

Abstract
Hepatocyte cell death and liver inflammation have been well recognized as central characteristics of nonalcoholic steatohepatitis (NASH), however, the underlying molecular basis remains elusive. The kinase receptor-interacting protein 1 (RIP1) is a multitasking molecule with distinct functions in regulating apoptosis, necroptosis, and inflammation. Dissecting the role of RIP1 distinct functions in different pathophysiology has absorbed huge research enthusiasm. Wild-type and RIP1 kinase-dead (Rip1K45A/K45A) mice were fed with high-fat diet (HFD) to investigate the role of RIP1 kinase activity in the pathogenesis of NASH. Rip1K45A/K45A mice exhibited significantly alleviated NASH phenotype of hepatic steatosis, liver damage, fibrosis as well as reduced hepatic cell death and inflammation compared to WT mice. Our results also indicated that both in vivo lipotoxicity and in vitro saturated fatty acids (palmitic acid) treatment were able to induce the kinase activation of RIP1 in liver macrophages. RIP1 kinase was required for mediating inflammasome activation, apoptotic and necrotic cell death induced by palmitic acid in both bone marrow-derived macrophage and mouse primary Kupffer cells. Results from chimeric mice established through lethal irradiation and bone marrow transplantation further confirmed that the RIP1 kinase in hematopoietic-derived macrophages contributed mostly to the disease progression in NASH. Consistent with murine models, we also found that RIP1 kinase was markedly activated in human NASH, and the kinase activation mainly occurred in liver macrophages as indicated by immunofluorescence double staining. In summary, our study indicated that RIP1 kinase was phosphorylated and activated mainly in liver macrophages in both experimental and clinical NASH. We provided direct genetic evidence that the kinase activity of RIP1 especially in hematopoietic-derived macrophages contributes to the pathogenesis of NASH, through mediating inflammasome activation and cell death induction. Macrophage RIP1 kinase represents a specific and potential therapeutic target for NASH.
Subject terms: Cell death and immune response, Chronic inflammation
Introduction
In the recent decades, the incidence of nonalcoholic fatty liver disease (NAFLD) increases rapidly and becomes a huge threat to public health of the whole world. Nonalcoholic steatohepatitis (NASH) is the aggressive subtype of NAFLD and is becoming the main cause for liver cirrhosis, hepatocellular carcinoma or liver transplantation in USA [1–3]. Regarding the mechanisms involved in the pathogenesis of NASH, it is now widely recognized that hepatic lipid accumulation and increased free fatty acids (FFA) cause lipotoxicity to hepatocytes and induces cell death, which is accompanied by the massive recruitment of monocytes into liver and the occurrence of chronic inflammation [4–6]. Accumulating studies suggest that hepatic cell death and inflammation play critical roles in the transformation of simple steatosis to steatohepatitis, promoting liver fibrosis and disease progression [7, 8]. However, the molecular basis regulating hepatic cell death and inflammation in steatohepatitis still remains unclear.
The serine/threonine kinase RIP1, as a key regulator determining the cell fate, is a multitasking molecule harboring distinct roles with its kinase activity and kinase-independent scaffolding function [9, 10]. In contrast to mediating NF-κB activation and cell survival through its scaffolding function, the kinase activity of RIP1 is critical in regulating apoptotic and necrotic cell death as well as inflammation [11]. Activation of RIP1 kinase promotes the occurrence of either apoptosis or necroptosis, depending on the cellular context and the molecules it interacts. If activated RIP1 interacts with caspase-8, it leads to apoptotic cell death. When caspase-8 is deficient or inhibited, RIP1 interacts with RIP3 and MLKL to initiate necroptosis [12, 13]. In addition to its key roles in mediating cell death, RIP1 kinase has also been found to mediate inflammatory pathways including inflammasome activation induced by different stimulus [14–16]. Post-translational modifications including phosphorylation, ubiquitination, and acetylation determine different function and activities of RIP1 [17]. Dissecting the roles of RIP1 distinct functions in different physiological and pathological context has emerged as a hot research topic and has gained intensive investigation recently. The kinase activity of RIP1 has been recently reported to play a critical role in mediating the development of different inflammatory diseases especially neurodegenerative disorders [9, 18], turning RIP1 kinase into a promising therapeutic target for human diseases. However, it still remains elusive regarding the role of RIP1 in chronic liver diseases, such as NASH.
In this study, we comprehensively investigated the role of RIP1 kinase in the pathogenesis of NASH and the underlying mechanisms by comparing RIP1 kinase-dead knock-in mice and wild-type mice in two different murine models of NASH. Results indicated that HFD feeding-induced liver damage, steatosis, fibrosis as well as macrophage infiltration and activation were significantly attenuated in RIP1 kinase-dead mice compared to WT control. More importantly, using irradiation and bone marrow transplantation (BMT), our results also revealed that it was the RIP1 kinase activity in hematopoietic-derived macrophages contributing to the inflammation and fibrosis progression in NASH. We further indicated that RIP1 kinase was obviously activated in liver macrophages in both human and mouse NASH. Our results collectively demonstrated the functional involvement of macrophage RIP1 kinase activity in NASH development.
Results
RIP1 kinase activity contributed to the pathogenesis of HFD-induced steatohepatitis
To investigate the role of RIP1 kinase in the development of steatohepatitis, RIP1 kinase-dead knock-in (Rip1K45A/K45A) mice, which contain a lysine point mutation (K45A) in the catalytic triad residues of the kinase domain as previously described [19, 20], were utilized (Fig. S1). Wild-type C57BL/6J and Rip1K45A/K45A mice were fed with normal diet (ND) or high-fat diet (HFD) for 24 weeks. Body weight, food and water consumption, fasting blood glucose, glucose tolerance test (GTT), and basic metabolism parameters were monitored during HFD feeding. There was no obvious difference regarding the food/water intake, oxygen consumption, CO2 production and energy expenditure features between WT and Rip1K45A/K45A mice (Fig. S2B&E-M), suggesting that RIP1 kinase inactivation did not affect the basic metabolism activity. For the body weight, although the initial body weight gain was lower in RIP1 KD mice than wild-type controls, there was no significant difference for the final body weight at the end of HFD feeding between WT and RIP1 KD mice (Fig. S2A). Moreover, fasting blood glucose levels as well as glucose tolerance were similar between WT and Rip1K45A/K45A mice (Fig. S2C, D), suggesting that RIP1 kinase inactivation did not affect the development of obesity and insulin resistance in this dietary model. In contrast, both serum alanine aminotransferase (ALT) and hepatic triglycerides (TG) levels were significantly reduced in Rip1K45A/K45A mice than WT following 24 weeks of HFD feeding (Fig. 1A, B). Reduction of steatosis in Rip1K45A/K45A mice was further confirmed by hematoxylin and eosin (H&E) and Oil Red O staining (Fig. 1C–E). Moreover, as indicated by Sirius Red staining, α-SMA immunohistochemistry analysis, hepatic hydroxyproline content as well as the mRNA levels of transforming growth factor-beta1 (Tgf-β1), Col3a1, and α-sma, all results consistently demonstrated that HFD feeding induced obvious liver fibrosis in WT mice, but not in Rip1K45A/K45A mice (Fig. 1F–J). These results suggested that the kinase activity of RIP1 contributes to pathology development especially fibrosis progression in HFD-induced steatohepatitis. We then wondered whether RIP1 kinase was activated in this model of NASH. Through utilizing phosphor-RIP1 (Ser166) antibody as a marker of RIP1 kinase activation, we found that HFD feeding induced more RIP1 phosphorylation in liver tissue than ND (Fig. 1K), suggesting that RIP1 kinase was activated during NASH.
