

Abstract
Stroke is an acute cerebrovascular disease caused by ruptured or blocked blood vessels. For the prevention of ischemic stroke, the coagulation state of blood and cerebrovascular protection should be considered. Our previous study has shown that salvianolic acid A (SAA), which is a water-soluble component from the root of Salvia Miltiorrhiza Bge, prevents thrombosis with a mild inhibitory effect on platelet aggregation. In this study we investigated the preventive effects of SAA on cerebrovascular endothelial injury caused by ischemia in vivo and oxygen-glucose deprivation (OGD) in vitro, and explored the underlying mechanisms. An autologous thrombus stroke model was established in SD rats by electrocoagulation. SAA (10 mg/kg) was orally administered twice a day for 5 days before the operation. The rats were sacrificed at 24 h after the operation. We showed that pretreatment with SAA significantly improved the neurological deficits, intracerebral hemorrhage, BBB disruption, and vascular endothelial dysfunction as compared with model group. In human brain microvascular endothelial cells (HBMECs), pretreatment with SAA (10 μM) significantly inhibited OGD-induced cell viability reduction and degradation of tight junction proteins (ZO-1, occludin, claudin-5). Furthermore, we found that SAA inhibited the upregulation of Src signaling pathway in vivo and vitro and reversed the increased expression of matrix metalloproteinases (MMPs) after ischemic stroke. In conclusion, our results suggest that SAA protects cerebrovascular endothelial cells against ischemia and OGD injury via suppressing Src signaling pathway. These findings show that pretreatment with SAA is a potential therapeutic strategy for the prevention of ischemic stroke.
Keywords: salvianolic acid A, ischemic stroke, cerebrovascular endothelial injury, blood brain barrier, Src, autologous thrombus stroke model, human brain microvascular endothelial cells
Introduction
Stroke is one of the leading causes of death, and the worldwide incidence, mortality and disability rates of stroke are high [1, 2]. Ischemic stroke, the most common kind of stroke, accounting for ~87% of all strokes, occurs when blood vessels in the neck or brain are blocked [3, 4]. Because of the potential of stroke to cause great harm, preventing this condition is of great significance. The World Health Organization reported that three-quarters of all deaths related to cardiovascular disease may be avoided by adequate coordinated preventive actions [5]. Many measures have been used for the prevention and control of cerebrovascular diseases over the past century, including changes in lifestyle habits and the application of preventive drugs [6, 7]. However, the dynamics of the epidemic of cerebrovascular diseases have remained unchanged [8].
The main strategy for preventing and treating ischemic stroke is inhibition of thrombosis formation [9]. At present, the prevention of thrombosis mainly focuses on the inhibition of excessive platelet activation [10]. However, the efficacy of antiplatelet drugs in stroke prevention needs to be further confirmed, and bleeding risk limits the application of antiplatelet drugs. Therefore, it is urgent to develop safe and effective drugs for the primary prevention of cerebrovascular diseases.
Currently, the application of antiplatelet [11] or anticoagulant treatments [12] is not sufficient to prevent thrombosis. Vascular protection strategies should also be considered [13, 14], including approaches for maintaining endothelial function and vascular integrity [15–17]. Cerebral endothelial cell function is associated with reduced resting blood flow (hypoperfusion) and impairment of vasodilator responses [18, 19]. Cerebrovascular protection against acute ischemic stroke mainly involves inhibition of vasoactive factors and vascular endothelial dysfunction, leading to a reduction in the permeability of the BBB [20]. Phosphorylation of the nonreceptor tyrosine kinase Src is associated with increased vascular permeability induced by vascular endothelial growth factor (VEGF) during acute ischemic stroke [21]. This process is triggered by the activation of the small GTPase Rac by VEGFA through the Src-dependent phosphorylation of vav guanine nucleotide exchange factor 2 (VAV2-Tyr172). Activation of Rac stimulates p21-activated kinase (PAK), which phosphorylates VE-cadherin, leading to its internalization and weakening of intercellular connections and the subsequent destruction of the intercellular junctions of the BBB [22–24].
Salvianolic acid A (SAA) is a water-soluble active compound extracted and purified from Salvia Miltiorrhiza Bge [25] (Fig. 1a). SAA has been shown to exert a variety of pharmacological effects, such as anti-inflammatory [26–28], antioxidant [29], antiapoptotic [30, 31], and neuroprotective activities [32–34]. Earlier studies have shown that SAA inhibits platelet activation via inhibition of phosphoinositide 3-kinase and elevation of cyclic guanosine monophosphate [35, 36]. Based on previous experimental evidence, we speculate that SAA may have good potential to prevent the occurrence of stroke. Our previous experiments suggested that unlike aspirin, SAA provides a mild inhibitory effect against platelet aggregation and protects against ischemic stroke induced by thrombus by decreasing the risk of bleeding [37]. However, the cerebrovascular protective effect of SAA, especially against acute ischemic stroke, has not been reported.
Fig. 1. Chemical structure of SAA and a schematic diagram of the experimental protocols.

a Chemical structure of SAA. b Schematic diagram of the experimental protocols.
In the present study, the preventive effect of SAA on cerebrovascular injury induced by ischemia was evaluated in vivo and in vitro, and the mechanism of Src pathway regulation underlying the effect cerebrovascular protective effect of SAA was investigated to evaluate whether SAA is a potential drug development candidate for the prevention of ischemic stroke that deserves further preclinical evaluation.
Materials and methods
Experimental animals and drug administration
Male Sprague-Dawley rats (220–260 g) were purchased from Beijing Vital River Experimental Animal Co., Ltd. (Beijing, China; certificate No. SCXK2016-0006). The animals were acclimatized for 3 days and randomly assigned to different groups. The animal care and experimental procedures were approved by the Ethics Committees of the Institute of Materia Medical, Chinese Academy of Medical Sciences and Peking Union Medical College. All experimental protocols were performed to minimize animal suffering and the number of animals used. The animals were randomly assigned to different groups (https://www.random.org/), and neurobehavioral outcome were scored in a blinded manner.
Autologous thrombus stroke model
The autologous thrombus stroke model was established according to a previously reported method with improvements [38]. Rats were anesthetized with chloral hydrate and fixed in the supine position. The common carotid artery (CCA), external carotid artery, and internal carotid artery (ICA) were obtusely separated. The right CCA was placed in the electric clamp of the YLS-14B thrombus formation tester (Jinan Yiyan Science and Technology Development Co., Ltd., Jinan, China). The CCA (1.00 mA) was prestimulated for 1 min and then stimulated for an additional 4 min. The thrombus was crushed with homemade soft tweezers several times. The arteriopalmus re-emerged when the thrombus pieces were crushed into homogeneous pieces. The artery clamp was removed from the CCA to flush the comminuted thrombus into the ICA, and the CCA was clamped for another 15 min to channel the thrombus into the middle cerebral artery. Then, the skin on the neck of each rat was sutured. The rats in the sham operation group were subjected to the same surgery as those in the operation group except electrocoagulation.
