chloroplast genome sequence is an ideal molecular marker for inferring the phylogenetic relationships of the Aquilegia genus.
The chloroplast genome is a closed‐loop structure approximately 115–210 kbp in size, and generally consists of four parts: two inverted repeat regions (IRA and IRB), a large single‐copy region (LSC), and a small single‐copy region (SSC) (Yurina and Odintsova, 1998; Park et al., 2018). Some plant groups have special chloroplast genome structures, such as species of the genus Erodium L’Hér., which lack the IR regions (Guisinger et al., 2010). Because of its stable genomic structure, identical gene content, and conserved sequence (Dong et al., 2012), the chloroplast genome is used as a molecular marker for the inference of phylogenetic relationships (Li et al., 2018; Liu et al., 2018; Lu et al., 2018; Mader et al., 2018; Xie et al., 2018) and adaptative evolution (Dong et al., 2018; Fan et al., 2018). In this study, we assembled and analyzed the chloroplast genomes of 14 columbine species from Asia, Europe, and North America, and constructed a phylogenetic tree of the genus to shed light on radiative speciation in Aquilegia and lay a foundation for inferring the evolutionary history of the columbines.
METHODS
Plant materials
Seeds of A. amurensis Kom., A. ecalcarata Maxim., A. oxysepala Trautv. & C. A. Mey. var. kansuensis Brühl, A. parviflora Ledeb., A. rockii Munz, A. viridiflora Pall., and A. yabeana Kitag. were collected from China (Appendix 1), and all voucher specimens were deposited in the Northeast Normal University Herbarium in Changchun, China (accession numbers NENU_Aq1001–NENU_Aq1007). Seeds were grown in the greenhouse of Northeast Normal University with 12 h of light at 25°C and 12 h of dark at 20°C.
DNA extraction and sequencing
Total genomic DNA was extracted from fresh leaves using a modified cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). Genomic library generation and sequencing were used to acquire 2 × 150‐bp paired reads generated on the Illumina Xten by Biomarker Technologies (Beijing, China). Furthermore, raw reads of A. aurea Janka, A. chrysantha A. Gray, A. formosa Fisch. ex DC., A. japonica Nakai & Hara, A. oxysepala var. oxysepala, A. sibirica Schur ex Nyman, and A. vulgaris L. previously published by Filiault et al. (2018) were downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive database (http://www.ncbi.nlm.nih.gov/sra [accessed December 2018]) to assemble the chloroplast genome (Appendix 2).
Chloroplast genome assembly and annotation
To obtain high‐quality genome sequences, all reads were filtered as follows: remove reads containing adapters, a content of more than 10% N, or more than 50% low‐quality bases (quality value <10). We then used the chloroplast_assembly_protocol pipeline to assemble the chloroplast genome (Sancho et al., 2018). Briefly, DUK (http://duk.sourceforge.net) was used to extract the chloroplast reads, which were filtered using FASTQC version 0.10.1 (Andrew, 2010) and Trimmomatic version 0.32 (Bolger et al., 2014). Next, the pass‐filtered reads were de novo assembled using Velvet version 1.2.07 (Zerbino, 2010), SSPACE Basic version 2.0 (Boetzer et al., 2011), and GapFiller version 1.11 (Boetzer and Pirovano, 2012; Nadalin et al., 2012), with annotation performed using the online program DOGMA (Wyman et al., 2004). Finally, the circular genome map of Aquilegia was illustrated using the Organellar Genome DRAW tool (Lohse et al., 2013) after manually checking the annotation results.
Repeat sequence characterization
The Perl script MISA (Thiel et al., 2003) was employed to identify the location of simple sequence repeat (SSR) loci in the complete chloroplast genome sequences. The thresholds used to detect the SSRs were 10, 5, 4, 3, 3, and 3 for mono‐, di‐, tri‐, tetra‐, penta‐, and hexanucleotides, respectively. The recognition results were checked manually, and the redundant results were removed. REPuter (Kurtz et al., 2001) was then used to identify repeat sequences in the chloroplast, including palindromic, forward, reverse, and complementary sequences. The parameters were set as follows: (1) Hamming distance of 3, (2) 90% or greater sequence identity, and (3) a minimum repeat size of 30 bp. The default settings were used for all other parameters.
Genetic divergence and phylogenetic analysis of Aquilegia
The homologous genes were extracted from 14 Aquilegia species using a Python script (available on GitHub, see Data Availability Statement), after which these homologous genes were aligned using MAFFT version 7.407 (Katoh and Standley, 2013) with the default settings. Furthermore, the nucleotide diversity (π) of these homologous genes was analyzed using DnaSP version 6.0 (Rozas et al., 2017). To avoid the effect of sequence redundancy when building the phylogenetic trees, we selected the LSC regions, IRB regions, and SSC regions as arrays. In addition, the published chloroplast genome sequences of A. rockii (MK573514.1, NC_033341.1), A. ecalcarata (NC_041528.1, MK569474.1), and A. coerulea (NC_041527.1, MK569492.1) in GenBank were used. Semiaquilegia adoxoides Makino (MH142265.2) was considered as the outgroup (Fior et al., 2013; Zhai et al., 2019). The array was aligned using MAFFT version 7.407 and was adjusted manually in CLC Sequence Viewer 8.0 (QIAGEN Digital Insights, Redwood City, California, USA). The maximum likelihood tree was generated using IQ‐TREE version 1.6.12 using 1000 bootstrap replicates (Nguyen et al., 2015). Meanwhile, the Bayesian inference trees were produced using MrBayes version 3.2 (Ronquist et al., 2012), based on Markov chain Monte Carlo analyses run for 1,000,000 generations. These trees were sampled every 1000 generations with the first 250 trees discarded in the burn‐in period. The program was stopped when the standard deviation was less than 0.01. The final tree was visualized in iTOL (https://itol.embl.de/itol.cgi) (Letunic and Bork, 2006).
Natural selection analysis
To identify genes under selection in Aquilegia, the genes of the chloroplast genomes were analyzed with the PAML package (Yang, 2007). First, all coding sequences (CDS) of the Aquilegia species and other Ranunculaceae species were extracted from the genome sequences using a Python script (Appendix 3). Each single‐copy sequence was aligned according to its codons using MEGA X (Kumar et al., 2018) and checked manually, and then used as input for CodeML in the PAML package. Moreover, the concatenated alignment was also used to construct phylogenetic relationships among species using IQ‐TREE version 1.6.12 (Nguyen et al., 2015). Finally, each CDS alignment was used to calculate the nonsynonymous (dN) and synonymous (dS) substitution rates, along with their ratio (ω = dN/dS). ω > 1 indicates positive selection, ω = 1 indicates neutral selection, and ω < 1 indicates negative selection (Yang and Nielsen, 2002). The branch‐site model (X. Yang et al., 1998; Z. Yang et al., 1998) was combined with the naive empirical Bayes (NEB) method, and the Bayesian empirical Bayes (BEB) method was used to identify potential positively selected genes using CodeML in the PAML package. The null hypothesis allows a ω for each clade (model = 2, NSsites = 2, fix ω = 1, and ω = 1), while the alternative hypothesis allows a ω for Aquilegia and another ω for other clades (model = 2, NSsites = 2, fix ω = 0, and ω = 2). A chi‐square test was completed with chi2 in the PAML package. A P value > 0.05 suggests the null hypothesis should be accepted; otherwise, the alternative hypothesis should be accepted and the site should be considered a positively selected gene.