Fig. 1. RIP1 kinase prompted the liver injury, steatosis, and fibrosis in HFD-induced steatohepatitis.

WT and Rip1K45A/K45A mice were fed with a control normal diet (ND) or a high-fat diet (HFD) for 24 weeks. Then A serum ALT and B hepatic TG levels were measured. Liver sections were analyzed with C H&E staining or D Oil Red O staining. Representative images of each group are presented. E Quantification of Oil Red O-stained area in (D). F Representative images of Sirius Red staining, G Hepatic mRNA levels of Tgf-β1, Col3a1, and α-sma and H hepatic hydroxyproline content in indicated groups. I Representative images of α-SMA protein expression and J the accordingly quantitative analysis of the α-SMA-positive signals as detected by immunohistochemistry in indicated groups. K HFD feeding induced the activation of RIP1 kinase as indicated by phosphor-RIP1 (Ser166) western blot in liver tissue of WT mice. Data are expressed as mean ± SEM (n = 6 or 9 per group). ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
We also used another mechanistically distinct model of steatohepatitis to further verify the role of RIP1 kinase activity in murine models of NASH. Wild-type C57BL/6J and Rip1K45A/K45A mice were fed with methionine-choline deficient (MCD) diet for 5 weeks to stimulate steatohepatitis rapidly. RIP1 Ser166 phosphorylation was also induced in the liver of MCD-fed mice (Fig. S3A). Rip1K45A/K45A mice exhibited significantly lower serum levels of ALT (p < 0.01) and hepatic TG concentrations (p < 0.05) than WT mice (Fig. S3B, C). Consistent with HFD-induced NASH model, hepatic steatosis and lipid accumulation as demonstrated by H&E staining and Oil Red O staining were also significantly alleviated in Rip1K45A/K45A mice (Fig. S3D–F) than WT control. Moreover, transcriptional expression level of α-smooth muscle actin (α-sma) was much higher in WT mice than Rip1K45A/K45A mice (p < 0.01) (Fig. S3G), suggesting that MCD feeding-induced hepatic fibrosis was also attenuated in RIP1 kinase-dead mice. These results suggest that the role of RIP1 kinase in NASH pathogenesis was not limited to HFD-induced model, but might be prevalent in different NASH models.
RIP1 kinase activity prompted hepatic cell death and liver inflammation in HFD-induced steatohepatitis
RIP1 kinase has been recognized as a critical regulator of apoptosis, necroptosis, and inflammation, which are all closely related to NASH pathology. To explore the underlying mechanisms, we examined these features in liver tissues of HFD-fed mice. As determined by TUNEL staining of liver sections, HFD feeding induced obvious cell death in WT liver tissue, and the cell death rate was significantly reduced in Rip1K45A/K45A mice (Figs. 2A, B and S2N). Both cleaved caspase-3 and phosphorylated MLKL were increased in WT liver, suggesting that both apoptotic and necroptotic cell death were induced during HFD-triggered steatohepatitis (Fig. 2C–E). RIP1 kinase activity was required for both cell death modes as the levels of cleaved caspase-3 and phosphorylated MLKL were significantly lower in Rip1K45A/K45A mice than WT control (Fig. 2C–E). HFD feeding also induced liver inflammation in WT mice (Fig. 2F–O). The mRNA levels of macrophage chemoattractant Mcp-1, and pro-inflammatory cytokines TNFα, IL-6, and IL-1β, were all significantly enhanced in the livers of HFD-fed WT mice, and the increase was efficiently inhibited in Rip1K45A/K45A mice (Fig. 2F). Consistent with transcriptional expression, the protein levels of IL-1β in liver tissue were also significantly higher in WT control than Rip1K45A/K45A mice (Fig. 2G, H). Immunohistochemistry analysis of F4/80, CD11b and CD68 indicated that HFD feeding not only induced the activation of liver resident macrophages, but also caused inflammatory monocytes/macrophages liver infiltration in WT mice, and these inflammatory responses were obviously suppressed in Rip1K45A/K45A mice (Fig. 2I–N). We used immunofluorescence double staining of CD11b and F4/80 to corroborate that hepatic recruitment of pro-inflammatory macrophages induced by HFD feeding was obviously reduced in Rip1K45A/K45A mice (Fig. 2O). Immunofluorescence dual-staining of phosphor-RIP1 (Ser166) and F4/80 further demonstrated that in HFD-induce NASH, RIP1 was significantly activated and phosphorylated in liver macrophages (Fig. 2P–R). In addition, RIP1 Ser166 phosphorylation signal was absent in Rip1K45A/K45A mice, confirming the kinase inactivation caused by K45A mutation in RIP1 KD mice (Fig. 2P). Taken together, these results demonstrated that RIP1 kinase was mainly activated in hepatic macrophages in HFD-fed mice. Both hepatic cell death and liver inflammation induced by HFD feeding were significantly suppressed in RIP1 kinase-dead mice, implying that the kinase activity of RIP1 might exert its role in NASH pathogenesis through regulating hepatic cell death induction, macrophage liver infiltration and activation. And the results obtained from MCD-induced NASH model further confirmed that the liver inflammation during dietary-induced steatohepatitis was significantly reduced in Rip1K45A/K45A mice compared to WT control (Fig. S3H–K).
Fig. 2. RIP kinase activity contributed to the hepatic cell death, inflammatory cell infiltration and liver inflammation in HFD-induced steatohepatitis.

