

Abstract
N-methyl-D-aspartate receptors (NMDARs) play critical roles in the physiological function of the mammalian central nervous system (CNS), including learning, memory, and synaptic plasticity, through modulating excitatory neurotransmission. Attributed to etiopathology of various CNS disorders and neurodegenerative diseases, GluN2B is one of the most well-studied subtypes in preclinical and clinical studies on NMDARs. Herein, we report the synthesis and preclinical evaluation of two 11C-labeled GluN2B-selective negative allosteric modulators (NAMs) containing N,N-dimethyl-2-(1H-pyrrolo[3,2-b]pyridin-1-yl)acetamides for positron emission tomography (PET) imaging. Two PET ligands, namely [11C]31 and [11C]37 (also called N2B-1810 and N2B-1903, respectively) were labeled with [11C]CH3I in good radiochemical yields (decay-corrected 28% and 32% relative to starting [11C]CO2, respectively), high radiochemical purity (>99%) and high molar activity (>74 GBq/μmol). In particular, PET ligand [11C]31 demonstrated moderate specific binding to GluN2B subtype by in vitro autoradiography studies. However, because in vivo PET imaging studies showed limited brain uptake of [11C]31 (up to 0.5 SUV), further medicinal chemistry and ADME optimization are necessary for this chemotype attributed to low binding specificity and rapid metabolism in vivo.
Keywords: ionotropic glutamate receptors (iGluRs), NMDARs, GluN2B subunit, positron emission tomography (PET), carbon-11
Introduction
L-Glutamate, a major endogenous excitatory neurotransmitter in the mammalian central nervous system (CNS), regulates a wide range of nervous system functions. The excitatory neurotransmitter signal is mediated through two types of L-glutamate receptors: ligand-gated ion channel ionotropic receptors (iGluRs) and G-protein-coupled metabotropic receptors (mGluRs). NMDARs are one class of iGluRs with three subunits (GluN1-GluN3), and the dysregulation of NMDARs has been identified in the pathophysiology of various brain diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), schizophrenia, and neuropathic pain [1–6]. The GluN2B subunit, one of four subunits of GluN2, has been found to play essential roles in these neurological and psychiatric disorders. This subunit encompasses polyamines as positive allosteric modulators (PAMs) and incorporates zinc ion-binding sites for docking negative allosteric modulators (NAMs), and these sites are also called ifenprodil-like sites [7–10]. The GluN2B subunit has high and widespread expression throughout the brain around birth; however, the distribution of this subunit is gradually restricted to the forebrain, including in the hippocampus, cerebral cortex, striatum, and thalamus, and it is rarely detected in the cerebellum in adult brains [11–14]. Although since the 1990s, several NMDAR antagonists, such as phencyclidine (PCP), ketamine, MK-801, memantine, and amantadine, have been developed and used in clinical applications, chronic glutamate stimulation to NMDARs may lead to excitotoxicity, along with many adverse CNS effects, including hallucinations, increasing blood pressure, and catatonia [15–18]. Therefore, GluN2B-selective NMDAR antagonists have attracted increasing attention because they have shown promising therapeutic potential by treating NMDAR-related diseases, yet they lack the same adverse reactions due to their unique activity [19–21].
Positron emission tomography (PET) is a noninvasive nuclear imaging technique that can help profile in vivo biological targets by detecting the uptake, distribution, and metabolism of radiotracers [22–27]. GluN2B-selective PET imaging radiotracers could provide a powerful tool for understanding the biological role of NMDAR subtypes in the pathophysiology of various brain diseases. Since several excellent reviews [1, 19–21] have discussed the current status of NMDAR radiotracers, we merely listed several representative GluN2B-selective PET radiotracers in Fig. 1 [28–41]. [11C]( ± )-Methoxy-CP-101606 (1, traxoprodil) [28, 29] was the first “prodil” PET radiotracer, and it was reported by Haradahira and coworkers in 2002. MK-0657 (10, rislenemdaz) is a new class of GluN2B-selected antagonists that has entered clinical trials, and trans-[18F]10 and cis-[18F]10 have been studied for imaging the GluN2B subunit in vitro and in vivo [31, 32]. In 2018, Kramer et al. developed a new type of PET ligand derived from sigma receptor modulators. For example, 11C-Me-NB1 ([11C]14) displayed high affinity for the GluN2B-NTD-binding site and enabled target engagement studies [35, 36]. Recently, we reported a novel GluN2B-selective PET radiotracer containing a triazole core with high in vitro-specific binding in brain sections [41]. However, clinical applications of the aforementioned radiotracers have not been demonstrated. Therefore, further studies on clinically relevant GluN2B-selective PET radiotracers are necessary.
Fig. 1.

Representative GluN2B-selective NMDAR PET radiotracers [28–41].
