

Abstract
Virus-induced asthma is prevalent among children, but its underlying mechanisms are unclear. Accumulated evidence indicates that early-life respiratory virus infection increases susceptibility to allergic asthma. Nonetheless, the relationship between systemic virus infections, such as enterovirus infection, and the ensuing effects on allergic asthma development is unknown. Early-life enterovirus infection was correlated with higher risks of allergic diseases in children. Adult mice exhibited exacerbated mite allergen-induced airway inflammation following recovery from EV-A71 infection in the neonatal period. Bone marrow-derived macrophages (BMDMs) from recovered EV-A71-infected mice showed sustained innate immune memory (trained immunity) that could drive naïve T helper cells toward Th2 and Th17 cell differentiation when in contact with mites. Adoptive transfer of EV-A71-trained BMDMs induced augmented allergic inflammation in naïve recipient mice, which was inhibited by 2-deoxy-D-glucose (2-DG) pretreatment, suggesting that trained macrophages following enterovirus infection are crucial in the progression of allergic asthma later in life.
Keywords: trained immunity, allergy, asthma
Subject terms: Innate immune cells, Mucosal immunology
Introduction
Bronchial asthma is a chronic airway inflammatory disorder characterized by bronchial epithelial inflammation, increased mucus secretion and bronchial hyperresponsiveness that is often exacerbated by environmental pollutants, allergen exposure and respiratory virus infection.1 The roles of viral infection in the development and acute exacerbation of pediatric asthma have been extremely scrutinized in various birth cohorts worldwide,2,3 with the results suggesting that early rhinovirus (RV)-induced wheezing is a major risk factor for asthma later in life, especially in children with atopic features.4,5 Moreover, young children who develop severe bronchiolitis due to respiratory syncytial virus (RSV) infection have a greater risk for ensuing asthma development.6 The typical features of virally induced asthma exacerbations are impaired synthesis of antiviral interferons and excess type 2 immune inflammation.7 Specifically, virus-infected bronchial epithelial cells in asthmatic patients induce epithelial-derived cytokines (alarmins), such as thymic stromal lymphopoietin (TSLP), interleukin (IL)-25 and IL-33. They initiate type 2 immune responses and can activate and trigger the production of IL-13 by type 2 innate lymphoid cells (ILC2s) and IL-4, IL-5, IL-9, and IL-17 from active T cells.8 Th2-dependent alternatively activated (M2) macrophages and Th17-dependent neutrophilic inflammation also worsen allergic inflammation.9 Nonetheless, the underlying mechanism that leads to the type 2 inflammation-prone nature of bronchial epithelial cells in asthma is still unclear. Furthermore, this mechanism cannot explain why virus infection in early life leads to asthma.
Apart from RV and RSV, which directly infect the bronchial epithelium in the lower respiratory tract and lead to wheezing and asthma exacerbation, enteroviruses, which target the gastrointestinal tract in children, also cause asthma. Taiwan has experienced various epidemics of enterovirus 71 (EV-A71) infections since 1998, with this virus causing outbreaks of hand, foot, and mouth disease (HFMD) and severe neurological involvement in young children.10 Three studies from the national health database in Taiwan11–13 indicated that enterovirus infection was substantially correlated with an elevated asthma incidence (hazard ratio [HR] = 1.65; 95% confidence interval [C.I.] = 1.60–1.71).12 The mean latency period between initial enterovirus infection and asthma onset was 2.77 years.13 The overall adjusted hazard ratio (AHR) of asthma was 1.48-fold higher in the enterovirus cohort than in the non-enterovirus cohort (95% C.I. = 1.45–1.50). The risk of asthma was increased with early-life (<1 year old) enterovirus infection, particularly in children with a higher frequency of asthma admission (>5 per year).13 However, the temporal effect between enterovirus infection and the development of asthma or other allergic disorders is still unknown. Furthermore, the underlying mechanism that links extrapulmonary virus infection and bronchial asthma development has not been studied.
In recent years, the innate immune system has been shown to build immunological memory of past infections and foreign encounters,14,15 leading to an augmented immune response to a secondary stimulus via metabolic and epigenetic reprogramming of innate immune cells. Importantly, trained immune cells adopt an activated phenotype that can respond strongly to a wide array of subsequent similar or unrelated stimuli for a prolonged period. Nonetheless, trained immunity is a double-edged sword. The uncontrolled hyperactivated immune response conferred by trained immunity can be harmful, leading to a range of chronic inflammatory disorders.16 Here, we described our population-based national health insurance data indicating that early-life enterovirus infection was correlated with higher risks of allergic diseases in children. We also developed a clinically relevant model of EV-A71 infection in neonatal mice (<2 weeks old) and showed that extrapulmonary EV-A71 infection in early life amplified allergen-induced airway inflammation when mice were sensitized and challenged with house dust mites, suggesting that trained immunity in myeloid cells following EV-A71 infection has significant roles in the development and progression of asthma later in life.
Results
Early-life enterovirus infection is correlated with the development of childhood eczema, asthma, and allergic rhinitis
A total of 61,225 live births from 1 January 2003 to 31 December 2005 were identified from two Longitudinal Health Insurance Databases (LHIDs) in Taiwan. The primary endpoint was whether enterovirus infection in the first 3 years was related to participants developing allergic diseases during years 4–8. The detailed flow chart of the inclusion and exclusion criteria for the study cohort is included in Fig. S1. The distributions of clinical and demographic characteristics between children with or without enterovirus infection in the first 3 years of life are outlined in Table S1. We found higher percentages of comorbid conditions and healthcare utilization in the group of children with enterovirus infection in early life than in the uninfected group. Table 1 outlines the temporal relationships between enterovirus infection in early life and the development of the three analyzed allergic diseases (atopic dermatitis, asthma, and allergic rhinitis) in childhood. Significant relationships were found between early-life enterovirus infection and asthma or allergic rhinitis development after adjusting for important covariates (AHR, 1.13 and 95% C.I., 1.07–1.20, for asthma; AHR, 1.13 and 95% C.I., 1.08–1.19, for allergic rhinitis). The cumulative incidence rates of these allergic disorders for the children with or without enterovirus infection in early life are outlined in Fig. 1. The cumulative incidence rates of atopic dermatitis, asthma and allergic rhinitis were substantially higher in the enterovirus infection group than in the uninfected group (plogrank < 6 × 10−3 for atopic dermatitis; plogrank < 10−3 for asthma; plogrank < 10−3 for allergic rhinitis). Table S2 outlines the findings of subgroup analyses on the three investigated allergic diseases stratified by sex, geographical area at birth, different comorbid physical conditions, acetaminophen use or healthcare system utilization. Comparable correlations between early-life enterovirus infection and the three allergic diseases across these strata were observed, showing that these factors did not alter the temporal relationships in our study cohort.
Table 1.
