

Abstract
The aim of the present study was to investigate the influence of butylphthalide on nerve cell apoptosis in rats with cerebral infarction through the c-Jun N-terminal kinase (JNK)/p38 mitogen-activated protein kinase (MAPK) signaling pathway. A total of 36 Sprague-Dawley rats were randomly divided into sham-operation group (n=12), model group (n=12) and butylphthalide group (n=12). Additionally, qPCR was performed to measure the mRNA expression of Bax and Bcl-2, and a TUNEL assay was conducted to investigate the cell apoptosis. Compared with the sham-operation group, the model group and the butylphthalide group had notably increased Zea-Longa scores (P<0.05), while the butylphthalide group exhibited a markedly decreased Zea-Longa score, compared with the model group (P<0.05). The positive expression of Bax was markedly higher (P<0.05), while that of Bcl-2 was notably lower in the model group and the butylphthalide group (P<0.05), compared with those in the sham-operation group. Furthermore, the positive expression of Bax was notably decreased (P<0.05), while that of Bcl-2 was markedly increased in the butylphthalide group in comparison with those in model group (P<0.05). The model group and the butylphthalide group had markedly higher relative protein expression levels of p-JNK and p-p38 MAPK than the sham-operation group (P<0.05), and the butylphthalide group displayed notably lower relative protein expression levels of p-JNK and p-p38 MAPK than the model group (P<0.05). The relative mRNA expression level of Bax was markedly increased (P<0.05), while that of Bcl-2 was notably decreased in the model group and the butylphthalide group (P<0.05), compared with those in the sham-operation group. Compared with those in the model group, the relative mRNA expression level of Bax decreased markedly (P<0.05), and that of Bcl-2 increased notably in the butylphthalide group (P<0.05). The apoptotic rate was markedly higher in the model group and the butylphthalide group than that in the sham-operation group (P<0.05), but it was notably lower in the butylphthalide group than that in the model group (P<0.05). In conclusion, butylphthalide may inhibit nerve cell apoptosis in rats with cerebral infarction to exert a protective effect, which may be associated with the JNK/p38 MAPK signaling pathway.
Keywords: butylphthalide, cerebral infarction, JNK/p38 MAPK signaling pathway, apoptosis
Introduction
Cerebral infarction is a relatively common critical disease in clinic, which may induce hypoxic-ischemic injury of brain tissues and neurons to cause serious damage to the nervous system. For patients with cerebral infarction, the central nervous system injury induced by the disease brings about severe consequences, and the patients often experience limb motor dysfunction, speech disorder and even the risk of death (1,2). Therefore, cerebral infarction seriously threatens the life, health and quality of life of human beings and brings heavy economic burdens to the patients' families and society. Therefore, it is very important to effectively treat cerebral infarction, promote repair of nervous system injury following cerebral infarction and ameliorate limb motor dysfunction, speech disorder and other complications of cerebral infarction (3,4).
At present, it is hypothesized that nerve cell apoptosis, a crucial pathological response, is associated with the repair of the nervous system following cerebral infarction injury and functional restoration of the patients. In particular, massive nerve cell apoptosis may severely affect the patients' limb motor function, sensory function and speech function (5,6). Decreasing nerve cell apoptosis following cerebral infarction is conducive to protecting the nerve cells and facilitating the repair of nervous system injury to a certain extent. Furthermore, it may aid in restoring relevant functions, including limb motor function and speech function following cerebral infarction. Therefore, efficacious anti-apoptosis of nerve cells is regarded as one of the keys to the treatment of cerebral infarction.
As a vital signal transduction pathway, the c-Jun N-terminal kinase (JNK)/p38 mitogen-activated protein kinase (MAPK) signaling pathway has correlations with multiple physiological and pathological responses, including cell apoptosis, proliferation and necrosis (7,8). It is argued at present that the JNK/p38 MAPK signaling pathway serves as an important signaling pathway regulating cell apoptosis. A large number of cytokines following cerebral infarction injury may significantly activate the JNK/p38 MAPK signaling pathway to promote the massive nerve cell apoptosis in the brain, thereby triggering nervous system damage. Butylphthalide, a brain protecting agent commonly used in clinic, has certain protective effects on the nerve cells following cerebral infarction, but its mechanism of action remains unclear. Therefore, the present study aims to investigate the influence of butylphthalide on nerve cell apoptosis in rats with cerebral infarction through the JNK/p38 MAPK signaling pathway.