RESULTS
Features of Aquilegia chloroplast genomes
The complete chloroplast genomes of the Aquilegia species from Asia, North America, and Europe displayed a typical quadripartite structure similar to the majority of land plant chloroplast genomes (Fig. 1). The sizes of the complete chloroplast genomes ranged from 157,689 to 161,387 bp. All complete chloroplast genomes were composed of four sections, including an LSC region (86,761–88,076 bp), an SSC region (17,466–18,879 bp), and two IR regions (25,612–28,015 bp). The GC content of the 14 species was very similar in both the whole chloroplast genome (38.94%–39.08%) and the corresponding regions (LSC [37.43%–37.71%], SSC [33.30%–33.91%], and IR [43.04%–43.41%]), with the IR regions having the highest GC contents (Table 1). These sequence data are available in GenBank (accession numbers MT919110–MT9191116 and MN809218–MN809224).
FIGURE 1.
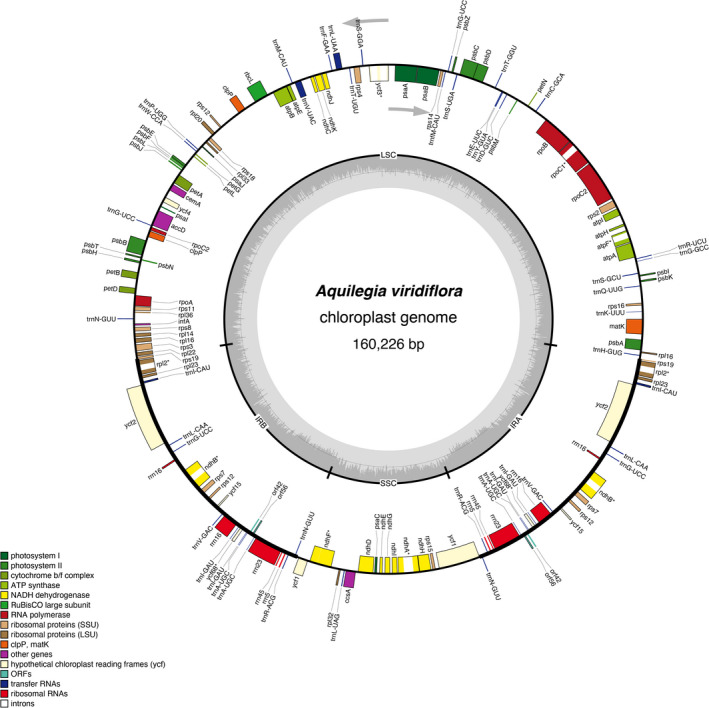
Gene maps of the Aquilegia viridiflora chloroplast genome. Genes inside the circle are transcribed clockwise, while genes outside are transcribed counterclockwise (as indicated by arrows). Different colors indicate different functional groups. The dark gray shading within the inner circle corresponds to the GC content and the light gray shading corresponds to the AT content. IRA and IRB, inverted repeat regions; LSC, large single‐copy region; ORF, open reading frame; SSC, small single‐copy region.
TABLE 1.
Summary of the complete Aquilegia chloroplast genomes sequenced in this study.
| Species | LSC | SSC | IRs | Total | NCBI no. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | GC (%) | Length (% of genome) | Length (bp) | GC (%) | Length (% of genome) | Length (bp) | GC (%) | Length (% of genome) | Length (bp) | GC (%) | ||
| A. aurea a | 87,724 | 37.54 | 54.80 | 18,879 | 33.30 | 11.79 | 26,735 | 43.37 | 16.70 | 160,073 | 39.00 | MT919114 |
| A. vulgaris a | 88,137 | 37.43 | 54.98 | 18,761 | 33.64 | 11.70 | 26,711 | 43.33 | 16.66 | 160,320 | 38.96 | MT919112 |
| A. japonica a | 87,986 | 37.52 | 55.13 | 18,169 | 33.47 | 11.38 | 26,723 | 43.37 | 16.74 | 159,601 | 39.00 | MT919110 |
| A. oxysepala var. oxysepala a | 87,651 | 37.43 | 55.08 | 18,474 | 33.91 | 11.61 | 26,503 | 43.31 | 16.65 | 159,131 | 38.96 | MT919111 |
| A. sibirica a | 88,053 | 37.44 | 54.56 | 17,466 | 33.36 | 10.82 | 27,934 | 43.04 | 17.31 | 161,387 | 38.94 | MT919115 |
| A. oxysepala var. kansuensis | 87,655 | 37.65 | 55.03 | 18,638 | 33.64 | 11.70 | 26,498 | 43.25 | 16.64 | 159,289 | 39.05 | MN809219 |
| A. yabeana | 88,030 | 37.60 | 54.86 | 18,744 | 33.59 | 11.68 | 26,845 | 43.29 | 16.73 | 160,464 | 39.04 | MN809218 |
| A. ecalcarata | 87,662 | 37.63 | 54.77 | 18,747 | 33.50 | 11.71 | 26,824 | 43.36 | 16.76 | 160,057 | 39.07 | MN809221 |
| A. rockii | 87,375 | 37.64 | 55.09 | 18,339 | 33.51 | 11.56 | 26,445 | 43.23 | 16.67 | 158,604 | 39.04 | MN809222 |
| A. viridiflora | 88,076 | 37.61 | 54.97 | 18,662 | 33.75 | 11.65 | 26,744 | 43.20 | 16.69 | 160,226 | 39.01 | MN809220 |
| A. amurensis | 87,865 | 37.71 | 55.72 | 18,600 | 33.62 | 11.80 | 25,612 | 43.41 | 16.24 | 157,689 | 39.08 | MN809224 |
| A. parviflora | 87,969 | 37.70 | 55.61 | 18,612 | 33.59 | 11.77 | 25,799 | 43.39 | 16.31 | 158,179 | 39.08 | MN809223 |
| A. chrysantha a | 87,371 | 37.52 | 54.72 | 18,724 | 33.50 | 11.73 | 26,786 | 43.34 | 16.78 | 159,667 | 38.96 | MT919113 |
| A. formosa a | 87,588 | 37.60 | 54.37 | 17,482 | 33.38 | 10.85 | 28,015 | 43.16 | 17.39 | 161,100 | 39.04 | MT919116 |
IRs = inverted repeat regions; LSC = large single‐copy region; NCBI = National Center for Biotechnology Information; SSC = small single‐copy region.
Raw data were downloaded from NCBI.
The chloroplast genomes of the Aquilegia species contained 154 genes (98 protein‐coding genes, 48 transfer RNA [tRNA] genes, and eight ribosomal RNA genes). Most of the genes located in the LSC and SSC regions were single copy, while 26 of the genes located in the IR regions were duplicated, including 11 protein‐coding genes (rps7, rps12, rps19, rpl2, rpl23, orf42, orf56, ycf2, ycf15, ycf68, and ndhB), 11 tRNA genes (trnI‐CAU [×3], trnL‐CAA, trnG‐UCC, trnV‐GAC, trnI‐GAU, trnA‐UGC [×2], trnR‐ACG, and trnN‐GUU), and four rRNA genes (rrn4.5, rrn5, rrn16, and rrn23). The LSC region comprises 63 protein‐coding genes and 25 tRNA genes, and the SSC region comprises 13 protein‐coding genes and a single tRNA gene. Among all the genes, seven protein‐coding genes (rpoC1, atpF, rpl2, ycf68, ndhB, ndhF, and ndhA) contained only one intron, while one protein‐coding gene (ycf3) contained two introns (Appendix S1).
