

Abstract
Chronic pain is a common and undertreated nonmotor symptom in Parkinson’s disease (PD). Although chronic pain is improved by L-dopa in some PD patients, the underlying mechanisms remain unclear. In this study, we established PD mice by unilateral microinjection of 6-OHDA in the medial forebrain bundle to investigate the contribution of spinal cord dopamine receptors to parkinsonian pain hypersensitivity. The von Frey filament tests and thermal pain tests revealed that these PD mice displayed decreased nociceptive thresholds in both hindpaws; intrathecal injection of L-dopa or apomorphine significantly increased the mechanical and thermal nociceptive thresholds, and the analgesic effect was mimicked by ropinirole (a D2 receptor agonist), but not SKF38393 (a D1/D5 receptor agonist), and blocked by sulpiride (a D2 receptor antagonist), but not SKF83566 (a D1/D5 receptor antagonist). Whole-cell recordings in lumber spinal cord slices showed that superficial dorsal horn (SDH) neurons in PD mice exhibited hyperexcitability, including more depolarized resting membrane potentials and more action potentials evoked by depolarizing current steps, which were mitigated by ropinirole. Furthermore, ropinirole inhibited the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in SDH neurons more strongly in PD mice than in control mice. However, sulpiride caused less disinhibition of sEPSCs in PD mice than in control mice. Taken together, our data reveal that pain hypersensitivity in PD mice is associated with hyperexcitability of SDH neurons, and both events are reversed by activation of spinal D2 receptors. Therefore, spinal D2 receptors can be promising therapeutic targets for the treatment of PD pain.
Keywords: Parkinson’s disease, pain hypersensitivity, spinal cord, dorsal horn neuron, hyperexcitability, dopamine D2 receptor, intrathecal administration, whole-cell recording
Introduction
Chronic pain is a common but overlooked nonmotor symptom in Parkinson’s disease (PD) [1–4]. Approximately 30%–83% of PD patients experience pain. In a subset of patients, pain is an early symptom that even precedes motor symptoms by several years. As pain symptoms substantially interfere with the quality of life of PD patients, they should not be neglected in the treatment of PD.
In general, pain in PD can be categorized into two types: PD-related pain, which is caused or exaggerated by PD, and non-PD-related pain [2, 3]. Both types of pain are responsive to dopamine compensation, but the underlying mechanisms are not clear. Studies have shown that levodopa (L-dopa) can increase the pain threshold in PD patients [5–7]. Both systemic administration of the neurotoxin MPTP (1-methyl‑4-phenyl‑1,2,3,6-tetrahydropyridine) and intracranial injection of 6-hydroxydopamine (6-OHDA) in rodents cause severe lesions of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc), mimicking the characteristic pathophysiology of PD. These animal models displayed reduced mechanical and thermal nociceptive thresholds in the hindpaw, and hypersensitivity could be relieved by systemic administration of dopamine receptor agonists [8, 9]. Therefore, dopamine depletion could contribute to changes in nociception processing in PD. However, the DA pathways and structures in pain processing that are involved in PD pain have not yet been identified.
The spinal cord dorsal horn is an important structure in pain processing because it not only receives nociceptive signals from primary nociceptors but is also subjected to modulation by local neurons and the supraspinal descending pathway. In the parkinsonian state, degeneration in DA pathways may alter the relay and processing of pain signals within the spinal cord [10, 11]. For example, activation of dopamine D2 receptors in the striatum increases nociceptive thresholds in animal models of spinal nerve ligation-induced neuropathic pain, which indicates that impairment of the striatal DA system modulates pain processing at the spinal level [12]. Furthermore, dopamine has dual effects on nociception by activating different receptors in the spinal cord dorsal horn: activation of D1-like receptors (D1Rs) exerts a pronociceptive effect, whereas activation of D2-like receptors (D2Rs) leads to an antinociceptive effect [13, 14]. In PD patients, electrical stimulation in the spinal cord has been shown to ameliorate not only motor symptoms but also analgesic-resistant chronic pain [15]. These data suggest that dysfunction of the nigrostriatal DA system in PD may directly or indirectly facilitate pain signaling at the spinal cord level. However, whether and how dopamine and its receptors in the spinal cord contribute to pain hypersensitivity in animal models of PD remain unclear.