Materials and methods
Laboratory animals
A total of 36 male one-month-old SPF Laboratory Sprague-Dawley (SD) rats, weighing 240-260 g, were purchased from Shanghai Laboratory Animal Co., Ltd. (license no. SCXK; 2014-0003). Rats were housed in a temperature-controlled room (21±2˚C) with a relative humidity range of 30-40% on a 12/12 h light/dark cycle (lights on at 06:00 a.m.). All rats had free access to water and food. The present study was approved by the Animal Ethics Committee of Zhejiang University School of Medicine Animal Center.
Experimental reagents and instruments
Butylphthalide injection was obtained from NBP Pharmaceutical Co. Ltd. The following primary antibodies: Anti-phosphorylated (p)-p38 MAPK (cat. no. ab170099), p38 MAPK (cat. no. ab31828), anti-p-JNK (cat. no. ab124956), anti-JNK (cat. no. ab76125), anti-Bcl-2 (cat. no. ab182858) and anti-Bax (cat. no. ab32503), and secondary antibodies, were obtained from Abcam. An immunohistochemistry kit and fluorescence quantitative polymerase chain reaction (qPCR) instrument were obtained from Applied Biosystems; Thermo Fisher Scientific, Inc. A light microscope was obtained from Leica Microsystems GmbH and qPCR-related kits (including TRIzol reagent (Thermo Fisher Scientific, Inc.), Primer Script real-time (RT) reagent kit with gDNA Eraser, reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and miRNA measurement kit were from Thermo Fisher Scientific, Inc.
Animal grouping, processing and modeling
The aforementioned 36 SD rats were randomly divided into a sham-operation group (n=12), a model group (n=12) and a butylphthalide group (n=12). The rats were fed in the Zhejiang University Animal Center. Rats were housed in a temperature-controlled room (21±2˚C) with a relative humidity range of 30-40% on a 12/12 h light/dark cycle (lights on at 06:00 a.m.). All rats had free access to water and food.
Only the common carotid artery, internal carotid artery and external carotid artery were exposed in the sham-operation group, while the model of cerebral infarction was established in the model group and the butylphthalide group. Following surgery, the rats in the butylphthalide group were intraperitoneally injected with butylphthalide (4.5 mg/kg) every day, and those in the sham-operation group and the model group were subjected to a daily intraperitoneal injection of an equal volume of normal saline. Specimens were obtained following 7 consecutive days of intervention.
Establishment of cerebral infarction model (9)
Pentobarbital sodium at a dose of 40 mg/kg was intraperitoneally injected for anesthesia. Subsequently, the rats were fixed in the supine position, and the neck was depilated and then covered with a sterile towel. Next, a longitudinal incision (~2 cm) was made at the anteromedian line of the neck, and the common carotid artery, external carotid artery and internal carotid artery were separated and exposed carefully. Subsequently, the common carotid artery and external carotid artery were ligated with silk sutures, and the internal carotid artery was clipped using vascular forceps. The suture was inserted into the ligated site of the common carotid artery, the vascular forceps at the internal carotid artery were released, and the suture was slowly pushed into the branch of middle cerebral artery. Later, the internal carotid artery was ligated again and fixed by the suture, and the incision was sutured following flushing with normal saline. Next, timing was commenced and the suture was withdrawn slowly at 90 min after vascular occlusion.
Specimen acquisition
Following successful anesthesia, specimens were directly obtained from 6 rats in each group. Specifically, the brain tissues were removed directly, washed with normal saline, put into EP tubes and stored at -80˚C for subsequent use. As for the remaining 6 rats in each group, the specimens were acquired via perfusion-fixation. Specifically, the thoracic cavity of the rat was cut open to expose the heart, and 400 ml 4% paraformaldehyde was perfused from the left atrial appendage. After 24 h, the brain tissues were removed and soaked in 4% paraformaldehyde solution at 20˚C for fixation for 24 h.
Zea-Longa score
At the beginning of feeding, 7 days after feeding, following modeling and at 2 weeks following intervention, the neurological deficits of the rats were assessed using the Zea-Longa score (10) on the basis of the symptoms and manifestations of the rats. The Zea-Longa scores are presented in Table I.
Table I.