Experimental groups and drugs
SAA (HPLC purity >99%) was prepared by the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS and PUMC, Beijing, China). Aspirin was purchased from Sigma-Aldrich (A2093-100G, St. Louis, MO, USA). The animals were randomly divided into the following group: the sham operation group, model group (0.5% CMC-Na, ig), aspirin + model (aspirin) group, and SAA + model (SAA) group. SAA was pre-administered by intragastric gavage every morning and evening for 5 consecutive days. The autologous thrombus stroke model was established on the 6th day. A schematic diagram of the experimental protocols is shown in Fig. 1b. All rats were sacrificed 24 h after ischemia. A total of 150 rats were used, of which 13 were excluded due to death and 17 were excluded for lack of neurological impairment, and 30 rats were added during the experiments.
Neurological function test
Neurological function tests were performed by an investigator blinded to the groups 24 h after ischemia.
Modified neurological severity score (mNSS) test
Neurological functions, i.e., sensory, reflexes and balance, were measured by the mNSS test, with scores ranging from 0 to 18 (normal score: 0, maximal deficit: score 18) [39]. Higher scores indicated greater severity of injury. Rat with a score equal to or greater than 8 at 24 h after ischemic stroke were used for further experiments.
Longa test
To evaluate ischemic stroke-induced neurobehavioral dysfunction, we used the Zea-Longa test [40]. One of five scores was assigned: 0 points: normal, no neurological deficits; 1 points: failure to extend the left forelimb fully, mild neurological deficits; 2 points: inability to walk in a straight line, moderate neurological deficits; 3 points: circling to the left (hemiplegic side), severe neurological deficits; and 4 points: unable to walk spontaneously, loss of consciousness.
Grasping ability test
The rats were suspended from a horizontal wire by the forelimbs and then released. The time that the rats remained suspended and their behavior were recorded and scored [41]. Scores were assigned as follows: 0 points: fell off the wire within 10 s; 1 point: lifted one of the hind limbs; 2 points: tried to climb the wire; 3 points: grasped the wire with the front claw and at least one rear claw; 4 points: wrapped the legs and tail around the wire; and 5 points: tried to escape to the end of the wire.
Assessment of intracerebral hemorrhage
The severity of hemorrhagic transformation (HT) was graded as follows: small petechial hemorrhagic infarction (HI1), confluent petechial hemorrhagic infarction (HI2), small parenchymal hemorrhage (PH1) (<30% of the infarct, mild mass effect), and large parenchymal hemorrhage (PH2, >30% of the infarct, marked mass effect) [42]. Twenty-four hours after ischemia, the brain was perfused, sectioned, photographed and scored, and then the tissue was sonicated for homogenization with 0.01 mol/L phosphate-buffered saline (PBS) at a proportion of 1:1. The homogenate was centrifuged at 4 °C 13,000 × g for 30 min. Then, 50 μL of the supernatant was added into a 96-well plate, 200 μL of reagent (QuantiChrom Hemoglobin Assay Kit, cat.DIHB-250, BioAssay Systems, NC, USA) was added, and the plate was incubated away from light for 5 min at room temperature. The OD value was measured at 400 nm.
Hematoxylin and eosin (H&E) staining
Twenty-four hours after ischemic stroke, rats were anesthetized and transcardially perfused with 0.9% normal saline followed by 4% paraformaldehyde until the limbs were stiff. The brains were removed immediately and postfixed in fixative for 24 h at 4 °C. The brain tissues were embedded in paraffin and cut into serial coronal sections (5 μm thick). The sections were then deparaffinized and rehydrated and stained with H&E.
Evaluation of BBB leakage
Twenty-four hours after ischemic stroke, 4% Evans blue dye (E2129–100G, Sigma-Aldrich Chemical Co., St. Louis, MO, USA) in saline was intravenously injected (0.25 mL/100 g) into the tail vein and allowed to circulate for 2 h. The left ventricles of the rats were then perfused with saline until colorless perfusion fluid was obtained from the right atrium. The brains were isolated, photographed, weighed and homogenized in 50% trichloroacetic acid (100 mg/300 μL). After centrifugation at 12,000 × g at 4 °C for 5 min, 200 μL of supernatant was collected, and the absorbance was measured at 620 nm. A standard curve was created with the brain homogenates of normal rats.
Immunohistochemical staining
Coronal sections were deparaffinized and rehydrated for immunohistochemical staining. Endogenous peroxidase activity was quenched using 3% H2O2 for 10 min at room temperature. Nonspecific staining was then blocked with 3% BSA in PBS for 30 min at room temperature, and the sections were incubated overnight at 4 °C with the primary antibody (rabbit anti-VWF, 1:300, Abcam, ab6994, Cambridge, UK). The slices were then washed with PBS and incubated with secondary antibodies for 50 min at room temperature. Then, the sections were incubated with 3,3′-diaminobenzidine peroxide (DAB) chromogenic substrate and counterstained with hematoxylin. Images were captured with a light microscope. Quantitative assessment of VWF density was performed by measuring the percentage of the positively stained area.
Enzyme-linked immunosorbent assay (ELISA)
Blood was collected from the abdominal aorta and stored at room temperature for 2 h. The serum was obtained by centrifugation at 1000 × g for 20 min, and then the supernatant was analyzed with different ELISA kits (Rat Von Willebrand Factor ELISA Kit, E-EL-R1079c, Elabscience®, Wuhan, China; Rat-like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13) ELISA Kit 9, NBP2–66441, Novus, Colorado, USA) according to the manufacturer’s instructions.
Human brain microvascular endothelial cell (HBMEC) culture
HBMECs (purchased from the Cell Center of the Basic College of Peking Union Medical College) were cultured in RPMI-1640 basic medium supplemented with 10% FBS (all purchased from the Gibco Life Technologies Inc., Grand Island, NY, USA), 100 μg/mL streptomycin and 100 U/mL penicillin. The cell culture conditions were maintained at 95% air and 5% CO2 with a humidified atmosphere in a 37 °C incubator (Sanyo, USA).
Oxygen-glucose deprivation (OGD) Model
An HBMEC model of OGD was established after plating for 24 h. Briefly, the cells were randomly divided into the control, OGD, SAA + OGD and aspirin + OGD groups. The cells in the control group were cultured in RPMI-1640 and the cells in the other groups were cultured in glucose-free RPMI-1640 (Gibco Life Technologies Inc., Grand Island, NY, USA). The cells were placed in a hypoxic incubator (Thermo Scientific, Waltham, USA) containing a mixture of 5% CO2 and 94% N2 for 6 h. The control cells were incubated under normal conditions and were not exposed to OGD.
Cell counting kit-8 (CCK-8) assay
HBMECs were seeded in 96-well culture plates at a density of 5000 cells per well. After the cells were exposed to OGD as mentioned above and preincubated with SAA/aspirin, the CCK-8 (Beyotime Biotech, Shanghai, China) assay was carried out to evaluate cell viability according to the manufacturer’s manual. Briefly, 10 μL of CCK-8 solution was added to a 96-well plate and incubated for 1 h at 37 °C. The absorbance at 450 nm was measured with a microplate reader (Molecular Devices, USA).
Western blot assay
HBMECs and brain tissues were both lysed in RIPA buffer containing protease inhibitors and phosphatase inhibitors. Total protein was extracted from cells and tissues using a Total Protein Extraction Kit following the manufacturer’s protocols. The brain tissues of rats were carefully and rapidly placed into centrifuge tubes, and 0.1 g of tissue was added to 1 mL of RIPA buffer and homogenized. The homogenate was then lysed on ice for 20 min. The cell culture medium was discarded, and the cells were washed twice with PBS, an appropriate amount of cell lysate was added, and the cells were incubated on ice for 15 min and then collected with a cell scraper. The tissue homogenates and cell lysates were subjected to centrifugation at 13,000 × g for 15 min at 4 °C.