Repeat analysis
We identified a range of 84–89 repeat sequences in the 14 Aquilegia chloroplast genomes, including 45–51 palindromic repeats and 33–44 forward repeats; reverse and complement repeats were not identified (Fig. 2A). In all species, the palindromic repeats were 56–398 bp in length and the forward repeats were 56–357 bp in length (Fig. 2B, C). The SSR analysis of the Aquilegia chloroplast genome identified a range of 69–84 microsatellites of six types; A. chrysantha and A. viridiflora had the lowest and highest numbers of microsatellites, respectively (Fig. 3A). Among all SSRs, the most abundant type was mononucleotide repeats, which accounted for 66.51% of the total SSRs, followed by dinucleotide (13.32%), tetranucleotide (7.22%), trinucleotide (5.91%), pentanucleotide (4.32%), and hexanucleotide (2.72%) repeats. AT repeats accounted for a larger proportion of mononucleotide repeats (92.95%) than GC repeats (7.05%). Similarly, the AT content (90.15%) accounted for a larger proportion than the GC content (9.85%) in dinucleotides (Fig. 3B, Appendix S2). Not surprisingly, all SSRs were detected in noncoding regions of the Aquilegia chloroplast genome.
FIGURE 2.
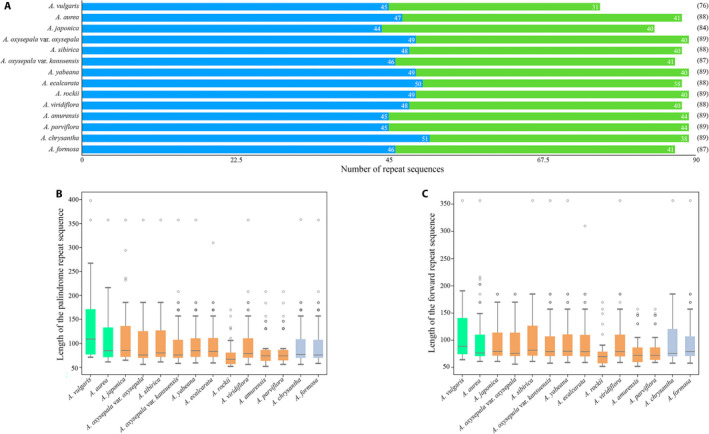
Analysis of repeat sequences in the Aquilegia chloroplast genomes, performed using REPuter. (A) Number of different repeat sequences detected in Aquilegia species. Blue and green represent palindrome repeat sequences and forward repeat sequences, respectively. (B) Length of the palindrome repeat sequences in Aquilegia species. (C) Length of the forward repeat sequence in Aquilegia species. In (B) and (C), green, orange, and purple represent European species, Asian species, and North American species, respectively.
FIGURE 3.
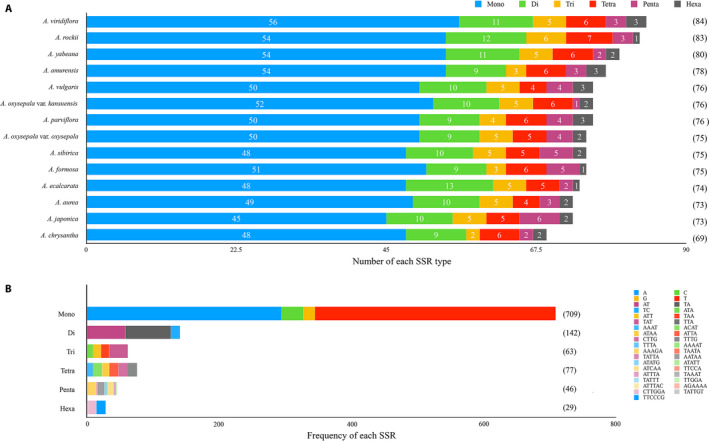
Analysis of simple sequence repeats (SSRs) in Aquilegia chloroplast genomes, performed using MISA (Thiel et al., 2003). (A) Number of various SSR types (mono‐, di‐, tri‐, tetra‐, penta‐, and hexanucleotides) detected in Aquilegia species. (B) Type and frequency of each SSR detected in the Aquilegia species analyzed.
Sequence divergence and phylogeny of Aquilegia
The π value was used to evaluate sequence divergence in Aquilegia chloroplast genomes. In genic regions, the range of variation in π was 0–0.00511, with a mean of 0.00061; π of the LSC region (0–0.00511, with a mean of 0.00055) was higher than in other regions (0–0.00453 in the IR regions, with a mean of 0.00041; 0–0.00252 in the SSC region, with a mean of 0.0013). Overall, these results demonstrated that the sequence divergence in Aquilegia chloroplast genomes was small, but some regions showed high genetic diversity, such as rpoC2, trnS‐GGA, and trnL‐CAA (π > 0.004) (Fig. 4, Appendix S3).
FIGURE 4.
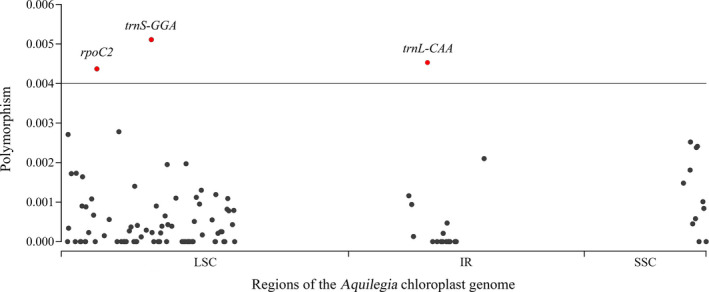
The nucleotide diversity of all chloroplast genes in Aquilegia. Red circles represent highly polymorphic genes.
To reveal the phylogeny of Aquilegia, aligned chloroplast genome sequences were used to construct phylogenetic trees using both maximum likelihood and Bayesian analyses. The two resulting trees showed identical topologies, and the bootstrap values and posterior probabilities were very high for each lineage. The Aquilegia species were divided into two clades: one clade contained A. aurea and A. vulgaris from Europe and A. sibirica, A. oxysepala var. oxysepala, A. japonica, A. ecalcarata, A. rockii, A. viridiflora, A. yabeana and A. oxysepala var. kansuensis from Asia; the other clade contained A. formosa, A. chrysantha, and A. coerulea from North America and A. amurensis and A. parviflora from Asia. All the topologies supported A. japonica and A. oxysepala var. oxysepala as sister clades, and A. sibirica shared a common ancestor with them. Interestingly, the A. ecalcarata sequence assembled by us clustered with A. rockii, while the A. ecalcarata sequence downloaded from GenBank was grouped with A. yabeana and A. oxysepala var. gansuensis. In addition, A. viridiflora formed a single clade with A. ecalcarata and A. rockii. Although A. oxysepala var. oxysepala and A. oxysepala var. kansuensis are considered varieties of the same species, they were found in two different clades. Similarly, A. japonica and A. amurensis, which are treated as a single species by the Flora of China (Li, 2007), were also found in two different clades (Fig. 5).
FIGURE 5.