Zea-Longa score.
| Score | Symptom |
|---|---|
| 0 point | No neurological deficits |
| 1 point | Mild: Unable to extend the right front paw completely when the tail is lifted |
| 2 points | Moderate: Circle to the right when walking |
| 3 points | Severe: Tumble to the right when walking |
| 4 points | Unable to walk spontaneously, with loss of consciousness |
Hematoxylin and eosin (H&E) staining
The tissues previously embedded in paraffin were sliced into 5 µm-thick sections, spread in warm water at 42˚C, collected and baked, so as to prepare the paraffin-embedded sections. Next, the sections obtained were soaked in xylene solution and a gradient of alcohol for routine deparaffinization until rehydration. Next, the sections were stained with hematoxylin dye for 5 min at 25˚C using the H&E staining kit and then placed in pure water for 10 min, followed by color separation in 95% ethanol for 5 sec, transparentizing with xylene for 10 sec and mounting in neutral balsam.
Immunohistochemistry
The process of obtaining paraffin-embedded tissue sections was the same as adorementioned. Subsequently, the aforementioned sections were immersed in citric acid buffer solution and heated repeatedly in a microwave oven for 3 times (heating for 3 min and braising for 5 min every time), so as to realize sufficient antigen retrieval. Following rinsing, 0.3% endogenous peroxidase blocker (hydrogen peroxide; Beyotime Institute of Biotechnology) was added dropwise onto the specimens and reacted for 10 min. Next, the specimens were rinsed and goat serum was added for sealing for 20 min. Anti-Bax primary antibody (dilution, 1:200) and anti-Bcl-2 primary antibody (dilution, 1:200) were added after the goat serum blocking buffer was shaken off, followed by culture in a refrigerator at 4˚C overnight. The next day, the specimens were rinsed and secondary goat anti-rabbit (HRP) IgG antibody (1:1,000; cat. no. ab6721; Abcam,) was added dropwise for 10 min. Following rinsing adequately, streptavidin-peroxidase solution was added for reaction for 10 min, followed by color development with 3,3'-diaminobenzidine added in drops, counterstaining of the nucleus at 25˚C for 5 min with hematoxylin, mounting and observation (magnification, x200). Images were visualized and captured using a light microscopy (Nikon Corporation).
Western blotting (WB) assay
The RIPA lysis buffer (Beyotime Institute of Biotechnology) was added to the cryopreserved brain tissues in an ice bath for 1 h. Next, the tissues were centrifuged at 4˚C, 14,000 x g for 10 min, and the protein was quantified using the bicinchoninic acid method (Pierce; Thermo Fisher Scientific, Inc.). Next, the absorbance and standard curve of the protein were obtained through a microplate reader and were applied to calculate the protein concentration in tissues. Subsequently, the proteins (20 mg/ml) in the tissue specimens were subjected to denaturation and separation via 10% SDS-PAGE. The position of the Marker proteins was observed, and the electrophoresis was stopped when the Marker proteins reached the bottom of glass plate in a straight line. Next, the proteins were transferred onto a polyvinylidene fluoride membrane (EMD Millipore) and reacted with blocking buffer (skimmed milk) at 25˚C for 1.5 h. Subsequently, the membranes were incubated overnight at 4˚C with anti-p-p38 MAPK (dilution, 1:1,000), anti-p38 MAPK (dilution, 1:1,000), anti-p-JNK (dilution, 1:1,000) and anti-JNK primary antibodies (dilution, 1:1,000), prior to incubation at 25˚C for 2 h with secondary goat anti-rabbit (HRP) IgG antibody (dilution: 1:1,000; cat. no. ab6721, Abcam). Finally, the image was fully developed using a chemiluminescent reagent (Beyotime Institute of Biotechnology) in the dark for 1 min after rinsing.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
The brain tissues stored for use were added into the RNA extraction reagent (TRIzol, Invitrogen; Thermo Fisher Scientific, Inc.) to extract the total RNA from the specimens. Next, total RNA was reverse transcribed into cDNA at 50˚C for 45 min by using PrimeScript RT reagent kit (Takara Biotechnology Co., Ltd.), according to the manufacturer's protocol. RT-qPCR was then performed based on the instructions of SYBR Premix Ex Taq™ (Takara Bio, Inc.), with 3 replicates in each group. Thermocycling conditions were as follows: Reaction at 53˚C for 5 min, pre-denaturation at 95˚C for 10 min, denaturation at 95˚C for 10 sec and annealing at 62˚C for 30 sec, for 35 cycles in total. ∆Cq was calculated first, and the difference in the expression of target genes was calculated (11). The detailed primer sequences are presented in Table II.