The protein concentration was determined based on the BCA method using the Protein Quantitative Analysis Kit. The protein samples were separated using sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a polyvinylidene fluoride membrane. The membrane was blocked with 5% bovine serum albumin (BSA) for 2 h at room temperature and then treated with the primary antibodies shown in Table 1 at 4 °C overnight. After the membrane was washed, it was incubated with HRP-conjugated rabbit (CWBIO, cat.CW0103S, Beijing, China) or mouse (CWBIO, cat.CW0102S, Beijing, China) IgG secondary antibody for 2 h at room temperature. The enhanced chemiluminescence (P1010–250, APPLYGEN, Beijing, China) method was used for image development. The band density was calculated with ImageJ software.
Table 1.
Primary antibodies used for Western blotting
| Primary antibody | Source species | Dilution | Company |
|---|---|---|---|
| Occludin (ab216327) | Rabbit | 1:1000 | Abcam (Cambridge, UK) |
| VEGFA (ab46154) | Rabbit | 1:1000 | Abcam (Cambridge, UK) |
| HIF-1α (ab51608) | Rabbit | 1:1000 | Abcam (Cambridge, UK) |
| Fibulin-5 (ab202977) | Rabbit | 1:1000 | Abcam (Cambridge, UK) |
| p-PAK (44–940G) | Rabbit | 1:1000 | Invitrogen (Carlsbad, CA) |
| MMP-2 (A11144) | Rabbit | 1:1000 | Abclonal (Wuhan, China) |
| Claudin-5 (YT0953) | Rabbit | 1:1000 | Immunoway (Plano, TX, USA) |
| MMP-9 (A0289) | Rabbit | 1:500 | Abclonal (Wuhan, China) |
| VAV2-Tyr172 (VP2641) | Rabbit | 1:1000 | ECM Biosciences (Versailles, KY, USA) |
| Src (11097–1-AP) | Rabbit | 1:1000 | Proteintech (Rosemont, IL, USA) |
| PAK (21401–1-AP) | Rabbit | 1:1000 | Proteintech (Rosemont, IL, USA) |
| Rac1 (24072–1-AP) | Rabbit | 1:1000 | Proteintech (Rosemont, IL, USA) |
| ZO-1 (21773–1-AP) | Rabbit | 1:1000 | Proteintech (Rosemont, IL, USA) |
| β-Actin (8H10D10) | Mouse | 1:1000 | Cell Signaling Technology (Beverly, MA, USA) |
| Scr-Tyr416 (D49G4) | Rabbit | 1:1000 | Cell Signaling Technology (Beverly, MA, USA) |
Statistical analysis
We used one-way ANOVA followed by Dunnett’s multiple comparison test for statistical analysis. All data in the experiment are presented as the mean ± standard error of the mean. A P value <0.05 was considered statistically significant. Statistical analysis was performed with GraphPad Prism software.
Results
SAA prevented neurological deficits in ischemic stroke rats
Neurological deficit scores were assessed 24 h after ischemic stroke. As shown in Fig. 2a, the Longa behavior test results showed that the neurological score of the SAA preventive treatment group was lower than that of the model group (2.30 ± 0.78 vs 1.50 ± 0.67; P < 0.05). This result was also confirmed by the mNSS test, which showed that the neurological score of the SAA preventive treatment group was lower than that of the model group (11.0 ± 1.41 vs 9.5 ± 1.02; P < 0.01) (Fig. 2b). Furthermore, an obvious improvement in neurobehavioral deficits in the grasping ability test was observed in the SAA preventive treatment group compared to the model group (Fig. 2c). Taken together, these results suggested that administration of SAA prevented neurological deficits in ischemic stroke rats. In addition, the body weight of the animals was not affected by the administration of SAA for 5 days, which proved that the drug itself had no adverse effect on the weight of the animals (Fig. 2d).
Fig. 2. Effects of SAA on neurobehavioral deficits in ischemic stroke rats and body weight.

a The Longa scores of the animals 24 h after ischemic stroke. b The mNSS scores of the animals 24 h after ischemic stroke. c The grasping ability scores of the ischemic stroke animals. d Body weight changes in the rats in each group following preventive administration. n = 10, mean ± SEM. ###P < 0.001 and ####P < 0.0001 vs the sham group, *P < 0.05 and **P < 0. 01 vs the model group.
SAA ameliorated intracerebral hemorrhage caused by ischemic stroke
Twenty-four hours after ischemic stroke, the occurrence of HT in the SAA preventive treatment group was lower than that in the model and aspirin groups (Fig. 3a). Analysis of brain slices showed slight hemorrhage in the model group and severe hemorrhage in the aspirin preventive treatment group (Fig. 3b). To confirm the occurrence of intracerebral hemorrhage, the cerebral hemoglobin content was measured to evaluate the severity of intracerebral hemorrhage. In the SAA preventive treatment group, the hemoglobin content was 45.04 ± 11.59 mg/dL, which was lower than that in the aspirin group (60.54 ± 9.29 mg/dL; P < 0.01) (Fig. 3c).
Fig. 3. Effect of SAA on cerebral hemorrhage caused by ischemic stroke.

a Incidence of HT (n = 64). b Occurrence of hemorrhagic transformation in the brain. c Content of cerebral hemoglobin. n = 10, mean ± SEM. #P < 0.05 and ##P < 0.01 vs the sham group, ^^P < 0.01 vs the ASP group. d Representative images of H&E staining of brain tissues from the different groups. The images are at ×100 magnification.
Furthermore, HE staining proved that severe intracerebral hemorrhage occurred in the aspirin group but not in the SAA preventive treatment group (Fig. 3d). These results demonstrated that preventive SAA administration reduced the incidence of intracerebral hemorrhage during ischemic stroke.
SAA attenuated BBB injury and TJ degradation caused by ischemic stroke
Loss of BBB integrity is one of the main mechanisms of intracerebral hemorrhage [43]. EB leakage was assessed to evaluate BBB permeability. Representative images of EB dye in brain tissue, which are shown in Fig. 4a, indicated that there was obvious EB dye extravasation in the ipsilateral hemisphere of the brain in model rats and a slight increase in EB dye extravasation in the aspirin group compared to the model group. Quantification showed that the EB content in the preventive SAA treatment group was dramatically lower than that in the model group (4.17 ± 1.59 μg/mL vs. 2.29 ± 0.88 μg/mL; P < 0.05) (Fig. 4b).
Fig. 4. SAA attenuated BBB injury and TJ degradation caused by ischemic stroke.

a Representative image of EB leakage. b Bar graph of EB content. EB content was measured 24 h after ischemic stroke. c–j Representative Western blot showing ZO-1, Occludin, Claudin-5, VE-cadherin, MMP-2, and MMP-9 expression. n = 6, mean ± SEM. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the sham group, *P < 0.05, **P < 0.01 and ***P < 0.001 vs the model group.
TJ proteins are essential components of the BBB and contribute to BBB integrity. Western blot analysis showed that the protein expression of ZO-1, claudin-5, occludin, and VE-cadherin was decreased in the model group compared to the control group and this decrease was reversed in the preventive SAA treatment group (Fig. 4c–g). In addition, because the degradation of TJ proteins is regulated by MMPs, the expression of MMPs was measured. The results indicated that the protein expression levels of MMP-9 and MMP-2 in the preventive SAA treatment group were much lower than those in the model group (Fig. 4h–j). Therefore, these results demonstrated that SAA preventive administration attenuated BBB injury and TJ degradation caused by ischemic stroke by inhibiting the expression of MMPs.