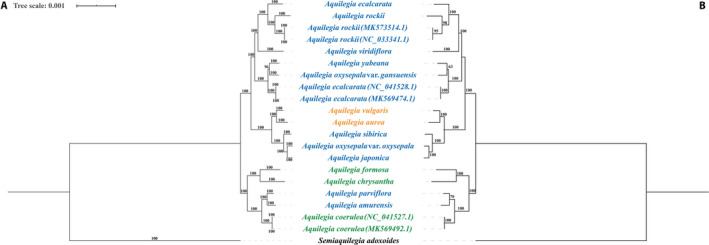
Phylogenetic relationships of Aquilegia. (A) Phylogeny of all chloroplast genome sequences built using Bayesian inference, with posterior probabilities (%) indicated above the branches. (B) Phylogeny of all chloroplast genome sequences using maximum likelihood, with bootstrap values indicated above the branches. Green, orange, and purple represent European species, Asian species, and North American species, respectively. Semiaquilegia adoxoides is included as the outgroup.
Positive selection analysis
Positive selection tests were performed on 54 CDS from Aquilegia and their related species using the PAML package. No significant selection was found to act on the chloroplast genes of Aquilegia (P > 0.05), but three genes with a higher posterior probability were detected using the BEB and NEB methods (atpB, petG, and rpl36). Therefore, atpB, petG, and rpl36 were considered to be genes potentially under positive selection (Table 2).
TABLE 2.
Analysis of the positive selection of all genes in the Aquilegia chloroplast genome based on the branch‐site model.
| Gene name | lnL0 | lnL1 | df | P | BEB | NEB |
|---|---|---|---|---|---|---|
| psbM | −293.77830 | −292.80279 | 1 | 0.08124 | NA | NA |
| psbL | −281.27366 | −281.27366 | 1 | 0.5 | NA | NA |
| ccsA | −6608.03557 | −6608.03529 | 1 | 0.5 | NA | NA |
| psaC | −927.93380 | −927.93380 | 1 | 0.14717 | NA | NA |
| psaB | −7228.74873 | −7228.74875 | 1 | 0.49748 | NA | NA |
| rpl33 | −957.68785 | −957.68785 | 1 | 0.5 | NA | NA |
| psbF | −281.15093 | −281.15093 | 1 | 0.5 | NA | NA |
| psaI | −404.70806 | −404.70806 | 1 | 0.5 | NA | NA |
| atpI | −2841.63957 | −2841.63959 | 1 | 0.49944 | NA | NA |
| atpH | −756.68830 | −756.68830 | 1 | 0.5 | NA | NA |
| rps19 | −1282.74781 | −1282.74782 | 1 | 0.49862 | NA | NA |
| rps18 | −331.72349 | −331.72349 | 1 | 0.5 | NA | NA |
| ndhK | −2320.17644 | −2320.17644 | 1 | 0.49831 | NA | NA |
| ndhJ | −1920.71103 | −1920.71103 | 1 | 0.5 | NA | NA |
| ndhA | −5351.81643 | −5351.81643 | 1 | 0.49944 | NA | NA |
| atpB a | −6085.06153 | −6085.06153 | 1 | 0.5 | 24 A 0.830 | NA |
| ycf4 | −2596.58913 | −2596.58915 | 1 | 0.49411 | NA | NA |
| rpoA | −5881.89224 | −5881.89227 | 1 | 0.49691 | NA | NA |
| rps14 | −1426.69915 | −1426.72524 | 1 | 0.49495 | NA | NA |
| ndhG | −2793.36061 | −2793.36063 | 1 | 0.49813 | NA | NA |
| atpE | −1748.33822 | −1748.33822 | 1 | 0.09889 | NA | NA |
| psbT | −407.74460 | −407.74460 | 1 | 0.5 | NA | NA |
| petN | −172.09765 | −172.09765 | 1 | 0.5 | NA | NA |
| ycf3 | −1481.19480 | −1481.19480 | 1 | 0.5 | NA | NA |
| psbJ | −349.00121 | −349.00121 | 1 | 0.5 | NA | NA |
| psbK | −764.88861 | −764.88862 | 1 | 0.4992 | NA | NA |
| ndhb | −3100.09672 | −3100.09649 | 1 | 0.49741 | NA | NA |
| ndhC | −1479.48370 | −1479.48370 | 1 | 0.49729 | NA | NA |
| atpA | −6298.17867 | −6298.17869 | 1 | 0.49767 | NA | NA |
| ndhH | −5676.37359 | −5676.37359 | 1 | 0.49171 | NA | NA |
| ndhI | −2058.27789 | −2058.27791 | 1 | 0.49831 | NA | NA |
| psbZ | −572.70301 | −572.70301 | 1 | 0.49887 | NA | NA |
| rps2 | −2861.59762 | −2861.59763 | 1 | 0.49822 | NA | NA |
| petA | −4229.69609 | −4229.69598 | 1 | 0.5 | NA | NA |
| psbD | −3394.40957 | −3394.4 0956 | 1 | 0.49831 | NA | NA |
| psbE | −724.90892 | −724.90892 | 1 | 0.49531 | NA | NA |
| rpoC2 | −21681.02490 | −21681.02489 | 1 | 0.49874 | NA | NA |
| psaJ | −520.11363 | −520.11362 | 1 | 0.5 | NA | NA |
| psbN | −365.09625 | −365.09625 | 1 | 0.5 | NA | NA |
| psaA | −6058.99244 | −6058.99242 | 1 | 0.49686 | NA | NA |
| rpl36 a | −480.99354 | −480.99353 | 1 | 0.17307 | NA | 0.996 b |
| psbC | −4629.80228 | −4629.80228 | 1 | 0.5 | NA | NA |
| psbB | −5837.08819 | −5837.08820 | 1 | 0.49652 | NA | NA |
| psbI | −326.55011 | −326.55011 | 1 | 0.5 | NA | NA |
| psbH | −1141.77124 | −1142.23505 | 1 | 0.49944 | NA | NA |
| rbcL | −5381.09986 | −5381.09986 | 1 | 0.5 | NA | NA |
| matK | −8587.46216 | −8587.46219 | 1 | 0.5 | NA | NA |
| ndhE | −1430.75023 | −1430.75023 | 1 | 0.5 | NA | NA |
| rpl20 | −2045.17646 | −2044.24507 | 1 | 0.49874 | NA | NA |
| atpF | −2329.18412 | −2329.18414 | 1 | 0.5 | NA | NA |
| petL | −364.79445 | −364.79445 | 1 | 0.5 | NA | NA |
| cemA | −3613.71602 | −3613.71602 | 1 | 0.5 | NA | NA |
| petG a | −345.98700 | −345.98700 | 1 | 0.49851 | NA | 0.997 b |
| rpoB | −13852.11743 | −13852.11743 | 1 | 0.49831 | NA | NA |
A = alanine (amino acid); BEB = Bayesian empirical Bayes; NA = not available; NEB = naive empirical Bayes.
Genes under positive selection.
P > 99%.
DISCUSSION
The structure of Aquilegia chloroplast genomes
In this study, we assembled and annotated the complete chloroplast genomes of 14 Aquilegia species, including 10 species from Asia, two from Europe, and two from North America. Based on these chloroplast genome sequences, we calculated polymorphism and inferred the phylogenetic relationships within Aquilegia.