Table II.
Primer sequences.
| Name | Primer sequence |
|---|---|
| Bax | F: 5'-GTGGCGGGACATAGTCAGCA-3' |
| R: 5'-CCCATTGGGACAGCTTGGA-3' | |
| Bcl-2 | F: 5'-AACCTTCCTTCCTGCTTGAG-3' |
| R: 5'-TGCTGTTCCTCTTCCAACG-3' | |
| GAPDH | F: 5'-ACGGCAAGTTCAACGGCACAG-3' |
| R: 5'-GAAGACGCCAGTAGACTCCACGAC-3' |
F, forward; R, reverse.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
Sections were fixed using 4% paraformaldehyde at 20˚C for 30 min. 50 µl TUNEL reagent (Beyotime Institute of Biotechnology; cat. no. C1088) was added to each section for incubation for 60 min at 37˚C. Nuclear staining reagent and mounting medium used were also from One Step TUNEL Apoptosis Assay Kit (Beyotime Institute of Biotechnology). The kit was used according to the manufacturer's protocols. Nuclear stain was added to each section for incubation for 5 min at 37˚C. After staining, a fluorescent microscope (magnification, x200) (IX70, Olympus Corporation) was used to observe the images (5 fields of view). The tissues previously embedded in paraffin were prepared into 5 µm-thick paraffin-embedded sections following spreading in warm water at 42˚C, collection and baking at 37˚C for 1 h, followed by soaking in xylene solution and a gradient of ethanol for routine deparaffinization until rehydration. Subsequently, the cell apoptosis in the brain tissues was detected according to the instructions of the TUNEL apoptosis kit.
Statistical analysis
SPSS 20.0 software (IBM Corp.) was used for statistical analysis, and the enumeration data are expressed as the mean ± standard deviation. Comparison between multiple groups was performed using one-way analysis of variance followed by the Bonferroni post hoc test. P<0.05 was used to indicate a statistically significant difference.
Results
Zea-Longa score
At the beginning of feeding and 7 days after feeding, there was no statistically significant difference in Zea-Longa score among the three groups. Following modeling, compared with the sham-operation group, the model group and the butylphthalide group had notably increased Zea-Longa scores (P<0.05), while there was no statistical difference between the model group and the butylphthalide group. At 2 weeks after intervention, compared with the sham-operation group, the model group and the butylphthalide group had significantly increased Zea-Longa scores (P<0.05), while the butylphthalide group exhibited a significantly decreased Zea-Longa score in comparison with the model group (P<0.05; Fig. 1).
Figure 1.
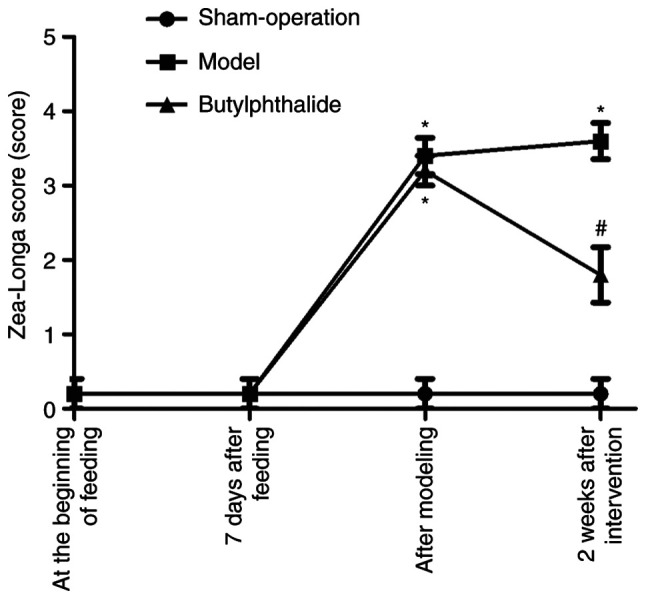
Zea-Longa score in each group. *P<0.05 vs. sham-operation group, #P<0.05 vs. model group.