A, B Representative images and quantification of TUNEL-stained liver sections. C Immunoblotting detection of cleaved caspase-3 in the liver homogenates. D, E Representative images and quantification of phospho-MLKL expression detected by immunofluorescence analysis. F Hepatic mRNA expression of the inflammatory molecules Mcp-1, IL-1β, Tnf-α, and IL-6 in indicated groups. G–N Representative images of IL-1β, F4/80, CD11b, and CD68 staining by immunohistochemistry analysis and quantification of the respective positive signals. O Representative images of immunofluorescence double staining of CD11b (green) and F4/80 (red). The cell nuclei were stained with DAPI (blue). Red arrow: F4/80+CD11b− cells, green arrow: F4/80−CD11b+ cells, yellow arrow: F4/80+CD11b+ cells. P Representative images of immunofluorescence double staining of phospho-RIP1 (green) and F4/80 (red). The cell nuclei were stained with DAPI (blue). White arrow: p-RIP1+/F4/80+ cells. Q, R Quantitative analysis of F4/80-positive signals or phosphor-RIP1-positive signals in (P). Data are expressed as mean ± SEM (n = 6 or 9 per group). ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Our results indicated that Rip1K45A/K45A mice exhibited significantly decreased hepatic steatosis compared to WT control upon both HFD and MCD feeding (Figs. 1B–E and S3C–F). To answer the questions whether steatosis is the real underlying driver of reduced inflammation and fibrosis and whether steatosis occurs before inflammation, we performed two sets of short-term experiments. WT, Rip1K45A/+, and Rip1K45A/K45A mice were fed with HFD for 3 days and then the hepatic TG levels was analyzed. As the results shown (Fig. S4A), 3 days of HFD feeding was able to increase the hepatic TG levels in WT, Rip1K45A/+, and Rip1K45A/K45A mice. There was no difference regarding the liver TG levels between WT and Rip1K45A/K45A mice, suggesting that RIP1 kinase inactivation did not affect dietary-induced steatosis at very early stage. And our results indicated that 3 days of HFD feeding did not induce obvious inflammation characteristics (data not shown). In addition, we performed another experiment that WT and Rip1K45A/K45A mice were fed with high-fat and high-cholesterol (HFHC) diet for 4 weeks. As the results demonstrated (Fig. S4B), serum TG levels were significantly reduced in Rip1K45A/K45A mice than WT following 4 weeks of HFHC feeding. Decreased steatosis in Rip1K45A/K45A mice was further confirmed by H&E and Oil Red O staining (Fig. S4C–E). In contrast to 3-day feeding, 4-week HFHC feeding induced the activation of hepatic macrophages as indicated by F4/80 IHC analysis (Fig. S4F), and this inflammatory feature were reduced in Rip1K45A/K45A mice than WT controls. These results suggest that in dietary-induced NASH models, liver steatosis was induced at very early stage once the HFD or HFHC feeding was initiated, and then followed by the activation of hepatic macrophages, recruiting circulating macrophages and neutrophils into liver to induce liver inflammation. Although RIP1 kinase inactivation did not affect the very early liver steatosis induction (3 days feeding), but it resulted in decreased liver steatosis at late stage (4-week and 24-week feeding). These results suggest that on one hand, steatosis occurs before inflammation and fibrosis, and steatosis could stimulate the inflammatory cell infiltration and macrophage activation; on the other hand, hepatic inflammation could also further aggravate steatosis, and therefore, steatosis and hepatic inflammation contribute to the progression of each other mutually.
RIP1 kinase activity in hematopoietic-derived macrophages was critical for the disease progression of steatohepatitis
Above results suggested that RIP1 kinase was not only activated in liver macrophages, but also involved in mediating the hepatic recruitment of CD11b+ monocytes/macrophages as well as activation of hepatic macrophages. And it is well known that liver macrophages, including resident Kupffer cells and monocyte-derived macrophages, play critical roles in triggering hepatic inflammation in chronic liver diseases [21]. Chronic liver injury induced by lipotoxicity, stimulates the activation of Kupffer cell and recruitment of circulating bone marrow-derived monocytes into the liver, leading to sustained inflammation and fibrosis progression [22, 23]. To further investigate the underlying mechanism and to distinguish the roles of hematopoietic or nonhematopoietic RIP1 kinase in NASH development, we used irradiation and BMT to create chimeric mice with RIP1 kinase inactivation only in hematopoietic cells (Fig. 3A). We first confirmed the efficiency of BMT (Fig. 3B). Following 5 weeks of MCD feeding, mice containing hematopoietic RIP1 K45A/K45A mutation exhibited attenuated liver injury and steatosis than WT→WT controls (Fig. 3C–G). Hepatic fibrosis was also significantly alleviated in KD→WT chimeric mice than WT→WT controls as determined by different endpoints analysis including Sirius Red staining, α-SMA immunohistochemical staining of liver sections, qPCR detection of fibrotic markers (Col3a1, Tgf-β, and α-sma), and hepatic hydroxyproline detection (Fig. 3H–L). We next examined whether RIP1 kinase inactivation only in hematopoietic cells affected the inflammatory characteristics associated with steatohepatitis. As shown in Fig. 4A, expression of inflammatory markers (Mcp-1, Il-1β, and Tnf-α) induced by MCD feeding was almost completely inhibited in KD→WT chimeric mice. Immunohistochemical analysis of CD68, F4/80, and CD11b implicated that RIP1 kinase activity in hematopoietic cells was not only required for the activation of resident CD68+ Kupffer cells, but also for the hepatic recruitment of CD11b+ bone marrow-derived macrophages (BMDMs) during steatohepatitis (Fig. 4B–G). Moreover, percentage of TUNEL-positive cells was significantly decreased in the liver of KD→WT chimeric mice than controls (Fig. 4H, I). Although there was no statistical significance, MLKL phosphorylation also exhibited a decreasing trend in the liver of KD→WT chimeric mice than controls (Fig. 4J, K). Taken together, these results suggest that the kinase activity of RIP1 in hematopoietic-derived macrophages determined its contribution to the hepatic inflammation, cell death, liver pathology, and fibrosis progression in experimental steatohepatitis.
Fig. 3. RIP1 kinase activity in hematopoietic-derived macrophages was crucial for the disease progression of steatohepatitis.

A The experimental design is illustrated. B Reconstitution of donor bone marrow cells were verified by DNA sequencing. C Serum ALT and AST, D hepatic TG levels were analyzed after 5-week MCD feeding. Liver sections were analyzed with E H&E staining, F Oil Red O staining, H Sirius Red staining, and I α-SMA immunohistochemistry staining. Representative photographs of each group are presented. G Quantification of Oil Red O-stained area in (F). J Quantitative scoring of α-SMA-positive signals in (I). K Hepatic mRNA levels of the fibrosis markers Tgf-β1, Col3a1, and α-sma in indicated groups. L Hepatic hydroxyproline content in indicated groups. Data are expressed as mean ± SEM (n = 6–9 per group). ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Fig. 4. RIP1 kinase activity in hematopoietic-derived macrophages contributed to the liver inflammation and hepatic cell death induction in steatohepatitis.