As a continuation of our previous work on GluN2B-selective PET radiotracers [41], we report two 11C-labeled compounds selected from a recently reported novel class of GluN2B-negative allosteric modulators based on their favorable pharmacological and physicochemical properties [42]. Compounds 31 and 37 demonstrated excellent in vitro GluN2B binding affinity (Ki = 11 nM and 4.3 nM, respectively) and possessed the desired GluN2B occupancy in the brain (peak occupancy for 31 = 83%, peak occupancy for 37 = 96%) in preliminary receptor occupancy studies. Pharmacological evaluation of 31 and 37 showed their potential for evaluation as PET ligands for GluN2B subunits. In this work, with an efficient N-methylation 11C-labeling strategy, radiosynthesis, and preliminary evaluation of [11C]31 and [11C]37 in the rodent brain were carried out. The brain permeability and binding specificity were evaluated by PET imaging in vivo, and autoradiography studies were conducted in vitro.
Materials and methods
The general procedure for the experimental section was described previously [43, 44] and was used with minor modifications in this work. All chemicals were purchased from commercial vendors and were used without further purification unless otherwise indicated. Thin-layer chromatography (TLC) was conducted with 0.25-mm silica gel plates (60F254) and visualized by exposure to UV light (254 nm) or by staining with potassium permanganate. Column chromatography separations were performed on silica gel (SiliCycle Inc., 230–400 mesh, 40–63 μm). 1H, 13C, and 19F NMR spectra were obtained at 300, 75, and 282 MHz, respectively, on a Bruker spectrometer in CDCl3 or d6-DMSO solutions at rt, and the chemical shifts are reported in δ values (parts per million, ppm) downfield relative to the internal TMS. The multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad signal, and dd = doublet of doublets. For LC-MS, the ionization method was ESI using Agilent 6430 Triple Quad LC/MS. The animal experiments were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital and National Institute of Radiological Sciences (Japan). CD1 (ICR) mice (female; 8 weeks, 20–25 g) and Sprague–Dawley rats (male; 7 weeks; 210–230 g) were kept on a 12 h light/12 h dark cycle and were allowed food and water ad libitum.
General procedure A for the synthesis of 30 and 31
A mixture of compound 27 or 28 (1 mmol), (4-fluoro-3-methylphenyl)boronic acid (29, 1.1 mmol, 1.1 equiv), Pd(dppf)Cl2 (0.2 mmol, 0.2 equiv) and Cs2CO3 (3 mmol, 3 equiv) in 11 mL of a mixture of 1,4-dioxane/H2O = 10/1 (v/v) was heated to 90 °C under an argon atmosphere for 4 h. After completion of the reaction, the solvent was removed, and the residue was dissolved in EtOAc (30 mL) and washed with water (20 mL) and brine (20 mL). After removing the solvent, 30 and 31 were purified by chromatography on silica gel (elution with EtOAc/n-hexane = 80/20, v/v).
General procedure B for the synthesis of 36 and 37
NaH (0.75 mmol, 1.5 equiv, 60 wt% in mineral oil) was carefully added to a solution of compound 35 (0.5 mmol) in anhydrous DMF (5 mL) in an ice bath. After stirring for 20 min, intermediate 24 or 25 (0.55 mmol, 1.1 equiv) was added to the reaction, and the mixture was stirred at rt for another 10 min. The reaction was quenched by cool water and extracted with EtOAc (20 mL × 3), and the combined organic layers were washed with water (20 mL × 2) and brine (20 mL). After removing the solvent, 36 and 37 were purified by chromatography on silica gel (elution with EtOAc/n-hexane = 80/20, v/v).
General procedure for 11C-methylation
[11C]CH3I was synthesized from cyclotron-produced [11C]CO2, which was produced by 14N(p,α)11C nuclear reaction. Briefly, [11C]CO2 was bubbled into a solution of LiAlH4 (0.4 M in THF, 300 μL). After evaporation, the remaining reaction mixture was treated with hydroiodic acid (57% aqueous solution, 300 μL). The resulting [11C]CH3I was transferred under helium gas with heating into a reaction vessel containing a solution of the precursor (1.0 mg) in presaturated anhydrous DMSO (300 μL) with NaOH powder (6.0 mg, 85 wt% powder). After the radioactivity reached a plateau during the transfer, the reaction vessel was warmed to 80 °C and maintained for 5 min. The mobile phase (2.5 mL) and H2O (1.5 mL) were added to the reaction mixture, which was then injected into a semipreparative HPLC system. HPLC purification was completed on a Triart C18 ExRS column (10 mm i.d. × 250 mm) using the appropriate mobile phases. The radioactive fraction corresponding to the desired product was collected into a flask containing polysorbate (80) (75 mL) and ethanol (150 mL), evaporated to dryness in vacuo, and redissolved in 3 mL of sterile normal saline. The radiochemical and chemical purities were measured by analytical HPLC (Triart C18 column 4.6 mm i.d. × 250 mm, UV at 254 nm; flow rate at 1.0 mL/min). The identities of [11C]31 and [11C]37 were confirmed by coinjection with unlabeled 31 and 37, respectively.
Measurement of lipophilicity
The general procedure for the lipophilicity measurement was described previously [43–45] and was used with minor modifications in this work. Briefly, the LogD values were measured by mixing [11C]31 or [11C]37 (radiochemical purity >99%) with n-octanol (3.0 g) and PBS (0.1 M, 3.0 g) in a test tube. Both n-octanol and PBS were presaturated with each other prior to use. The sample was first vortexed for 5 min and then centrifuged (~3500–4000 rpm) for an additional 5 min. PBS and n-octanol were aliquoted and weighed, and the radioactivity in each component was measured using a Cobra Model 5002/5003 autogamma counter. The LogD was determined from the Log of the ratio of radioactivity between the n-octanol and PBS solutions (n = 3).