Incidence rates and hazard ratio of allergic diseases in children with or without enterovirus infection in early life
| Non-enterovirus-infected group | Enterovirus-infected group | Adjusted HRa (95% CI) | |||||
|---|---|---|---|---|---|---|---|
| Number of events | Person-years | Incidence rate (per 1000 person-years) | Number of events | Person-years | Incidence rate (per 1000 person-years) | ||
| Atopic dermatitis | 558 | 80242.6 | 6.95 | 717 | 88169.99 | 8.13 | 1.09 (0.97–1.23) |
| Asthma | 2262 | 70297.48 | 32.18 | 2940 | 70364.48 | 41.78 | 1.13 (1.07–1.20)* |
| Allergic rhinitis | 3835 | 64142.21 | 59.79 | 4910 | 62676.57 | 78.34 | 1.13 (1.08–1.19)* |
Bold values indicate statistical significance p < 0.05
aAdjusted for age, sex, urbanisation level, upper respiratory tract infection, lower respiratory tract infection, otitis media, bronchitis, congenital anomalies, infectious diarrhoea, infectious colitis, enteritis and gastroenteritis, acetaminophen, outpatient visits, and inpatient visit
p < 0.0001
Fig. 1.

Early-life enterovirus infection is correlated with childhood atopic dermatitis, asthma, and allergic rhinitis development. Kaplan–Meier analysis of the cumulative incidence rates of atopic dermatitis, asthma and allergic rhinitis in children with or without early-life enterovirus infection
EV-A71 infection in newborn mice causes typical symptoms in the first week and resolves in the second week following infection
To establish an early-life EV-A71 infection model in mice, 14-day-old BALB/c mice underwent either mock infection or EV-A71 infection with 3 × 106 plaque forming units (pfu) via intraperitoneal injection and were followed up for another 14 days (protocol 1, Fig. 2a). The low inoculum utilized for the current study was based on our earlier reports on EV-71A-infected mice.17,18 The disease scores largely increased at 4 days postinfection (d.p.i.), and disease symptoms slowly improved until 14 d.p.i. (Fig. 2b). The body weights of the infected mice were reduced to the baseline weight at 4 d.p.i. but increased at 14 d.p.i. (Fig. 2c). Overall, the survival rate of the low-dose EV-A71 infection mouse model was ~80% at 14 d.p.i. (Fig. 2d). Infectious virus was detected in the blood at 3 × 103, 1 × 103, and 2 × 102 pfu/ml at 1, 3, and 5 d.p.i., respectively (data not shown). As expected, the viral titers in central nervous tissues (the brain without the brainstem region) were higher than those in the lungs (peripheral organ), but they were almost undetectable at 14 d.p.i. in both brain tissue and lung tissue (Fig. 2e). Lung function measurements were performed with mice at 1, 3, and 5 days following EV-A71 infection, and mock-infected mice of the same age were used as controls. No significant difference in airway resistance (Rrs) following EV-A71 infection was observed between the infected and mock-infected mice (Fig. 2f). In contrast, lung tissue elastane (Ers) levels were significantly increased in the infected mice at 5 d.p.i. in comparison with the mock-infected mice in response to acetyl-β-methylcholine chloride (methacholine) challenge (Fig. 2g). In bronchoalveolar lavage fluid (BALF) samples, the levels of the cytokines IFN-γ and TNF-α were apparently increased following EV-A71 infection (Fig. 2h). In lung tissues from EV-A71–infected mice, the inflammatory cell infiltration level was increased in the peribronchial and alveolar regions. Mucus hypersecretion and EV-A71 antigens detected by an anti-EV-A71 antibody (brown staining) were observed in the bronchial epithelium starting from 1 to 5 d.p.i. (Fig. 2i), suggesting that low-dose EV-A71 infection in the newborn period causes typical EV-A71 neurological symptoms and mild lung inflammation for 1 week. EV-A71 infection slowly resolved, and the EV-A71 antigen was barely detected during the second week following infection.
Fig. 2.

Early-life infection with EV-A71 causes typical inflammation but resolves within 2 weeks following infection. A Protocol 1 experimental scheme. Fourteen-day-old newborn mice underwent mock infection or EV-A71 infection through intraperitoneal injection. B The disease scores, C body weights, and D survival rates of mice were monitored every 2 days for 2 weeks following viral infection. Disease scores were defined as follows: 0, healthy; 1, fur loss, wasting, or ruffled fur; 2, limb weakness; 3, paralysis in only 1 limb; 4, paralysis in 2 to 4 limbs; and 5, death. The findings represent pooled data from two independent experiments. E Viral titers in mouse brain and lung tissues were monitored at 1, 3, 5, and 14 days postinfection (d.p.i.) (n = 6–8 mice). F–I Samples were collected from mice that underwent mock infection or EV-A71 infection at 1, 3, and 5 d.p.i. F Airway resistance (Rrs) and G elastane (Ers) levels in response to increasing doses of aerosolized methacholine were calculated using a flexiVent FX system (n = 6–10 mice, **p < 0.01, compared with the mock group by two-way ANOVA with the Bonferroni posttest). H TNF-α and IFN-γ levels in the BALF were determined using ELISA (n = 6–10 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). I Lung histology. Lung sections were stained with H&E, PAS, or an anti-EV-A71 antibody. The findings represent pooled data from two independent experiments
EV-A71–infected mice exhibit augmented airway inflammation following HDM sensitization and challenge
At 35 days of age (3 weeks following virus infection), EV-A71–infected mice and mock-infected mice were subjected to HDM intranasal (i.n.) sensitization and intratracheal (i.t.) challenge (protocol 2, Fig. 3a). Total IgE (Fig. 3b) and HDM-specific IgE (Fig. 3c) concentrations were significantly increased in the EV-A71-infected and HDM-sensitized mice (EV-A71 + HDM) in comparison with the mock-infected and HDM-sensitized (Mock + HDM) mice and EV-A71-infected and PBS-treated (EV-A71 + PBS) control mice. Airway resistance (Rrs) (Fig. 3d) and elastance (Ers) (Fig. 3e) levels were notably and dramatically increased in response to methacholine challenge in EV-A71 + HDM mice compared to Mock + HDM and EV-A71 + PBS mice. In BALF samples collected 2 days after HDM i.t. challenge, there were substantially higher numbers of total inflammatory cells and eosinophils in the samples from EV-A71 + HDM mice than in those from Mock + HDM mice (Fig. 3f). By lung histological examination, substantially increased inflammatory cell infiltration, exacerbated eosinophilia, and heavy staining for mucus production in the bronchial epithelium and peribronchial areas were observed in Mock + HDM and EV-A71 + HDM mice compared to Mock + PBS and EV-A71 + PBS mice (Fig. 3g). Remarkably, higher levels of TARC were observed in EV-A71 + HDM mice than in Mock + HDM mice (Fig. 3h). By analysis of single-cell suspensions generated from the lungs, we discovered significantly increased levels of Th2 and Th17 cell infiltration in the lungs of EV-A71 + HDM mice compared with those of Mock + HDM mice (Fig. 3i), suggesting that mice with EV-A71 infection in early life exhibit amplified allergen-induced airway inflammation in comparison with mock-infected mice.
Fig. 3.