Immunohistochemistry
The neuronal structure was clear and intact in the sham-operation group, with abundant Nissl bodies. In the model group, there was disturbance in the neuronal structure, fewer Nissl bodies and partial cleavage. The neuronal morphology was improved distinctly in the butylphthalide group compared with that in model group, with a larger number of Nissl bodies and improved morphology. The cells with positive expression were sepia. There was low positive expression of Bax and high positive expression of Bcl-2 in the sham-operation group. By contrast, the model group had more positive expression of Bax and less positive expression of Bcl-2. Compared with the model group, the positive expression of Bax was weakened and the positive expression of Bcl-2 was enhanced in the butylphthalide group. The statistical results (Fig. 2) indicated that the average optical density of the positive expression of Bax was significantly increased, while that of Bcl-2 was significantly decreased in the model group and the butylphthalide group, compared with those in sham-operation group (P<0.05). In comparison with the model group, the butylphthalide group exhibited significantly decreased average optical density of the positive expression of Bax and significantly increased average optical density of the positive expression of Bcl-2 (P<0.05).
Figure 2.
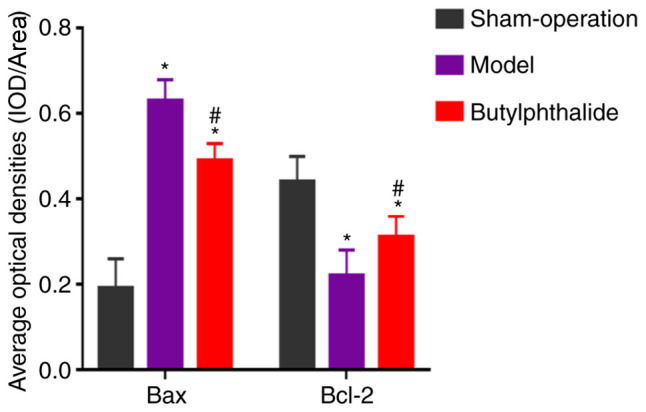
Average optical densities of positive expression of Bax and Bcl-2 in each group. *P<0.05 vs. sham-operation group, #P<0.05 vs. model group.
Relative expression of associated proteins detected via WB
The protein expression of p-p38 MAPK and p-JNK were low in the sham-operation group, but high in the remaining groups. However, they were decreased in the butylphthalide group, compared with those in the model group (Fig. 3A). The statistical results (Fig. 3B) demonstrated that the model group and the butylphthalide group had significantly higher relative protein expression levels of p-p38 MAPK and p-JNK than those in the sham-operation group (P<0.05). Furthermore, the butylphthalide group displayed notably lower relative protein expression levels of p-JNK and p-p38 MAPK than the model group (P<0.05).
Figure 3.
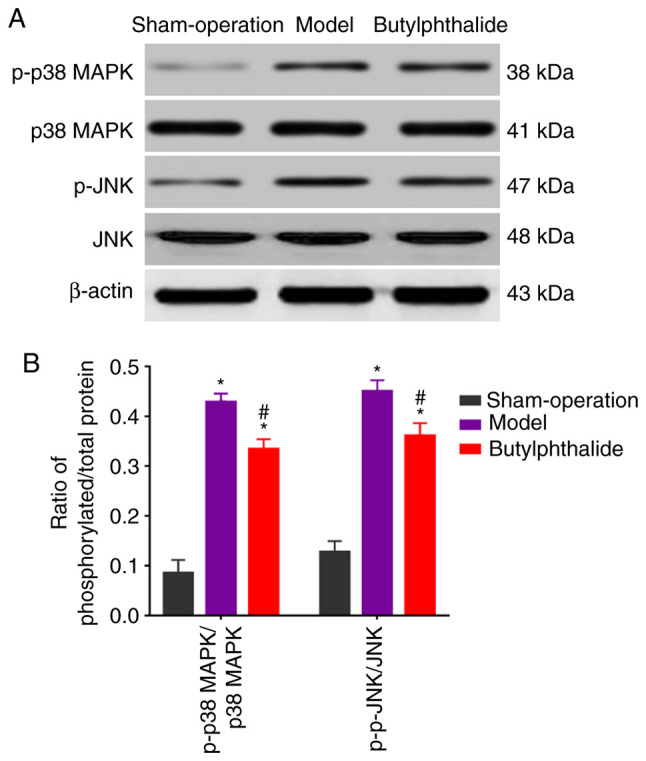
Protein expression detected via WB. (A) Protein expression of p-p38 MAPK, p38 MAPK, p-JNK and JNK determined by WB. (B) Ratio of phosphorylated/total protein in each group. *P<0.05 vs. sham-operation group, #P<0.05 vs. model group. WB, western blotting.
mRNA expressions detected via qPCR
The relative mRNA expression level of Bax was signficiantly increased, while that of Bcl-2 was significantly decreased in the model group and the butylphthalide group, compared with those in the sham-operation group (P<0.05; Fig. 4). Additionally, compared with those in the model group, the relative mRNA expression level of Bax decreased significantly, and that of Bcl-2 increased significantly in the butylphthalide group (P<0.05).