In vitro autoradiography
The general procedure for in vitro autoradiography was described previously [43, 44] and was used with minor modifications in this work. Briefly, rat brains were cut into 20 mm sections and stored at −80 °C until they were used for the experiment. The rat brain sections were preincubated with Tris·HCl buffer (pH 7.4, 50 mM), MgCl2 (1.2 mM), and CaCl2 (2 mM) solution for 20 min at ambient temperature and then incubated with [11C]31 (5.21 MBq/50 mL, 4.37 × 10−4 μg/mL) or [11C]37 (6.07 MBq/50 mL, 5.46 × 10−4 μg/mL) for 30 min at ambient temperature. For blocking studies, the blocking reagent (two known GluN2B inhibitors, BMT-108908 (10 μM) and (+)-CP101606 (10 μM), and unlabeled 31 or 37 (10 μM)) was added to the incubation solution in advance to determine the specificity of the radioligand binding. After incubation, brain sections were washed twice with Tris buffer for 2 min and dipped in cold distilled water for 10 s. The brain sections were dried with cold air and then placed on imaging plates (BASMS2025, GE Healthcare, NJ, USA) for 60 min. Autoradiograms were obtained, and ROIs were carefully drawn based on observation by the naked eye. The radioactivity is expressed as photostimulated luminescence values per unit area (PSL/mm2), measured by a Bio-Imaging analyzer system (BAS5000, Fujifilm) and normalized to a % of radioactivity vs the control.
PET imaging studies
The general procedure for PET studies was described previously [43, 44] and was used with minor modifications in this work. Briefly, PET scans were carried out with an Inveon PET scanner (Siemens Medical Solutions, Knoxville, TN, USA). Sprague–Dawley male rats were kept under anesthesia with 1%–2% (v/v) isoflurane during the scan. [11C]31 or [11C]37 (37 MBq/100 µL,1.55 μg/mL for [11C]31 and 1.66 μg/mL for [11C]37, respectively) was injected via a preinstalled catheter via the tail vein. A dynamic scan was acquired for 90 min in three-dimensional list mode. For pretreatment studies, unlabeled 31 or 37 (1 mg/kg rat weight) dissolved in 300 μL of saline containing 10% ethanol and 5% Tween® 80 was injected via the pre-embedded tail vein catheter 5 min before the injection of [11C]31 or [11C]37. The PET dynamic images were reconstructed using ASIPro VW software (Analysis Tools and System Setup/Diagnostics Tool, Siemens Medical Solutions). Volumes of interest, including the hippocampus, thalamus, pons, striatum, cerebral cortex, and cerebellum, were placed using ASIPro VM. The radioactivity was decay-corrected, and is expressed as the standardized uptake value. SUV = (radioactivity per mL tissue)/(injected radioactivity) × body weight.
Whole-body ex vivo biodistribution studies
The general procedure for the ex vivo biodistribution studies was described previously [43, 44] and was used with minor modifications in this work. Briefly, a solution of [11C]31 or [11C]37 (1.85 MBq/100 µL) was injected into CD1 mice via the tail vein. These mice (each time point n = 3–4) were sacrificed at 5, 15, 30, and 60 min post radiotracer injection. The major organs, including the whole brain, heart, liver, lungs, spleen, kidneys, small intestine (including contents), muscles, pancreas, stomach, and blood, were quickly harvested and weighed. The radioactivity present in these tissues was measured using a Cobra Model 5002/5003 autogamma counter, and all the radioactivity measurements were automatically decay-corrected based on the half-life of carbon-11. The results are expressed as the percentage of the injected dose per gram of wet tissue (% ID/g).
Radiometabolite analysis
After intravenous injection of [11C]31 through the tail vein, mice (n = 2) were sacrificed at 30 min post injection. Blood samples were collected and centrifuged at 14,000 rpm for 3 min at 4 °C to separate the plasma. The supernatant was collected and added to an ice-cooled test tube containing 100 μL of CH3CN. After vortexing for 10 s, the mixture was centrifuged at 14,000 rpm for 3 min at 4 °C for deproteinization. The supernatant was collected, and the process was repeated until no precipitate was observed when CH3CN was added. The mouse brains were immediately dissected, homogenized with 400 μL of ice-cooled CH3CN, and then centrifuged at 14,000 rpm for 5 min at 4 °C. The supernatant was collected into a test tube containing 100 μL of ice-cooled CH3CN, and the process (vortex and centrifuge) was repeated until no precipitate was observed when CH3CN was added. The resulting supernatants of the plasma and brain samples were mixed with 50 μL of a solution of cold compound 31 and then analyzed on a radio-HPLC system using a Phenomenex C18 column (5 μm, 10 × 250 mm, 5 mL/min, elution: CH3CN/50 mM AcONH4 = 55%/45%). The radioactivity was collected and counted using a 1480 Wizard gamma counter (PerkinElmer, USA). The radioactivity overlapping with 31 in the UV signal corresponded to that of [11C]31 (tR = 6.50 min). The percentage of [11C]31 to the total radioactivity was calculated as (counts for [11C]31/total counts) × 100.