The structure and gene order of chloroplast genomes are highly conserved in the angiosperms (Choi et al., 2016). In our study, the chloroplast genomes of 14 Aquilegia species showed a typical quadripartite structure (Fig. 1), and the gene composition and gene order were similar in each species. The expansion or contraction of IR regions plays an important role in the length of the chloroplast genome (Raubeson et al., 2007; Wang et al., 2008; Yang et al., 2010). In the Aquilegia chloroplast genomes, the total length of the complete sequence was directly proportional to the length of the IR region (Table 1). Insertion/deletion polymorphisms (indels) in these sequences resulted in variations in the length of the Aquilegia chloroplast genome, which is a common phenomenon found in Camellia L. (Huang et al., 2014), Quercus L. (Yin et al., 2018), Amaranthus L. (Chaney et al., 2016), and the other angiosperms (Jiang et al., 2017). Compared with the other two regions, the GC content was the highest in the IR regions in Aquilegia. This effect may be caused by the presence of more rDNA in the IR regions, which has a higher GC content (approximately 50%) (Xie et al., 2018).
Both long repetitive sequences and SSRs with high copy‐number diversity are valuable and useful molecular markers in studies of plant population genetics, phylogenetic reconstruction, and plant evolution at the intraspecific level (Wu et al., 2015; Ivanova et al., 2017). Here, long repeat sequences and SSRs of different lengths were found in each species (Figs. 2, 3), indicating that they can both be used as molecular markers for research on Aquilegia. Among these regions, the SSC region had the highest nucleotide polymorphism level, followed by the LSC region; the IR regions had the lowest nucleotide polymorphism level, indicating that the IR regions were most conserved. This result is likely due to the high conservation of the rDNA in the IR regions (Hershkovitz and Zimmer, 1996). The nucleotide polymorphisms of chloroplast genes in Aquilegia were smaller than those of other genera, such as Populus L. (Gao et al., 2019), Camellia (Li et al., 2019a), and Anguinum Fourr. (Jin et al., 2019); however, some variable genes were identified, including rpoC2, trnS‐GGA, and trnL‐CAA (Fig. 4). These regions with high levels of polymorphism are also a good resource for studying the phylogeny and population genetics of Aquilegia, especially rpoC2, which has the highest levels of polymorphism (Walker et al., 2019).
The phylogeny of Aquilegia based on chloroplast genomes
Biogeographic and phylogenetic analyses have indicated that Aquilegia had a common ancestor from eastern Asia, and later adaptive radiations took place independently in North America and Western Europe (Bastida et al., 2010; Fior et al., 2013). Aquilegia amurensis is restricted to the northern Greater Khingan Mountains, while A. parviflora is distributed in the northern Greater Khingan Mountains and Siberia. Despite this, we found these species were phylogenetically close to Aquilegia species from North America, whereas the remaining Asian species were phylogenetically close to Aquilegia species from Europe.
The phylogeny based on the chloroplast genome was not completely consistent with that of the study by Fior et al. (2013). In our study, A. oxysepala var. oxysepala, A. japonica, and A. sibirica fell within a single clade; however, Filiault et al. (2018) had concluded that A. oxysepala var. oxysepala was located at the base of the phylogenetic tree, and A. japonica and A. sibirica shared a most recent common ancestor (MRCA). Li et al. (2014) used a combination of morphological characteristics, habitat type, and nuclear and chloroplast phylogenies (Bastida et al., 2010; Fior et al., 2013; Li et al., 2014) of these three species to propose that A. sibirica diverged first from the MRCA, and A. oxysepala var. oxysepala and A. japonica then differentiated into new species (Li et al., 2019b) containing more individuals. Our results also support the research of Li et al. (2019b). In addition, the position of A. viridiflora in this study was inconsistent with the phylogeny based on chloroplast genes by Fior et al. (2013) and the phylogeny by Lu et al. (2019). The inconsistency may be caused by incomplete lineage sorting and introgression in species undergoing rapid adaptive radiation (Meyer et al., 2017; Cai et al., 2020); therefore, the taxonomic status of A. viridiflora is worthy of further study. In addition, according to the Flora of China (Li, 2007), A. oxysepala var. kansuensis is considered a variety of A. oxysepala var. oxysepala, although their morphological characteristics, distribution ranges, and habitats all differ from each other. In both the present and previous studies (Fior et al., 2013), A. oxysepala var. oxysepala and A. oxysepala var. kansuensis showed distant genetic relationships; therefore, we suggest that A. oxysepala var. kansuensis should be considered as a separate species rather than a variety. The phylogenetic tree shows that A. ecalcarata sequences were present on two different branches, providing further evidence to the previous report that A. ecalcarata is not monophyletic with a single origin and may have a complicated evolutionary history (Huang et al., 2018). In the future, to infer the phylogenetic relationships of rapidly evolving species within Aquilegia, we should collect more varieties and a greater number of species to construct the phylogeny.
Adaptative evolution of Aquilegia
Synonymous and nonsynonymous nucleotide substitution patterns play an important role in adaptive evolution. In Aquilegia, no significant positive selection was detected for the majority of genes, with only three genes (petG, rpl36, and atpB) showing possible positive selection; these may have played an important role in adaptive evolution in Aquilegia. Based on annotation information from the UniProtKB database (https://www.uniprot.org), in Arabidopsis thaliana (L.) Heynh., petG controls the components of the cytochrome bf6‐f complex subunit 5, which mediates electron transfer between photosystem II (PSII) and PSI, cyclic electron flow around PSI, and state transitions (Sato et al., 1999; Kandlbinder et al., 2004); the rpl36 gene encodes the 50S ribosomal protein L36, which serves as a structural component of the ribosome (Sato et al., 1999; Koia et al., 2013); and the atpB gene controls the ATP synthase subunit beta, which produces ATP from ADP in the presence of a proton gradient across the membrane (Sato et al., 1999; Friso et al., 2004). Previous studies showed that rpl36 was under positive selection in the Araceae and Sophora tonkinensis Gagnep. (Fan et al., 2020; Henriquez et al., 2020), while atpB was under positive selection in Urophysa Ulbr. and the Liliaceae (sensu lato) (Xie et al., 2018; She et al., 2020). These genes are highly correlated with physiological processes such as photosynthesis and disease resistance; thus, their positive selection may assist Aquilegia species in rapid adaptation to various environments and enable their wide global distribution.
AUTHOR CONTRIBUTIONS
X.H. and W.H. designed the study and evaluated the results; W.H. and Z.W. collected the materials; Z.W., D.J., and Z.T. participated in the data analysis; Z.W. and W.H. prepared the manuscript; and all authors read and approved the final manuscript.
Supporting information
APPENDIX S1. List of genes encoded by the Aquilegia chloroplast genomes.
APPENDIX S2. Number of each type of simple sequence repeat in Aquilegia species.
APPENDIX S3. The nucleotide diversity of all genes of Aquilegia.
Acknowledgments
The authors thank the reviewers for their valuable suggestions and comments and acknowledge Mingzhou Sun and Xiaoxue Fang (Key Laboratory of Molecular Epigenetics, Northeast Normal University, Ministry of Education) for their help in collection of material. This research was supported by the National Natural Science Foundation of China (32070244).