Figure 4.
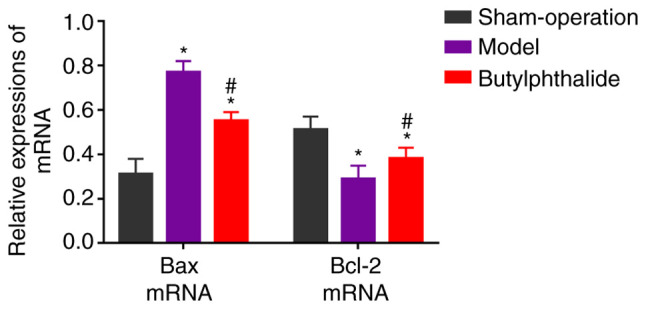
Relative expression of mRNAs in each group. *P<0.05 vs. sham-operation group, #P<0.05 vs. model group.
Cell apoptosis detected via TUNNEL assay
There were fewer TUNEL-positive cells in the sham-operation group and more TUNEL-positive cells in the remaining groups. The apoptotic rate was significantly higher in the model group and the butylphthalide group than that in the sham-operation group (P<0.05), but it was significantly lower in the butylphthalide group than that in the model group (P<0.05; Fig. 5).
Figure 5.
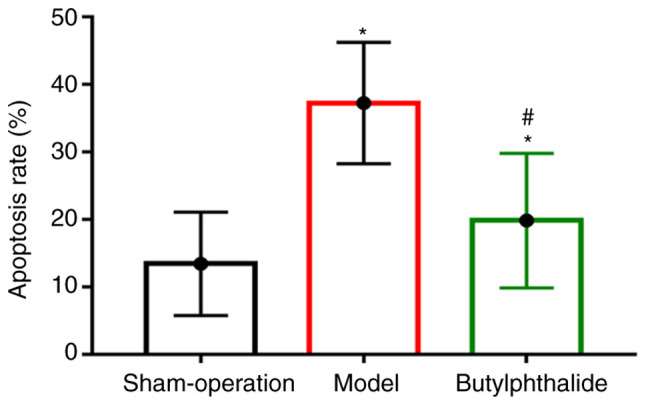
Cell apoptotic rate in each group. *P<0.05 vs. sham-operation group, #P<0.05 vs. model group.
Discussion
Cerebral infarction, a clinically common cerebrovascular disease, usually occurs in elderly patients and leads to nervous system damage, including motor and sensory dysfunctions of limbs, thereby triggering neurological deficits and resulting in the death of patients in severe cases. Therefore, it is a ubiquitous critical disease in clinic (12,13). It is essential to effectively treat cerebral infarction, accelerate the rehabilitation of nervous system injury and neurological deficits following cerebral infarction and relieve the limb motor dysfunction, sensory disturbance and speech disorder, thereby significantly improving the quality of life of patients with cerebral infarction and reducing the economic burdens on their family and society. The nerve cell apoptosis, one of the critical pathological responses and processes following cerebral infarction, is considered an important action pathway that regulates the repair of nervous system following the disease (14,15). Studies have demonstrated that injury-induced numerous inflammatory factors and cytokines may stimulate the abnormal expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 in the brain tissues under hypoxic and ischemic conditions, and the proportion imbalance between the two factors will ultimately lead to the nerve cell apoptosis (16,17). A great amount of nerve cell apoptosis may further destroy the nervous system, thereby causing neurological deficits and even death. Therefore, efficiently alleviating the nerve cell apoptosis following cerebral infarction may protect the nerve cells and aid in the repair of the nervous system following cerebral infarction (18,19).