SAA alleviated ischemia-induced vascular endothelial dysfunction
VWF is an important marker of vascular endothelial damage [44]. As shown in Fig. 5a, the content of VWF in the serum in the preventive SAA treatment group was appreciably lower than that in the model group after ischemic stroke (374.22 ± 59.79 ng/mL vs 215.27 ± 66.31 ng/mL; P < 0.001). Immunohistochemistry assays further verified that compared with that in the model group, the VWF level in rat brain tissue was obviously decreased in the preventive SAA treatment group (Fig. 5b, c). The content of ADAMTS13 [45] in the preventive SAA treatment group, which can cleave VWF in the serum, was dramatically higher than that in the model group (351.41 ± 47.71 pg/mL vs 616.42 ± 110.71 pg/mL; P < 0.001) (Fig. 5d). The protein expression of Fibulin-5 [46], which is associated with vascular injury, was evidently increased in the model group compared to the control group and downregulated in the preventive SAA treatment group (Fig. 5e). These results indicated that preventive SAA administration alleviated vascular endothelial dysfunction during ischemic stroke.
Fig. 5. SAA alleviated vascular endothelial dysfunction during ischemic stroke.

a The effects of SAA (10 mg/kg) on VWF concentrations in rat serum. b, c Representative images of immunohistochemical staining and the pixel area of VWF (n = 3); the images are at ×200 magnification. d The effect of SAA (10 mg/kg) on ADAMTS13 concentrations in the serum. e Representative Western blot showing Fibulin-5 expression. n = 6, mean ± SEM. #P < 0.05, ###P < 0.001 and ####P < 0.0001 vs the sham group. *P < 0.05, **P < 0.01 and ***P < 0.001 vs the model group.
SAA inhibited Src activation during ischemic stroke
To investigate the mechanism underlying the protective effect of SAA on the BBB, we evaluated the expression of proteins in the VEGFA-Src signaling pathway, which participates in vascular permeability regulation, by Western blot analysis (Fig. 6a). Compared with that in the preventive SAA treatment group, the expression of VEGFA and its transcription factor HIF-1α was markedly increased in the model group (Fig. 6b–d). During ischemic stroke, the ratio of Src-Tyr416/Src was notably increased, indicating that the Src pathway was activated, while SAA inhibited the activation of Src (Fig. 6e, f). The expression of Rac1, which is the downstream protein of Src, was not changed during ischemia. However, the phosphorylation of VAV2-Tyr172 was dramatically increased in the model group, which indicates that Rac1 was activated (Fig. 6g–i) and was downregulated in the preventive SAA treatment group (Fig. 6g–i). Moreover, activated Rac1 stimulates PAK. The ratio of p-PAK/total PAK was markedly increased in the model group compared to the control group and was lower in the preventive SAA treatment group than that in the model group (Fig. 6j, k). These observations indicated that preventive SAA administration protected the BBB by inhibiting VEGFA-Src-VAV2-Rac1-PAK signaling pathways during ischemic stroke.
Fig. 6. SAA inhibited the activation of the VEGFA-Src signaling pathways induced by ischemic injury.

a Schematic diagram of the VEGF-Src signaling pathway. b–k Representative Western blot showing HIF-1α, VEGFA, total Src, Src-Tyr416, VAV2-Tyr172, Rac1, total PAK, and p-PAK expression. n = 6, mean ± SEM. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the sham group. *P < 0.05 and **P < 0.01 vs the model group.
SAA pretreatment inhibited Src activation in HBMECs after OGD
We further verified the results by inducing OGD injury in HBMECs in vitro. OGD stimulation resulted in severe cell damage and a loss of cell viability (P < 0.001), while preincubation with SAA concentration-dependently inhibited OGD injury-triggered cell viability loss (Fig. 7a). Furthermore, preincubation with SAA (10 μmol/L) reversed the OGD-induced decrease in TJ protein (ZO-1, Occludin, and Claudin-5) expression (Fig. 7b–e).
Fig. 7. Effects of SAA pretreatment on loss of HBMEC viability and the protective effects on TJ proteins after OGD injury.

a Effects of different concentrations of SAA (1, 3, and 10 μmol/L) on cell viability after OGD injury (n = 4). b–d Effects of SAA on ZO-1, Occludin, and Claudin-5 protein expression after OGD. e–l Representative Western blot showing HIF-1α, VEGF, total Src, Src-Tyr416, VAV2-Tyr172, Rac1, total PAK, and p-PAK protein expression. n = 6, mean ± SEM. #P < 0.05, ###P < 0.001 vs the sham group, *P < 0.05 and **P < 0.01 vs the model group.
In addition, we verified the mechanism of the protective effect of SAA in HBMECs after OGD injury in vitro. The levels of HIF-1α and VEGFA as well as the ratio of Src-Tyr416/Src were increased after OGD, while these increases were notably attenuated by SAA (10 μmol/L) preincubation (Fig. 7f–j). As shown in Fig. 7k–m, there was no obvious difference in total Rac1 protein expression after OGD injury. However, the phosphorylation levels of VAV2-Tyr172 were upregulated after OGD, which indicated that Rac1 was activated. Moreover, SAA preincubation downregulated the expression of VAV2-Tyr172 (Fig. 7k–m). PAK is the downstream protein of Rac1, and the ratio of p-PAK/PAK was increased by OGD. SAA preincubation inhibited the upregulation of p-PAK/PAK (Fig. 7n, o). These data suggested that SAA pretreatment inhibited the activation of Src signaling pathways in HBMECs after OGD injury, which was consistent with the in vivo results.
Discussion
It has been reported that cerebrovascular protection is very important for the treatment of acute ischemic stroke. Currently, the most effective treatment for acute ischemic stroke is thrombolytic therapy with tissue plasminogen activator (tPA), but tPA can cause vascular damage, leading to edema and HT [47], which is related to the disruption of the BBB [48]. However, the role of the cerebrovascular protection in the prevention of ischemic stroke is ignored. Although several kinds of drugs have been used in the clinic for the prevention of cerebrovascular diseases, including antiplatelet aggregation drugs [49], antihypertensive drugs [50], antioxidants [51], anticoagulants [52], and statins [53], none of these drugs specifically regulates the cerebrovascular injury.
In the present study, SAA pretreatment markedly prevented neurological deficits, intracerebral hemorrhage, BBB disruption and vascular endothelial dysfunction during ischemic injury and protected HBMECs against OGD-induced the loss of cell viability and degradation of TJ proteins. The protective effects of SAA were achieved via the inhibition of the Src pathway in vivo and in vitro. This is the first study to reveal that SAA has a preventive effect against cerebrovascular injury during ischemic stroke. Our results suggest that cerebrovascular protection is very important for the prevention of acute ischemic stroke, which provides a theoretical basis for the prevention of ischemic cerebrovascular diseases.