Statistical analysis
Statistical analysis was performed by Student’s two-tailed t test, and asterisks are used to indicate statistical significance: *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P < 0.0001.
Results and discussion
Chemistry
Two GluN2B-selective NAMs, compounds 31 and 37, and their corresponding 11C-labeling precursors were obtained following the synthetic routes in Scheme 1. Amides 24 and 25 were obtained through amidation of 2-bromoacetic acid (23) with the corresponding amines. The SN2 reactions between amides 24 or 25 and 6-bromo-1H-pyrrolo[3,2-b]pyridine (26) in the presence of NaH gave intermediates 27 and 28 in 52% and 61% yields, respectively. Desired standard compound 31 and the corresponding precursor 30 were obtained via palladium-catalyzed cross-coupling reactions between 28 or 27 and substituted aromatic boronic acid 29 in 79% and 61% yields, respectively (Scheme 1a). Unfortunately, an analogous synthetic strategy was not suitable for the preparation of 36 and 37, which bear a thiophene fragment. The existence of N–H functional groups in 27 and 26 hampered their cross-coupling reactions with thiophene borate 33 (Scheme 1c). To our delight, palladium-catalyzed cross-coupling reactions between Boc-protected intermediate 32 and thiophene borate 33 provided key intermediate 35 and Boc-protected product 34 (converted to 35 under the treatment with 3 N aqueous HCl) in high efficiency. Specifically, compound 35 was synthesized from 32 in 66% overall yield over two steps. In all, the final synthesis was completed with the SN2 reactions between 35 and α-bromo amide 25 or 24 to give standard compound 37 and the corresponding precursor 36 in 36% and 29% yields, respectively (Scheme 1b).
Scheme 1.

Synthesis of GluN2B-selective NMDAR antagonists and corresponding precursors. Reagents and conditions: a (i) (1) 1.5 equiv (COCl)2, DCM, DMF, rt, 30 min; (2) 1.2 equiv H2NMe (2 M in THF) for 24 or HNMe2 (2 M in THF) for 25, DCM, 0 °C to rt, 4 h, 23% for 24, 43% for 25; (ii) 1 equiv of 26, 1.5 equiv NaH, DMF, rt, 2 h, 52% for 27, 61% for 28; (iii) 1.1 equiv of 29, 0.2 equiv Pd(dppf)Cl2, 3 equiv Cs2CO3, 1,4-dioxane/H2O = 10/1, 90 °C, 4 h, 61% for 30, 79% for 31; b (iv) 1.2 equiv Boc2O, 1 equiv DMAP, 1 equiv Et3N, DCM, rt, overnight, 82%; (v) 1 equiv 33, 0.2 equiv Pd(dppf)Cl2, 3 equiv Cs2CO3, 1,4-dioxane/H2O = 5/1, 90 °C, overnight; (vi) 20 equiv 3 N HCl, THF, reflux, 1 h, 66% overall for 35 from 32 and 34; (vii) 1.1 equiv 24 or 25, 1.5 equiv NaH, DMF, 0 °C to rt, 10 min, 29% for 36, 36% for 37; c (viii) 1.1 equiv of 33, 0.2 equiv Pd(dppf)Cl2, 3 equiv Cs2CO3, 1,4-dioxane/H2O = 10/1, 90°C, 4 h, 0% for 35; (ix) 1.1 equiv of 33, 0.2 equiv Pd(dppf)Cl2, 3 equiv Cs2CO3, 1,4-dioxane/H2O = 10/1, 90°C, 4 h, 0% for 36; DCM dichloromethane, DMF N,N-dimethylformamide, THF tetrahydrofuran, Pd(dppf)Cl2 [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), Boc2O di-tert-butyl dicarbonate, DMAP 4-dimethylaminopyridine.
Radiochemistry
As shown in Scheme 2, the N-methyl amide moieties of precursors 30 and 36 were selected as the most convenient sites for labeling with [11C]CH3I [46]. Initially, [11C]CH3I was transferred into a reaction vessel containing precursor 30 in a mixed solvent of DMSO and aqueous KOH (45 wt%), followed by heating at 130 °C for 5 min. However, only trace amounts of [11C]31 were obtained by analytic radio-HPLC (radiochemical conversion, RCC < 5%). In our modified method, a mixture of DMSO and aqueous KOH was replaced with presaturated anhydrous DMSO with NaOH powder at rt. The reaction mixture was heated to 80 °C and maintained for 5 min after the completion of the [11C]CH3I transfer (Scheme 2a). The resulting mixture was purified by semiprep radio-HPLC to provide desired ligand [11C]31 in an average of 28% (n = 2) decay-corrected radiochemical yield (RCY) relative to starting [11C]CO2 at the end of the synthesis, and the obtained material showed high radiochemical purity (>99%) and high molar activity (>74 GBq/μmol). Following a similar protocol, [11C]37 was obtained in 32% (n = 2) decay-corrected RCY with a high radiochemical purity (>99%) and high molar activity (>74 GBq/μmol). No radiolysis was observed within 90 min of formulation. The lipophilicities of [11C]31 and [11C]37 were measured through the “shake flask” method [45], and the results indicated that both radiotracers possessed favorable logD values (2.81 ± 0.01 and 3.18 ± 0.03, respectively) for brain penetration [47].
