

Abstract
Myeloid cells, such as neutrophils, are produced in the bone marrow in high quantities and are important in the pathogenesis of vascular diseases such as pulmonary hypertension (PH). Although neutrophil recruitment into sites of inflammation has been well studied, the mechanisms of neutrophil egress from the bone marrow are not well understood. Using computational flow cytometry, we observed increased neutrophils in the lungs of patients and mice with PH. Moreover, we found elevated levels of IL-6 in the blood and lungs of patients and mice with PH. We observed that transgenic mice overexpressing Il-6 in the lungs displayed elevated neutrophil egress from the bone marrow and exaggerated neutrophil recruitment to the lungs, resulting in exacerbated pulmonary vascular remodeling, and dysfunctional hemodynamics. Mechanistically, we found that IL-6-induced neutrophil egress from the bone marrow was dependent on interferon regulatory factor 4 (IRF-4)-mediated CX3CR1 expression in neutrophils. Consequently, Cx3cr1 genetic deficiency in hematopoietic cells in Il-6-transgenic mice significantly reduced neutrophil egress from bone marrow and decreased neutrophil counts in the lungs, thus ameliorating pulmonary remodeling and hemodynamics. In summary, these findings define a novel mechanism of IL-6-induced neutrophil egress from the bone marrow and reveal a new therapeutic target to curtail neutrophil-mediated inflammation in pulmonary vascular disease.
Keywords: neutrophil, IL-6, pulmonary hypertension, CX3CR1, inflammation
Subject terms: Inflammation, Chemokines, Interleukins
Introduction
Pulmonary hypertension (PH) is a progressive cardiopulmonary disease characterized by elevated mean pulmonary arterial pressure at rest that leads to right ventricular failure, multiorgan dysfunction, and often death. The vascular manifestations of PH are associated with cellular and soluble inflammatory mediators.1,2 Inflammation plays an important role in the establishment of PH.3,4 We have recently shown that in PH, circulating monocytes are recruited to the lungs, differentiate into inflammatory interstitial macrophages, and participate in local lung inflammation and vascular remodeling.5,6 Notably, two molecules, soluble interleukin-6 (IL-6)7,8 and the fractalkine receptor C-X3-C motif chemokine receptor 1 (CX3CR1),6,9,10 which is expressed on the surface of monocytes and macrophages, have been found to be central effectors that regulate the recruitment of these cells to vascular sites of inflammation in PH,11–13 as well as other diseases.14,15
Neutrophils are a vital myeloid subset that are important in vascular remodeling16–18 and are the first cells to infiltrate sites of inflammation.19,20 The neutrophil/lymphocyte ratio in the peripheral blood is a marker of subclinical inflammation and is associated with poor prognosis in patients with pulmonary arterial hypertension (PAH)21,22 and other diseases.16,23,24 Once recruited to sites of inflammation, neutrophils create neutrophil extracellular traps and exacerbate angiogenesis25 and overall PH pathogenesis through different mechanisms, such as myeloperoxidase and elastase production.25–28
Certain aspects of the role of IL-6 in neutrophil trafficking are well documented.29,30 Neutrophil-mediated inflammation, particularly in vascular diseases such as PH, consists of at least three prerequisite steps: (a) exaggerated neutrophil production in the bone marrow or spleen, (b) neutrophil release from the sites of their production into the blood, and (c) the recruitment of neutrophils at sites of injury from the blood.29 Neutrophil production31,32 and recruitment at the sites of inflammation33,34 have been thoroughly studied. In contrast to our relatively advanced understanding of neutrophil production and recruitment, the molecular control of neutrophil egress from the bone marrow into the blood circulation in various vascular disease conditions has been largely understudied. Furthermore, while neutrophils express CX3CR1,35 any neutrophil-specific functions of this molecule have yet to be defined. In PAH, both the present study and published studies13,36,37 suggest that IL-6 is deleterious in PH pathogenesis. Using comprehensive molecular analyses of samples from PAH patients and genetically engineered rodent models of PH, our data identified IL-6 as a crucial trigger that promotes neutrophil egress, which is a process that is dependent upon CX3CR1. Consequently, by defining a crucial mechanism of neutrophil mobilization, this study identified multiple therapeutic targets that are specifically involved in neutrophil-mediated inflammation in pulmonary vascular disease.
Results
Neutrophils are increased in the blood and lungs of hypoxic mice and PH patients
To enumerate neutrophils in the blood of patients suffering from PAH, we used multicolor flow cytometry (Fig. S1A). The number and percentage of neutrophils in PAH blood were increased compared to those in healthy controls (Figs. 1A, S1B, Table S1). However, no significant difference in neutrophil numbers or frequencies was observed between idiopathic and scleroderma-associated PAH patients (Table S1, Fig. S1C). These data were confirmed by computational flow analysis of leukocyte populations in PAH blood vs. control blood (Figs. 1B, C, S2, S3). Furthermore, flow cytometry showed that the frequency and numbers of neutrophils were increased in samples representing multiple subtypes of PH, as defined by the World Symposium on Pulmonary Hypertension (WSPH)38 (Fig. S4). Of note, Group 3 PH patients suffering from lung disease displayed, on average, the highest levels of neutrophils in the blood. Circulating neutrophils in PAH patients consisted of at least three subpopulations phenotypically characterized by the expression of CD16, CD24, and CD14 (Figs. 1D, S5A). These three neutrophil populations were identified as CD16hi CD24lo (Neutro 1), CD16hi CD24hi CD14lo (Neutro 2), and CD16hi CD24hi CD14hi (Neutro 3) (Fig. S5A). All three populations were expanded in frequency and in numbers in the blood of PH patients compared to controls (Fig. S5B). Additionally, varying levels of elastase were found in the three populations of neutrophils (Fig. S5C). Furthermore, these three subpopulations had different inflammatory properties, as shown by gene expression analysis (Fig. S5D). Neutrophil Group 1 (Neutro 1) expressed the highest levels of the chemokine receptors CXCR1, CXCR2, and CXCR4 and cytokines such as IFNB, IL1B, LTB4R, and MPO. Spanning tree progression analysis confirmed the heterogeneity of neutrophils in the blood (Figs. 1E, S6A). These three neutrophil subpopulations were morphologically heterogeneous (Fig. S6B), with neutrophil group 1 exhibiting the lowest number of nuclear lobules and the highest nucleus-to-cytoplasm ratio (Fig. S6C).