In the present study, we established a mouse model of parkinsonism by microinjection of 6-OHDA in the medial forebrain bundle (MFB) in one hemisphere. We observed that these mice developed pain hypersensitivity, which was relieved by intrathecal administration of dopamine agonists through activation of D2Rs. Our spinal cord slice patch-clamp recording data revealed that 6-OHDA lesion-induced pain hypersensitivity may be relevant to the elevated intrinsic excitability in superficial dorsal horn (SDH) neurons. We also observed that D2R function in both the glutamatergic terminals and the SDH neurons was enhanced following 6-OHDA lesion, and activation of D2Rs reversed the hyperexcitability in the SDH neurons. Our data indicate that presynaptic and postsynaptic D2Rs in the SDH of the spinal cord may contribute to the antinociceptive effect of L-dopa in PD-related pain. Therefore, D2Rs in the spinal cord may be promising therapeutic targets for the treatment of PD-related pain.
Materials and methods
C57BL/6 male mice (23–28 g, 8–16 weeks old) were purchased from Jinan Pengyue Laboratory Animal Breeding Co,. Ltd., and were group-housed (2–5 mice per cage) on a 12-h light/dark cycle. The mice had free access to water and food. Efforts were made to minimize the suffering of the mice and to reduce the number of mice used. The care and use of animals and the experimental protocols of this study were approved by the Institutional Animal Care and Use Committee and the Office of Laboratory Animal Resources of Xuzhou Medical University.
Unilateral 6-hydroxydopamine (OHDA) lesion of SNc DA neurons
In brief, mice were intraperitoneally (i.p.) administered with desipramine (Sigma-Aldrich, MO, USA) (20 mg/kg) 30 min prior to surgery to protect the noradrenergic nerve terminals and then were deeply anesthetized with sodium pentobarbital (40 mg/kg, i.p.). A small craniotomy was performed above the right MFB. The coordinate for the MFB is 1 mm anterior to the bregma, 1.2 mm lateral to the midline, and 4.8 mm deep relative to the bregma. Then, 0.3 µL of 6-OHDA (Tocris, Bristol, UK) (12 µg/µL in 0.2% vitamin C/normal saline) was stereotaxically injected at a rate of 0.1 µL/min. Control mice were injected with 0.3 µL of normal saline containing 0.2% vitamin C. Postsurgical care was carried out similarly in both the control and 6-OHDA-injected mice.
Behavioral tests
Open field test
Individual mice were placed in a cylinder with a diameter of 30 cm. The motor behavior of each mouse was recorded in a 20 min session through a video camera controlled by Ethovision XT 9 software (Noldus, Waningen, Netherland). The video was analyzed either online or offline with Ethovision software.
von Frey filament test
The mechanical pain threshold was measured in acclimated mice with the up-down method [16], using von Frey filaments (Bio-VF-M, Bioseb, USA). In brief, the first von Frey filament (0.41 g) was applied to the plantar surface of the hindpaw. If a withdrawal response was observed within 2 s, the filament with the next lower force was used. Conversely, if the filament failed to elicit a withdrawal response, the next filament with a higher force was applied. After the first withdrawal response occurred, this paradigm continued until a total of six responses, starting from the one before the first withdrawal response, were recorded. A score ranging between 0.01 and 3.0 g was assigned in the case of four consecutive positive responses to filaments with decreasing force or three consecutive negative responses to filaments with increasing forces. The 50% paw withdrawal threshold (PWT) was determined using these responses as described previously [16].
Thermal pain test
Mice were acclimatized for at least 30 min in a test compartment placed on the glass surface of a plantar anesthesia tester (Boerni, Tianjin, China). A heat and light source was positioned under the glass surface pointing to the plantar surface of the hindpaw. Paw withdrawal latency (PWL) was then recorded. The cutoff time of heating was set to 20 s to prevent tissue damage.
In the above experiments, the investigators were blind to the treatment of the mice.