Early-life infection with EV-A71 enhances allergic asthma responses to HDM allergen. A Protocol 2 experimental scheme. Thirty-five-day-old EV-A71–infected or mock-infected mice were intranasally and intratracheally challenged with PBS or HDM allergen. B Total IgE and C HDM-specific IgE serum concentrations were determined using ELISA (n = 15 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). D Airway resistance (Rrs) and E elastane (Ers) levels in response to increasing doses of aerosolized methacholine were calculated using a flexiVent FX system (n = 15 mice, *p < 0.05 and **p < 0.01, two-way ANOVA with the Bonferroni posttest). F Total cell, eosinophil, neutrophil, lymphocyte, and macrophage counts in the BALF were determined (n = 15 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). G Lung histology. Lung sections were stained with H&E, PAS, or an anti-EV-A71 antibody. H TARC production in the BALF was evaluated using ELISA (n = 15 mice, *p < 0.05, one-way ANOVA with the Bonferroni multiple-comparison test). I IL-4+, IL-5+, IL-13+, and IL-17A+CD4+ T cell percentages in lung single-cell suspensions were determined using FACS (n = 15 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). The findings represent pooled data from three independent experiments
Innate immune memory (trained immunity) involves EV-A71–infected macrophages
To investigate why relatively early EV-A71 infection can augment HDM-induced airway inflammation in the lungs, we speculated that systemic virus infection may confer innate immune memory mediated by mucosal and bone marrow myeloid cells. Mouse BMDMs were inoculated with various concentrations of EV-A71 for 24 h. After resting for 6 days, the infected BMDMs were restimulated with 100 µg/ml HDM allergen (protocol 3, Fig. 4a). The canonical characteristics of trained immunity were observed in these cells, as indicated by increases in IL-6 and TNF-α cytokine production levels in a virus dose-dependent manner upon exposure to the same dose of HDM (Fig. 4b). Glycolytic levels were also determined to be increased based on increased lactate levels and glucose consumption in the culture supernatants of infected BMDMs at day 3, and the effect was sustained at day 7 following HDM allergen stimulation (Fig. 4c). Furthermore, when EV-A71–infected BMDMs were pretreated with different doses of inhibitors of trained immunity such as 2-DG (Figs. 4d), 5’-deoxy-5’-(methylthio) adenosine (MTA, Fig. S2A), and metformin (Fig. S2B), the concentrations of the cytokines IL-6 and TNF-α in culture supernatants were reduced in a dose-dependent manner following HDM stimulation. EV-A71-induced trained immunity in mucosal and systemic macrophages was also observed in an ex vivo study. Alveolar macrophages and BMDMs were isolated from mock-infected and EV-A71-infected mice at 35 days of age (3 weeks after virus infection). Following stimulation with HDM allergen for 48 or 72 h, alveolar macrophages from the EV-A71-infected group demonstrated increased IL-6 and TNF-α cytokine production in comparison with those from the mock-infected group (Fig. 5a). Furthermore, the EV-A71-infected group also demonstrated significantly reduced M1 but increased M2 cell percentages in the alveolar macrophage population after culture with HDM for 72 h (Fig. 5b). The BMDM culture system produced the same findings as those for alveolar macrophages. IL-6 and TNF-α cytokine levels were considerably increased in the EV-A71-infected group following HDM stimulation (Fig. 5c). These infected BMDMs also had reduced M1 and increased M2 cell percentages by flow cytometry analysis (Fig. 5d). When BMDMs from groups of EV-A71-infected asthmatic experimental study mice (protocol 2, Fig. 3a) were cocultured with naïve T cells with or without HDM, we observed that the percentages of activated Th17 and Th2 lymphocytes were markedly increased in the presence of HDM in comparison with the absence of HDM in the coculture (Fig. 5e). Furthermore, activated Th17 and Th2 cell percentages were highest when naïve T cells were cocultured with BMDMs from EV-A71 + HDM mice (Fig. 5e), revealing that EV-A71-trained macrophages were polarized into the M2 phenotype and maintained innate immune memory. Moreover, these trained myeloid cells could drive naïve T cell differentiation into Th2 and Th17 lymphocytes following allergen stimulation.
Fig. 4.

EV-A71-trained macrophages augment HDM-induced inflammation. A Trained immunity experimental scheme. BMDMs were preinfected with EV-A71 with or without inhibitors for 24 h. Following rest for 6 days, the cells were restimulated with HDM for 24 h. B Relative IL-6 and TNF-α production was determined for mock-infected and EV-A71-trained BMDMs (n = 6, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). C Kinetic changes in lactate production and glucose consumption in cells collected on days 1, 4, and 8 (n = 6, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). D Relative IL-6 and TNF-α production by cells incubated with increasing concentration of 2-DG (glycolysis inhibitor) was determined (n = 9, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). The findings represent pooled data from three independent experiments
Fig. 5.

Trained immunity in myeloid cells following EV-A71 infection induces type 2 inflammation upon stimulation with HDM allergen. A–D Alveolar macrophages and BMDMs were harvested from mock-infected and EV-A71–infected mice. The isolated cells were restimulated with HDM for 48 or 72 h ex vivo. A Relative IL-6 and TNF-α production and B M1 and M2 proportions in HDM-stimulated alveolar macrophages from mock-infected and EV-A71–infected mice were determined (n = 6–8 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). C Relative IL-6 and TNF-α production and D M1 and M2 proportions in HDM-stimulated BMDMs from mock-infected and EV-A71–infected mice were determined (n = 6–8 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). E Th2 and Th17 cells were evaluated in a coculture system containing naïve T cells and BMDMs from EV-A71–infected asthmatic experimental (protocol 2) study mice (n = 6 mice, *p < 0.05 and **p < 0.01, one-way ANOVA with the Bonferroni multiple-comparison test). The findings represent pooled data from two independent experiments
Adoptive transfer of EV-A71–trained BMDMs induces allergic inflammation and is blocked by 2-DG pretreatment
To investigate the role of the innate immune memory mediated by EV-A71-infected macrophages in HDM-induced airway inflammation, BMDMs underwent mock infection (EV−) or infection with EV-A71 (EV+) for 24 h; then, the cells were extensively washed with medium to remove infectious virus and rested for another 6 days. These cells were i.t. instilled into naïve mouse lungs, and the recipient mice were then treated with HDM allergen as described in the Methods section (protocol 4, Fig. 6a). We found that mock-infected BMDMs could not induce airway inflammation or asthma attack, while EV-A71-trained BMDMs could induce airway hyperresponsiveness with increases in Rrs (Fig. 6b) and Ers (Fig. 6c) levels in response to methacholine inhalation. Moreover, the findings also showed increased total IgE (Fig. 6d) and HDM-specific IgE (Fig. 6e) concentrations in the serum and elevated eosinophil (Fig. 6f) and TARC cytokine levels (Fig. 6g) in the BALF of the EV+ group. By lung histological examination, increased mucus production and inflammatory cell infiltration were observed in the bronchial epithelium and peribronchial regions of the EV+ group following HDM challenge (Fig. 6h). In contrast, pretreatment with 2-DG, a glucose molecule that is taken up by glucose transporters and blocks further glycolysis in cells participating in trained immunity, inhibited the innate immune memory of BMDMs induced by EV-A71 infection and led to a failure to illicit allergen-induced airway inflammation in HDM-sensitized and HDM-challenged mice (2-DG/EV+ group). These mice had no AHR elevation in response to methacholine (Fig. 6b, c). Furthermore, reduced total IgE (Fig. 6d) and HDM-specific IgE (Fig. 6e) concentrations in the serum were also observed. Reduced inflammatory cell infiltration and eosinophil (Fig. 6f), and TARC cytokine levels (Fig. 6g) were found in the BALF of mice in the 2-DG/EV+ group. Lung histological examination also demonstrated reduced mucus production and inflammatory cell infiltration levels in the bronchial epithelium and peribronchial regions of 2-DG/EV+ group mice following HDM challenge (Fig. 6h), suggesting that the inhibition of trained immunity to EV-A71 can relieve the development of allergen-induced asthma and weaken airway inflammation in this disease.