Fig. 1.

The number of neutrophils is increased in the blood and lungs of PAH patients and hypoxic mice. Lungs and blood were collected from PAH patients and healthy controls (n = 5 per group). A Quantification of neutrophils in patient blood. Computational flow cytometric analysis of the expression of different cell surface markers on blood leukocytes of patients was performed. B The bh-SNE plot shows various leukocyte populations in the blood of control patients. C The bh-SNE plots depict the abundance of leukocyte populations in the blood of control and PAH patients. D The circled gate in the PhenoGraph defines the neutrophil subpopulations. The different cell populations are color coded. E Spanning-tree progression analysis of density-normalized events (SPADE) was performed to determine the heterogeneity of circulating leukocytes in PAH patients. The size of the dots represents the relative abundance of a given cell population. F Quantification of neutrophils in the lungs of PAH patients versus controls was performed by flow cytomertry. G The number of lung neutrophils in C57BL/6 mice placed in chronic hypoxia (10% O2) or normoxia (n = 5 per group) to induce PH was quantified by flow cytometry. H The bh-SNE plot shows various leukocyte populations in the lungs of normoxic and hypoxic mice. I The bh-SNE plot shows the abundance of leukocyte populations in the lungs of normoxic and hypoxic mice. J The circled gate in the PhenoGraph defines the neutrophil population among other leukocyte populations. K Lung remodeling scores and neutrophil frequencies were quantified using confocal microscopy. Arrows indicate lung vasculature-infiltrating neutrophils. The data are shown as the mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001
Flow cytometry also revealed increased neutrophil accumulation in the lungs of PH patients (Figs. 1F, S7A, B, Supplementary Table 1) and mice with chronic hypoxia-induced PH (Figs. 1G–K, S8A, B). Of note, the 3-week hypoxia time point was found to be associated with the maximum number and frequency of blood and lung-infiltrating neutrophils, as shown by a time course experiment (Fig. S9A, B). These data are congruent with those of ours and others that revealed increased numbers of other myeloid cells, such as monocytes, in the lungs of hypoxic mice starting at day 3 and accumulating up to day 21 of hypoxia exposure.5,6 Similar to circulating neutrophils, we found three neutrophil populations in the lungs of PH patients (Fig. S10A). qPCR showed that these three neutrophil subpopulations expressed varying levels of CX3CR1 (Fig. S10B), with Neutro 1 cells expressing the highest levels. All three neutrophil subpopulations demonstrated increased CX3CR1 expression in PH. In summary, these data suggest increased mobilization of neutrophils from the bone marrow into blood circulation and recruitment to the lungs in PH.
IL-6 levels are elevated in the blood and lungs of hypoxic mice and PH patients
To determine the molecular cues responsible for neutrophil egress from the bone marrow and their subsequent infiltration in the lungs, we measured the levels of inflammatory cytokines and chemokines in the blood and lungs of PAH patients and mice with PH. We observed significantly increased Il-6 mRNA and protein levels in the lungs of hypoxic mice (Fig. 2A) and PAH patients (Fig. 2B) compared to controls. Additionally, elevated levels of IL-6 were found in the blood of hypoxic mice and PAH patients (Fig. 2C). A time course experiment revealed that IL-6 mRNA and protein levels peaked in mice at days 3 and 12 of hypoxia, respectively (Fig. S11).
Fig. 2.
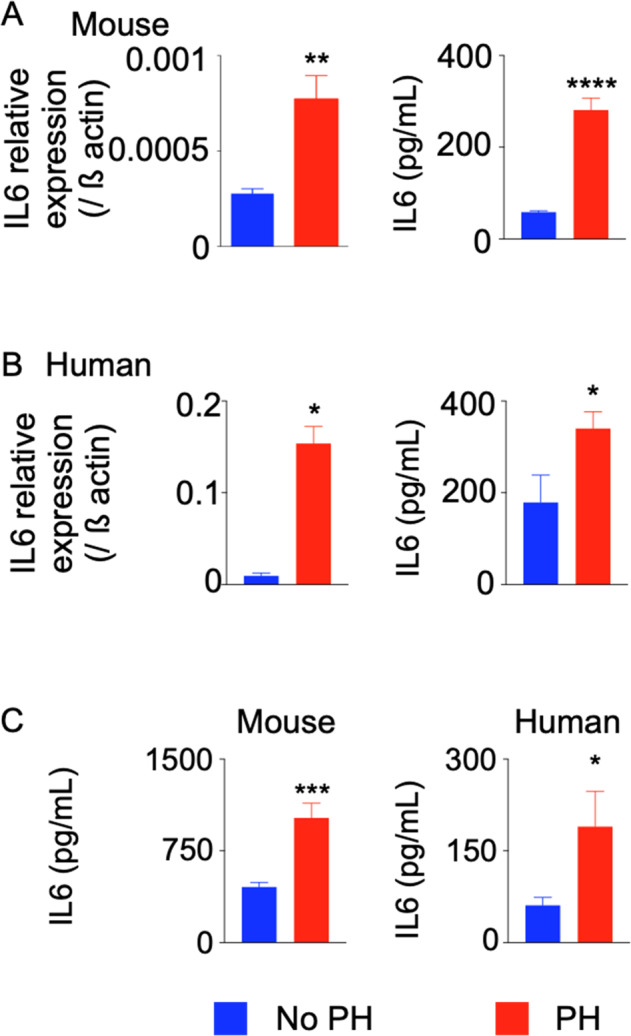
IL-6 levels are increased in patients and mice with PH. C57BL/6 mice were placed in a hypoxic chamber (10% O2) (n = 5 per group) for 3 weeks to induce PH. Lungs were collected from PAH patients and healthy controls (n = 5 for PAH patients and n = 4 for healthy controls). IL6 expression in the lungs of mice (A) (n = 6 per group) and patients (B) (n = 7–8 per group) was assessed by RT-qPCR and ELISA. C IL-6 protein levels in the blood of PAH patients and hypoxic mice were quantified by ELISA. The data are shown as the mean ± s.e.m. *P < 0.05; **P < 0.01; ****P < 0.001
Il-6 overexpression in mice triggers neutrophilia and worsens PH pathogenesis
To investigate the causative role of IL-6 in neutrophil egress from the bone marrow, we used a transgenic mouse strain that expresses pulmonary-specific Il-6, leading to secondary systemic increases in Il-6.36 Flow cytometric analysis revealed substantial increases in neutrophil numbers and frequencies in the lungs of these transgenic mice (Figs. 3A, S12A, B), which was confirmed by confocal imaging (Fig. 3B). As previously reported,36 these mice displayed increased pulmonary vascular histologic remodeling (Fig. 3B), as well as worsened hemodynamic manifestation of PH, as evidenced by increased right ventricular systolic pressure (RVSP) and right ventricular remodeling (RV/(LV+S) mass ratio or the Fulton index) (Fig. 3C). Additionally, Il-6-transgenic mice displayed increased numbers and frequencies of neutrophils in the blood (Figs. 3D, S12C). Moreover, significant decreases were observed in neutrophil numbers and frequencies in the bone marrow of these mice compared to wild-type control mice (Figs. 3E, S12D), indicating accelerated egress of this granulocyte subset from the bone marrow. Collectively, these data indicate that IL-6 is one of the driving forces of neutrophil egress from the bone marrow and recruitment to the lungs in PH.