In this article, we used a novel rat thromboembolic stroke model to evaluate the cerebrovascular protective effect of SAA. At present, there are limited ideal animal models for studying stroke prevention besides the stroke-prone spontaneously hypertensive model [54]. The model we used is different from the existing embolization model [55–57], and thrombus and infarction development in our model is more similar to that of human ischemic stroke than the development of these pathological features in the embolization model [45, 54]. Twenty-four hours after ischemic stroke, focal neurological deficits occur [55]. Zhang et al. previously reported that intravenous injection of SAA (5, 10, or 20 mg/kg) improved neurological deficits in ischemia/reperfusion rats [56]. Our results demonstrated that preventive administration of SAA (10 mg/kg) prevented neurological deficits in ischemic stroke rats, which indicated that SAA had a neuroprotective effect.
Disruption of the BBB plays an extremely important role in the development of neurological dysfunction and intracerebral hemorrhage in ischemic stroke [57, 58]. The degradation of TJ proteins, including claudins, occludin, zonula occludens and junction adhesion molecules [56, 59, 60], leads to the disruption of the BBB and the occurrence of hemorrhage [61, 62]. In our experiment, intracerebral hemorrhage was observed in the model and aspirin groups after ischemic stroke, while preventive SAA administration reduced the occurrence of hemorrhage and ameliorated intracerebral hemorrhage. Our preliminary results also showed that pretreatment with SAA appreciably reduced the cerebral infarction area and cerebral edema after acute ischemic stroke [37]. In the present study, BBB permeability and TJ protein expression were evaluated by assessing Evans Blue leakage and by Western blotting. The data clearly showed that pretreatment with SAA maintained BBB permeability and dramatically reversed the degradation of TJ proteins during ischemia and OGD injury.
The endothelium is the central unit of the BBB [63]. Endothelial dysfunction not only represents a marker of ischemic cerebrovascular disease [64] but also may exacerbate disease and impact the ultimate degree of tissue damage in acute ischemic stroke [65]. Therefore, protecting the cerebrovascular endothelium is very important for cerebrovascular protection. Previous studies have shown that SAA has a protective effect against diabetic vascular endothelial dysfunction [66] and inhibits endothelial dysfunction and vascular remodeling in spontaneously hypertensive rats [67]. Our study demonstrated that SAA could regulate the protein expression of VWF, ADAMTS13, and Fibulin-5, which indirectly reflect the functional changes in the cerebrovascular endothelium [68–70]. These results indicated that preventive SAA administration protected the cerebrovascular endothelium during acute ischemic stroke. However, the protective effect of SAA on the cerebrovascular endothelium is not clear.
VEGF is a multifunctional cytokine that regulates diverse biological functions in endothelial cells [71]. The nonreceptor tyrosine kinases of the Src family are involved in the increase in BBB permeability induced by VEGF, as shown in Fig. 8 [72, 73]. Yang et al. reported that SAA inhibited Ang II-induced proliferation of human umbilical vein endothelial cells by reducing the expression levels of phospho-Src and phospho-Akt [74]. However, the specific mechanism underlying the protective effect of SAA on the BBB through Src signaling has not yet been clarified. Our study showed that SAA pretreatment protected BBB integrity by inhibiting the Src signaling pathway in vivo and in vitro. The regulatory mechanism of SAA is related to the protein–protein interactions mediated by the SH2 domains of the Src family kinases Src and Lck [75]. In addition, MMPs are closely associated with BBB disruption [76] and hemorrhage [77] in acute ischemic stroke. Jiang et al. reported that the activity of MMP-2/-9 is related to the Src signaling pathway [78]. Our results also indicated that preventive SAA administration obviously alleviated the BBB injury induced by MMP-9/MMP-2 upregulation via the Src signaling pathway. In short, our results indicated that SAA protected the BBB by inhibiting the activation of the VEGF-Src-MMP pathway during ischemic stroke and OGD injury (Fig. 8).
Fig. 8. Schematic diagram of the protective effect of SAA on vascular permeability via inhibition of VEGFA-Src-MMP signaling pathway activation during acute ischemic stroke.
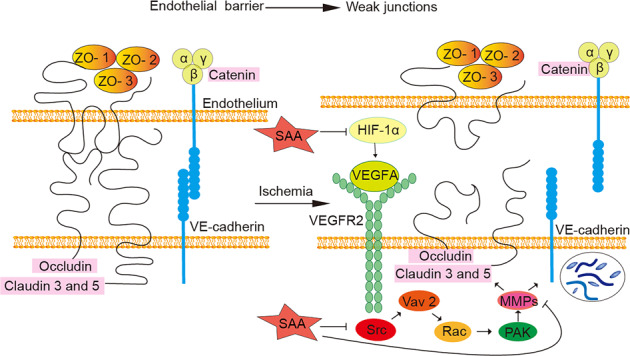
The endothelial barrier is maintained by the integrity of TJs and adherens junctions. During acute ischemic stroke, the expression of VEGFA increases obviously, and VEGFA binds to its receptor VEGFR-2. This receptor causes the sequential activation of VAV2-Tyr172, Rac, and PAK through Src. Activation of PAK increases the activity of MMPs, leading to VE-cadherin internalization and a subsequent increase in vascular permeability (VE-cadherin is an important factor in the formation of the vascular endothelial cell adhesion junction complex, which regulates the opening and closing of the endothelial barrier [23]). Upregulation of MMP protein expression also induces the degradation of TJ proteins. SAA protects the BBB by inhibiting the activation of the VEGFA-Src-MMP signaling pathway during acute ischemic stroke.
In addition, the blood and the functional status of blood vessels should be considered after preventive treatment. After ischemic stroke, the blood is in a high viscosity and agglutination state, and cerebrovascular function is disrupted. Thrombosis- and vasoconstriction-related molecules are also disordered in ischemic brain tissue [79, 80]. Pretreatment with SAA had a mild effect on vascular function and the formation of blood clots.
Aspirin is a classic antiplatelet drug that exerts antithrombotic effects and is widely used in clinical practice [49, 79]. The benefit of aspirin for the secondary prevention of cerebrovascular diseases is well established [80]. However, due to serious adverse reactions, such as cerebral hemorrhage and gastrointestinal bleeding, the use of aspirin for primary prevention remains controversial [81–84]. Aspirin has a strong inhibitory effect on platelets, especially AA-induced platelet aggregation [9]. However, in terms of cerebrovascular protection, aspirin could not reverse neurobehavioral deficits or BBB damage and increased the risk of bleeding after ischemic stroke, as proven in our experiment.
Unlike aspirin treatment, pretreatment with SAA not only has a mild inhibitory effect on platelet aggregation [36, 37] but also has a protective effect against cerebrovascular endothelial cell damage caused by ischemia, which may be related to alleviation of hemorrhage. Our results suggest that the prevention of cerebrovascular injury is as important as inhibiting thrombosis for the prevention of ischemic stroke and that SAA improves cerebrovascular function and alleviates thrombosis. In summary, SAA is a potential drug development candidate that should undergo further preclinical evaluation for the prevention of ischemic cerebrovascular disease.
Conclusions
In conclusion, our present study showed that SAA prevents cerebrovascular endothelial injury caused by acute ischemic stroke by inhibiting the Src signaling pathway. Pretreatment with SAA is a potential therapeutic strategy for the prevention of ischemic stroke.
Acknowledgements
This study was supported by National Key Research and Development Plan (2016YFC1000905); Beijing Municipal Natural Science Foundation (7182113); National Major Scientific and Technological Special Project for “Significant New Drugs Development” (2018ZX09711001–009–009); and CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-3–007, 2017-I2M-1–010).