APPENDIX 1. Aquilegia sampling information.
| Species | Latitude (°N) | Longitude (°E) | Distribution region | Size (Gbp) | Raw reads | Chloroplast reads | Depth | Voucher specimen |
|---|---|---|---|---|---|---|---|---|
| A. viridiflora | 40.954 | 111.672 | Asia | 13 | 18,729,599 | 9,936,532 | 4774× | NENU_Aq1001 |
| A. oxysepala var. kansuensis | 31.815 | 109.009 | Asia | 11 | 16,161,175 | 3,273,451 | 1519× | NENU_Aq1002 |
| A. ecalcarata | 37.160 | 102.223 | Asia | 11 | 16,159,854 | 3,721,439 | 1875× | NENU_Aq1003 |
| A. parviflora | 50.422 | 121.476 | Asia | 9.6 | 14,222,775 | 3,179,153 | 1517× | NENU_Aq1004 |
| A. amurensis | 52.672 | 123.870 | Asia | 9.9 | 14,758,620 | 6,110,285 | 2874× | NENU_Aq1005 |
| A. rockii | 29.951 | 101.964 | Asia | 11 | 15,337,263 | 3,696,958 | 1664× | NENU_Aq1006 |
| A. yabeana | 33.9125 | 112.041 | Asia | 13 | 18,296,460 | 3,276,927 | 1523× | NENU_Aq1007 |
APPENDIX 2. Information about the Aquilegia sequence data previously published by Filiault et al. (2018) and downloaded from the National Center for Biotechnology Information Sequence Read Archive (SRA). a
| Species | SRA no. | Size (Gbp) | Chloroplast reads | Depth | Distribution region |
|---|---|---|---|---|---|
| A. aurea | SRR405095 | 25.9 | 15,526,578 | 8520× | Europe |
| A. vulgaris | SRR404349 | 27.5 | 48,865,464 | 26,870× | Europe |
| A. sibirica | SRR405090 | 25.2 | 28,912,821 | 16,384× | Asia |
| A. formosa | SRR408554 | 28.4 | 11,593,572 | 7209× | North America |
| A. chrysantha | SRR408559 | 26.8 | 11,964,708 | 7209× | North America |
| A. japonica | SRR413499 | 26.6 | 28,881,079 | 16,384× | Asia |
| A. oxysepala var. oxysepala | SRR413921 | 28.0 | 41,390,034 | 24,248× | Asia |
Sequencing was performed on the Illumina platform.
APPENDIX 3. Chloroplast genome sequences downloaded from GenBank.
| Species | GenBank accession no. |
|---|---|
| Aconitum brachypodum | NC_041579.1 |
| Actaea vaginata | MK253451.1 |
| Adonis coerulea | MK253469.1 |
| Anemoclema glaucifolium | MH205609.1 |
| Anemone raddeana | NC_041526.1 |
| Anemonopsis macrophylla | NC_041527.1 |
| Aquilegia coerulea | NC_041528.1 |
| Aquilegia coerulea | MK569474.1 |
| Aquilegia ecalcarata | NC_041529.1 |
| Aquilegia ecalcarata | MK569475.1 |
| Aquilegia rockii | NC_046738.1 |
| Aquilegia rockii | MK573514.1 |
| Asteropyrum cavaleriei | NC_041530.1 |
| Beesia calthifolia | NC_041531.1 |
| Calathodes oxycarpa | NC_041475.1 |
| Callianthemum taipaicum | NC_041476.1 |
| Caltha palustris | MK253465.1 |
| Ceratocephala falcata | MK253464.1 |
| Clematis terniflora | KJ956785.1 |
| Consolida ajacis | NC_041534.1 |
| Coptis chinensis | MK569483.1 |
| Delphinium anthriscifolium | MK253461.1 |
| Dichocarpum dalzielii | MK253459.1 |
| Enemion raddeanum | NC_041535.1 |
| Eranthis stellata | NC_041536.1 |
| Glaucidium palmatum | MK569492.1 |
| Gymnaconitum gymnandrum | NC_033341.1 |
| Halerpestes sarmentosa | MK253457.1 |
| Helleborus thibetanus | NC_041540.1 |
| Hydrastis canadensis | MK569495.1 |
| Isopyrum manshuricum | NC_041541.1 |
| Leptopyrum fumarioides | NC_041542.1 |
| Megaleranthis saniculifolia | FJ597983.1 |
| Naravelia pilulifera | NC_039542.1 |
| Nigella damascena | NC_041537.1 |
| Oxygraphis glacialis | NC_041538.1 |
| Paraquilegia anemonoides | NC_041479.1 |
| Pulsatilla chinensis | MK569491.1 |
| Ranunculus macranthus | DQ359689.1 |
| Semiaquilegia adoxoides | MK569498.1 |
| Staphisagria macrosperma | MN648404.1 |
| Thalictrum thalictroides | NC_039433.1 |
| Trollius chinensis | NC_031849.1 |
| Trollius ranunculoides | MK253447.1 |
| Urophysa rockii | MK569502.1 |
Zhang, W. , Wang H., Dong J., Zhang T., and Xiao H.. 2021. Comparative chloroplast genomes and phylogenetic analysis of Aquilegia . Applications in Plant Sciences 9(3): e11412.
Data Availability
Raw sequence data is available from the National Center for Biotechnology Information Sequence Read Archive under the accession number PRJNA666554. Chloroplast sequence data are available in GenBank (accession numbers MT919110–MT9191116 and MN809218–MN809224). The Python script used for extraction of homologous genes is available on GitHub (https://github.com/zhangw348/NENU_plant‐systems‐and‐evolution).
LITERATURE CITED
- Andrews, S. 2010. FastQC: A quality control tool for high throughput sequence data. Website http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [accessed 19 January 2021].