Fig. 3.

Il6 overexpression in pneumocytes triggers neutrophilia in the blood and lungs. Il6-transgenic (Il6tg) and C57BL/6 wild-type (WT) mice were placed in a hypoxic chamber (10% O2) (n = 5 per group) for 3 weeks to induce PH. A The number of neutrophils in the lungs of hypoxic mice was quantified by flow cytometry. B The percentage of neutrophils among DAPI-stained cells and the vasculature remodeling scores of the lungs of hypoxic mice were quantified by confocal microscopy. Arrows indicate lung vasculature-infiltrating neutrophils. C Right ventricular systolic pressure and the Fulton index (RV/LV + S mass ratio) were assessed. The number of neutrophils was also assessed in the blood (D) and bone marrow (E) of hypoxic mice by flow cytometry. The data are shown as the mean ± s.e.m. *P < 0.05; **P < 0.01; ****P < 0.001
Fractalkine receptor (CX3CR1) is necessary for neutrophil egress from the bone marrow
To understand the mechanisms of IL-6-driven neutrophil egress from the bone marrow, we measured the expression of CX3CR1, a chemokine receptor that is important for monocyte recruitment into inflamed tissues and PH,6 in neutrophils. To measure Cx3cr1 expression in neutrophils, we used Cx3cr1 reporter (Cx3cr1GFP/+) mice and measured the GFP mean fluorescence intensity by flow cytometry. We observed that neutrophils expressed Cx3cr1 (Fig. 4A). Computational flow cytometric analysis of lung leukocytes demonstrated that neutrophils (Ly-6ghigh, left panel of Fig. 4B) expressed Cx3cr1 (right panel) at a lower level than monocytes. Additionally, hypoxic mice exhibited increased numbers of Cx3cr1high neutrophils in the lungs after 3 weeks of hypoxia (Fig. 4C). Correspondingly, we found increased proportions of Cx3cr1high neutrophils in the lungs of Il-6-transgenic mice compared to wild-type control mice by confocal imaging (Fig. 4D). Thus, these data support the notion that Cx3cr1-expressing neutrophils may preferentially exit the bone marrow in PH.
Fig. 4.

Neutrophils require CX3CR1 for egress from the bone marrow. Wild-type and Cx3cr1GFP/+ mice were placed in a hypoxic chamber for 3 weeks (n = 5 per group). The expression of Cx3cr1 in bone marrow neutrophils from these mice was evaluated by flow cytometry (A) and computational analyses (B). The left and right bh-SNE plots show the expression of Ly-6G and Cx3cr1 in bone marrow leukocytes, respectively. Total and Cx3cr1high neutrophils in the lungs of wild-type mice exposed to normoxia (No PH) or hypoxia (PH) were enumerated by flow cytometry (C) and wild-type and Il6 transgenic (Il6tg) mice exposed to hypoxia were examined using confocal microscopy (D). Arrows indicate Cx3cr1+ neutrophils. E Lethally irradiated Cx3cr1+/+ or Cx3cr1−/− mice were transplanted with bone marrow isolated from Cx3cr1+/+ or Cx3cr1−/− mice. Four months after transplantation, the chimeric mice were placed in hypoxic chambers for 3 weeks (10% O2) (n = 10 per group). The numbers and percentages of neutrophils among myeloid cells in the blood (F) and bone marrow (G) were assessed by flow cytometry. H Schematic showing the strategy to generate mixed bone marrow chimeras by transplanting Cx3cr1−/− mice with Cx3cr1+/+ bone marrow (n = 10 per group). I Bar graph showing the ratio of blood and bone marrow neutrophils of either donor or host origin. The data are shown as the mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.005
To further investigate the role of Cx3cr1 in neutrophil migration from the bone marrow, we generated mice that lacked Cx3cr1 in hematopoietic cells by bone marrow transplantation of wild-type mice with Cx3cr1-null cells, followed by the induction of PH by chronic hypoxia exposure (Fig. 4E). We observed that global or hematopoietic-specific Cx3cr1-deficient mice exhibited decreased frequencies and numbers of neutrophils in the blood compared to those of mice with Cx3cr1-null hematopoietic cells (Fig. 4F). Additionally, the frequency and number of bone marrow neutrophils in Cx3cr1-deficient mice significantly increased, but the ratio of blood to bone marrow neutrophils in these mice substantially decreased (Fig. 4G). To examine neutrophil egress more specifically, we generated mixed chimeric mice by transferring Cx3cr1+/+ bone marrow cells into sublethally irradiated Cx3cr1−/− mice (Fig. 4H) and exposed these mice to hypoxia for 3 weeks. The frequency of donor-derived neutrophils (Cx3cr1+/+) in the blood was significantly higher than that of recipient-derived neutrophils lacking this chemokine receptor (Fig. 4I). Collectively, these data strongly indicate that neutrophils express Cx3cr1, which is necessary for their egress from the bone marrow in hypoxic PH.