Intrathecal injection of drugs
Mice were tightly restrained. Individual drugs or a combination of two drugs in a total volume of 10 μL was injected intrathecally (i.t.) in the vertebrae between L4 and L5 with a 30-gauge needle. Tail flick was indicative of successful performance. The control group received an i.t. injection of vehicle or drugs, including L-dopa, apomorphine hydrochloride, sulpiride, SKF38393, Ropinirole, and SKF83566, were purchased from Tocris Bioscience (Bristol, UK) and dissolved in normal saline.
Patch-clamp recordings of spinal cord slices
Patch-clamp recordings of spinal cord slices were performed according to a previously described protocol [16–19]. In brief, the mice were deeply euthanized with CO2 and then decapitated. The spinal cord was extracted, and the lumbar spinal cord was cut into 300 μm thick slices with a vibratome (VT-1200S, Leica, Germany) in ice-cold modified sucrose-based artificial cerebral spinal fluid (sACSF) saturated with 95% O2/5% CO2 (carbogen) containing the following (mM): 85 NaCl, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 4.0 MgCl2, 0.5 CaCl2, 24 NaHCO3, and 25 glucose, pH 7.3. Lumber spinal cord slices were allowed to recover at 32 ± 1 °C for a minimum of 1 h in carbogenated sACSF. The spinal cord slices were then transferred into carbogenated normal ACSF (in mM: 125 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 glucose, pH 7.3) and were kept at room temperature.
Patch-clamp recordings were performed on SDH neurons in spinal cord slices, stabilized in a recording chamber with a U-shaped anchor. The neurons were identified using infrared differential interference contrast on an upright FN-1 Nikon microscope equipped with a CCD camera (Flash 4.0 LTE, Hamamatsu, Japan). The data were acquired and amplified via a MultiClamp 700B amplifier (Molecular Devices, CA, USA) and digitized at 10 kHz via a Digidata 1550B analog-to-digital converter (Molecular Devices, CA, USA) under the control of pClamp 10.7 software (Molecular Devices, CA, USA). During recordings, the temperature of the recording chamber was maintained at 32 ± 0.5 °C with a temperature controller (Warner Instruments, CT, USA), and the flow rate of the carbogenated ACSF was set to 1.5–2.0 mL/min. Patch pipettes had a resistance of 4–6 MΩ when filled with intrapipette solution with an osmolarity of 300 mOsm/L (in mM: 135 K gluconate, 5 KCl, 0.2 EGTA, 0.5 CaCl2, 10 HEPES, 2 Mg-ATP, and 0.1 GTP, pH 7.2).
The junction potential between the patch pipette and bath solution was nulled just before gigaseal formation. Series resistance was monitored without compensation throughout the experiment, and the data were discarded if the series resistance (11–20 MΩ) changed by >20% during whole-cell recordings.
Immunohistochemistry
Animals were sacrificed in a CO2 chamber and subjected to transcardial perfusion with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Brains were removed and postfixed in PFA for 12 h at 4 °C. Brain sections (100 μm) were made with a vibratome and mounted onto glass slides. For antibody staining, brain sections were washed in PBS for 10 min, blocked in 5% donkey serum and 0.1% Triton in PBS for 90 min, followed by incubation in primary antibody (mouse anti-TH, Santa Cruz, 1:500) in 1% donkey serum and 0.1% Triton in PBS for 24 h at 4 °C. After three washes (10 min each) in PBS, the sections were subsequently incubated with secondary antibodies (donkey anti-mouse Cy2, Jackson ImmunoResearch, 1:500) for 90 min at room temperature. The sections were washed three times (10 min each) in PBS, dried in the dark, and then coverslipped in mounting medium.
The stained sections were imaged with a Zeiss LSM 880 confocal microscope (Zeiss, Wetzlar, Germany), equipped with a 10× Plan Apochromat air objective (NA, 0.45) and a 20× Plan Apochromat objective (NA, 0.8). The images were processed with ImageJ [20].
Statistics
GraphPad Prism 7.0 was used for all statistical analyses. Data are shown as the mean ± SEM in all graphs. Two-tailed paired t-test, one-way ANOVA or two-way ANOVA was used as indicated in the figure legends. Values of P < 0.05 were considered statistically significant.