Scheme 2.
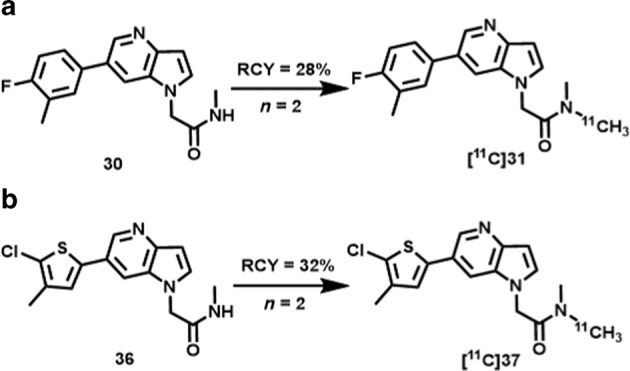
Radiosynthesis of GluN2B-selective radiotracers [11C]31 (a) and [11C]37 (b).
2-(6-(4-Fluoro-3-methylphenyl)-1H-pyrrolo[3,2-b]pyridin-1-yl)-N-(λ1-methyl-11C)-N-methylacetamide ([11C]31): According to the general procedure for 11C-methylation, the retention time for [11C]31 was 10.0 min. The synthesis time was ∼40 min from the end of the bombardment. HPLC purification was completed on a Triart C18 ExRS column (10 mm i.d. × 250 mm) using a mobile phase of CH3CN/50 mM AcONH4 = 45/55 at a flow rate of 4.0 mL/min. The radiochemical and chemical purities were measured by analytical HPLC (Triart C18 column, 4.6 mm i.d. × 250 mm, UV at 254 nm; CH3CN/50 mM AcONH4 = 50/50 at a flow rate of 1.0 mL/min). The decay-corrected radiochemical yield was 27.8% ± 7.2% (n = 2) based on [11C]CO2 with >99% radiochemical purity and greater than 74 GBq/μmol molar activity.
2-(6-(5-Chloro-4-methylthiophen-2-yl)-1H-pyrrolo[3,2-b]pyridin-1-yl)-N-(λ1-methyl-11C)-N-methylacetamide ([11C]37): According to the general procedure for 11C-methylation, the retention time for [11C]37 was 10.2 min. The synthesis time was ∼40 min from the end of the bombardment. HPLC purification was completed on a Triart C18 ExRS column (10 mm i.d. × 250 mm) using a mobile phase of CH3CN/50 mM AcONH4 = 50/50 at a flow rate of 5.0 mL/min. The radiochemical and chemical purities were measured by analytical HPLC (Triart C18 column, 4.6 mm i.d. × 250 mm, UV at 254 nm; CH3CN/50 mM AcONH4 = 50/50 at a flow rate of 1.0 mL/min). The decay-corrected radiochemical yield was 31.6% ± 4.7% (n = 2) based on [11C]CO2 with >99% radiochemical purity and greater than 74 GBq/μmol molar activity.
Autoradiography
With two 11C-labeled radioligands in hand, in vitro autoradiography studies were carried out in brain sections of Sprague–Dawley rats to evaluate the binding specificity of the radiotracers towards the GluN2B subunit (Fig. 2). As shown in Fig. 2a, [11C]31 exhibited a heterogeneous distribution of radioactivity in rat brain sections at the baseline, in which the uptakes decreased in the order of hippocampus, striatum, thalamus, and cortex. The weakest signal was detected in the cerebellum, which is in accordance with the expression pattern of GluN2B subunits in rodents [11–14]. In the blocking studies (Fig. 2b), pretreatment with “cold” (nonradioactive) compound 31 substantially reduced the uptake of [11C]31. Blocking studies with commercially available GluN2B inhibitors, BMT-108908 and (+)-CP101606, showed a similar reduction in bound activity, which confirmed the specific binding of [11C]31 to GluN2B. In in vitro ARG studies of [11C]37 (Fig. 2c, d) showed 34%, 30%, and 37% reduced binding in the hippocampus by pretreatment with 37, BMT-108908 and (+)-CP101606, respectively. In addition, up to a 30% reduced uptake of [11C]37 in the cerebral cortex compared with that of the baseline was detected in the blocking studies. Encouraged by our in vitro experimental results, we performed an in vivo evaluation of these radiotracers by PET imaging studies.
Fig. 2. In vitro autoradiography results of [11C]31 and [11C]37 in rat brain sections.

a [11C]31 (baseline), pretreatment with 31 (self-blocking, 10 μM), BMT-108908 (10 μM) and (+)-CP101606 (10 μM). b Quantitative analysis of the baseline and blocking experiments (n = 4, see quantitative data in Supporting Information Table). c [11C]37 (baseline), pretreatment with 37 (self-blocking, 10 μM), BMT-108908 (10 μM) and (+)-CP101606 (10 μM). d Quantitative analysis of the baseline and blocking experiments (n = 4, see quantitative data in Supporting Information Table). The value is expressed as the ratio of the radioactivity of different brain regions to that of the cerebellum (reference region). Hi hippocampus, Cx cerebral cortex, St striatum, Th thalamus, Cb cerebellum. Asterisks indicate statistical significance: ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, and N.S. = no statistics.