IL-6 increases neutrophil-specific CX3CR1 expression via IRF-4
We found that Cx3cr1 mRNA expression was increased in the lungs of Il-6-transgenic mice compared to wild-type control mice (Fig. 5A). Additionally, the levels of Cx3cr1 protein in the bone marrow neutrophils of Il-6-transgenic mice expressing GFP under the Cx3cr1 promoter were increased compared to those of control mice (Fig. 5B), suggesting a role of Il-6 in increasing Cx3cr1 expression. To determine a causative link between these molecules, we differentiated HL-60 cells, a human promyeloblast cell line, into neutrophils and exposed these cells to recombinant IL-6 (Fig. S13A). Consistent with the increased Cx3cr1 expression in neutrophils in Il-6-transgenic mice, increased doses of IL-6 increased CX3CR1 expression at the mRNA and protein levels in the human neutrophil cell line (Fig. 5C), demonstrating that IL-6 augments CX3CR1 expression. These data indicate that neutrophils are capable of responding to IL-6-mediated signaling. To this end, we quantified pSTAT3 and pJAK, which are key factors in IL-6-mediated signaling, especially in cancer biology,39,40 in HL-60 cells treated with IL-6 (Fig. 5D) and pulmonary perivascular neutrophils from mice (Fig. 5E, F) and patients (Fig. 5G) with PH. We observed increased pSTAT3 and JAK expression in these cells.
Fig. 5.

IL-6 increases CX3CR1 expression in neutrophils by increasing IRF4 expression. Il6-transgenic (Il6tg) and wild-type mice were placed in a hypoxic chamber (10% O2) (n = 5 per group) for 3 weeks to induce PH. A Relative Cx3cr1 expression in the lungs was quantified by RT-qPCR and standardized to beta actin expression. B Histogram and bar graph showing the expression of Cx3cr1 in wild-type and Il-6tg mice expressing GFP under the Cx3cr1 promoter. C Differentiated HL-60 cells were treated with various concentrations of recombinant IL-6 (0, 100, and 400 ng/mL). CX3CR1 expression was determined by RT-qPCR and flow cytometry (n = 5 per group). One of three representative experiments is shown. pSTAT3 and JAK levels in HL-60 cells treated with IL-6 (D) and neutrophils from the lungs of mice (E, F) and patients (G) with PH were quantified using immunoblotting and confocal microscopy. H Ingenuity pathway analysis (IPA) software was used to identify possible transcription factors that mediate IL-6-induced CX3CR1 expression. I, J Differentiated HL-60 cells were treated with recombinant IL6 (400 ng/mL) and control or IRF4 siRNA (n = 5 per group). A Representative experiment is shown. IRF4 expression was determined by RT-qPCR (I). CX3CR1 expression was determined by RT-qPCR (J) and flow cytometry (K). L Bar graph showing IRF4 expression in HL-60 cells treated with siControl or siSTAT3 and recombinant IL6. The data are shown as the mean ± s.e.m., *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001
Ingenuity pathway analysis suggested that Il-6 was associated with Cx3cr1 through two different transcription factors: Stat1 and Irf4 (Fig. 5H). We showed that exposure of differentiated HL-60 cells to IL-6 had a negligible effect on the activation of STAT-1 (p-STAT1) (Fig. S13B). Therefore, we concluded that the regulation of CX3CR1 by IL-6 must occur through a different transcription factor, such as IRF4. Consistently, IL-6 treatment induced the expression of IRF4 in HL-60 cells (Fig. 5I). To discern whether IL-6-induced CX3CR1 expression in neutrophils was IRF4-dependent, we treated differentiated human neutrophils with siRNA against IRF4 and exposed these cells to recombinant IL-6. siIRF4 efficiently inhibited IRF4 expression (Fig. S13C). IL-6-mediated CX3CR1 expression was abolished by siIRF4 treatment (Fig. 5J, K). Ingenuity pathway analysis indicated that STAT3 mediates IL-6-induced IRF4 upregulation. Consistently, knocking down STAT3 expression in HL-60 cells treated with recombinant IL-6 resulted in decreased IRF4 expression in these cells (Fig. 5L). Taken together, these data indicate that IL-6 increases CX3CR1 expression by activating IRF4 expression in neutrophils.
Cx3cr1 deficiency ameliorates IL-6-driven PH
Since Cx3cr1 deficiency decreased neutrophil egress from the bone marrow (Fig. 4) and neutrophils are known to drive PH pathogenesis,2,25,41 we sought to determine whether Cx3cr1 was essential for mediating the effects of IL-6 on PH. Thus, we generated Cx3cr1-deficient Il-6-transgenic mice. When exposed to hypoxia, these mice exhibited significantly fewer neutrophils in the lungs and blood than Il-6-transgenic mice expressing Cx3cr1 (Fig. 6A). Conversely, bone marrow neutrophil levels increased (Fig. 6A), and the proportions of neutrophils in the lungs, blood and bone marrow followed a similar pattern (Fig. S14). These data demonstrate that the absence of Cx3cr1 limits the recruitment of neutrophils to the lungs by increasing their retention in the bone marrow. Hemodynamic parameters of PH, such as elevated RVSP and the Fulton index (Fig. 6B), as well as pulmonary vascular histologic remodeling (Fig. 6C), were also attenuated in Cx3cr1-deficient Il-6-transgenic mice. These alterations were accompanied by decreases in proinflammatory cytokines, such as Ifnb, Il-1b, and Il-18, in the lungs (Fig. 6D). To assess the impact of Cx3cr1 deficiency on neutrophil recruitment in mice that do not overexpress Il6 in pneumocytes, we used Cx3cr1−/− mice. These mice exhibited reduced neutrophil recruitment to the lungs compared that of to Cx3cr1+/+ mice (Fig. S15). This result is consistent with the observation that Cx3cr1 deficiency decreases lung vascular remodeling in hypoxic conditions.6 To further understand the effect of IL-6 on neutrophil recruitment and PH pathogenesis, we housed Il6−/− mice under hypoxic conditions. Hypoxic mice that were deficient in Il6 exhibited fewer blood and lung neutrophils than hypoxic Il6+/+ mice despite similar neutrophil abundances in the bone marrow (Fig. S16A). Correspondingly, Il6−/− mice also exhibited attenuated features of PH, as shown by decreased RVSP and reduced lung remodeling compared to those of Il6+/+ mice (Fig. S16B). Taken together, these data indicate that Cx3cr1 is critical for Il-6-mediated neutrophil egress from the bone marrow and subsequent recruitment into the lungs of mice with PH, resulting in histologic and hemodynamic manifestations of PH in vivo.