A Hepatic mRNA expression of the inflammatory molecules Mcp-1, IL-1β, Tnf-α, and IL-6. B–G Representative images of F4/80, CD11b, and CD68 staining by immunohistochemistry analysis and quantification of the positive signals, respectively. H, I Representative images and quantification of TUNEL-stained liver sections. J, K Representative images and quantification of immunofluorescence staining for phosphorylated MLKL. Data are expressed as mean ± SEM (n = 6– 9 per group). ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
RIP1 kinase was required for saturated fatty acid-induced cell death and inflammasome activation in hepatic macrophages
Above in vivo results suggested that the kinase activity of RIP1 especially in hematopoietic-derived macrophages contributes to the pathogenesis of dietary-induced NASH. Considering its key function in mediating apoptotic and necrotic cell death and inflammation, we postulated that RIP1 kinase might exert its role via regulating hepatic cell death and inflammatory response induced by lipotoxicity burden in steatohepatitis. To model this in vitro, we used palmitic acid (PA) or oleic acid (OA), which are dominant saturated or unsaturated FFA in human plasma, to treat BMDMs, or primary mouse Kupffer cells isolated from mouse liver respectively. Serum FFA especially PA levels are elevated in patients with NASH [24, 25] and have been shown to cooperate with gut-derived endotoxin (LPS) to contribute to the pathogenesis of NASH [26]. Our results indicated that saturated fatty acid PA, other than unsaturated fatty acid OA, induced obvious cell death in BMDMs (Fig. S5A), and the cell death was significantly reduced but not completely blocked in Rip1K45A/K45A BMDMs or by RIP1 kinase inhibitor pretreatment (Figs. 5A and S5B). Pan-caspase inhibitor z-VAD greatly aggravated the cell death induced by PA, while combinational pretreatment with Nec-1s was able to prevent the cell death amplified by caspase inhibition (Fig. 5B). These results suggested that caspase inhibition sensitized WT BMDMs to PA-induced necroptosis, which demanded the involvement of RIP1 kinase. Moreover, PA induced the activation of caspase-8, cleavage of caspase-3 and phosphorylation of MLKL, which were all reduced in Rip1K45A/K45A BMDMs (Fig. 5C, D), suggesting that RIP1 kinase mediated the induction of both apoptosis and necroptosis triggered by fatty acids and this correlated with our in vivo data. In agreement with previous studies [26, 27], PA also induced inflammasome activation, which led to the secretion of IL-1β in a dose-dependent manner in LPS-primed BMDMs (Fig. S5C, D). Secretion of TNF-α which is controlled by NF-κB pathway (“signal 1”) was not influenced by RIP1 kinase inactivation or inhibition (Fig. S5F), consistent with previous studies that the kinase activity of RIP1 was dispensable for NF-κB pathway [28]. In contrast, IL-1β release and cleavage of caspase-1 were significantly reduced in Rip1K45A/K45A BMDMs or Nec-1s-pretreated BMDMs (Figs. 5E, F and S5E, G), suggesting that RIP1 kinase contributed to the inflammasome activation through mediating “signal 2” and this result is in line with previous findings [16]. PA-induced IL-1β secretion was completely blocked in NLRP3−/− BMDMs (Fig. S5H) and this result was consistent with previous reports by Wen et al. that PA stimulated the activation of NLRP3 inflammasome and IL-1β release [27]. Together, these results indicated that the kinase activity of RIP1 contributed to the saturated fatty acid-induced apoptosis, necroptosis, and inflammasome activation. At the meantime, we also hypothesized that PA could activate RIP1 kinase in macrophages. As expected, PA treatment induced the Ser166 phosphorylation of RIP1 in BMDMs (Fig. 5G), which was consistent with our in vivo results (Fig. 1K), suggesting that RIP1 kinase can be activated by either saturated fatty acid in vitro or lipotoxicity in vivo.
Fig. 5. RIP1 kinase mediated saturated fatty acid PA-induced cell death and inflammasome activation in both bone marrow-derived macrophages and primary Kupffer cells.

A Bone marrow-derived macrophages (BMDMs) isolated from WT or Rip1K45A/K45A mice were treated with different concentrations of PA for 24 h and then the cell death was evaluated by LDH release assay. B With pretreatment by pan-Caspase inhibitor z-VAD-fmk, or RIP1 kinase inhibitor Nec-1s, or both for 1 h, WT BMDMs were then stimulated by PA and the cell death rate was measured. C, D WT or Rip1K45A/K45A BMDMs were treated as indicated for different time points. Activation of caspase-8, cleavage of caspase-3, and phosphorylation of MLKL were analyzed. E, F WT or Rip1K45A/K45A BMDMs were primed with LPS and then treated with different concentrations of PA. Then the secretion of IL-1β cytokine was detected by ELISA E and the cleaved caspase-1 (p20) in supernatants (SN) or pro-caspase-1 and β-actin in cell lysates (Input) were analyzed using immunoblot F. G WT BMDMs were treated with PA (400 μM) for indicated time. The Ser166 phosphorylation and expression of RIP1 were detected by immunoblot. H Primary Kupffer cells isolated from WT or Rip1K45A/K45A mice were treated with PA (400 μM) for indicated time. The Ser166 phosphorylation and expression of RIP1 were detected by immunoblot. I, J With or without Nec-1s pretreatment, WT primary Kupffer cells were stimulated as indicated, and the cell death rate or the secretion of IL-1β cytokine were measured respectively. Figures are representative of three independent experiments. Data are expressed as mean ± SEM. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
We further confirmed these results using mouse primary Kupffer cells. Similar as BMDMs, PA treatment stimulated the phosphorylation of RIP1 Ser166 in WT primary Kupffer cells, but not in Kupffer cells isolated from RIP1 kinase-dead mice (Fig. 5H), confirming the kinase inactivation of Rip1K45A/K45A mice. PA also induced obvious cell death and IL-1β release in primary Kupffer cells, both of which was significantly reduced by Nec-1s pretreatment (Fig. 5I, J). Taken together, these results suggest that RIP1 kinase activity played a key role in mediating the cell death and inflammasome activation induced by saturated fatty acid PA in both resident hepatic macrophages and bone marrow-derived macrophages, which have been recognized as important players in NASH pathogenesis.
In human NASH, RIP1 kinase was activated in liver macrophages
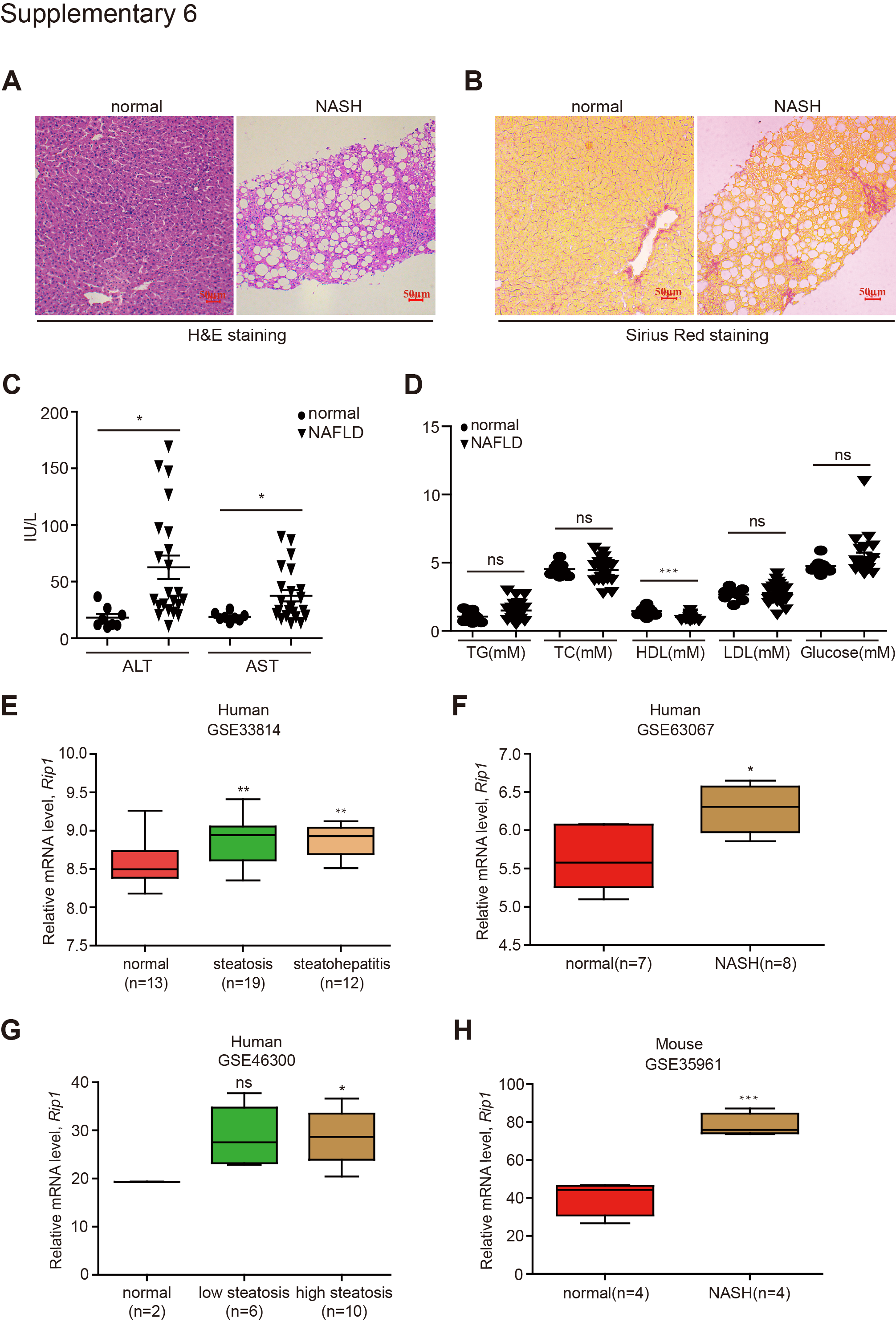