Previous studies have proven that the JNK/p38 MAPK signaling pathway serves as a crucial cell signal transduction pathway to serve important regulatory roles in such physiological and pathological processes as cell proliferation, differentiation, apoptosis and necrosis (20-23). Furthermore, it was also manifested through research that with the presence of injury factors and hypoxic and ischemic conditions, massive inflammatory factors and cytokines are able to phosphorylate JNK in the JNK/p38 MAPK signaling pathway to activate p-JNK. Additionally, p-JNK may further interact with p38 MAPK to generate p-p38 MAPK via phosphorylation, activating the JNK/p38 MAPK signaling pathway and modulating the expression of downstream apoptosis-related molecules Bax and Bcl-2. The results of this study also confirmed that the apparently aberrant expression of Bax and Bcl-2, namely high expression of Bax and low expression of Bcl-2, were observed at the injury site following cerebral infarction, suggesting that the aggravation of nerve cell apoptosis is associated with the abnormal expression of Bax and Bcl-2. Meanwhile, the expression of p-JNK and p-p38 MAPK were increased following the occurrence of cerebral infarction, illustrating that the JNK/p38 MAPK signaling pathway in the body was activated and enhanced nerve cell apoptosis following cerebral infarction.
The results of the present study indicated that the Zea-Longa score was significantly decreased following intervention with butylphthalide for the rats with cerebral infarction in the butylphthalide group, illustrating that butylphthalide may favorably ameliorate neurological deficits and cognitive impairment following cerebral infarction. It was also demonstrated that the neuronal morphology was significantly improved in the butylphthalide group via HE staining, indicating that butylphthalide has a protective effect on neurons in cerebral infarction. Additionally, butylphthalide group had decreased expression of Bax, p-JNK and p-p38 MAPK, a decreased apoptotic rate and increased Bcl-2 expression following intervention with butylphthalide after the onset of cerebral infarction. These results suggested that butylphthalide has an anti-apoptotic effect in cerebral infarction, and this may be related to fact that butylphthalide represses the activation of the JNK/p38 MAPK signaling pathway. There are certain limitations to the present study. For example, there was only one time point to determine the mechanism; therefore, whether different time points have influence on the experimental results requires further research to confirm. In addition, no blockers or other methods were used to further confirm the effect of the JNK/p38 MAPK signaling pathway on cerebral infarction. Therefore, the specific mechanism of butylphthalide on cerebral infarction and its association with the JNK/p38 MAPK signaling pathway require further study to clarify in the future.
Therefore, it may be concluded that butylphthalide may inhibit the nerve cell apoptosis in rats with cerebral infarction to exert a protective effect, which may be associated with the JNK/p38 MAPK signaling pathway.
Acknowledgements
Not applicable.
Funding Statement
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
XB and YG designed the study and performed the experiments, XB and WX collected the data, XW and SL analyzed the data, XB and YG prepared the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Animal Ethics Committee of Zhejiang University School of Medicine Animal Center.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wang WT, Li YY, Lin WC, Chen JY, Lan KM, Sun CK, Hung KC. Bilateral visual loss and cerebral infarction after spleen embolization in a trauma patient with idiopathic thrombocytopenic purpura: A case report. Medicine (Baltimore) 2018;97(e332) doi: 10.1097/MD.0000000000010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tao L, ShiChuan W, DeTai Z, Lihua H. Evaluation of lipoprotein-associated phospholipase A2, serum amyloid A, and fibrinogen as diagnostic biomarkers for patients with acute cerebral infarction. J Clin Lab Anal. 2020;34(e23084) doi: 10.1002/jcla.23084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu SS, Ge S, Su CQ, Xie J, Mao J, Shi HB, Hong XN. MRI of plaque characteristics and relationship with downstream perfusion and cerebral infarction in patients with symptomatic middle cerebral artery stenosis. J Magn Reson Imaging. 2018;48:66–73. doi: 10.1002/jmri.25879. [DOI] [PubMed] [Google Scholar]