Results
Unilateral parkinsonian mice exhibit mechanical and thermal pain hypersensitivity
We microinjected 6-OHDA (3.6 µg in 0.3 µL) or the same volume of saline into the right MFB to establish unilateral 6-OHDA-lesioned mice and saline control mice (see Materials and Methods). In this experiment, all 6-OHDA-lesioned and control mice survived. Midbrain slices were collected 1, 2, 3, and 4 weeks after the 6-OHDA and saline injection, and DA neurons in the substantia nigra pars compacta (SNc) were quantified by immunostaining for tyrosine hydroxylase (TH) antibody to assess DA neuron loss on the ipsilesional side (Fig. 1a). Relative to the contralesional SNc, the ipsilesional SNc had an ~60% loss in DA neurons (Fig. 1b). The loss of DA neurons occurred one week after surgery and remained at a similar level thereafter. However, the saline control mice showed no DA neuron loss within 5 weeks (Fig. 1b).
Fig. 1. Establishment of unilateral 6-OHDA-lesioned parkinsonian mice.

First, 6-OHDA (3.6 µg/0.3 μL) and the same volume of saline were microinjected into the right MFB of two groups of mice. Mice were sacrificed at each time point shown in the panel (b), and brain sections containing the SNc were subjected to immunostaining with TH antibody to label the DA neurons. a Representative immunofluorescence images showing a robust loss of DA neurons in the ipsilesional SNc (right panel) relative to the contralesional SNc (left panel) 4 weeks after 6-OHDA injection. Scale bar, 100 µm. b Percentage of TH-positive neurons in the ipsilesional SNc relative to those in the contralesional SNc at different time points after saline or 6-OHDA injection. Two-way ANOVA, n = 15 brain sections from 5 mice for quantification of TH-positive neurons; saline vs 6-OHDA: F (1, 20) = 55.18, P < 0.0001; Week: F (4, 20) = 4.855, P = 0.007; Interaction: F (4, 20) = 4.44, P = 0.01. **P < 0.01, compared with baseline, ##P < 0.01, compared with the respective saline group. c Representative movement tracks in 20 min sessions of a mouse 1 day prior to and 1 week and 2 weeks after unilateral 6-OHDA injection. d Average velocity of saline and 6-OHDA mice in an open field arena for a 20 min session. Two-way ANOVA, n = 7 in each group; Group: F (1, 47) = 36.04, P < 0.0001; Week: F (3, 47) = 5.71, P = 0.002; Interaction: F (3, 47) = 8.18, P = 0.0002. *P < 0.05, **P < 0.01, compared with baseline; #P < 0.01, ##P < 0.01, compared with the respective saline group.
Locomotor activity in the 6-OHDA-lesioned mice was also evaluated with an open field test one week prior to surgery and 1, 2, 3, and 4 weeks after surgery (Fig. 1c, d). We observed that the moving velocity of the 6-OHDA mice was significantly decreased by 50% one week after surgery but gradually improved 2–4 weeks after surgery (Fig. 1c, d).
PWTs and paw withdrawal latencies in response to punctuate mechanical and thermal stimuli, respectively, were measured 1 week prior to surgery and 1, 2, 3, and 4 weeks after surgery. Before surgery, the 6-OHDA-lesioned mice exhibited baseline paw withdrawal thresholds and latencies similar to those of the saline-injected mice (control mice) (Fig. 2a–d). However, at all of the time points after surgery, the 6-OHDA-lesioned mice displayed significantly lower mechanical paw withdrawal thresholds and shorter thermal paw withdrawal latencies in both hindpaws than the saline control mice (Fig. 2a–d). The above-mentioned hypersensitivity to mechanical and thermal stimulation was observed at postsurgical week 1 and remained stable from week 1 throughout the duration of the study (4 weeks post-surgery) (Fig. 2a–d).
Fig. 2. Unilateral 6-OHDA-lesioned mice exhibit mechanical and thermal pain hypersensitivity.