Above results indicated that RIP1 kinase was activated and contributed to the disease progression in experimental steatohepatitis. We then wondered whether it was also involved in human NASH development. First, analysis of the public GEO database suggested that the expression of RIP1 was significantly elevated in liver tissues of NASH or NAFLD patients, similar as in murine models of NASH (Fig. S6E–H). We also examined whether RIP1 was phosphorylated and activated in the liver tissues of NAFLD or NASH patients. The patients’ information are summarized in Supplementary Table 1. As determined by immunofluorescence staining analysis, although the protein expression of RIP1 was not obviously altered, S166 phosphorylation of RIP1, which represented as the marker of RIP1 kinase activation, was significantly triggered in patients with NAFLD/NASH, but not in healthy controls (Fig. 6A, C). The increased phosphorylation of RIP1 was correlated with the steatosis, liver injury and fibrosis in the NASH livers (Figs. 6D and S6A–D), suggesting that during the pathology development of NASH, RIP1 kinase was activated in liver tissue. We next employed immunofluorescence double staining to further examine in which cell type RIP1 was activated in human NASH. As demonstrated in Fig. 6B, E, cells with positive phosphor-RIP1 staining were also positive for CD68 staining, suggesting that in human NASH, RIP1 was mainly phosphorylated and activated in hepatic macrophages and this was in agreement with our results obtained from murine models.
Fig. 6. RIP1 kinase was significantly activated in hepatic macrophages in human NASH.

A Representative images of liver tissue sections from control or NASH patients analyzed for the expression and phosphorylation of RIP1 at Ser166 by immunofluorescence staining. B Representative images of immunofluorescence double staining of phosphor-RIP1 (Ser166) (red) and CD68 (green). The nuclei were stained with DAPI (blue). C–E Quantitative analysis of RIP1-positive, phospho-RIP1-positive signals or CD68-positive signals in (A) or (B). Data are expressed as mean ± SEM. ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Our study for the first time demonstrated the critical contribution of RIP1 kinase in hematopoietic-derived macrophages to the pathology of steatohepatitis. Our study also for the first time demonstrated that during both experimental and clinical NASH, RIP1 kinase was mainly phosphorylated and activated in hepatic macrophages. Our results not only provide direct compelling evidence to support recent work by Majdi et al. for a critical role of RIP1 kinase in pathogenesis of NASH [29], but also further suggest a special and key function of macrophages to the NASH development via RIP1 kinase activity. Several recent human genetic studies have reported that dysregulation of RIP1 was closely related to the pathogenesis of inflammatory and neurodegenerative diseases [30–32]. RIP1 has therefore become an important drug target absorbing the attention of pharmaceutical industry. In addition, Tao et al. recently reported on Nature journal that different human cell types exhibit opposite phenotypes regarding RIP1 kinase-mediated cell death and inflammation [31]. Combined with our results, it suggested that when developing pharmaceutical strategy targeting RIP1 kinase to treat inflammatory diseases, it should take the specialized cell type to target into account and it would be more safe and beneficial to target RIP1 kinase in specialized cell type(s), for example, in macrophages and monocytes.
In our study, we have established two entirely different murine models of steatohepatitis, both of which have been extensively used in NASH research but emphasizing different characteristics and mechanisms regarding steatohepatitis [33]. MCD model exhibits hallmarks of NASH pathology, including steatosis, significant lobular inflammation, and liver fibrosis, as often observed in human NASH. But mice in MCD model lose body weight and this does not mimic human NASH. In contrast, HFD model is an overnutrition model in which NASH accompanies the development of obesity and related metabolic syndromes, including insulin resistance and glucose intolerance, which are invariable characteristics in human NASH [33, 34]. RIP1 kinase-dead (Rip1K45A/K45A) mice exhibited significantly attenuated liver injury, inflammation as well as fibrosis in both models, suggesting that the involvement of RIP1 kinase in NASH pathogenesis is not limited to specific model, but might generally apply to models induced by different factors or conditions.
Hepatocellular death represents a central feature in chronic liver disease [35], contributing to the recruitment of immune cells, activation of hepatic stellate cells and the development of liver fibrosis, cirrhosis and even cancer. In terms of the cell death mode in steatohepatitis, earlier studies suggested that hepatocytes died mainly by apoptosis, a classical programmed and non-inflammatory mode of cell death [36]. Cleaved caspase-3 and other apoptosis markers like CK18 were detected in samples from NASH patients [37, 38]. Genetic or pharmaceutical inhibition of caspases has been shown to attenuate liver injury and liver fibrosis in experimental NASH models or in clinical trials [39]. However, along with the advances in knowledge regarding different forms of cell death and the relevant molecular mechanisms, growing evidences suggest that the mode of hepatocellular death during steatohepatitis is not limited to apoptosis, and another programmed and inflammatory cell death mode, necroptosis, is also involved [8]. The critical regulator and executioner of necroptosis, RIP3 and MLKL, have been demonstrated to play roles in NASH development, despite that there were controversies regarding the role of RIP3 in NASH [40–42]. As an important regulator of both apoptosis and necroptosis, RIP1 has been intensively studied in different liver diseases, including acute liver failure and hepatocellular carcinoma [43–45]. In Concanavalin A (ConA)-induced liver injury model, the mice with RIP1 deficiency in liver parenchymal cells (Ripk1LPC-KO) showed opposite phenotypes with the RIP1 kinase-dead (Rip1K45A/K45A) mice, suggesting that the kinase activity of RIP1 play distinct roles than its scaffolding function in liver injury [46]. Using specific RIP1 kinase-dead mice, our study provided direct genetic evidence that the kinase activity of RIP1 is important for promoting the pathological progression in NASH. Regarding the underlying mechanisms, our results indicated that HFD feeding increased the cleaved caspase-3 and phosphorylated MLKL in WT mice other than Rip1K45A/K45A mice, suggesting that RIP1 kinase mediated both apoptotic and necrotic cell death during steatohepatitis. In addition, expression of inflammatory molecules including IL-1β, TNFα, and Mcp-1 as well as macrophage markers CD68 and F4/80 induced by HFD feeding were all downregulated in Rip1K45A/K45A mice, implying that RIP1 kinase also regulated the inflammatory pathways during the pathogenesis of NASH. Dr. Kanneganti recently reported that there are remarkable crosstalk and redundancies between pyroptosis, apoptosis, and necroptosis, and hence proposed the concept of PANoptosis, which was defined as the integration of pyroptosis, apopotosis and necroptosis pathways [47, 48]. PANoptosis has been found to contribute to auotinflammation, neuroinflammation, and metabolic inflammation, and RIP1 kinase belonged to one of the master regulators mediating PANoptosis. Here we showed that the kinase activity of RIP1 mediated the apoptosis, necroptosis and inflammasome activation induced by lipotoxicity during NASH or by saturated FFA treatment, suggesting that during the progression of steatohepatitis, PANoptosis was induced which required the involvement of key regulators like RIP1 kinase.