PET study
Dynamic PET scans (0-90 min) were carried out with [11C]31 and [11C]37 in Sprague–Dawley rats. Representative PET images (coronal and sagittal, summed images for [11C]31 in Fig. 3a and [11C]37 in Fig. 3c) in the whole brain and time–activity curves of six representative brain regions (baseline) are shown in Fig. 3b and d, respectively. Unfortunately, both ligands displayed homogenous distributions, inconsistent with the expression pattern of GluN2B subunits in previously reported works [11–14], and the uptakes of [11C]31 and [11C]37 in rat brain were limited to a peak SUV of 0.5–0.7, respectively, at ca. 2 min. Although [11C]31 and [11C]37 possess favorable logD values (2.81 ± 0.01 and 3.18 ± 0.03, respectively) for brain penetration, high plasma protein binding (reported as 95.53% for 31 and 96.68% for 37 in rats [42]) could potentially reduce the available radiotracers in the plasma that can traverse the BBB. The self-blocking studies by pretreatment with the corresponding “cold” compounds showed no substantial reductions in brain uptake, indicating low levels of binding specificity (Supporting Information).
Fig. 3. Representative PET images and time–activity curves of [11C]31 and [11C]37 in rat brain.

a Baseline PET images (summed 0–20 min, 20-40 min, 40–60 min, and 60–90 min) of [11C]31. b Time–activity curves in different brain regions at baseline. c Baseline PET images (summed 0–20 min, 20-40 min, 40–60 min, and 60–90 min) of [11C]37. d Time–activity curves in different brain regions at baseline.
Biodistribution and radiometabolite analysis
The whole-body ex vivo biodistributions of [11C]31 and [11C]37 were assessed at four different time points (5, 15, 30, and 60 min) post radiotracer injection (Fig. 4; Supporting Information Tables 1 and 2). An initial high uptake of [11C]31 (>3% ID/g) was detected in multiple organs, including the heart, lungs, spleen, pancreas, liver, kidneys, stomach, and small intestine, at 5 min. A similar uptake pattern was observed for ligand [11C]37. While [11C]31 exhibited relatively low uptake (<2% ID/g) in the brain and low brain/plasma (B/P) ratio (<0.5) at 5 min, high brain uptake (>4% ID/g), and B/P ratio (>1) of [11C]37 were observed. In general, the radioactivity of bound [11C]31 and [11C]37 in the spleen, heart, lungs, pancreas, kidneys, and liver washed out rapidly from 5 to 60 min. It is worth noting that uptake in the stomach and small intestine remained high (>3% ID/g) until 60 min post injection. A radiometabolism study of [11C]31 in mice (n = 2) was also performed. Only 5.0% ± 0.4% and 2.6% ± 0.2% of the parent fraction of compound [11C]31 were found in the brain and plasma extracts at 30 min post injection (see Supplementary Fig. S4, Supplementary Information). Therefore, rapid in vivo metabolism of radioligand [11C]31 may be responsible for the moderate brain uptake and low specific binding.
Fig. 4. Whole-body ex vivo biodistribution in mice at four different time points (5, 15, 30, and 60 min, n = 3–4) post injection.

a [11C]31 and b [11C]37. Asterisks indicate statistical significance: ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05.
In summary, two GluN2B-targeted PET ligands bearing key N,N-dimethyl-2-(1H-pyrrolo[3,2-b]pyridin-1-yl)acetamide scaffolds (compounds [11C]31 and [11C]37, also called N2B-1810 and N2B-1903, respectively) and their corresponding 11C-labeling precursors were efficiently synthesized and characterized. The 11C-methylation labeling strategy was successfully applied and provided the two radiotracers with good RCYs (decay-corrected 28%–32%), high radiochemical purities (>99%) and high molar activities (>74 GBq/μmol). While the two radiotracers failed to demonstrate high in vivo-specific binding to GluN2B, probably because of rapid metabolism and limited brain permeability, the in vitro specificity of [11C]31 in autoradiography studies highlights its potential as a chemotype for the further development of GluN2B PET radiotracers.
Acknowledgements
We thank Drs. Thomas J. Brady and Lei Zhang for their helpful discussion. This work was financially supported by NSFC (No. 81901802, No. 81701751, No. 81871383), Postdoctoral Fund of the First Affiliated Hospital, Ji-nan University (No. 801328) to JYS, the Science and Technology Program of Guangzhou (201804010440), and the Project of Innovative Team for the Guangdong Universities (2018KCXTD001).The characterization of small molecules in this work was performed (in part) using the JEOL 500 MHz NMR Spectrometer that was purchased with funding from a National Institutes of Health SIG grant (S10OD025234).
Supporting information
Author contributions
HX, MRZ, and SHL designed the research; JYS, KK, ZC, YDZ, JHC, AH, HLF, JR, XYD, TY, LX, KH, MF, QZY, TS, and TLC performed research; JYS analyzed the data; JYS and SHL wrote the paper; LJ, YHS, YFD, LW, HX, MRZ, and SHL reviewed the paper.
Competing interests
The authors declare no competing interests.