- Bastida, J. M. , Alcántara J. M., Rey P. J., Vargas P., and Herrera C. M.. 2010. Extended phylogeny of Aquilegia: The biogeographical and ecological patterns of two simultaneous but contrasting radiations. Plant Systematics and Evolution 284: 171–185. [Google Scholar]
- Boetzer, M. , Henkel C. V., Jansen H. J., Butler D., and Pirovano W.. 2011. Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics 27(4): 578–579. [DOI] [PubMed] [Google Scholar]
- Boetzer, M. , and Pirovano W.. 2012. Toward almost closed genomes with GapFiller. Genome Biology 13(6): R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, L. , Xi Z., Lemmon E. M., Lemmon A. R., Mast A., Buddenhagen C. E., Liu L., and Davis C. C.. 2020. The perfect storm: Gene tree estimation error, incomplete lineage sorting, and ancient gene flow explain the most recalcitrant ancient angiosperm clade, Malpighiales. Systematic Biology 10.1093/sysbio/syaa083. [DOI] [PubMed] [Google Scholar]
- Chaney, L. , Mangelson R., Ramaraj T., Jellen E. N., and Maughan P. J.. 2016. The complete chloroplast genome sequences for four Amaranthus species (Amaranthaceae). Applications in Plant Sciences 4(9): 1600063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K. S. , Chung M. G., and Park S.. 2016. The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): Comparative analysis and highly divergent regions. Frontiers in Plant Science 7: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curci, P. L. , De Paola D., Danzi D., Vendramin G. G., and Sonnante G.. 2015. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 10(3): e0120589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, W. , Liu J., Yu J., Wang L., and Zhou S.. 2012. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7(4): e35071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, W.‐L. , Wang R.‐N., Zhang N.‐Y., Fan W.‐B., Fang M.‐F., and Li Z.‐H.. 2018. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. International Journal of Molecular Sciences 19(3): 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie, S. R. , and Jansen R. K.. 2015. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Systematic Botany 40(1): 336–351. [Google Scholar]
- Doyle, J. J. , and Doyle J. L.. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- Erst, A. S. , Wang W., Yu S.‐X., Xiang K., Wang J., Shaulo D. N., Smirnov S. V., et al. 2017. Two new species and four new records of Aquilegia (Ranunculaceae) from China. Phytotaxa 316(2): 121–137. [Google Scholar]
- Erst, A. S. , Pendry C. A., Erst T. V., Ikeda H., Xiang K., and Wang W.. 2020. Two new taxa and one new record of Aquilegia (Ranunculaceae) from India and Pakistan. Phytotaxa 439(2): 108–118. [Google Scholar]
- Fan, W.‐B. , Wu Y., Yang J., Shahzad K., and Li Z.‐H.. 2018. Comparative chloroplast genomics of Dipsacales species: Insights into sequence variation, adaptive evolution, and phylogenetic relationships. Frontiers in Plant Science 9: 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, W. , Tang D., Kunhua W., Fang Q., Li L., Yang L., Yanxia Z., et al. 2020. The complete chloroplast genome sequence of the medicinal plant Sophora tonkinensis . Scientific Reports 10(1): 12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiault, D. L. , Ballerini E. S., Mandakova T., Aköz G., Derieg N. J., Schmutz J., Jenkins J., et al. 2018. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLife 7: e36426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fior, S. , Li M., Oxelman B., Viola R., Hodges S. A., Ometto L., and Varotto C.. 2013. Spatiotemporal reconstruction of the Aquilegia rapid radiation through next‐generation sequencing of rapidly evolving cp DNA regions. New Phytologist 198(2): 579–592. [DOI] [PubMed] [Google Scholar]
- Friso, G. , Giacomelli L., Ytterberg A. J., Peltier J.‐B., Rudella A., Sun Q., and Van Wijk K. J.. 2004. In‐depth analysis of the thylakoid membrane proteome of Arabidopsis thaliana chloroplasts: New proteins, new functions, and a plastid proteome database. The Plant Cell 16(2): 478–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, K. , Li J., Khan W. U., Zhao T., Yang X., Yang X., Guo B., and An X.. 2019. Comparative genomic and phylogenetic analyses of Populus section Leuce using complete chloroplast genome sequences. Tree Genetics & Genomes 15(3): 32. [Google Scholar]
- Guisinger, M. M. , Kuehl J. V., Boore J. L., and Jansen R. K.. 2010. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Molecular Biology and Evolution 28(1): 583–600. [DOI] [PubMed] [Google Scholar]
- Henriquez, C. L. , Mehmood F., Shahzadi I., Ali Z., Waheed M. T., Croat T. B., Poczai P., and Ahmed I.. 2020. Comparison of chloroplast genomes among species of unisexual and bisexual clades of the monocot family Araceae. Plants 9(6): 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershkovitz, M. A. , and Zimmer E. A.. 1996. Conservation patterns in angiosperm rDNA ITS2 sequences. Nucleic Acids Research 24(15): 2857–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges, S. A. , and Arnold M. L.. 1994. Columbines: A geographically widespread species flock. Proceedings of the National Academy of Sciences, USA 91(11): 5129–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges, S. A. , and Derieg N. J.. 2009. Adaptive radiations: From field to genomic studies. Proceedings of the National Academy of Sciences, USA 106(Suppl 1): 9947–9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H. , Shi C., Liu Y., Mao S.‐Y., and Gao L.‐Z.. 2014. Thirteen Camellia chloroplast genome sequences determined by high‐throughput sequencing: Genome structure and phylogenetic relationships. BMC Evolutionary Biology 14(1): 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. , Geng F. D., Fan J. J., Xue C., Zhang X. Y., Kang J. Q., Zhang J. Q., and Ren Y.. 2018. Genetic diversity and evolutionary history of four closely related Aquilegia species revealed by 10 nuclear gene fragments. Journal of Systematics and Evolution 56(2): 129–138. [Google Scholar]
- Ivanova, Z. , Sablok G., Daskalova E., Zahmanova G., Apostolova E., Yahubyan G., and Baev V.. 2017. Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Frontiers in Plant Science 8: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, D. , Zhao Z., Zhang T., Zhong W., Liu C., Yuan Q., and Huang L.. 2017. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in scutellaria. Genes 8(9): 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, F. , Xie D., Li H., Yu Y., Zhou S., and He X.. 2019. Comparative complete chloroplast genome analyses and contribution to the understanding of chloroplast phylogeny and adaptive evolution in subgenus Anguinum . Russian Journal of Genetics 55(7): 872–884. [Google Scholar]
- Kandlbinder, A. , Finkemeier I., Wormuth D., Hanitzsch M., and Dietz K. J.. 2004. The antioxidant status of photosynthesizing leaves under nutrient deficiency: Redox regulation, gene expression and antioxidant activity in Arabidopsis thaliana . Physiologia Plantarum 120(1): 63–73. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30(4): 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koia, J. , Moyle R., Hendry C., Lim L., and Botella J. R.. 2013. Pineapple translation factor SUI1 and ribosomal protein L36 promoters drive constitutive transgene expression patterns in Arabidopsis thaliana . Plant Molecular Biology 81(4–5): 327–336. [DOI] [PubMed] [Google Scholar]
- Kubo, T. , Nishizawa S., Sugawara A., Itchoda N., Estiati A., and Mikami T.. 2000. The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNACys (GCA). Nucleic Acids Research 28(13): 2571–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher G., Li M., Knyaz C., and Tamura K.. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution 35(6): 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, S. , Choudhuri J. V., Ohlebusch E., Schleiermacher C., Stoye J., and Giegerich R.. 2001. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research 29(22): 4633–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , and Bork P.. 2006. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 23(1): 127–128. [DOI] [PubMed] [Google Scholar]
- Li, J. 2007. Flora of China. Harvard Papers in Botany 13(2): 301–303. [Google Scholar]
- Li, L. F. , Wang H. Y., Pang D., Liu Y., Liu B., and Xiao H. X.. 2014. Phenotypic and genetic evidence for ecological speciation of Aquilegia japonica and A. oxysepala . New Phytologist 204(4): 1028–1040. [DOI] [PubMed] [Google Scholar]