In addition to cell death, chronic hepatic inflammation also plays an important role during the disease progression of steatohepatitis. Upon inflammation induction, massive monocytes are recruited to the sites of liver injury. Monocyte-derived macrophages thereby turn to be the dominant population among hepatic macrophages, which further aggravate hepatic inflammation, activate HSCs and prolong the survival of activated HSCs, thus leading to the progression of liver fibrosis [22, 23]. Macrophage has been proposed as a potential therapeutic target for developing novel strategies to treat chronic liver diseases [21]. Krenkel et al. showed that inhibition of monocyte recruitment could efficiently reduce steatohepatitis and fibrosis [23]. In line with these findings, our study indicated that RIP1 kinase was mainly activated in hepatic macrophages and the kinase activity of RIP1 in monocyte-derived macrophage plays a critical role in promoting the fibrosis in NASH. The chimeric mice with kinase-inactive RIP1 only in hematopoietic cells exhibited similar protection against NASH as whole-body Rip1K45A/K45A mice. We also confirmed the clinical relevance of RIP1 kinase activation and found that RIP1 kinase was markedly activated in the liver macrophages of NASH patients, but not in healthy controls. The phosphorylation of RIP1 at Ser166 correlated with the disease progression of NAFLD or NASH. Recently, Majdi et al. just reported that inhibitor of RIP1 kinase (RIPA-56) was able to reduce the liver injury, inflammation, and fibrosis in experimental NASH models [29]. Combined with our results, it suggests that macrophage RIP1 kinase might be a potential therapeutic target for NASH and it will be interesting and worthy to investigate the clinical potential usage of RIP1 kinase inhibitor to treat NASH or other related diseases.
In summary, our study provided direct genetic evidence that RIP1 kinase plays a key role in the pathogenesis of NASH, supporting the potential application of RIP1 kinase inhibitor in treating steatohepatitis. Both mouse and clinical results indicated that macrophage was the main cell type in which RIP1 kinase was activated in liver tissue. The kinase activity of RIP1 in hematopoietic-derived macrophages contributed significantly to the disease progression of NASH, via mediating the induction of apoptotic and necrotic cell death as well as inflammatory pathways. Our study suggest that pharmaceutical targeting RIP1 kinase activity in specialized cell type such as monocyte-derived macrophage might be more beneficial.
Materials and methods
Reagents
Necrostatin-1 (Nec-1) and Necrostatin-1s (Nec-1s) were purchased from Enzo Lifesciences (Switzerland). Anti-Caspase-1 (p20) (mouse) mAb (Casper-1) (AG-20B-0042-C100) was from Adipogen (Germany). Anti-RIP1 (mouse) mAb was from BD Biosciences (America). Phospho-RIP (Ser166) ((D8I3A) Rabbit mAb #44590)/(Antibody (Rodent Specific) #31122), α-Smooth Muscle Actin ((D4K9N) XP® Rabbit mAb #19245), F4/80 ((D2S9R) XP® Rabbit mAb #70076) and IL-1β ((3A6) Mouse mAb #12242), CD68 ((E3O7V) Rabbit mAb #97778)/((D4B9C) XP® Rabbit mAb #76437), CD11b/ITGAM ((D6X1N) Rabbit mAb #49420) and cleaved Caspase-3 ((Asp175) Antibody #9661) were all purchased from Cell Signaling Technology. Anti-MLKL (phospho S345) (ab196436) was from Abcam. Anti-RIPK1/ RIP1 phospho (Ser166) antibody (YJY-1-5) was from Arigobio. Lipopolysaccharides (LPS) was from Sigma-Aldrich. Nigericin (481990) was from Merck-Millipore (Germany). Penicillin–streptomycin, liquid (15140122) were from GIBCO (America). Hifair® II 1st Strand cDNA Synthesis Kit (11119ES60) was from Yeasen (Shanghai, China). KAPA SYBR® Fast (KK4601) was from KAPA BIOSYSTEMS (America). IL-1β ELISA kit (Catalog #88-7013-22) and TNFα ELISA kit (Catalog #88-7324-88) were from Invitrogen (America). LDH Cytotoxicity Assay Kit (C00170) was from Beyotime (Shanghai, China). Alanine transaminase (ALT) detection kit (C009-2-1), aspartate transaminase (AST) detection kit (C010-2-1), Triglyceride (TG) detection kit (A110-1-1) or (BC0625), cholesterol (TC) detection kit (A111-1-1), hydroxyproline detection kit (A030-2-1) and Percoll (P8370) were all purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) or Solarbio Science & Technology (Beijing, China). Type 4 Collagenase (LS004209) was obtained from Worthington.
Mouse models
C57BL/6J mice were purchased from Model Animal Research Center of Nanjing University (Nanjing, China). Rip1K45A/K45A mice on a C57BL/6J background as previously described [20] were kindly provided by HZ (Chinese Academy of Sciences, Shanghai, China). Rip1K45A/K45A mice were crossed with WT C57BL/6J mice to generate Rip1K45A/+ heterozygous mice. Then male and female heterozygous mice were set up to give birth to WT and Rip1K45A/K45A littermates, which were utilized in different diet-induced NASH models. All animals were maintained under standard laboratory conditions, with free access to food and water. All animal experiments were performed under protocols approved by the Animal Care and Use Committee at Nanjing University of Science & Technology.
Eight-week-old male Rip1K45A/K45A mice (C57BL/6J background) and male wild-type C57BL/6J controls were randomly grouped and fed either with a MCD (TP3006, Trophic Animal Feed, China) for 5 weeks, or with a high-fat and high-cholesterol diet (HFHC, 42% fat, 15% protein, 43% carbohydrate and 0.2% cholesterol, TP26303; Trophic Animal Feed, China) for 4 weeks to induce steatohepatitis.
Separate groups of Rip1K45A/K45A and WT mice were fed either with a HFD (60.9% fat, 18.3% protein, 20.8% carbohydrate, TP23520; Trophic Animal Feed, China) or control diet (Trophic Animal Feed, China) for 3 days or 24 weeks. During the 24-week HFD feeding period, body weight gain, food and water consumption, fasting blood glucose were monitored or detected. At the end of HFD feeding, metabolic parameters including O2 consumption, CO2 production, respiratory exchange ratio (RER) as well as heat production were measured by metabolic cage detection (Comprehensive Lab Animal Monitoring System). GTT was also performed as previously described [49].