- 4.Tajima H, Kasai H, Sugiura T, Tatsumi K. Pulmonary arteriovenous fistula complicated by venous thromboembolism and paradoxical cerebral infarction during early pregnancy. BMJ Case Rep. 2018;2018(bcr2017222519) doi: 10.1136/bcr-2017-222519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao J, Mao Y, Qi J. Expression of cytoskeleton and apoptosis related genes after cerebral infarction. Neurol Res. 2006;28:71–75. doi: 10.1179/016164106X91906. [DOI] [PubMed] [Google Scholar]
- 6.Li C, Che LH, Ji TF, Shi L, Yu JL. Effects of the TLR4 signaling pathway on apoptosis of neuronal cells in diabetes mellitus complicated with cerebral infarction in a rat model. Sci Rep. 2017;7(43834) doi: 10.1038/srep43834. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Kim SH, Yoo ES, Woo JS, Han SH, Lee JH, Jung SH, Kim HJ, Jung JY. Antitumor and apoptotic effects of quercetin on human melanoma cells involving JNK/P38 MAPK signaling activation. Eur J Pharmacol. 2019;860(172568) doi: 10.1016/j.ejphar.2019.172568. [DOI] [PubMed] [Google Scholar]
- 8.Kwon HC, Kim TY, Lee CM, Lee KS, Lee KK. Active compound chrysophanol of Cassia tora seeds suppresses heat-induced lipogenesis via inactivation of JNK/p38 MAPK signaling in human sebocytes. Lipids Health Dis. 2019;18(135) doi: 10.1186/s12944-019-1072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kodata T, Kamata K, Fujiwara K, Kano M, Yamakawa T, Yuki I, Murayama Y. A new infarction detection method based on heart rate variability in rat middle cerebral artery occlusion model. Conf Proc IEEE Eng Med Biol Soc. 2017;2017:3061–3064. doi: 10.1109/EMBC.2017.8037503. [DOI] [PubMed] [Google Scholar]
- 10.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Liu D, Tang ZY, Hu ZJ, Li WW, Yuan WN. miR-940 regulates angiogenesis after cerebral infarction through VEGF. Eur Rev Med Pharmacol Sci. 2018;22:7899–7907. doi: 10.26355/eurrev_201811_16416. [DOI] [PubMed] [Google Scholar]
- 13.Wang D, Wang L, Bai L, Du Y, Liu L, Chen X. Effects of inhibition of miR-155-5p in neural stem cell subarachnoid transplant on rats with cerebral infarction. Hum Gene Ther Methods. 2019;30:184–193. doi: 10.1089/hgtb.2019.118. [DOI] [PubMed] [Google Scholar]
- 14.Xu XH, Zhang SM, Yan WM, Li XR, Zhang HY, Zheng XX. Development of cerebral infarction, apoptotic cell death and expression of X-chromosome-linked inhibitor of apoptosis protein following focal cerebral ischemia in rats. Life Sci. 2006;78:704–712. doi: 10.1016/j.lfs.2005.05.080. [DOI] [PubMed] [Google Scholar]
- 15.Ji JF, Ma XH. Effect of baculovirus P35 protein on apoptosis in brain tissue of rats with acute cerebral infarction. Genet Mol Res. 2015;14:9353–9360. doi: 10.4238/2015.August.10.16. [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, Xu Y, Geng L. Caspase-3 inhibitor prevents the apoptosis of brain tissue in rats with acute cerebral infarction. Exp Ther Med. 2015;10:133–138. doi: 10.3892/etm.2015.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie YL, Zhang B, Jing L. MiR-125b blocks Bax/Cytochrome C/Caspase-3 apoptotic signaling pathway in rat models of cerebral ischemia-reperfusion injury by targeting p53. Neurol Res. 2018;40:828–837. doi: 10.1080/01616412.2018.1488654. [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, Zhang J, Wang C, Sun Q. Tanshinone inhibits neuronal cell apoptosis and inflammatory response in cerebral infarction rat model. Int J Immunopathol Pharmacol. 2017;30:123–129. doi: 10.1177/0394632017703274. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Lee JB, Woo HG, Chang Y, Jin YM, Jo I, Kim J, Song TJ. Plasma Klotho concentrations predict functional outcome at three months after acute ischemic stroke patients. Ann Med. 2019;51:262–269. doi: 10.1080/07853890.2019.1617434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu N, Gu C, Gu H, Hu H, Han Y, Li Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma. 2011;58:482–490. doi: 10.4149/neo_2011_06_482. [DOI] [PubMed] [Google Scholar]
- 21.Su JC, Lin KL, Chien CM, Lu CM, Chen YL, Chang LS, Lin SR. Novel indoloquinoline derivative, IQDMA, induces G(2)/M phase arrest and apoptosis in A549 cells through JNK/p38 MAPK signaling activation. Life Sci. 2009;85:505–516. doi: 10.1016/j.lfs.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Osone S, Hosoi H, Kuwahara Y, Matsumoto Y, Iehara T, Sugimoto T. Fenretinide induces sustained-activation of JNK/p38 MAPK and apoptosis in a reactive oxygen species-dependent manner in neuroblastoma cells. Int J Cancer. 2004;112:219–224. doi: 10.1002/ijc.20412. [DOI] [PubMed] [Google Scholar]
- 23.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.