The averaged paw withdrawal threshold (PWT) (a, b) and paw withdrawal latency (PWL) (c, d) of contralesional and ipsilesional hindpaws of the saline- and 6-OHDA-injected mice. Two-way ANOVA was used for data analysis. a Group: F (1, 92) = 73.47, P < 0.0001; Week: F (4, 92) = 0.0006; Interaction: F (4, 92) = 9.54, P < 0.0001). b Group: F (1, 92) = 57.74, P < 0.0001; Week: F (4, 92) = 5.44, P = 0.0006; Interaction: F(4, 92) = 11.14, P < 0.0001. c Group: F (1, 79) = 66.68, P < 0.0001; Week: F (4, 79) = 4.02, P = 0.0051; Interaction: F (4, 79) = 4.08, P = 0.0046. d Group: F (1, 79) = 25.8, P < 0.0001; Week: F(4, 79) = 9.27, P < 0.0001; Interaction: F (4, 79) = 5.69, P = 0.0006. **P < 0.01, compared with pre-6-OHDA, ##P < 0.01, compared with the respective saline group.
The time courses of DA neuron loss, locomotion dysfunction, and hypersensitive nociception show that the mechanical allodynia and thermal hyperalgesia in the 6-OHDA-lesioned mice may be associated with dopamine depletion but is not relevant to minor motor deficit.
Membrane excitability of the SDH neurons is enhanced in the mice with unilateral parkinsonism
We demonstrated antinociceptive effects of L-dopa and apomorphine on mechanical allodynia in the 6-OHDA-lesioned mice under our experimental conditions. The SDH is an important region for pain processing, and growing evidence suggests that increased sensitivity in spinal cord dorsal horn neurons plays a critical role in the development and maintenance of chronic pain [16, 21]. We investigated whether lesions of SNc DA neurons led to hyperexcitability of the SDH neurons with spinal slice patch-clamp techniques.
We injected steps of depolarizing current (0–260 pA with duration of 1 s) in SDH neurons to evoke action potentials in the saline control and 6-OHDA-lesioned mice. The number of evoked action potentials was used to assess the membrane excitability. In either the saline control or 6-OHDA-lesioned mice, the contralesional and ipsilesional SDH neurons exhibited similar membrane properties, including resting membrane potential, rheobase, action potential threshold, and membrane resistance (Table 1). Based on these data, we pooled all data from the ipsilesional and contralesional SDH neurons together.
Table 1.
Comparison of electrophysiological parameters of SDH neurons between non-injection and injection sides in saline control mice and 6-OHDA lesioned mice.
| Saline | t | P | 6-OHDA | t | P | |||
|---|---|---|---|---|---|---|---|---|
| Non-injection side (n = 9) | Injection side (n = 9) | Contralesional (n = 9) | Ipsilesional (n = 9) | |||||
| Rheobase (pA) | 35.0 ± 6.70 | 37.14 ± 8.08 | 0.21 | 0.84 | 20.00 ± 6.17 | 25.71 ± 5.71 | 0.68 | 0.51 |
| RMP (mV) | −56.44 ± 2.02 | −56.03 ± 1.11 | 0.13 | 0.90 | −48.65 ± 2.06 | −51.36 ± 1.05 | 0.93 | 0.36 |
| Rm (MΩ) | 623.4 ± 90.4 | 544.8 ± 38.84 | 0.80 | 0.44 | 627.4 ± 96.09 | 509.9 ± 43.31 | 1.12 | 0.28 |
| AP threshold (mV) | −30.93 ± 0.67 | −30.44 ± 0.59 | 0.55 | 0.59 | −30.69 ± 0.68 | −31.38 ± 0.58 | 0.76 | 0.46 |
| sEPSC frequency (Hz) | 4.83 ± 0.91 | 5.03 ± 0.90 | 0.15 | 0.88 | 4.59 ± 0.69 | 5.87 ± 1.29 | 0.89 | 0.38 |
| sEPSC amplitude (pA) | 10.7 ± 1.0 | 11.19 ± 1.11 | 0.33 | 0.75 | 10.7 ± 1.08 | 10.26 ± 0.78 | 0.34 | 0.74 |
Data are shown as mean ± SEM.
Rheobase the minimal depolarizing current triggers neuronal firing, RMP resting membrane potential, Rm membrane resistance, AP action potential, sEPSC spontaneous excitatory postsynaptic current.