For BMT, the recipient wild-type C57BL/6J mice were lethally irradiated (9 Gy) and then injected through tail veins with bone marrow cells (5 × 106 cells) harvested from wild-type or Rip1K45A/K45A donor mice. Six weeks after irradiation and BMT, the chimeric mice were fed with either MCD diet or ND for another 5 weeks to induce NASH. Bone marrow cells and blood cells of the chimeric mice were also collected and analyzed by DNA sequencing to verify the bone marrow reconstitution efficiency.
Human samples
Human liver tissue samples and corresponding clinical information were provided by Nanjing Drum Tower Hospital (Nanjing, China). Paraffin-embedded human liver tissue sections were analyzed by H&E staining or Sirius Red staining. Expression and phosphorylation (S166) of RIP1 were detected by immunofluorescence staining using specific antibodies. All individuals gave written informed consent before joining this study. All research procedures were approved by the Clinical Research Ethics Committee of the Nanjing Drum Tower Hospital with the approval number 2008022.
Cell culture and treatments
Mouse BMDMs were derived by maturing bone marrow cells harvested from adult wild-type, Rip1K45A/K45A, or Nlrp3−/− mice in the presence of M-CSF containing supernatant from L929 cells as previously described [50]. Mouse primary Kupffer cells were isolated by liver perfusion as previously described [51]. Freshly isolated hepatocytes or Kupffer cells were separated by 50% percoll or 25% and 50% percoll centrifugation respectively to remove debris and other non parenchymal cells.
Some cells were primed with LPS (100 ng/mL) for 3 h and then cells were treated with different concentrations of saturated fatty acid PA or unsaturated fatty acid OA for 24 h. Then cell supernatant was harvested and the cytokine IL-1β (Catalog #88-7013-22, Invitrogen) and TNFα (Catalog #88-7324-88, Invitrogen) in cell supernatants were measured using Enzyme-linked Immunosorbent Assay (ELISA) kits according to the manufacturer’s instructions. Cell lysates were also harvested and analyzed by immunoblotting. In addition, cell death rate was quantified by detecting the lactate dehydrogenase (LDH) release using LDH Cytotoxicity Assay Kit (C00170; Beyotime, China) following the manufacturer’s instruction. And caspase-8 activity was measured using Caspase-Glo 8 assay kit (Promega, USA) according to the manufacturer’s instructions.
Immunoblotting
Mouse liver tissues, or BMDMs were lysed in RIPA cell lysis buffer with protease inhibitor (P1005; Beyotime, China) and phosphatase inhibitor (P1081; Beyotime, China). Equal amounts of proteins were separated by SDS-PAGE (Acryl/Bis 30% solution (29:1)) (B546017; Sangon Biotech, China), transferred to 0.22 μm PVDF membranes, blocked with 5% skim milk powder in TBS-T (0.1% TWEEN-20). The membranes were incubated with primary antibodies at 4 °C overnight, washed with TBS-T and incubated with secondary antibodies at room temperature for 1–2 h. Chemiluminescent Substrate System from KPL was utilized for final detection.
TUNEL assay
Apoptotic cells in livers were measured by TUNEL Apoptosis Assay Kits. Paraffin-embedded liver tissue sections were pretreated by proteinase K for 20 min at 25 °C. Then tissue sections were washed three times with PBS and then incubated in the mixture of reaction buffer with TdT enzyme in dark for 60 min at 37 °C. Meanwhile the cell nucleuses were stained with DAPI. The tissue sections were observed and photographed using the fluorescence microscope (NIKON ECLIPSE 80i). The apoptotic cell rate was calculated with the formula: (TUNEL-positive cell number/total area cell number) ×100%.
mRNA isolation and qPCR analysis
TRIzol (Invitrogen) reagent was added to cells or tissues and total RNA was extracted according to the manufacturer’s instructions. Next cDNA was synthesized using Hifair® II 1st Strand cDNA Synthesis Kit. PCR reactions were performed on the ABI 7300 real-time PCR system using KAPA SYBR® Fast. GAPDH mRNA was used as an internal control to normalize mRNA expression. The sequences of primers for qPCR were as follows:
| Name | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| Mcp-1 | AGGTCCCTGTCATGCTTCTG | TCTGGACCCATTCCTTCTTG |
| Il-1β | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG |
| Tnf-ɑ | ACGGCATGGATCTCAAAGAC | CGGACTCCGCAAAGTCTAAG |
| Il-6 | CATGTTCTCTGGGAAATCGTGG | GTACTCCAGGTAGCTATGGTAC |
| Nlrp3 | ACTTGCAGAAGCTGGGGTTG | AGTTTACAGTCCGGGTGCAG |
| Tgf-β1 | GTTTGCCGAGTAGATCTC | ATGCTAAAGAGGTCACCC |
| Col3a1 | ACGTAGATGAATTGGGATGCAG | GGGTTGGGGCAGTCTAGTG |
| ɑ-sma | GTACCACCATGTACCCAGGC | GCTGGAAGGTAGACAGCGAA |
| Gapdh | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
Histological analysis
Mouse liver tissues were fixed in 4% paraformaldehyde and then embedded in paraffin and sectioned. Paraffin-embedded liver sections were stained with H&E to evaluate the gross morphology or with Sirius Red to evaluate liver fibrosis. At the same time, frozen sections of liver tissues were stained by Oil Red O working solution to determine the steatosis in liver tissues.
Immunohistochemistry and immunofluorescence analysis
Paraffin-embedded liver sections were firstly deparaffinized and then boiled in citrate buffer (pH 6.0) for antigen retrieval, followed by hydrogen peroxide/PBS blocking of endogenous peroxidase. Slides were pre-blocked and then incubated with primary antibodies at 4 °C overnight. Primary antibodies, including the anti-F4/80, anti-ɑ-SMA, anti-CD68, anti-CD11b, and anti-IL-1β antibodies were used respectively. The slides were then washed with PBS and incubated with secondary antibodies at 37 °C for 50 min, and then stained with DAB substrate after 20 min with streptavidin-HRP. The cell nuclei were stained with hematoxylin. For immunofluorescence, after being incubated with primary antibodies (anti-F4/80, anti-CD11b, anti-phospho-MLKL (S345), anti-RIP1 and anti-phospho-RIP1 (Ser166)) at 4 °C overnight, slides were washed with PBS for three times and then incubated with fluorescent secondary antibody in dark for 60 min. The cell nuclei were stained by DAPI. At last, the slides were observed and photographed using the fluorescence microscope (NIKON ECLIPSE 80i).
Biochemical analysis
Serum levels of ALT, AST, TG and TC, and hepatic hydroxyproline content were analyzed by respective detection kits according to the manufactures’ protocols.
Sequencing data analysis
Several datasets which were published in public database were utilized for human or murine Rip1 expression analyses, including GSE33814 [52], GSE63067 [53], GSE46300 [54], GSE35961 [55], all of which can be downloaded from GEO database.
Statistical analysis
All the data are expressed as the mean ± standard error of the mean (SEM). The statistical analysis of the results was performed using GraphPad Prism® 7.01 software (San Diego, USA). Unpaired Student’s t test or one-way ANOVA (for more than 2 groups) analysis were used to calculate the differences in mean values. p < 0.05 was considered to be a statistically significant difference.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This study was supported by the National Natural Science Foundation of China under Grant 31970897 and 21677076, Outstanding Youth Foundation of Jiangsu Province (BK20190069), the Fundamental Research Funds for the Central Universities No. 30919011102, Qing Lan Project of Jiangsu Province.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by A. Ashkenazi
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41418-020-00668-w) contains supplementary material, which is available to authorized users.