Fig. 6.

Adoptive transfer of EV-A71-trained BMDMs induces augmented airway allergic inflammation, which is blocked by pretreatment with 2-DG. A Protocol 3 experimental scheme. Mock-infected BMDMs (EV- group), 2-DG–treated mock-infected BMDMs (2-DG/EV- group), EV-A71–trained BMDMs (EV+ group) or 2-DG–treated EV-A71-trained BMDMs (2-DG/EV+ group) were intratracheally transferred into naïve mice. One day after adoptive transfer, allergic asthma was induced in these mice via intranasal and intratracheal challenge with HDM. B Airway resistance (Rrs) and C elastane (Ers) levels in response to increasing doses of aerosolized methacholine were measured using a flexiVent FX system (n = 6–8 mice, **p < 0.01, two-way ANOVA with the Bonferroni posttest). D Total IgE and E HDM-specific IgE serum concentrations were calculated using ELISA (n = 6–8 mice, *p < 0.05, one-way ANOVA with the Bonferroni multiple-comparison test). F Total cell, eosinophil, neutrophil, lymphocyte, and macrophage counts in the BALF were determined (n = 6–8 mice, *p < 0.05, one-way ANOVA with the Bonferroni multiple-comparison test). G TARC production in the BALF was evaluated using ELISA (n = 6–8 mice, *p < 0.05, one-way ANOVA with the Bonferroni multiple-comparison test). H Lung histology. Lung sections were stained with H&E (Scale bars = 100 μm). The findings represent pooled data from two independent experiments
Discussion
In this study, we discovered that enterovirus infections via the fecal–oral route in early life are correlated with allergic disease development in children. We also demonstrated that extrapulmonary EV-A71 infection at a young age could increase the effect of allergen-induced airway inflammation in the lungs of previously virus-infected and recovered mice. These findings are different (infection via the fecal–oral route) from previous results for experimental virus-induced asthma established by directly instilling respiratory viruses,19 such as influenza, RSV and RV, that bind to specific viral receptors in the bronchial epithelium and replicate in bronchial epithelial cells, as the earlier work with respiratory viruses suggests that the disruption of the bronchial epithelial barrier and reduced gap junction integrity are due to respiratory virus infection leading to an inflammatory cascade with exposure of tissue Toll-like receptors to pathogen-associated molecular patterns or environmental allergens in asthma.19 Reports of enterovirus infection-induced asthma are rare, although enteroviruses can also infect the respiratory tract. Respiratory enterovirus D68 (EV-D68) has been reported to be related to virus-induced asthma exacerbation. Unlike all other enteroviruses, EV-D68 displays acid lability and a lower optimum growth temperature, both characteristic features of human rhinoviruses.20 On the other hand, the respiratory system is usually spared in EV-A71 infection except in extremely complicated cases of autonomic nervous system dysregulation in the brainstem, resulting in pulmonary edema.10,21 In our animal study, EV-A71 infection of the bronchial epithelium and lungs was also present at low levels in EV-A71-infected newborn mice, despite the mice showing the classic neurological manifestations of a hunched posture and hindlimb paralysis (Fig. 1). The symptoms started at 1 d.p.i. and resolved within 2 weeks following infection in surviving mice. Furthermore, the Rrs against methacholine was still low, and bronchial epithelial structures were intact, with mock-infected mice and surviving EV-A71–infected mice appearing similar. EV-A71 was also not detectable in the lungs or blood of the surviving infected mice following 2 weeks of infection. Thus, the supplemented Th2 and Th17 immune responses to HDM allergen and allergic airway inflammation in surviving EV-A71–infected mice cannot be elucidated by the epithelial barrier damage theory of asthma due solely to respiratory viruses. In fact, our findings indicated functional and phenotypic changes in macrophages in EV-A71-infected mice, which increased mouse susceptibility to allergen stimulation without predisposition to bronchial epithelial cell dysfunction or inflammation.
The mechanism of enhanced allergic responsiveness following early childhood respiratory virus infection is still unclear.22 Various groups have established mouse models of virus-enhanced responses to later allergen exposure using adult or neonatal mice.23–26 Infection of neonatal mice (7–8 days old) with RSV,24 HRV,25 or influenza26 induced rapid IL-13, IL-4, and TSLP production in the lungs, which preceded enhanced responses to allergens administered 7–10 days following infection. One of the theories for the enhanced response to inhaled allergens is that allergen-associated molecular patterns (AAMPs)27 may stimulate pattern recognition receptors on virus-induced alternatively activated macrophages (AAMs, M2 phenotype).28 AAMs amplify allergic airway inflammation and the asthma response by generating different interleukins and chemokines that activate dendritic cells and polarize T cells. Along with epithelial cell-related inflammatory cytokines, type 2 innate lymphoid cells (ILC2s) are recruited and activated to produce IL-13; eosinophil infiltration also occurs to establish the positive amplification loop of type 2 inflammation.29 In fact, AAMs induced by RSV or influenza virus persist in the lungs of infected mice for up to 90 days following infection, which is longer than the period of viral clearance.14 In this study, BMDMs and alveolar macrophages isolated from surviving EV-A71-infected mice expressed high levels of IL-6 and TNF-α and exhibited the M2 macrophage phenotype. Moreover, we found that EV-A71-induced M2 macrophages maintained innate memory and strongly responded to subsequent allergen stimulation in various ways, such as enhanced IL-6 and TNF-α cytokine production, an increased immune response and polarization of naïve T cells into Th2 and Th17 effector lymphocytes in the presence of HDM allergens (Fig. 5a–e). In vitro studies and an adoptive transfer mouse model, which involved transfer of BMDMs pretreated with the trained immunity inhibitor 2-DG to allergic recipient mice, validated our hypothesis that trained immunity in myeloid cells following viral infection has an important role in allergic asthma development.