Footnotes
Thease authors contributed equally: Ji-yun Sun, Katsushi Kumata
Contributor Information
Hao Xu, Email: txh@jnu.edu.cn.
Ming-rong Zhang, Email: zhang.ming-rong@qst.go.jp.
Steven H Liang, Email: liang.steven@mgh.harvard.edu.
Supplementary information
The online version of this article (10.1038/s41401-020-0456-9) contains supplementary material, which is available to authorized users.
References
- 1.Fu H, Chen Z, Josephson L, Li Z, Liang SH. Positron emission tomography (PET) ligand development for ionotropic glutamate receptors: challenges and opportunities for radiotracer targeting N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA), and kainate receptors. J Med Chem. 2019;62:403–19. doi: 10.1021/acs.jmedchem.8b00714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–96. [DOI] [PMC free article] [PubMed]
- 3.Fuchigami T., Nakayama M., Yoshida S. Development of PET and SPECT probes for glutamate receptors. Sci World J. 2015;2015:716514. [DOI] [PMC free article] [PubMed]
- 4.Majo VJ, Prabhakaran J, Mann JJ, Kumar JS. PET and SPECT tracers for glutamate receptors. Drug Discov Today. 2013;18:173–84. doi: 10.1016/j.drudis.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez J, Jurado-Coronel JC, Avila MF, Sabogal A, Capani F, Barreto GE. NMDARs in neurological diseases: a potential therapeutic target. Int J Neurosci. 2015;125:315–27. [DOI] [PubMed]
- 6.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu S, Paoletti P. Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr Opin Pharmacol. 2015;20:14–23. doi: 10.1016/j.coph.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–20. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karakas E, Simorowski N, Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–53. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mony L, Zhu S, Carvalho S, Paoletti P. Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J. 2011;30:3134–46. doi: 10.1038/emboj.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–40. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 12.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–7. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 13.Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994;347:150–60. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport. 1992;3:1138–40. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- 15.Chazot PL. The NMDA receptor NR2B subunit: a valid therapeutic target for multiple CNS pathologies. Curr Med Chem. 2004;11:389–96. doi: 10.2174/0929867043456061. [DOI] [PubMed] [Google Scholar]
- 16.Ogden KK, Traynelis SF. New advances in NMDA receptor pharmacology. Trends Pharmacol Sci. 2011;32:726–33. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5:1039–42. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- 18.Mony L, Kew JN, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol. 2009;157:1301–17. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurons. J Physiol. 1996;497(Pt 3):761–72. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stroebel D, Buhl DL, Knafels JD, Chanda PK, Green M, Sciabola S, et al. A novel binding mode reveals two distinct classes of NMDA receptor GluN2B-selective antagonists. Mol Pharmacol. 2016;89:541–51. [DOI] [PMC free article] [PubMed]
- 21.Burger PB, Yuan H, Karakas E, Geballe M, Furukawa H, Liotta DC, et al. Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol Pharmacol. 2012;82:344–59. [DOI] [PMC free article] [PubMed]
- 22.Lee CM, Farde L. Using positron emission tomography to facilitate CNS drug development. Trends Pharmacol Sci. 2006;27:310–6. doi: 10.1016/j.tips.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Fowler JS, Wolf AP. Working against time: rapid radiotracer synthesis and imaging the human brain. Acc Chem Res. 1997;30:181–8. doi: 10.1021/ar960068c. [DOI] [Google Scholar]
- 24.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Disco. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Yang Y, Sun M, Cui M, Fu Yi, Lin Y, et al. Highly specific noninvasive photoacoustic and positron emission tomography of brain plaque with functionalized croconium dye labeled by a radiotracer. Chem Sci. 2017;8:2710–6. [DOI] [PMC free article] [PubMed]
- 26.Yu Q, Huang S, Wu Z, Zheng J, Chen X, Nie L. Label-free visualization of early cancer hepatic micrometastasis and intraoperative image-guided surgery by photoacoustic imaging. J Nucl Med. 2019; 10.2967/jnumed.119.233155. [DOI] [PMC free article] [PubMed]
- 27.Hong H, Zhang L, Xie F, Zhuang R, Jiang D, Liu H, et al. Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors. Nat Commun. 2019;10:989–96. [DOI] [PMC free article] [PubMed]
- 28.Chazot PL. CP-101606 Pfizer Inc. Curr Opin Investig Drugs. 2000;1:370–4. [PubMed] [Google Scholar]
- 29.Yurkewicz L, Weaver J, Bullock MR, Marshall LF. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J Neurotrauma. 2005;22:1428–43. doi: 10.1089/neu.2005.22.1428. [DOI] [PubMed] [Google Scholar]