Fig. 6.

CX3CR1 deficiency alleviates IL-6-driven PH. Cx3cr1+/+ Il6tg and Cx3cr1−/– Il6tg mice were placed in hypoxic chambers (10% O2) (n = 6 per group) for 3 weeks to induce PH. A The numbers of neutrophils in the lungs, bone marrow and blood of hypoxic mice were quantified by flow cytometry. B Right ventricular systolic pressure (RVSP) and the Fulton index (RV/LV + S mass ratio) were assessed. C Pulmonary vasculature remodeling scores of the lungs of hypoxic mice were quantified by confocal microscopy. D Ifnb, Il1b, and Il18 expression levels were quantified by RT-qPCR. E Schematic showing the mechanisms of neutrophil egress from the bone marrow (adapted from Medical Servier Art). The data are shown as the mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001
Discussion
In summary, our study defines a mechanism of neutrophil egress from the bone marrow in PH that functionally link IL-6 to CX3CR1-dependent activity (Fig. 6E). While the pathogenic role of CX3CR1 has canonically been studied in monocytes and macrophages in PH,2,42–45 our findings identify a unique role for this molecule in neutrophils, significantly extending our mechanistic understanding of how these cells are produced, migrate, and home to sites of pulmonary vascular inflammation in PH. These findings also emphasize the deleterious role of IL-6 in PH pathogenesis by contributing to inflammatory myeloid cell recruitment to the lungs. In contrast, Mickael et al. reported that global genetic ablation of IL-6 and smooth muscle-specific IL-6 receptor deficiency was detrimental to S. mansoni-induced PH.46 A molecular explanation underlying this discrepancy has not fully been elucidated but could be associated with differences in IL-6-dependent neutrophil recruitment. Given that neutrophils appear to play a major role in PH pathogenesis, our findings offer valuable mechanistic insight into the mobilization of these cells during PH pathogenesis, paralleling their importance in other vascular diseases such as atherosclerosis47,48 and rheumatoid arthritis.13,49
Our findings provide important insights into the roles of IL-6 in PH. IL-6 levels have been correlated with right ventricular dysfunction in PH patients.12 In accordance with these results, a recent study showed that upregulation of the IL-6 receptor on the surface of smooth muscle cells promotes their proliferation and contributes to overall vascular remodeling in PAH,13 rendering classic IL-6 signaling a potential therapeutic target for treating patients with PAH.50 As such, a phase II clinical trial using an IL-6 receptor antagonist in patients with PAH is underway.37 Our study adds substantially to these findings by describing the importance of IL-6 in inducing CX3CR1 expression in neutrophils, thus driving egress of these cells from bone marrow. Although it has been reported that neutrophils express IL-6R,51,52 the current study does not investigate the importance of this receptor in neutrophil egress.
Moreover, our work adds to ongoing studies of the pleiotropic role of CX3CR1 in PH pathogenesis. CX3CR1 is the only known receptor of CX3CL1 and is highly expressed by monocytes and macrophages.5,6,53–55 Hypoxic mice lacking Cx3cr1 exhibited less monocyte-mediated inflammation and lung remodeling than hypoxic wild-type mice.6 Our study demonstrated that neutrophils also express CX3CR1, albeit at low levels. Mice deficient in CX3CR1 in hematopoietic cells displayed diminished neutrophil numbers in the lungs. However, it is not clear whether this decreased neutrophil recruitment in the lungs is solely dependent upon neutrophil egress from bone marrow or mediated by the direct reduction in CX3CR1-mediated neutrophil recruitment in the lungs. Nonetheless, we cannot rule out the contribution of other CX3CR1-expressing hematopoietic cells, such as monocytes, to IL-6-mediated exacerbation of PH pathogenesis. Additionally, even though our data reveal that neutrophils greatly depend on Cx3cr1 to exit the BM in the context of PH, other chemokine receptors might be needed for neutrophil egress, as shown by the number of Cx3cr1−/− neutrophils present in the lungs. Future experiments utilizing conditional Cx3cr1 deficiency in specific leukocyte populations will be required to address this question.
In addition to defining the fundamental molecular axis controlling neutrophil egress, our data suggest the potential of characterizing neutrophil subtypes as a diagnostic tool in PH. Within small cohorts of patients recruited at a single center, we found distinct alterations in the number and profile of neutrophils among WSPH PH subtypes, with Group 3 PH patients demonstrating the highest levels of neutrophils in the blood. However, while prior studies have corroborated our findings and shown elevations in blood neutrophil counts in certain PH populations,56,57 it is notable that steady-state neutrophilia is not typical. Future studies encompassing larger cohorts of PH patients (WSPH PH subtypes 1–4) are warranted to define any time-dependence of neutrophil egress during the course of PH development. Nevertheless, our in vivo findings on hypoxic WT mice (representative of WSPH group 3) and Il6tg mice (WSPH group 1) were consistent with previously found results showing elevated neutrophil counts in PH patients. Moreover, given our findings of alterations in neutrophil inflammatory states, coupling neutrophil counts with gene expression profiling could offer an even greater diagnostic or prognostic discernment in PH and should be explored.