Trained immunity, or innate immune cell memory, is mediated by epigenetic, metabolic and functional reprogramming of innate immune cells, such as myeloid and NK cells.14 It is systemically induced at the level of bone marrow progenitors.30,31 It is also related to various autoimmune diseases and inflammatory disorders. Recently, Machiels et al.32 reported that respiratory infection with herpesvirus induced persistent changes in alveolar macrophages that protected against HDM-induced asthma. They also discovered that resident alveolar macrophages were replaced by BMDMs following virus infection. Monocytes maintained a regulatory profile for at least 3 weeks following herpesvirus infection, showing that the virus-induced long-term phenotypic changes in monocytes prior to their infiltration into the lungs. The ensuing infiltrating population of BMDMs rapidly differentiated into alveolar macrophages and was sufficient to inhibit HDM-induced sensitization. In contrast, our findings indicated that alveolar macrophages and BMDMs isolated from recovered EV-A71–infected mice showed both an EV-A71-trained M2 phenotype following stimulation with HDM ex vivo (Fig. 5a–d) and enhanced HDM-induced airway inflammation (Fig. 6b–h) following adoptive transfer into naïve mice. The various outcomes of trained immunity in macrophages following different viral infections are currently unclear. Yao et al.33 showed that alveolar macrophages develop a trained immunity phenotype following respiratory virus infection. These trained alveolar macrophages have typical properties of trained immunity, such as a defense-ready signature, high glycolytic rates and increased chemokine release levels following restimulation. Importantly, the induction of the mentioned macrophages results in strong host defense against heterologous bacterial infection. It is interesting to note that myeloid cells are not the only cells that exhibit features of trained immunity. ILC2s in the lungs proliferate and secrete cytokines following allergen and IL-33 stimulation. Some allergen-experienced ILC2s persist longer than the period of airway inflammation resolution. They also respond to unrelated allergens more potently than naïve ILC2s, which amplifies extreme allergic inflammation.34 Thus, our study findings are consistent with Yao’s report on trained immunity in alveolar macrophages (Fig. 5a, b) but via a different route. To the best of our knowledge, we are the first to show that extrapulmonary viral infection can lead to trained immunity in bone marrow progenitor myeloid cells, amplifying mucosal allergic inflammation in response to sensitization and challenge by airborne allergens such as HDM.
There are still numerous unanswered questions regarding the memory of innate immune cells and associated involvement in the development of allergic disorders and asthma, despite numerous clinical reports on increased AAM levels in the blood and BALF of asthmatic patients in comparison with normal subjects and early-life respiratory virus infection being correlated with the development of allergies and asthma in childhood.35 Although we have no clear answers yet, Fig. 7 provides the possible roles for trained macrophages in virus-induced enhancement of the response to allergen exposure and development of allergic asthma. Our findings also provide a new preventive and therapeutic pathway for allergic asthma.
Fig. 7.
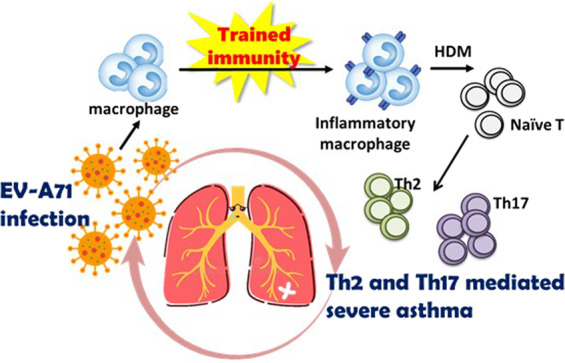
Early-life EV-A71 infection augments HDM-induced allergic airway inflammation via trained immunity in macrophages. Early-life EV-A71 infection causes sustained innate immune memory (trained immunity). When EV-A71-trained macrophages are exposed to HDM allergen, they can drive naïve T cells into Th2 and Th17 effector cells, indicating that EV-A71 infection in early life produces exacerbated features of allergen-induced airway inflammation in asthma
Materials and methods
National health data source
This study utilized data collected from LHIDs, which were from the National Health Insurance Research Database (NHIRD) in Taiwan. In brief, the National Health Insurance Program (NHIP) in Taiwan was launched in 1995 as a single-payer insurance system operated by the government. The NHIRD, a medical claims database (including information such as inpatient and outpatient services), was established and has been utilized for research purposes since 2000. The NHIRD includes demographic characteristics, disease diagnoses, prescription records, and medical expenditures. Therefore, the NHIRD covers almost 99% of Taiwan’s population and provides a valuable source to address the objectives of this study. Two LHIDs, LHID2005, and LHID2010, were constructed by randomly choosing 1 million enrollees in 2005 and 2010, separately, from the NHIP. A total of 2 million participants were enrolled in this study. The study protocol was approved by the Institutional Review Board of the National Health Research Institutes in Taiwan.
Study population
Definition of exposure
Here, children in the exposure group were defined as those born between 2003 and 2005 with a diagnosed enteroviral infection (International Classification of Diseases [ICD]-9-CM codes: 008.67, 047, 048, 074, 079.1, and 079.2) from either inpatient or outpatient medical claim records during the first 3 years of life. A detailed flow chart of the inclusion and exclusion criteria for the study cohort is provided in Fig. S1. The exclusion criteria were as follows: (1) subjects aged less than 8 years or greater than 10 years in 2012; and (2) study children who developed the examined allergic diseases during the first 4 years of life.
Definition of the three examined allergic diseases
The outcomes of interest in this study were three pediatric allergic diseases: atopic dermatitis, asthma, and allergic rhinitis. We studied whether enterovirus infection in the first 3 years was related to subjects developing allergic diseases in years 4–8. The following were defined:
-
(i)
Atopic dermatitis (ICD-9 code = 691.8): at least one inpatient claim record or two outpatient claim records within a year;
-
(ii)
Asthma (ICD-9 code = 493): at least one inpatient claim record or two outpatient claim records within a year; and
-
(iii)
Allergic rhinitis (ICD-9 code = 477): at least one inpatient claim record or two outpatient claim records within a year.
We utilized the date of the one inpatient claim record or the date of the second outpatient claim record as the index date. The ICD-9 codes utilized for disease diagnosis here identified atopic dermatitis, asthma, and allergic rhinitis.36–38
Potential confounding factors
We included pertinent confounding factors, such as comorbid physical illness, medication use, and healthcare utilization within 1 year prior to the index date. In detail, the following comorbid physical illnesses were included: upper respiratory tract infection, lower respiratory tract infection, otitis media, bronchitis, congenital anomalies, infectious diarrhea, infectious colitis, enteritis, and gastroenteritis. Acetaminophen use was a confounding factor here. We calculated healthcare system utilization as the sum of the number of clinic visits and hospitalizations, separately, within 1 year prior to the index date.
Cells, the virus, and the allergen
The human rhabdomyosarcoma (RD) cell line and mouse-adapted EV-A71 strain MP4 were kindly provided by Dr. Shih-Min Wang. RD cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies Gibco, CA, USA) supplemented with heat-inactivated 10% fetal bovine serum (FBS) (Corning, VA, USA). Viruses were prepared following the method from an earlier study. Viruses were propagated in RD cells with DMEM supplemented with 2% heat-inactivated FBS for one additional passage to prepare virus stocks. Virus stocks were collected following two freeze-thaw cycles and centrifuged at 1800 × g and 4 °C for 20 min. The reagents used included the following: HDM (Dermatophagoides pteronyssinus, Der p, Allergon, Engelholm, Sweden) was dissolved in pyogenic-free isotonic saline, filtered through a 0.22-μm filter and stored at −80 °C before use.