We observed that depolarizing current steps evoked more action potentials in the SDH neurons in the 6-OHDA-lesioned mice than in the saline control mice (Fig. 3a). The rheobase, the minimal depolarizing current that evokes firing, was significantly lower in the SDH neurons of the 6-OHDA-lesioned mice than in those of the saline control mice (Fig. 3b). We then normalized the frequencies of firing evoked by the depolarizing current steps to the frequency of firing triggered by rheobase. As illustrated in Fig. 3c, the depolarizing currents evoked higher frequency firing in the SDH neurons of the 6-OHDA-lesioned mice than in those of the saline control mice. We also observed a significant rightward shift (becoming more depolarized) in the distribution of the resting membrane potentials of the SDH neurons in the 6-OHDA-lesioned mice relative to that of the saline control mice (Fig. 3d). The Pearson correlation coefficient between the rheobase and the resting membrane potential (−0.67) in the SDH neurons was statistically significant (P = 0.0002) (data not shown). However, there was no significant difference in the action potential threshold and the membrane resistance between these two groups (Fig. 3e, f). The results suggest that the SDH neurons in the 6-OHDA-lesioned mice have higher excitability than those in the saline control mice.
Fig. 3. Membrane excitability of SDH neurons is enhanced in the unilateral 6-OHDA lesioned mice.

a Representative current-clamp recordings showing responses of SDH neurons from saline (left panels) and 6-OHDA (right panels) mice to injection of 80 pA (upper panels) and 140 pA (lower panels) depolarizing currents. b Rheobase of SDH neurons in the saline mice and 6-OHDA mice. Saline (n = 20) vs 6-OHDA (n = 20), t = 3.09, P = 0.01, unpaired t-test. c The frequencies of firing evoked by depolarizing current steps were normalized to the frequency of firing triggered by rheobase stimulation in the SDH neurons in the saline and 6-OHDA mice. Two-way ANOVA: data were from 11 neurons in each group; Group: F (1, 245) = 91.15, P < 0.0001; Current injection: F (13, 245) = 16.21, P < 0.0001; Interaction: F (13, 245) = 2.68, P = 0.001. d Histogram showing the distribution of the resting membrane potentials of SDH neurons in the saline control mice (black solid bars) and the 6-OHDA mice (red open bars). Kolmogorov–Smirnov (K–S) test: P = 0.03, n = 20 in each group. e Action potential (AP) threshold of the SDH neurons in the saline mice (black) and the 6-OHDA mice (red). Saline (n = 20) vs 6-OHDA (n = 16), t = 0.56, P = 0.57, unpaired t-test. f Membrane resistance (Rm) of the SDH neurons in the saline mice (black) and the 6-OHDA mice (red). Saline (n = 20) vs 6-OHDA (n = 16), t = 0.24, P = 0.81, unpaired t-test. g Examples of sEPSCs recorded in an SDH neuron from a saline control mouse (left panels) and an SDH neuron from a 6-OHDA mouse (right panels). h–i Summarized data showing the frequency (h) and amplitude (i) of the sEPSCs in the saline and 6-OHDA groups. Saline (n = 24) vs 6-OHDA (n = 28), (h) t = 0.02, P = 0.98; (i) t = 0.47, P = 0.63, unpaired t-test. *P < 0.05, compared with the saline mice.
We then recorded spontaneous excitatory postsynaptic currents (sEPSCs) in the SDH neurons in lumbar spinal cord slices. In both the saline control and 6-OHDA-lesioned mice, no difference was found in the frequency and amplitude of the sEPSCs between the contralesional and ipsilesional SDH neurons (Table 1). After we pooled the data from the contralesional and ipsilesional SDH neurons together, we observed that neither the frequency nor the amplitude of the sEPSCs in the SDH neurons differed between the 6-OHDA-lesioned mice and the saline control mice (Fig. 3g, h, i).
The data suggest that the alteration of depolarization-evoked firing in the SDH neurons of the 6-OHDA-lesioned mice may not result from the change in spontaneous glutamatergic synaptic transmission.