Our findings also offer new therapeutic strategies for PH, such as developing neutralizing antibodies against CX3CR1 or IL6-R and/or targeted depletion of IRF4. IRF4 is a transcription factor that belongs to the interferon regulatory factor family (IRF). IRFs are potent regulators of interferon production in response to various infections and inflammation and modulate the production of interferon-inducible genes.58 The interaction between IL6, IRF4, and CX3CR1 has been reported in the literature.59,60 Similar to our observation, Balbanian et al. demonstrated that CD4+ T lymphocytes expressing high levels of CX3CR1 were recruited to the lungs to pulmonary ECs with CX3CL1 expression.61 The mechanism by which leukocyte CX3CR1 expression is increased remains unclear. IL6/IRF4-mediated elevations in CX3CR1 expression, as shown in our study, may be common in both neutrophils and T lymphocytes. Further experiments are warranted to delineate the importance of IL-6 in T lymphocyte recruitment to the lungs in PH. Although a recent report showed alterations in IRF4 expression in PAH patients,62 mechanistic data demonstrating the involvement of IRF4 in PH have been lacking until now. Our study identifies IRF4 as another potential therapeutic target to reduce PH severity, but future preclinical work will be needed to ascertain the efficacy of pharmacologically targeting IRF4 to alleviate PH pathogenesis.
Materials and methods
Human samples and cell storage
Lungs and peripheral blood from PH patients and healthy donors were collected and processed as previously described.63 To define the clinical PH subtype, after hemodynamic identification, third-party expert clinicians reviewed clinical notes and relevant studies to determine the WSPH classification.38 Leukocytes were separated from total blood by a Ficoll gradient as previously described.6
Animals
All animal experiments were conducted according to NIH guidelines under protocols approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Adult male and female C57BL/6 wild-type, Cx3cr1GFP/GFP and lung-specific Il-6-overexpressing transgenic mice (10–12 weeks old) were obtained from Jackson Lab. As previously described,6 to induce PH, mice were placed in normobaric hypoxic chambers with 10% O2 for 3 weeks. RVSP was recorded via right heart catheterization as previously described.64 The heart was flushed with 10 mL of PBS, and the right ventricle (RV) was separated from the left ventricle (LV). Both ventricles were weighed, and RV/LV + S (Fulton index) mass ratios were calculated.
Bone marrow reconstitution
To assess the importance of Cx3cr1 expression in neutrophil egress, C57BL/6 wild-type and Cx3cr1GFP/GFP mice were lethally irradiated (10 Gy). Subsequently, each mouse was injected with approximately 1 million bone marrow hematopoietic cells from C57BL/6 wild type or Cx3cr1GFP/GFP mice by i.v. under anesthesia.
Organ harvesting and flow cytometry
Organs were harvested, and single-cell suspensions were prepared as previously reported.6,63 The following panel of antibodies were used to analyze the myeloid cell population in mice: anti-CD45.2 (104), Siglec F (SH2.1), CD11c (N418), CD11b (M1/70), CD115 (AFS98), and Ly6G (1A8). Neutrophils were identified as CD11b+, Ly6G+, and CD115−. The following panel of antibodies was used to analyze the myeloid cell population in humans: anti-CD45 (HI30), CD206 (19.2), CD14 (61D3), CD16 (3G8), and CD24 (ML5). Neutrophils were identified as CD14− CD24low, and CD16+. A Fortessa flow cytometer (BD) was used to acquire the data, which were analyzed with FlowJo software (Tree Star).
Cell sorting, cytospin, and Giemsa staining
Human lung and blood neutrophils were sorted using a FACS Aria II directly in RNA extraction buffer or suspended in FACS buffer. The following panel of antibodies was used to stain the neutrophil population in human samples: CD14 (61D3), CD45 (HI30), CD16 (3G8), and CD24 (ML5). Three neutrophil populations were sorted as CD16hi CD24lo (Neutro 1), CD16hi CD24hi CD14lo (Neutro 2), and CD16hi CD24hi CD14hi (Neutro 3). These neutrophil subpopulations were collected in FACS tubes with caps and centrifuged for 10 min at 1000 rpm. The cells (~50,000) were resuspended in 200 µl of cold 2% FBS-PBS in a 1.5 ml Eppendorf tube. The cells were deposited onto poly-l-lysine-coated “+” slides using a cytospin (800 rpm for 3 min). The cells were then processed for Giemsa staining.
Computational flow cytometry
bh-SNE and PhenoGraph plots depicting murine and human leukocyte subpopulations in the lungs and blood were generated with cyt3 (MATLAB) as previously described.65 Spanning-tree progression analysis of density-normalized events (SPADE) was performed to determine the heterogeneity of circulating leukocytes in PAH patients as previously described.66
Immunofluorescence
Lung sections were prepared and stained as previously described.6 Remodeling scores were assessed in α-SMA–stained vessels (<100 mm in diameter) using ImageJ software by measuring the arterial wall thickness divided by the inner diameter (Fiji), as previously described.64 All measurements were performed in a blinded manner.
Ingenuity pathway analysis
Guided by IPA, we focused on gene sets that were targets of IL-6 signaling. Briefly, we selected IL6 from the list of upstream regulators and CX3CR1 from the gene list and added them to My Pathway. We then selected CX3CR1 and grew a network involving transcriptional regulators. This network displayed all transcription factors that modulated CX3CR1 expression. We then selected all of these transcription factors and IL6 and connected these factors together.
HL-60 cell differentiation and transcription factor inhibition
All-trans retinoic acid (ATRA) (Sigma Aldrich) was dissolved in 100% dimethyl sulfoxide (DMSO) to obtain a 5 mM stock solution. This solution was further diluted to a working concentration of 1 µM. HL-60 cells underwent ATRA-mediated differentiation for 5 days. After 5 days, the medium containing 1 µM ATRA was replaced, and the cells were treated with siRNA against IRF4. Concomitantly, HL-60 cells were treated with 100 ng/mL or 400 ng/mL IL-6. The cells were then incubated at 37 °C for 24 h, after which gene expression was analyzed and apoptosis assays were performed.