Isolation of single lung cells, BMDMs, and naïve Th cells
For single lung cell harvest, mouse lungs were first perfused with 15 ml of sterile ice-cold PBS via the heart to remove blood prior to being dissected from the trachea and cut into small pieces. Following incubation in digestion medium containing 0.7 mg/ml collagenase (Sigma, MO, USA) and 0.03 mg/ml deoxyribonuclease (DNase) I (Sigma, MO, USA) for 90 min at 37 °C, the digested tissues were passed through 70-μm cell strainers. For BMDM differentiation, femurs and tibias were isolated from BALB/c mice, and the muscle was removed. Bone marrow was flushed out with RPMI 1640 medium (Life Technologies Gibco, CA, USA) using a 25-gauge needle. The cells were then seeded at a density of 4 × 105/ml in RPMI 1640 medium containing 10% FBS and 10 ng/ml M-CSF (PeproTech, CI, USA). Cell culture medium was subsequently replaced every 3 days with fresh complete medium. After 7 days in culture, adherent BMDMs were harvested. Naïve T helper cells were isolated using the Naïve CD4+ T Cell Isolation Kit, an LS column and a MidiMACS™ separator (Miltenyi Biotec, Germany) in accordance with the manufacturer’s instructions. Spleens from 6- to 8-week-old BALB/c mice were homogenized into single-cell suspensions. Following the removal of RBCs, non-CD4+ cells were depleted using the LS columns with a cocktail of biotinylated antibodies and anti-biotin microbeads. Thus, the flow-through containing unlabeled cells was enriched with naïve CD4+ T cells.
Animal experimental models and protocols
All mouse experimental protocols were approved by the Institutional Animal Care and Use Committee of National Cheng Kung University (approved IACUC nos. 105274 and 107286). BALB/c mice were bought, bred and maintained under specific pathogen-free conditions in the Laboratory Animal Center of National Cheng Kung University and in accordance with national animal guidelines and regulations. For EV-A71–infected mouse experiments, 14-day-old newborn mice underwent either mock infection or EV-A71 infection with 3 × 106 pfu through intraperitoneal injection. All mice were monitored every 2 days to measure weight, clinical score and survival rate until 14 days following inoculation. Clinical scores were defined as follows: 0, healthy; 1, fur loss, wasting, or ruffled fur; 2, limb weakness; 3, paralysis in only 1 limb; 4, paralysis in 2 to 4 limbs; and 5, death. At day 14 after infection (28 days old), mouse AHR was determined before sacrifice for examination of lung pathology (protocol 1; Fig. 2a). For EV-A71–infected asthmatic mouse experiments, 35-day-old mock-infected or EV-A71–infected mice were intranasally sensitized with PBS or 50 μg of HDM daily for 14 days and intratracheally challenged with 50 μg of HDM. Mouse AHR was evaluated, and mice were sacrificed 48 h after the last HDM exposure (52 days old) (protocol 2; Fig. 3a). For the adoptive transfer mouse model, macrophages were differentiated from the bone marrow of naïve BALB/c mice. BMDMs underwent mock infection or EV-A71 infection (MOI = 10) or were stimulated with 5 mM 2-DG (Sigma, MO, USA) along with EV-A71 infection for 24 h. Following the removal of virus and resting for 6 days, the cells were collected and intratracheally transferred at a density of 1 × 105 cells/50 μl into naïve mice. Beginning one day following adoptive transfer, these mice were intranasally sensitized with HDM for 2 weeks and intratracheally challenged once on day 15 to induce asthma. The AHR of the mice was then determined, and they were sacrificed 48 h after the last HDM exposure (protocol 3; Fig. 6a).
Virus plaque assay
At 1, 3, and 5 d.p.i., brain and lung tissues were collected from infected mice, homogenized and centrifuged at 1000 × g and 4 °C for 20 min. The collected supernatants were sterilized using filtration through a 0.22-μm filter. Serial decimal dilutions were plated onto preseeded RD cells (6 × 105 cells in 6-well plates) and incubated at 37 °C for 45 min. Subsequently, medium containing 2% methylcellulose was added and incubated at 37 °C for 72 h. The overlay medium was discarded, and the plates were stained with a solution containing 4% formaldehyde and 1% crystal violet in PBS at room temperature for 45 min. The plates were then washed with flowing water and dried to count plaques.
Lung function measurements
Rrs and Ers measurements were conducted with the use of a flexiVent FX system (SCIREQ Inc., Montreal, Quebec, Canada). Mice were anaesthetized with an intraperitoneal injection of pentobarbital sodium (110 mg/kg body weight) and then tracheostomized for i.t. microtube measurement. AHR was determined in response to increasing doses of aerosolized methacholine (Sigma, MO, USA).
Measurement of total and HDM-specific IgE concentrations in the serum
Serum was collected from mice on the day prior to sensitization, the day before challenge and the second day after the last challenge. Total IgE levels in the serum were assayed by ELISA (Bethyl Laboratories, Inc., Montgomery, TX, USA; Thermo Fisher Scientific, MA, USA). For HDM-specific IgE, 96-well plates were coated with 10 μg of HDM at 4 °C for 18 h. After blocking, diluted serum samples (1:2 dilution) were added and incubated overnight at 4 °C. The plates were incubated with a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgE detection antibody (Bethyl Laboratories, Inc., Montgomery, TX, USA) for 3 h. Finally, the plates were washed and developed with 3,3’,5,5’-tetramethylbenzidine (TMB). The absorbance of samples was determined at 450 nm.
Lung histology and immunohistochemistry
The entire lung was removed and fixed in 3.7% neutral buffered formalin (pH 7.4) for 2 days. Specimens were embedded in paraffin and sectioned at a thickness of 4 μm. Hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), and combined eosinophil-mast cell staining were performed in accordance with the manufacturer’s protocols (ScyTek Laboratories, Inc., UT, USA). Lung sections were deparaffinized for EV-A71 particle staining. Following antigen retrieval and blocking of nonspecific binding, samples were incubated with anti-EV-A71 antibodies (Merck, Germany). Immunoreactivity was visualized using the diaminobenzidine immunoperoxidase methodology with a Novolink™ Polymer Detection Systems kit (Leica, UK).
BALF collection and cell infiltration analysis
BALF was acquired by inserting a catheter into the trachea, through which cold PBS (1 ml × 2) was instilled into the bronchioles. The supernatant and cells were separated by centrifugation at 300 × g for 5 min. The total number of cells in two collections was counted using a hemocytometer. Differential cellular analysis was conducted using cytocentrifugation. A Liu’s stain solution was used for staining for microscopic examination, and 300 cells were counted.
Trained immunity experiments
In vitro trained immunity or inhibition experiments and ex vivo experiments were conducted based on the earlier studies of Cheng et al.39 and Arts et al.40 In short, BMDMs were preseeded at a density of 5 × 105 in 12-well plates for 18 h and incubated with serial dilutions of EV-A71 for 24 h. The cells were washed two times with PBS and rested for 6 days in culture medium supplemented with 2% FBS. The medium was replaced once on day 4 of incubation. On day 7, the cells were restimulated for 24 h with serial dilutions of HDM (Fig. 4a). Supernatants were collected on days 1, 4, and 8 and stored at −80 °C until measurement of cytokine and metabolite concentrations. For in vitro trained immunity inhibition experiments, cells were preincubated with serial dilutions of 2-DG, MTA (Sigma, MO, USA) or metformin (Sigma, MO, USA) along with EV-A71 (MOI 10) for 24 h. After resting for 6 days, the cells were restimulated with 100 μg/ml HDM for 24 h. Supernatants were collected on day 8 and stored at −80 °C until cytokine measurement. For ex vivo trained immunity experiments, alveolar macrophages and BMDMs were harvested from 35-day-old mock-infected or EV-A71–infected mice (3 weeks following viral infection). Isolated cells were stimulated with 100 μg/ml HDM for 48 or 72 h. The cultured cells and supernatants were collected for flow cytometry (macrophage panel) and cytokine measurement, respectively.