- Li, X. , Li Y., Zang M., Li M., and Fang Y.. 2018. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima . International Journal of Molecular Sciences 19(8): 2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Zhang C., Guo X., Liu Q., and Wang K.. 2019a. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE 14(5): e0216645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.‐R. , Wang H.‐Y., Ding N., Lu T., Huang Y.‐C., Xiao H.‐X., Liu B., and Li L.‐F.. 2019b. Rapid divergence followed by adaptation to contrasting ecological niches of two closely related columbine species Aquilegia japonica and A. oxysepala . Genome Biology and Evolution 11(3): 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Kong H., Zhou J., Fritsch P., Hao G., and Gong W.. 2018. Complete chloroplast genome of Cercis chuniana (Fabaceae) with structural and genetic comparison to six species in Caesalpinioideae. International Journal of Molecular Sciences 19(5): 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse, M. , Drechsel O., Kahlau S., and Bock R.. 2013. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Research 41(W1): W575–W581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Q. , Ye W., Lu R., Xu W., and Qiu Y.. 2018. Phylogenomic and comparative analyses of complete plastomes of Croomia and Stemona (Stemonaceae). International Journal of Molecular Sciences 19(8): 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, T. , Li M.‐R., Ding N., Wang Z.‐H., Lan L.‐Z., Gao X., and Li L.‐F.. 2019. Genetic and epigenetic signatures associated with the rapid radiation of Aquilegia species. bioRxiv: 782821 [Preprint] [published 7 December 2019]. Available at 10.1101/782821 [accessed 19 January 2021]. [DOI] [Google Scholar]
- Luo, Y. , Erst A. S., Yang C.‐X., Deng J.‐P., and Li L.. 2018. Aquilegia yangii (Ranunculaceae), a new species from western China. Phytotaxa 348(4): 289–296. [Google Scholar]
- Mader, M. , Pakull B., Blanc‐Jolivet C., Paulini‐Drewes M., Bouda Z., Degen B., Small I., and Kersten B.. 2018. Complete chloroplast genome sequences of four Meliaceae species and comparative analyses. International Journal of Molecular Sciences 19(3): 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, B. S. , Matschiner M., and Salzburger W.. 2017. Disentangling incomplete lineage sorting and introgression to refine species‐tree estimates for Lake Tanganyika cichlid fishes. Systematic Biology 66(4): 531–550. [DOI] [PubMed] [Google Scholar]
- Munz, P. A. . 1946. Aquilegia: The cultivated and the wild columbines. Gentes Herbarum 7: 1–50. [Google Scholar]
- Nadachowska‐Brzyska, K. , Li C., Smeds L., Zhang G., and Ellegren H.. 2015. Temporal dynamics of avian populations during Pleistocene revealed by whole‐genome sequences. Current Biology 25(10): 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadalin, F. , Vezzi F., and Policriti A.. 2012. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13(S14): S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt H. A., Von Haeseler A., and Minh B. Q.. 2015. IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution 32(1): 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, M. , Park H., Lee H., Lee B.‐H., and Lee J.. 2018. The complete plastome sequence of an Antarctic bryophyte Sanionia uncinata (Hedw.) Loeske. International Journal of Molecular Sciences 19(3): 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raubeson, L. A. , Peery R., Chumley T. W., Dziubek C., Fourcade H. M., Boore J. L., and Jansen R. K.. 2007. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus . BMC Genomics 8(1): 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko M., Van Der Mark P., Ayres D. L., Darling A., Höhna S., Larget B., et al. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61(3): 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J. , Ferrer‐Mata A., Sánchez‐DelBarrio J. C., Guirao‐Rico S., Librado P., Ramos‐Onsins S. E., and Sánchez‐Gracia A.. 2017. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution 34(12): 3299–3302. [DOI] [PubMed] [Google Scholar]
- Sancho, R. , Cantalapiedra C. P., López‐Alvarez D., Gordon S. P., Vogel J. P., Catalán P., and Contreras‐Moreira B.. 2018. Comparative plastome genomics and phylogenomics of Brachypodium: Flowering time signatures, introgression and recombination in recently diverged ecotypes. New Phytologist 218(4): 1631–1644. [DOI] [PubMed] [Google Scholar]
- Sato, S. , Nakamura Y., Kaneko T., Asamizu E., and Tabata S.. 1999. Complete structure of the chloroplast genome of Arabidopsis thaliana . DNA Research 6(5): 283–290. [DOI] [PubMed] [Google Scholar]
- She, R. , Zhao P., Zhou H., Yue M., Yan F., Hu G., Gao X., and Zhang S.. 2020. Complete chloroplast genomes of Liliaceae (s.l.) species: Comparative genomic and phylogenetic analyses. Nordic Journal of Botany 38(1). 10.1111/njb.02477. [DOI] [Google Scholar]
- Taylor, R. J. 1967. Interspecific hybridization and its evolutionary significance in the genus Aquilegia . Brittonia 19(4): 374–390. [Google Scholar]
- Thiel, T. , Michalek W., Varshney R., and Graner A.. 2003. Exploiting EST databases for the development and characterization of gene‐derived SSR‐markers in barley (Hordeum vulgare L.). Theoretical and Applied Genetics 106(3): 411–422. [DOI] [PubMed] [Google Scholar]
- Walker, J. F. , Walker‐Hale N., Vargas O. M., Larson D. A., and Stull G. W.. 2019. Characterizing gene tree conflict in plastome‐inferred phylogenies. PeerJ 7: e7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R.‐J. , Cheng C.‐L., Chang C.‐C., Wu C.‐L., Su T.‐M., and Chaw S.‐M.. 2008. Dynamics and evolution of the inverted repeat‐large single copy junctions in the chloroplast genomes of monocots. BMC Evolutionary Biology 8(1): 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z. , Tembrock L. R., and Ge S.. 2015. Are differences in genomic data sets due to true biological variants or errors in genome assembly: An example from two chloroplast genomes. PLoS ONE 10(2): e0118019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman, S. K. , Jansen R. K., and Boore J. L.. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20(17): 3252–3255. [DOI] [PubMed] [Google Scholar]
- Xie, D.‐F. , Yu Y., Deng Y.‐Q., Li J., Liu H.‐Y., Zhou S.‐D., and He X.‐J.. 2018. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. International Journal of Molecular Sciences 19(7): 1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. , Zhang X., Liu G., Yin Y., Chen K., Yun Q., Zhao D., Al‐Mssallem I. S., and Yu J.. 2010. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 5(9): e12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Vologodskii A. V., Liu B., Kemper B., and Seeman N. C.. 1998. Torsional control of double‐stranded DNA branch migration. Biopolymers 45(1): 69–83. [DOI] [PubMed] [Google Scholar]
- Yang, Z. 2007. PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24(8): 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, Z. , and Nielsen R.. 2002. Codon‐substitution models for detecting molecular adaptation at individual sites along specific lineages. Molecular Biology and Evolution 19(6): 908–917. [DOI] [PubMed] [Google Scholar]
- Yang, Z. , Nielsen R., and Hasegawa M.. 1998. Models of amino acid substitution and applications to mitochondrial protein evolution. Molecular Biology and Evolution 15(12): 1600–1611. [DOI] [PubMed] [Google Scholar]
- Yin, K. , Zhang Y., Li Y., and Du F.. 2018. Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: Evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. International Journal of Molecular Sciences 19(4): 1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurina, N. , and Odintsova M.. 1998. Comparative structural organization of plant chloroplast and mitochondrial genomes. Russian Journal of Genetics 34(1): 1–16. [Google Scholar]
- Zerbino, D. 2010. Using the columbus extension to Velvet. Available at http://gensoft.pasteur.fr/docs/velvet/1.1.02/Columbus_manual.pdf [accessed 19 January 2021].
- Zhai, W. , Duan X., Zhang R., Guo C., Li L., Xu G., Shan H., et al. 2019. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Molecular Phylogenetics and Evolution 135: 12–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. List of genes encoded by the Aquilegia chloroplast genomes.
APPENDIX S2. Number of each type of simple sequence repeat in Aquilegia species.
APPENDIX S3. The nucleotide diversity of all genes of Aquilegia.
Data Availability Statement
Raw sequence data is available from the National Center for Biotechnology Information Sequence Read Archive under the accession number PRJNA666554. Chloroplast sequence data are available in GenBank (accession numbers MT919110–MT9191116 and MN809218–MN809224). The Python script used for extraction of homologous genes is available on GitHub (https://github.com/zhangw348/NENU_plant‐systems‐and‐evolution).