Statistical analysis
The data were compiled with Prism software (GraphPad). Statistics were generated and are presented as the mean ± SEM. The normality of the data distribution was determined by Shapiro–Wilk tests. For normally distributed data, statistical significance between two categories of analyzed samples was calculated using two-tailed Student’s t tests. For multiple category comparisons, one-way ANOVA was used with a post hoc Bonferroni test. Differences with P values <0.05 were considered statistically significant.
Ethical approval
All experimental procedures involving the use of human lung tissue included the relevant receipt of written informed consent and were approved by the Committee for Oversight of Research and Clinical Training Involving Decedents (no. 101) at the University of Pittsburgh, as well as the Institutional Review Board of the University of Pittsburgh (no. REN17020169/IRB020810) and the Institutional Review Board at Boston Children’s Hospital. All experimental procedures involving the use of human peripheral blood included the relevant receipt of written informed consent and were approved by the Institutional Review Board of the University of Pittsburgh (no. REN16070123/PRO11070366), as well as the Institutional Review Board of the University of Pittsburgh (no. REN17030011/IRB0306040). Ethical approval for this study and informed consent conformed to the standards of the Declaration of Helsinki.
Supplementary information
Acknowledgements
This work was supported by National Institute of Health grants R00HL12076, R01HL143967, and R01HL142629 to P.D.; NIH grants R01 HL124021, HL 122596, HL 138437, and UH2/UH3 TR002073; and the American Heart Association Established Investigator Award 18EIA33900027 to S.Y.C.; the AHA Transformational Project Award (19TPA34910142), AHA Innovative Project Award (19IPLOI34760566) and ALA Innovation Project Award (IA-629694) to P.D.; the VMI Postdoctoral Training Program in Translational Research and Entrepreneurship in Pulmonary and Vascular Biology T32 funded by the National, Heart, Lung and Blood Institute (NHLBI) to J.F.; the AHA postdoctoral fellowship award 20POST35210088 to S.B.V.; the American Heart Association Grant 19CDA34730030 to R.K.; and NIH Grants and R01HL135872 to B.B.G. We thank the NIH-supported microscopy resources at the Center for Biologic Imaging (NIH grant 1S10OD019973-01). We thank the Center for Organ Recovery & Education (CORE) as well as organ donors and their families for the generous donation of tissues used in this study.
Author contributions
J.F. conducted experiments, analyzed the data, and wrote the paper. J.Z., Y.Y.T., R.K., L.S., B.K., and B.B.G. conducted experiments. S.B.V., S.P.O.N., A.A., G.C.B., L.S., and B.K. conducted experiments and analyzed the data. A.W., J.S., and M.R. recruited patients and provided peripheral blood and lung samples from healthy donors and PAH patients. S.Y.C. and P.D. designed the research study, analyzed the data, and composed the paper.
Competing interests
S.Y.C. has served as a consultant for Zogenix, Aerpio, and United Therapeutics. S.Y.C. holds research grants from Actelion and Pfizer. S.Y.C. has filed patent applications regarding the targeting of metabolism in PH. The authors declare no other competing interests.
Footnotes
These authors contributed equally: Stephen Y. Chan, Partha Dutta.
Contributor Information
Stephen Y. Chan, Email: chansy@pitt.edu
Partha Dutta, Email: duttapa@pitt.edu.
Supplementary information
The online version of this article (10.1038/s41423-020-00608-1) contains supplementary material.
References
- 1.Sawada H, et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014;211:263–80. doi: 10.1084/jem.20111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frid MG, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006;168:659–69. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mouraret N, et al. Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation. 2013;127:1664–76. doi: 10.1161/CIRCULATIONAHA.113.002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vergadi E, et al. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123:1986–95. doi: 10.1161/CIRCULATIONAHA.110.978627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amsellem, V. et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 chemokine systems in hypoxic pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 56, 597–608 (2017). [DOI] [PubMed]
- 6.Florentin, J. et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J. Immunol. 200, 3612–25 (2018). [DOI] [PMC free article] [PubMed]
- 7.Romano M, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–25. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto-Kataoka T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl Acad. Sci. U.S.A. 2015;112:E2677–E86. doi: 10.1073/pnas.1424774112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tacke F, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Combadiere C, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 11.Graham BB, et al. Transforming growth factor-beta signaling promotes pulmonary hypertension caused by Schistosoma mansoni. Circulation. 2013;128:1354–64. doi: 10.1161/CIRCULATIONAHA.113.003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prins KW, et al. Interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension. J. Heart Lung Transplant. 2018;37:376–84. doi: 10.1016/j.healun.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamura Y, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Investig. 2018;128:1956–70. doi: 10.1172/JCI96462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol. Rev. 2014;22:147–51. doi: 10.1097/CRD.0000000000000021. [DOI] [PubMed] [Google Scholar]
- 15.Qu D, Liu J, Lau CW, Huang Y. IL-6 in diabetes and cardiovascular complications. Br. J. Pharmacol. 2014;171:3595–603. doi: 10.1111/bph.12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012;110:875–88. doi: 10.1161/CIRCRESAHA.111.257535. [DOI] [PubMed] [Google Scholar]
- 17.Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F. Pathophysiological role of neutrophils in acute myocardial infarction. Thromb. Haemost. 2013;110:501–14. doi: 10.1160/TH13-03-0211. [DOI] [PubMed] [Google Scholar]
- 18.Taylor S, Dirir O, Zamanian RT, Rabinovitch M, Thompson AAR. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front. Med. 2018;5:217. doi: 10.3389/fmed.2018.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008;14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 20.De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin. Cancer Res. 2004;10:4895–900. doi: 10.1158/1078-0432.CCR-03-0760. [DOI] [PubMed] [Google Scholar]
- 21.Yildiz A, et al. Association between neutrophil to lymphocyte ratio and pulmonary arterial hypertension. Turk Kardiyol. Dern. Ars. 2013;41:604–9. doi: 10.5543/tkda.2013.93385. [DOI] [PubMed] [Google Scholar]
- 22.Harbaum L, et al. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC Pulm. Med. 2017;17:72. doi: 10.1186/s12890-017-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horckmans M, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017;38:187–97. doi: 10.1093/eurheartj/ehw002. [DOI] [PubMed] [Google Scholar]