Coculture system
Twenty-four-well plates were precoated with anti-CD3/CD28 antibodies in PBS at 37 °C for 24 h and washed once with PBS prior to cell plating. Differentiated BMDMs from protocol 2 mice were incubated with naïve Th cells at a 1:10 ratio (BMDMs: naïve T cells = 5 × 104:5 × 105). These cells were costimulated with or without 10 μg/ml HDM allergen at 37 °C for 3 days. Next, the cells were collected for flow cytometry.
Cytokine and metabolite measurement by ELISA
The levels of IFN-γ, TNF-α, and TARC in the BALF of experimental animals were calculated using ELISA (R&D Systems, MN, USA). Trained macrophage culture supernatants were collected on day 8, and the levels of TNF-α and IL-6 were determined using commercial ELISA kits (R&D Systems, MN, USA). Trained macrophage culture supernatants were collected on days 1, 4, and 8 to measure lactate and glucose concentrations using fluorometric quantification assay kits (BioVision, CA, USA).
Flow cytometry (FACS) analysis
For analysis of Th cells in the lungs, isolated lung cells were stimulated with a leukocyte activation cocktail plus GolgiPlug (BD Bioscience, NJ, USA) at 37 °C for 4 h. Isolated single cells were harvested, and surface markers were stained with FITC-conjugated anti-CD45 (30-F11) and PerCP-conjugated anti-CD4 (RM4-5) (BD Bioscience, NJ, USA) antibodies. The cells were fixed and permeabilized with a Foxp3 intracellular staining kit (Invitrogen, CA, USA). Following incubation with anti-CD16/CD32 (2.4G2) monoclonal antibodies (BD Bioscience, NJ, USA) to block Fc receptors, the cells were intracellularly stained with PE-conjugated anti-IL-4 (11B11), anti-IL-5 (TRFK5), anti-IL-13 (eBio13A) or anti-IL-17A (eBio17B7) antibodies and eFluor660-conjugated anti-GATA3 (TWAJ) and APC-conjugated anti-RORγt (AFKJS-9) antibodies (Invitrogen, CA, USA and BD Bioscience, NJ, USA). The cells were then fixed in PBS containing 1% paraformaldehyde (Sigma–Aldrich, MO, USA), and 10,000 gated events were analyzed using a CytoFLEX S V4-B4-R3-I2 flow cytometer (Beckman Coulter, CA, USA). Th2 and Th17 cells were identified as CD45+CD4+GATA3+ and CD4+CD4+ RORγt+ cells, respectively. Surface markers expressed by macrophages in the lungs and bone marrow were determined with the following mAbs: FITC-conjugated anti-F4/80 (BM8), PerCP-Cy5.5-conjugated anti-CD45 (30-F11), APC-Cy7-conjugated anti-Ly6c (AL-21) and BV605-conjugated anti-CD11b (M1/70) (Invitrogen, CA, USA and BD Bioscience, NJ, USA). M1 and M2 cells were identified as F4/80lowCD11blowLy6chi and F4/80hiCD11bhiLy6clow using a gating strategy adjusted from Capote J et al.41 and Carlson S et al.42 The examination of Th cell markers in the coculture system was conducted using the following mAbs: FITC-conjugated anti-CD4 (GK1.5); PE-conjugated anti-IL-4 (11B11), anti-IL-5 (TRFK5), anti-IL-13 (eBio13A) or anti-IL-17A (eBio17B7); eFluor660-conjugated anti-GATA3 (TWAJ); and APC-conjugated anti-RORγt (AFKJS-9) (Invitrogen, CA, USA and BD Bioscience, NJ, USA).
Statistics
For analysis of data from the NHIRD, we calculated and compared the distributions of clinical, demographic and healthcare characteristics between children with and without enterovirus infection. Next, we did not include children who developed allergic diseases in the first 4 years of life or with enterovirus infection after the first 3 years of life. We employed Cox proportional hazard models to assess the temporal relationship between enterovirus infection in the first 3 years of life and the development of allergic diseases in years 4–8 with or without adjustment for the above-listed covariates. We followed the development of examined allergic diseases among the study children until they developed the examined allergic diseases or were 8 years old. Children without enterovirus infection in the first 3 years of life were treated as the reference group. We also performed subgroup analyses to study whether the temporal relationship between enterovirus infection and the development of allergic disease was altered by different clinical, demographic or healthcare characteristics among the study children. Kaplan–Meier plots and log-rank tests were used to compare the cumulative incidence rates of the three allergic diseases between children with or without enterovirus infection in early life. P values < 0.05 were considered statistically significant. All data from mouse studies are presented as the mean ± SEM and were analyzed by two-way ANOVA or one-way ANOVA with the Bonferroni multiple comparison using GraphPad Prism 8 software.
Supplementary information
Acknowledgements
We are grateful to S.H.C. and S.M.W. for providing EV-A71, incubation cell lines and experimental instructions for EV-A71 infection. This study was based, in part, on data from the NHIRD provided by the Bureau of National Health Insurance and Department of Health and managed by the National Health Research Institutes. The interpretation and conclusions herein do not represent those of the Bureau of National Health Insurance, Department of Health or National Health Research Institutes. This study was, in part, supported by the Centre of Allergy and Mucosal Immunity, Headquarters of University Advancement at the National Cheng Kung University, Ministry of Education, Taiwan. H.J.T is supported in part by a grant from the National Health Research Institutes (PI: Tsai, PH-101-PP-14, PH-101-SP-14, and PH-108-PP-08).
Author contributions
P.C.C. and Y.T.S. designed the original model. S.H.C. and S.M.W. provided EV-A71, incubation cell lines, and experimental instructions for EV-A71 infection. P.C.C., Y.T.S., and M.H. designed and conducted all experiments with guidance from H.F.K., W.S.K., R.V., Z.G.L., and J.Y.W. The national health database analysis was conducted by H.J.T. L.S.H.W. fit the statistical models for the analysis of experimental data, and P.C.C., M.H.H., H.F.K., R.V., H.J.T., and J.Y.W. analyzed all the data, wrote the original manuscript, and prepared the final figures. H.J.T. and J.Y.W. secured funding for this project. J.Y.W. authored the final manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version of this article (10.1038/s41423-020-00621-4) contains supplementary material.