Author contributions
GHD, LLK, and CDL designed the research; CDL and NNL performed the research; HGY and GDM contributed new reagents or analytic tools; CDL and SZ analyzed the data; CDL wrote the manuscript; and CDL, LLK, and GHD finalized the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Ling-lei Kong, Email: konglinglei@imm.ac.cn.
Guan-hua Du, Email: dugh@imm.ac.cn.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics-2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao B, Yuan Q, Hou JB, Xia ZY, Zhan LY, Li M, et al. Inhibition of HDAC3 ameliorates cerebral ischemia reperfusion injury in diabetic mice in vivo and in vitro. J Diabetes Res. 2019;2019:8520856. doi: 10.1155/2019/8520856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Randolph SA. Ischemic stroke. Workplace Health Saf. 2016;64:444. doi: 10.1177/2165079916665400. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation. 2017;135:e146–603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishida C, Uauy R, Kumanyika S, Shetty P. The joint WHO/FAO expert consultation on diet, nutrition and the prevention of chronic diseases: process, product and policy implications. Public Health Nutr. 2004;7:245–50.. doi: 10.1079/phn2003592. [DOI] [PubMed] [Google Scholar]
- 6.Kotseva K, Wood D, De Backer G, De Bacquer D, Pyörälä K, Keil U. Cardiovascular prevention guidelines in daily practice: a comparison of EUROASPIRE I, II, and III surveys in eight European countries. Lancet. 2009;373:929–40.. doi: 10.1016/S0140-6736(09)60330-5. [DOI] [PubMed] [Google Scholar]
- 7.Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren M, et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The fifth joint task force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of nine societies and by invited experts) Eur Heart J. 2012;33:1635–701.. doi: 10.1093/eurheartj/ehs092. [DOI] [PubMed] [Google Scholar]
- 8.Kim J, Thayabaranathan T, Donnan GA, Howard G, Howard VJ, Rothwell PM, et al. Global stroke statistics 2019. Int J Stroke. 2020;15:819–38. [DOI] [PubMed]
- 9.Ma N, Liu XW, Yang YJ, Shen DS, Zhao XL, Mohamed I, et al. Evaluation on antithrombotic effect of aspirin eugenol ester from the view of platelet aggregation, hemorheology, TXB2/6-keto-PGF1α and blood biochemistry in rat model. BMC Vet Res. 2016;12:108. doi: 10.1186/s12917-016-0738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu C, Qi D, Lian W, Li QZ, Li HJ, Fan HY. Effects of danshensu on platelet aggregation and thrombosis: in vivo arteriovenous shunt and venous thrombosis models in rats. PLoS One. 2014;9:e110124. doi: 10.1371/journal.pone.0110124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohr JP, Thompson JL, Lazar RM, Levin B, Sacco RL, Furie KL, et al. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med. 2001;345:1444–51.. doi: 10.1056/NEJMoa011258. [DOI] [PubMed] [Google Scholar]
- 12.Mtwesi V, Amit G. Stroke prevention in atrial fibrillation: the role of oral anticoagulation. Med Clin North Am. 2019;103:847–62.. doi: 10.1016/j.mcna.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Tomaiuolo M, Brass LF, Stalker TJ. Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Inter Cardiol Clin. 2017;6:1–12. doi: 10.1016/j.iccl.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmalbach B, Stepanow O, Jochens A, Riedel C, Deuschl G, Kuhlenbaumer G. Determinants of platelet-leukocyte aggregation and platelet activation in stroke. Cerebrovasc Dis. 2015;39:176–80.. doi: 10.1159/000375396. [DOI] [PubMed] [Google Scholar]
- 15.Stein-Merlob AF, Hara T, McCarthy JR, Mauskapf A, Hamilton JA, Ntziachristos V, et al. Atheroma susceptible to thrombosis exhibit impaired endothelial permeability in vivo as assessed by nanoparticle-based fluorescence molecular imaging. Circ Cardiovasc Imaging. 2017;10:e005813. [DOI] [PMC free article] [PubMed]
- 16.Hu X, De Silva TM, Chen J, Faraci FM. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ Res. 2017;120:449–71.. doi: 10.1161/CIRCRESAHA.116.308427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernstein DL, Gajghate S, Reichenbach NL, Winfield M, Persidsky Y, Heldt NA, et al. let-7g counteracts endothelial dysfunction and ameliorating neurological functions in mouse ischemia/reperfusion stroke model. Brain Behav Immun. 2020;87:543–55. [DOI] [PMC free article] [PubMed]
- 18.Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008;7:476–84.. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joutel A, Monet-Lepretre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120:433–45.. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke. 2004;35:2220–5. doi: 10.1161/01.STR.0000138023.60272.9e. [DOI] [PubMed] [Google Scholar]
- 21.Zan L, Zhang X, Xi Y, Wu H, Song Y, Teng G, et al. Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience. 2014;262:118–28.. doi: 10.1016/j.neuroscience.2013.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–94.. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–34.. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Sun L, Ding GB, Chen L, Jiang L, Wang J, et al. Oxygen-glucose deprivation/reoxygenation induces human brain microvascular endothelial cell hyperpermeability via ve-cadherin internalization: roles of RhoA/ROCK2. J Mol Neurosci. 2019;69:49–59. doi: 10.1007/s12031-019-01326-8. [DOI] [PubMed] [Google Scholar]
- 25.Lian-Niang L, Rui T, Wei-Ming C. Salvianolic acid A, a new depside from roots of Salvia miltiorrhiza. Planta Med. 1984;50:227–8. doi: 10.1055/s-2007-969684. [DOI] [PubMed] [Google Scholar]
- 26.Chien MY, Chuang CH, Chern CM, Liou KT, Liu DZ, Hou YC, et al. Salvianolic acid A alleviates ischemic brain injury through the inhibition of inflammation and apoptosis and the promotion of neurogenesis in mice. Free Radic Biol Med. 2016;99:508–19.. doi: 10.1016/j.freeradbiomed.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Mao K, Shu W, Qiu Q, Gu Q, Wu X. Salvianolic acid A protects retinal pigment epithelium from OX-LDL-induced inflammation in an age-related macular degeneration model. Discov Med. 2017;23:129–47. [PubMed] [Google Scholar]
- 28.Yuan X, Xiang Y, Zhu N, Zhao X, Ye S, Zhong P, et al. Salvianolic acid A protects against myocardial ischemia/reperfusion injury by reducing platelet activation and inflammation. Exp Ther Med. 2017;14:961–6. doi: 10.3892/etm.2017.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Song JK, Yan R, He GR, Zhang X, Zhou QM, et al. Salvianolic acid A alleviate the brain damage in rats after cerebral ischemia-reperfusion through Nrf2/HO-1 pathway. Yao Xue Xue Bao. 2016;51:1717–23. [PubMed] [Google Scholar]