- 30.Fuchigami T, Fujimoto N, Haradahira T, Nojiri Y, Okauchi T, Maeda J, et al. Synthesis and characterization of 11C‐labeled benzyl amidine derivatives as PET radioligands for GluN2B subunit of the NMDA receptors. J Label Compd Radiopharm. 2018;61:1095–105. [DOI] [PubMed]
- 31.Ibrahim L, Diaz Granados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, et al. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32:551–7. [DOI] [PMC free article] [PubMed]
- 32.Garner R, Gopalakrishnan S, McCauley JA, Bednar RA, Gaul SL, Mosser SD, et al. Preclinical pharmacology and pharmacokinetics of CERC-301, a GluN2B-selective N-methyl-D-aspartate receptor antagonist. Pharmacol Res Perspect. 2015;3:e00198. [DOI] [PMC free article] [PubMed]
- 33.Jakobsson JE, Gourni E, Khanapur S, Brito B, Riss PJ. Synthesis and characterization in rodent brain of the subtype-selective NR2B NMDA receptor ligand [11C]Ro04-5595 as a potential radiotracer for positron emission tomography. ACS Omega. 2019;4:9925–31. doi: 10.1021/acsomega.9b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Aart J, Yaqub M, Kooijman EJM, Bakker J, Langermans JAM, Schuit RC, et al. Evaluation of the novel PET tracer [11C]HACH242 for imaging the GluN2B NMDA receptor in non-human primates. Mol Imaging Biol. 2019;21:676–85. [DOI] [PubMed]
- 35.Kramer SD, Betzel T, Mu L, Haider A, Herde AM, Boninsegni AK, et al. Evaluation of 11C-Me-NB1 as a potential PET radioligand for measuring GluN2B-containing NMDA receptors, drug occupancy, and receptor cross talk. J Nucl Med. 2018;59:698–703. [DOI] [PubMed]
- 36.Szermerski M, Borgel F, Schepmann D, Haider A, Betzel T, Ametamey SM, et al. Fluorinated GluN2B receptor antagonists with a 3-benzazepine scaffold designed for PET studies. ChemMedChem . 2018;13:1058–68. [DOI] [PubMed]
- 37.Ahmed H, Haider A, Varisco J, Stanković M, Wallimann R, Gruber S, et al. Structure–affinity relationships of 2,3,4,5-tetrahydro-1H-3-benzazepine and 6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amine analogues and the discovery of a radiofluorinated 2,3,4,5-tetrahydro-1H-3-benzazepine congener for imaging GluN2B subunit-containing N-methyl-D-aspartate receptors. J Med Chem. 2019;62:9450–70. [DOI] [PubMed]
- 38.Haider A, Herde AM, Krämer SD, Varisco J, Keller C, Frauenknecht K, et al. Preclinical evaluation of benzazepine-based PET radioligands (R)- and (S)-11C-Me-NB1 reveals distinct enantiomeric binding patterns and a tightrope walk between GluN2B- and σ1-receptor–targeted PET imaging. J Nucl Med. 2019;60:1167–73. [DOI] [PMC free article] [PubMed]
- 39.Haider A, Iten I, Ahmed H, Herde AM, Gruber S, Krämer SD, et al. Identification and preclinical evaluation of a radiofluorinated benzazepine derivative for imaging the GluN2B subunit of the ionotropic NMDA receptor. J Nucl Med. 2019;60:259–66. [DOI] [PMC free article] [PubMed]
- 40.Cai L, Liow JS, Morse CL, Telu S, Davies R, Frankland MP, et al. Evaluation of 11C-NR2B-SMe and its enantiomers as PET radioligands for imaging the NR2B subunit within the NMDA receptor complex in rats. J Nucl Med. 2020. 10.2967/jnumed.119.235143. [DOI] [PMC free article] [PubMed]
- 41.Fu H, Tang W, Chen Z, Belov VV, Zhang G, Shao T, et al. Synthesis and preliminary evaluations of a triazole-cored antagonist as a PET imaging probe ([18F]N2B-0518) for GluN2B subunit in the brain. ACS Chem Neurosci. 2019;10:2263–75. [DOI] [PMC free article] [PubMed]
- 42.Chrovian CC, Soyode-Johnson A, Wall JL, Rech JC, Schoellerman J, Lord B, et al. 1H-Pyrrolo[3,2-b]pyridine GluN2B-selective negative allosteric modulators. ACS Med Chem Lett. 2019;10:261–6. [DOI] [PMC free article] [PubMed]
- 43.Cheng R, Mori W, Ma L, Alhouayek M, Hatori A, Zhang Y, et al. In vitro and in vivo evaluation of 11C-labeled azetidinecarboxylates for imaging monoacylglycerol lipase by PET imaging studies. J Med Chem. 2018;61:2278–91. [DOI] [PMC free article] [PubMed]
- 44.Zhang X, Kumata K, Yamasaki T, Cheng R, Hatori A, Ma L, et al. Synthesis and preliminary studies of a novel negative allosteric modulator, 7-((2,5-dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro-4-[11C]methoxyphenyl) quinoline-2-carboxamide, for imaging of metabotropic glutamate receptor 2. ACS Chem Neurosci. 2017;8:1937–48. [DOI] [PMC free article] [PubMed]
- 45.Organisation for Economic Co-operation and Development (OECD). Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method, OECD Guidelines for the Testing of Chemicals, Section 1, OECD Publishing, Paris, 1995. 10.1787/9789264069626-en.
- 46.Deng X, Rong J, Wang L, Vasdev N, Zhang L, Josephson L, et al. Chemistry for positron emission tomography: recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions. Angew Chem Int Ed Engl. 2019;58:2580–605. [DOI] [PMC free article] [PubMed]
- 47.Waterhouse R. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376–89. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]