References
- 1.Boonpiyathad T, Sozener ZC, Satitsuksanoa P, Akdis C. Immunologic mechanisms in asthma. Semin. Immunol. 2019;46:101333. doi: 10.1016/j.smim.2019.101333. [DOI] [PubMed] [Google Scholar]
- 2.Jackson DJ, Gern JE, Lemanske RF., Jr. Lessons learned from birth cohort studies conducted in diverse environments. J. Allergy Clin. Immunol. 2017;139:379–386. doi: 10.1016/j.jaci.2016.12.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Custovic A, et al. Cytokine responses to rhinovirus and development of asthma, allergic sensitization, and respiratory infections during childhood. Am. J. Respir. Crit. Care Med. 2018;197:1265–1274. doi: 10.1164/rccm.201708-1762OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukkarinen M, Jartti T. The first rhinovirus-wheeze acts as a marker for later asthma in high-risk children. J. Allergy Clin. Immunol. 2016;138:313. doi: 10.1016/j.jaci.2016.01.040. [DOI] [PubMed] [Google Scholar]
- 5.Su YT, et al. High correlation between human rhinovirus type C and children with asthma exacerbations in Taiwan. J. Microbiol. Immunol. Infect. 2020;53:561–568. doi: 10.1016/j.jmii.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Shi T, et al. Association between respiratory syncytial virus-associated acute lower respiratory infection in early life and recurrent wheeze and asthma in later childhood. J. Infect. Dis. 2020;222:S628–S633. doi: 10.1093/infdis/jiz311. [DOI] [PubMed] [Google Scholar]
- 7.Farne HA, Johnston SL. Immune mechanisms of respiratory viral infections in asthma. Curr. Opin. Immunol. 2017;48:31–37. doi: 10.1016/j.coi.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 8.Kumar RK, Foster PS, Rosenberg HF. Respiratory viral infection, epithelial cytokines, and innate lymphoid cells in asthma exacerbations. J. Leukoc. Biol. 2014;96:391–396. doi: 10.1189/jlb.3RI0314-129R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70:1189–1196. doi: 10.1136/thoraxjnl-2015-207020. [DOI] [PubMed] [Google Scholar]
- 10.Huang CC, et al. Neurologic complications in children with enterovirus 71 infection. N. Engl. J. Med. 1999;341:936–942. doi: 10.1056/NEJM199909233411302. [DOI] [PubMed] [Google Scholar]
- 11.Lee ZM, Huang YH, Ho SC, Kuo HC. Correlation of symptomatic enterovirus infection and later risk of allergic diseases via a population-based cohort study. Medicine. 2017;96:e5827. doi: 10.1097/MD.0000000000005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeh JJ, Lin CL, Hsu WH. Effect of enterovirus infections on asthma in young children: a national cohort study. Eur. J. Clin. Invest. 2017;47:12. doi: 10.1111/eci.12784. [DOI] [PubMed] [Google Scholar]
- 13.Wang YC, et al. Association between enterovirus infection and asthma in children: a 16-year nationwide population-based cohort study. Pediatr. Infect. Dis. J. 2018;37:844–849. doi: 10.1097/INF.0000000000001918. [DOI] [PubMed] [Google Scholar]
- 14.Netea MG, Latz E, Mills KH, O’Neill LA. Innate immune memory: a paradigm shift in understanding host defense. Nat. Immunol. 2015;16:675–679. doi: 10.1038/ni.3178. [DOI] [PubMed] [Google Scholar]
- 15.Netea MG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Włodarczyk M, Druszczyńska M, Fol M. Trained innate immunity not always amicable. Inter. J. Mol. Sci. 2019;20:10. doi: 10.3390/ijms20102565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang SW, et al. Exogenous interleukin-6, interleukin-13, and interferon-γ provoke pulmonary abnormality with mild edema in enterovirus 71-infected mice. Respir. Res. 2011;12:147. doi: 10.1186/1465-9921-12-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen FH, Shen TJ, Chang TM, Su IJ, Chen SH. Early dexamethasone treatment exacerbates enterovirus 71 infection in mice. Virology. 2014;464-465:218–227. doi: 10.1016/j.virol.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 19.Siegle JS, et al. Early-life viral infection and allergen exposure interact to induce an asthmatic phenotype in mice. Respir. Res. 2010;11:14. doi: 10.1186/1465-9921-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blomqvist S, Savolainen C, Råman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J. Clin. Microbiol. 2002;40:4218–4223. doi: 10.1128/JCM.40.11.4218-4223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang SM, et al. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J. Infect. Dis. 2003;188:564–570. doi: 10.1086/376998. [DOI] [PubMed] [Google Scholar]
- 22.Edwards MR, et al. Viral infections in allergy and immunology: how allergic inflammation influences viral infections and illness. J. Allergy Clin. Immunol. 2017;140:909–920. doi: 10.1016/j.jaci.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 2017;140:895–906. doi: 10.1016/j.jaci.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cormier SA, You D, Honnegowda S. The use of a neonatal mouse model to study respiratory syncytial virus infections. Expert Rev. Anti Infect. Ther. 2010;8:1371–1380. doi: 10.1586/eri.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider D, et al. Neonatal rhinovirus infection induces mucous metaplasia and airways hyperresponsiveness. J. Immunol. 2012;188:2894–2904. doi: 10.4049/jimmunol.1101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Garawi A, et al. Influenza A facilitates sensitization to house dust mite in infant mice leading to an asthma phenotype in adulthood. Mucosal Immunol. 2011;4:682–694. doi: 10.1038/mi.2011.35. [DOI] [PubMed] [Google Scholar]
- 27.Pali-Scholl I, Jensen-Jarolim E. The concept of allergen-associated molecular patterns (AAMP) Curr. Opin. Immunol. 2016;42:113–118. doi: 10.1016/j.coi.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Keegan AD, et al. Enhanced allergic responsiveness after early childhood infection with respiratory viruses: Are long-lived alternatively activated macrophages the missing link? Pathog. Dis. 2016;74:5. doi: 10.1093/femspd/ftw047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl. Res. 2018;191:1–14. doi: 10.1016/j.trsl.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufmann E, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 2018;172:176–190.e119. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Mitroulis I, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018;172:147–161.e112. doi: 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Machiels B, et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat. Immunol. 2017;18:1310–1320. doi: 10.1038/ni.3857. [DOI] [PubMed] [Google Scholar]
- 33.Yao Y, et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell. 2018;175:1634–1650. doi: 10.1016/j.cell.2018.09.042. [DOI] [PubMed] [Google Scholar]
- 34.Halim TY, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 2016;17:57–64. doi: 10.1038/ni.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Ramon S, et al. Trained immunity-based vaccines: a new paradigm for the development of broad-spectrum anti-infectious formulations. Front. Immunol. 2018;9:2936. doi: 10.3389/fimmu.2018.02936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marra F, et al. Antibiotic use in children is associated with increased risk of asthma. Pediatrics. 2009;123:1003–1010. doi: 10.1542/peds.2008-1146. [DOI] [PubMed] [Google Scholar]
- 37.Suh DC, et al. Economic burden of atopic manifestations in patients with atopic dermatitis–analysis of administrative claims. J. Manag. Care Pharm. 2007;13:778–789. doi: 10.18553/jmcp.2007.13.9.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hankin CS, et al. Allergy immunotherapy among Medicaid-enrolled children with allergic rhinitis: patterns of care, resource use, and costs. J. Allergy Clin. Immunol. 2008;121:227–232. doi: 10.1016/j.jaci.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 39.Cheng SC, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arts RJW, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016;17:2562–2571. doi: 10.1016/j.celrep.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Capote J, et al. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol. 2016;213:275–288. doi: 10.1083/jcb.201510086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlson S, et al. Cardiac macrophages adopt profibrotic/M2 phenotype in infarcted hearts: role of urokinase plasminogen activator. J. Mol. Cell Cardiol. 2017;108:42–49. doi: 10.1016/j.yjmcc.2016.05.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.