- 30.Qian W, Wang Z, Xu T, Li D. Anti-apoptotic effects and mechanisms of salvianolic acid A on cardiomyocytes in ischemia-reperfusion injury. Histol Histopathol. 2019;34:223–31. doi: 10.14670/HH-18-048. [DOI] [PubMed] [Google Scholar]
- 31.Xu T, Wu X, Chen Q, Zhu S, Liu Y, Pan D, et al. The anti-apoptotic and cardioprotective effects of salvianolic acid a on rat cardiomyocytes following ischemia/reperfusion by DUSP-mediated regulation of the ERK1/2/JNK pathway. PLoS One. 2014;9:e102292. doi: 10.1371/journal.pone.0102292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du G, Zhang J. Protective effects of salvianolic acid A against impairment of memory induced by cerebral ischemia-reperfusion in mice. Chin Med J. 1997;110:65–8. [PubMed] [Google Scholar]
- 33.Jiao CX, Zhou H, Yang CX, Ma C, Yang YX, Mao RR, et al. Protective efficacy of a single salvianolic acid A treatment on photothrombosis-induced sustained spatial memory impairments. Neuropsychiatr Dis Treat. 2017;13:1181–92.. doi: 10.2147/NDT.S127094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahmood Q, Wang GF, Wu G, Wang H, Zhou CX, Yang HY, et al. Salvianolic acid A inhibits calpain activation and eNOS uncoupling during focal cerebral ischemia in mice. Phytomedicine. 2017;25:8–14. doi: 10.1016/j.phymed.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Huang ZS, Zeng CL, Zhu LJ, Jiang L, Li N, Hu H. Salvianolic acid A inhibits platelet activation and arterial thrombosis via inhibition of phosphoinositide 3-kinase. J Thromb Haemost. 2010;8:1383–93.. doi: 10.1111/j.1538-7836.2010.03859.x. [DOI] [PubMed] [Google Scholar]
- 36.Fan HY, Fu FH, Yang MY, Xu H, Zhang AH, Liu K. Antiplatelet and antithrombotic activities of salvianolic acid A. Thromb Res. 2010;126:e17–22. doi: 10.1016/j.thromres.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Wang HG, Kong LL, Wang R, Chen YX, Yang SL, Zhao XY, et al. Comparative study on antithrombotic effects of Salvianolic acid A and aspirin. Yao Xue Xue Bao. 2019;54:301–7. [Google Scholar]
- 38.Ma YZ, Li L, Song JK, Niu ZR, Liu HF, Zhou XS, et al. A novel embolic middle cerebral artery occlusion model induced by thrombus formed in common carotid artery in rat. J Neurol Sci. 2015;359:275–9. doi: 10.1016/j.jns.2015.09.362. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–8. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 40.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 41.Barez-Lopez S, Bosch-Garcia D, Gomez-Andres D, Pulido-Valdeolivas I, Montero-Pedrazuela A, Obregon MJ, et al. Abnormal motor phenotype at adult stages in mice lacking type 2 deiodinase. PLoS One. 2014;9:e103857. doi: 10.1371/journal.pone.0103857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fiorelli M, Bastianello S, von Kummer R, del Zoppo GJ, Larrue V, Lesaffre E, et al. Hemorrhagic transformation within 36 h of a cerebral infarct: relationships with early clinical deterioration and 3-month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke. 1999;30:2280–4. doi: 10.1161/01.str.30.11.2280. [DOI] [PubMed] [Google Scholar]
- 43.Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–99.. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denorme F, Martinod K, Vandenbulcke A, Denis CV, Lenting PJ, Deckmyn H, et al. The von willebrand factor A1 domain mediates thromboinflammation, aggravating ischemic stroke outcome in mice. Haematologica. 2020; 10.3324/haematol.2019.241042. [DOI] [PMC free article] [PubMed]
- 45.Denorme F, Langhauser F, Desender L, Vandenbulcke A, Rottensteiner H, Plaimauer B, et al. ADAMTS13-mediated thrombolysis of t-PA-resistant occlusions in ischemic stroke in mice. Blood. 2016;127:2337–45.. doi: 10.1182/blood-2015-08-662650. [DOI] [PubMed] [Google Scholar]
- 46.Spencer JA, Hacker SL, Davis EC, Mecham RP, Knutsen RH, Li DY, et al. Altered vascular remodeling in fibulin-5-deficient mice reveals a role of fibulin-5 in smooth muscle cell proliferation and migration. Proc Natl Acad Sci U S A. 2005;102:2946–51.. doi: 10.1073/pnas.0500058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishrat T, Soliman S, Guan W, Saler M, Fagan SC. Vascular protection to increase the safety of tissue plasminogen activator for stroke. Curr Pharm Des. 2012;18:3677–84.. doi: 10.2174/138161212802002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kastrup A, Gröschel K, Ringer TM, Redecker C, Cordesmeyer R, Witte OW, et al. Early disruption of the blood-brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke. 2008;39:2385–7. doi: 10.1161/STROKEAHA.107.505420. [DOI] [PubMed] [Google Scholar]
- 49.Duceppe E, Mrkobrada M, Thomas S, Devereaux PJ. Role of aspirin for prevention and treatment of perioperative cardiovascular events. J Thromb Haemost. 2015;13(Suppl 1):S297–303. doi: 10.1111/jth.12975. [DOI] [PubMed] [Google Scholar]
- 50.Julius S, Nesbitt SD, Egan BM, Weber MA, Michelson EL, Kaciroti N, et al. Feasibility of treating prehypertension with an angiotensin-receptor blocker. N Engl J Med. 2006;354:1685–97.. doi: 10.1056/NEJMoa060838. [DOI] [PubMed] [Google Scholar]
- 51.Tousoulis D, Antoniades C, Tountas C, Bosinakou E, Kotsopoulou M, Toutouzas P, et al. Vitamin C affects thrombosis/ fibrinolysis system and reactive hyperemia in patients with type 2 diabetes and coronary artery disease. Diabetes Care. 2003;26:2749–53.. doi: 10.2337/diacare.26.10.2749. [DOI] [PubMed] [Google Scholar]
- 52.Dalle Carbonare L, Mottes M, Brunelli A, Deiana M, Cheri S, Suardi S, et al. Effects of oral anticoagulant therapy on gene expression in crosstalk between osteogenic progenitor cells and endothelial cells. J Clin Med. 2019;8:329. [DOI] [PMC free article] [PubMed]
- 53.Żółciński M, Cieśla-Dul M, Potaczek DP, Undas A. Atorvastatin favorably modulates proinflammatory cytokine profile in patients following deep vein thrombosis. Thromb Res. 2013;132:e31–5. doi: 10.1016/j.thromres.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 54.Lan C, Chen X, Zhang Y, Wang W, Wang WE, Liu Y, et al. Curcumin prevents strokes in stroke-prone spontaneously hypertensive rats by improving vascular endothelial function. BMC Cardiovasc Disord. 2018;18:43. 10.1186/s12872-018-0768-6. [DOI] [PMC free article] [PubMed]
- 55.Neuhaus AA, Couch Y, Hadley G, Buchan AM. Neuroprotection in stroke: the importance of collaboration and reproducibility. Brain. 2017;140:2079–92.. doi: 10.1093/brain/awx126. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, Song JK, Zhang X, Zhou QM, He GR, Xu XN, et al. Salvianolic acid A attenuates ischemia reperfusion induced rat brain damage by protecting the blood brain barrier through MMP-9 inhibition and anti-inflammation. Chin J Nat Med. 2018;16:184–93.. doi: 10.1016/S1875-5364(18)30046-3. [DOI] [PubMed] [Google Scholar]