- 24.Talukdar S, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012;18:1407–12. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldabbous L, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016;36:2078–87. doi: 10.1161/ATVBAHA.116.307634. [DOI] [PubMed] [Google Scholar]
- 26.Doring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ. Res. 2017;120:736–43. doi: 10.1161/CIRCRESAHA.116.309692. [DOI] [PubMed] [Google Scholar]
- 27.Klinke, A. et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight.3, e97530 (2018). [DOI] [PMC free article] [PubMed]
- 28.Warnatsch, A., Ioannou, M., Wang, Q. & Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science349, 316–20 (2015). [DOI] [PMC free article] [PubMed]
- 29.Fielding CA, et al. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 2008;181:2189–95. doi: 10.4049/jimmunol.181.3.2189. [DOI] [PubMed] [Google Scholar]
- 30.McLoughlin RM, et al. Interplay between IFN-γ and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J. Clin. Investig. 2003;112:598–607. doi: 10.1172/JCI17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagareddy PR, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engblom, C. et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science358, eaal5081 (2017). [DOI] [PMC free article] [PubMed]
- 33.Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood. 2005;105:2543–8. doi: 10.1182/blood-2004-08-3193. [DOI] [PubMed] [Google Scholar]
- 34.Del Fresno C, et al. DNGR-1 in dendritic cells limits tissue damage by dampening neutrophil recruitment. Science. 2018;362:351–6. doi: 10.1126/science.aan8423. [DOI] [PubMed] [Google Scholar]
- 35.Jung S, et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steiner MK, et al. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009;104:236–44. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez-Sanchez J, et al. Clinical trial protocol for TRANSFORM-UK: A therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm. Circ. 2018;8:2045893217735820. doi: 10.1177/2045893217735820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galiè, N., McLaughlin, V. V., Rubin, L. J. & Simonneau, G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur. Resp. J.53, 802148 (2019). [DOI] [PMC free article] [PubMed]
- 39.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018;15:234–48. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jung IH, et al. Predominant activation of JAK/STAT3 pathway by interleukin-6 is implicated in hepatocarcinogenesis. Neoplasia. 2015;17:586–97. doi: 10.1016/j.neo.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010;62:726–59. doi: 10.1124/pr.110.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994;144:275–85. [PMC free article] [PubMed] [Google Scholar]
- 43.Stenmark KR, Davie NJ, Reeves JT, Frid MG. Hypoxia, leukocytes, and the pulmonary circulation. J. Appl. Physiol. 2005;98:715–21. doi: 10.1152/japplphysiol.00840.2004. [DOI] [PubMed] [Google Scholar]
- 44.Pugliese SC, et al. A Time- and compartment-specific activation of lung macrophages in hypoxic pulmonary hypertension. J. Immunol. 2017;198:4802–12. doi: 10.4049/jimmunol.1601692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014;115:165–75. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mickael C, et al. IL-6Ra in smooth muscle cells protects against schistosoma- and hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2019;61:123–6. doi: 10.1165/rcmb.2018-0277LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- 48.Ionita MG, et al. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler. Thromb. Vasc. Biol. 2010;30:1842–8. doi: 10.1161/ATVBAHA.110.209296. [DOI] [PubMed] [Google Scholar]
- 49.Sur Chowdhury C, et al. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014;16:R122. doi: 10.1186/ar4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pullamsetti SS, Seeger W, Savai R. Classical IL-6 signaling: a promising therapeutic target for pulmonary arterial hypertension. J. Clin. Investig. 2018;128:1720–3. doi: 10.1172/JCI120415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chalaris A, et al. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–55. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 52.Farahi N, et al. Neutrophil-mediated IL-6 receptor trans-signaling and the risk of chronic obstructive pulmonary disease and asthma. Hum. Mol. Genet. 2017;26:1584–96. doi: 10.1093/hmg/ddx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imai T, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–30. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 54.Landsman L, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113:963–72. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 55.Panek CA, et al. Differential expression of the fractalkine chemokine receptor (CX3CR1) in human monocytes during differentiation. Cell. Mol. Immunol. 2015;12:669–80. doi: 10.1038/cmi.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Balta S, Demirkol S, Aparci M, Celik T, Ozturk C. The neutrophil lymphocyte ratio in coronary heart disease. Int. J. Cardiol. 2014;176:267. doi: 10.1016/j.ijcard.2014.06.098. [DOI] [PubMed] [Google Scholar]
- 57.Benites-Zapata VA, et al. Usefulness of neutrophil-to-lymphocyte ratio in risk stratification of patients with advanced heart failure. Am. J. Cardiol. 2015;115:57–61. doi: 10.1016/j.amjcard.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paun A, Pitha PM. The IRF family, revisited. Biochimie. 2007;89:744–53. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–87. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Refaat A, et al. Distinct roles of transforming growth factor-beta-activated kinase 1 (TAK1)-c-Rel and interferon regulatory factor 4 (IRF4) pathways in human T cell lymphotropic virus 1-transformed T helper 17 cells producing interleukin-9. J. Biol. Chem. 2011;286:21092–9. doi: 10.1074/jbc.M110.200907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balabanian K, et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002;165:1419–25. doi: 10.1164/rccm.2106007. [DOI] [PubMed] [Google Scholar]
- 62.Lenna S, et al. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum. 2013;65:1357–66. doi: 10.1002/art.37891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vasamsetti SB, et al. Sympathetic neuronal activation triggers myeloid progenitor proliferation and differentiation. Immunity. 2018;49:93–106. doi: 10.1016/j.immuni.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertero T, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016;126:3313–35. doi: 10.1172/JCI86387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saeys Y, Van Gassen S, Lambrecht BN. Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016;16:449–62. doi: 10.1038/nri.2016.56. [DOI] [PubMed] [Google Scholar]
- 66.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011;29:886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.