References
- 1.Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 2018;69:896–904. doi: 10.1016/j.jhep.2018.05.036. [DOI] [PubMed] [Google Scholar]
- 2.Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411–28.. doi: 10.1038/s41575-019-0145-7. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152:1090–9 e1091. doi: 10.1053/j.gastro.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noureddin M, Sanyal AJ. Pathogenesis of NASH: the impact of multiple pathways. Curr Hepatol Rep. 2018;17:350–60.. doi: 10.1007/s11901-018-0425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13:316–27.. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 7.Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68:280–95.. doi: 10.1016/j.jhep.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15:738–52.. doi: 10.1038/s41575-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci USA. 2019;116:9714–22.. doi: 10.1073/pnas.1901179116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129–50.. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- 11.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–36.. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 12.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–53. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol. 2014;15:1126–33.. doi: 10.1038/ni.3015. [DOI] [PubMed] [Google Scholar]
- 15.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci USA. 2014;111:7391–6. doi: 10.1073/pnas.1403477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao L, Lin H, Wen J, Sun Q, Gao Y, Xu X, et al. The kinase receptor-interacting protein 1 is required for inflammasome activation induced by endoplasmic reticulum stress. Cell Death Dis. 2018;9:641–54.. doi: 10.1038/s41419-018-0694-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang K, Lee SR, Piao X, Hur GM. Post-translational modification of the death receptor complex as a potential therapeutic target in cancer. Arch Pharm Res. 2019;42:76–87. doi: 10.1007/s12272-018-01107-8. [DOI] [PubMed] [Google Scholar]
- 18.Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in ageing. Cell. 2018;174:1477–91 e1419. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–80.. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Fan C, Zhang Y, Yu X, Wu X, Zhang X, et al. RIP1 kinase activity-dependent roles in embryonic development of Fadd-deficient mice. Cell Death Differ. 2017;24:1459–69.. doi: 10.1038/cdd.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300–12.. doi: 10.1016/j.jhep.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 22.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–73.. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67:1270–83.. doi: 10.1002/hep.29544. [DOI] [PubMed] [Google Scholar]
- 24.de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr. 2002;21:219–23.. doi: 10.1054/clnu.2001.0529. [DOI] [PubMed] [Google Scholar]
- 25.Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50:1827–38.. doi: 10.1002/hep.23229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–44.. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15.. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newton K. Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb Perspect Biol 2020;12:a036368. [DOI] [PMC free article] [PubMed]
- 29.Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F, et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J Hepatol. 2020;72:627–35.. doi: 10.1016/j.jhep.2019.11.008. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Fuhrer M, Bahrami E, Socha P, Klaudel-Dreszler M, Bouzidi A, et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc Natl Acad Sci USA. 2019;116:970–5. doi: 10.1073/pnas.1813582116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577:109–14.. doi: 10.1038/s41586-019-1830-y. [DOI] [PubMed] [Google Scholar]
- 32.Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577:103–8. doi: 10.1038/s41586-019-1828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, Schuppan D. Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology. 2019;69:2241–57.. doi: 10.1002/hep.30333. [DOI] [PubMed] [Google Scholar]
- 34.Haczeyni F, Yeh MM, Ioannou GN, Leclercq IA, Goldin R, Dan YY, et al. Mouse models of non-alcoholic steatohepatitis: A reflection on recent literature. J Gastroenterol Hepatol. 2018;33:1312–20.. doi: 10.1111/jgh.14122. [DOI] [PubMed] [Google Scholar]
- 35.Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov. 2016;2:16089. doi: 10.1038/cddiscovery.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H, et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. 2018;24:2661–72.. doi: 10.3748/wjg.v24.i25.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boege Y, Malehmir M, Healy ME, Bettermann K, Lorentzen A, Vucur M, et al. A dual role of caspase-8 in triggering and sensing proliferation-associated DNA damage, a key determinant of liver cancer development. Cancer Cell. 2017;32:342–59.e310. doi: 10.1016/j.ccell.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 39.Ratziu V, Sheikh MY, Sanyal AJ, Lim JK, Conjeevaram H, Chalasani N, et al. A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology. 2012;55:419–28.. doi: 10.1002/hep.24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology. 2016;64:1518–33.. doi: 10.1002/hep.28676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062–74.. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Investig. 2016;126:4346–60.. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vucur M, Schneider AT, Gautheron J, Luedde T. The enigma of RIPK1 in the liver: more than just a kinase. Mol Cell Oncol. 2017;4:e1304191. doi: 10.1080/23723556.2017.1304191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kondylis V, Pasparakis M. RIP kinases in liver cell death, inflammation and cancer. Trends Mol Med. 2019;25:47–63. doi: 10.1016/j.molmed.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Van TM, Polykratis A, Straub BK, Kondylis V, Papadopoulou N, Pasparakis M. Kinase-independent functions of RIPK1 regulate hepatocyte survival and liver carcinogenesis. J Clin Investig. 2017;127:2662–77.. doi: 10.1172/JCI92508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Filliol A, Piquet-Pellorce C, Le Seyec J, Farooq M, Genet V, Lucas-Clerc C, et al. RIPK1 protects from TNF-alpha-mediated liver damage during hepatitis. Cell Death Dis. 2016;7:e2462. doi: 10.1038/cddis.2016.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malireddi RKS, Tweedell RE, Kanneganti TD. PANoptosis components, regulation, and implications. Aging. 2020;12:11163–4. doi: 10.18632/aging.103528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis) Front Cell Infect Microbiol. 2020;10:237. doi: 10.3389/fcimb.2020.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao P, Wang L, Yang N, Wen J, Zhao M, Su G, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) activation and metabolism disturbance induced by bisphenol A and its replacement analog bisphenol S using in vitro macrophages and in vivo mouse models. Environ Int. 2020;134:105328. doi: 10.1016/j.envint.2019.105328. [DOI] [PubMed] [Google Scholar]
- 50.Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aparicio-Vergara M, Tencerova M, Morgantini C, Barreby E, Aouadi M. Isolation of Kupffer cells and hepatocytes from a single mouse liver. Methods Mol Biol. 2017;1639:161–71.. doi: 10.1007/978-1-4939-7163-3_16. [DOI] [PubMed] [Google Scholar]
- 52.Starmann J, Falth M, Spindelbock W, Lanz KL, Lackner C, Zatloukal K, et al. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS ONE. 2012;7:e46584. doi: 10.1371/journal.pone.0046584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frades I, Andreasson E, Mato JM, Alexandersson E, Matthiesen R, Martinez-Chantar ML. Integrative genomic signatures of hepatocellular carcinoma derived from nonalcoholic fatty liver disease. PLoS ONE. 2015;10:e0124544. doi: 10.1371/journal.pone.0124544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wruck W, Kashofer K, Rehman S, Daskalaki A, Berg D, Gralka E, et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci Data. 2015;2:150068. doi: 10.1038/sdata.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kita Y, Takamura T, Misu H, Ota T, Kurita S, Takeshita Y, et al. Metformin prevents and reverses inflammation in a non-diabetic mouse model of nonalcoholic steatohepatitis. PLoS ONE. 2012;7:e43056. doi: 10.1371/journal.pone.0043056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.