- 57.Yang C, Hawkins KE, Doré S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2019;316:C135–53.. doi: 10.1152/ajpcell.00136.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu X, Fu S, Liu Y, Luo H, Li F, Wang Y, et al. NDP-MSH binding melanocortin-1 receptor ameliorates neuroinflammation and BBB disruption through CREB/Nr4a1/NF-κB pathway after intracerebral hemorrhage in mice. J Neuroinflammation. 2019;16:192. doi: 10.1186/s12974-019-1591-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anasooya Shaji C, Robinson BD, Yeager A, Beeram MR, Davis ML, Isbell CL, et al. The tri-phasic role of hydrogen peroxide in blood-brain barrier endothelial cells. Sci Rep. 2019;9:133. 10.1038/s41598-018-36769-3. [DOI] [PMC free article] [PubMed]
- 60.Xia ZY, Luo C, Liu BW, Bian XQ, Li Y, Pang AM, et al. Shengui Sansheng Pulvis maintains blood-brain barrier integrity by vasoactive intestinal peptide after ischemic stroke. Phytomedicine. 2020;67:153158. 10.1016/j.phymed.2019.153158. [DOI] [PubMed]
- 61.Turner RJ, Sharp FR. Implications of mmp9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci. 2016;10:56. 10.3389/fncel.2016.00056. [DOI] [PMC free article] [PubMed]
- 62.Fredriksson L, Lawrence DA, Medcalf RL. tPA modulation of the blood-brain barrier: a unifying explanation for the pleiotropic effects of tPA in the CNS. Semin Thrombosis Hemost. 2017;43:154–68.. doi: 10.1055/s-0036-1586229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keaney J, Campbell M. The dynamic blood-brain barrier. FEBS J. 2015;282:4067–79.. doi: 10.1111/febs.13412. [DOI] [PubMed] [Google Scholar]
- 64.Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011;37:3–23. doi: 10.1111/j.1365-2990.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- 65.Cosentino F, Rubattu S, Savoia C, Venturelli V, Pagannonne E, Volpe M. Endothelial dysfunction and stroke. J Cardiovasc Pharmacol. 2001;38:S75–8. doi: 10.1097/00005344-200111002-00018. [DOI] [PubMed] [Google Scholar]
- 66.Yang XY, Qiang GF, Zhang L, Zhu XM, Wang SB, Sun L, et al. Salvianolic acid A protects against vascular endothelial dysfunction in high-fat diet fed and streptozotocin-induced diabetic rats. J Asian Nat Products Res. 2011;13:884–94.. doi: 10.1080/10286020.2011.598457. [DOI] [PubMed] [Google Scholar]
- 67.Teng F, Yin Y, Cui Y, Deng Y, Li D, Cho K, et al. Salvianolic acid A inhibits endothelial dysfunction and vascular remodeling in spontaneously hypertensive rats. Life Sci. 2016;144:86–93. doi: 10.1016/j.lfs.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 68.Banarjee R, Sharma A, Bai S, Deshmukh A, Kulkarni M. Proteomic study of endothelial dysfunction induced by AGEs and its possible role in diabetic cardiovascular complications. J Proteom. 2018;187:69–79. doi: 10.1016/j.jprot.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 69.Buchtele N, Schwameis M, Gilbert JC, Schorgenhofer C, Jilma B. Targeting von willebrand factor in ischaemic stroke: focus on clinical evidence. Thrombosis Haemost. 2018;118:959–78.. doi: 10.1055/s-0038-1648251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo J, Cheng C, Chen CS, Xing X, Xu G, Feng J, et al. Overexpression of fibulin-5 attenuates ischemia/reperfusion injury after middle cerebral artery occlusion in rats. Mol Neurobiol. 2016;53:3154–67.. doi: 10.1007/s12035-015-9222-2. [DOI] [PubMed] [Google Scholar]
- 71.Zachary I, Mathur A, Yla-Herttuala S, Martin J. Vascular protection: a novel nonangiogenic cardiovascular role for vascular endothelial growth factor. Arteriosclerosis Thrombosis Vasc Biol. 2000;20:1512–20.. doi: 10.1161/01.atv.20.6.1512. [DOI] [PubMed] [Google Scholar]
- 72.He YX, Liu J, Guo B, Wang YX, Pan X, Li D, et al. Src inhibitor reduces permeability without disturbing vascularization and prevents bone destruction in steroid-associated osteonecrotic lesions in rabbits. Sci Rep. 2015;5:8856. doi: 10.1038/srep08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han YW, Liu XJ, Zhao Y, Li XM. Role of Oleanolic acid in maintaining BBB integrity by targeting p38MAPK/VEGF/Src signaling pathway in rat model of subarachnoid hemorrhage. Eur J Pharmacol. 2018;839:12–20. doi: 10.1016/j.ejphar.2018.09.018. [DOI] [PubMed] [Google Scholar]
- 74.Yang LL, Li DY, Zhang YB, Zhu MY, Chen D, Xu TD. Salvianolic acid A inhibits angiotensin II-induced proliferation of human umbilical vein endothelial cells by attenuating the production of ROS. Acta Pharmacol Sin. 2012;33:41–8. doi: 10.1038/aps.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sperl B, Seifert MH, Berg T. Natural product inhibitors of protein-protein interactions mediated by Src-family SH2 domains. Bioorg Medicinal Chem Lett. 2009;19:3305–9. doi: 10.1016/j.bmcl.2009.04.083. [DOI] [PubMed] [Google Scholar]
- 76.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–95.. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 77.Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–6. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- 78.Jiang Q, Pan Y, Cheng Y, Li H, Liu D, Li H. Lunasin suppresses the migration and invasion of breast cancer cells by inhibiting matrix metalloproteinase-2/-9 via the FAK/Akt/ERK and NF-kappaB signaling pathways. Oncol Rep. 2016;36:253–62.. doi: 10.3892/or.2016.4798. [DOI] [PubMed] [Google Scholar]
- 79.Walker J, Cattaneo M, Badimon L, Agnelli G, Chan AT, Lanas A, et al. Highlights from the 2019 International Aspirin Foundation Scientific Conference, Rome, 28 June 2019: benefits and risks of antithrombotic therapy for cardiovascular disease prevention. Ecancermedicalscience. 2020;14:998. 10.3332/ecancer.2020.998. [DOI] [PMC free article] [PubMed]
- 80.Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–236.. doi: 10.1161/STR.0000000000000024. [DOI] [PubMed] [Google Scholar]
- 81.Patrono C, Baigent C. Role of aspirin in primary prevention of cardiovascular disease. Nat Rev Cardiol. 2019;16:675–86.. doi: 10.1038/s41569-019-0225-y. [DOI] [PubMed] [Google Scholar]
- 82.Zheng SL, Roddick AJ. Association of aspirin use for primary prevention with cardiovascular events and bleeding events: a systematic review and meta-analysis. JAMA. 2019;321:277–87. doi: 10.1001/jama.2018.20578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whitlock EP, Burda BU, Williams SB, Guirguis-Blake JM, Evans CV. Bleeding risks with aspirin use for primary prevention in adults: a systematic review for the U.S. preventive services task force. Ann Intern Med. 2016;164:826–35. doi: 10.7326/M15-2112. [DOI] [PubMed] [Google Scholar]
- 84.Xie W, Luo Y, Liang X, Lin Z, Wang Z, Liu M. The efficacy and safety of aspirin as the primary prevention of cardiovascular disease: an updated meta-analysis. Ther Clin Risk Manag. 2019;15:1129–40.. doi: 10.2147/TCRM.S198403. [DOI] [PMC free article] [PubMed] [Google Scholar]