Intrathecal injection of dopamine agonists relieves pain hypersensitivity in mice with parkinsonism
To test the effect of dopamine agonists on pain hypersensitivity in 6-OHDA lesioned mice, we administered the dopamine agonists, L-dopa (10 nmol/10 µL) and apomorphine (25 nmol/10 µL) by intrathecal injection in the 6-OHDA-lesioned mice and the saline control mice 3 weeks after surgery, and mechanical and thermal pain thresholds were measured 0.5 and 1 h after drug administration. A single bolus of L-dopa elevated the mechanical paw withdrawal threshold (Fig. 4a) and prolonged the thermal PWL (Fig. 4b) on both the ipsilesional and contralesional hindpaws 1 h after administration. Similarly, 0.5–1 h after intrathecal administration, apomorphine resulted in an increased mechanical paw withdrawal threshold (Fig. 4c) and prolonged thermal PWL (Fig. 4d) in both iplilesional and contralesional hindpaws in 6-OHDA-lesioned mice. The effect of L-dopa and apomorphine on both the mechanical and thermal pain thresholds recovered 24 h after drug administration. Notably, intrathecal injection of either L-dopa or apomorphine in the control mice had no significant impact on either paw withdrawal threshold or PWL (Fig. 4a–d); intrathecal injection of saline did not change the mechanical and thermal pain thresholds in the 6-OHDA-lesioned mice (Fig. 4e, f). These results reveal that activation of spinal cord dopamine receptors relieves pain hypersensitivity in mice with parkinsonism.
Fig. 4. Dopamine receptor agonists attenuate mechanical and thermal pain hypersensitivity in the unilateral 6-OHDA-lesioned mice.
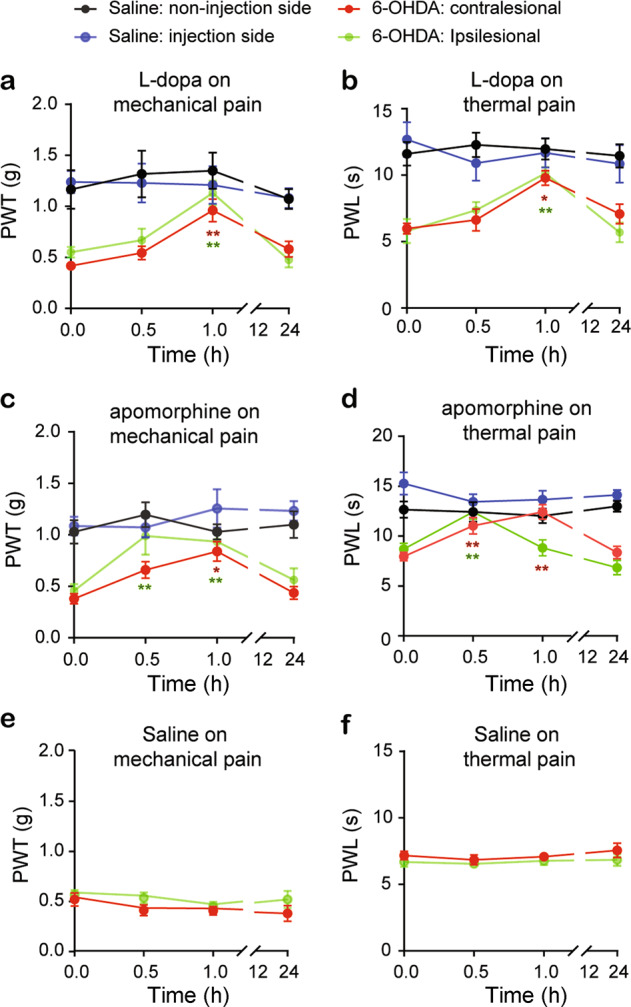
A single intrathecal injection of L-dopa (a, b), apomorphine (c, d) or saline (e, f) in the saline mice or unilateral 6-OHDA-lesioned mice. PWT and PWL in both hindpaws were measured in separate experiments before (0) and 0.5, 1 and 24 h after injection. Two-way ANOVA was used to analyze the data in (a–f). a n = 8 mice; Group: F (3, 109) = 24.23, P < 0.0001; Time point: F (3, 109) = 6.49, P = 0.0004; Interaction: F (9, 109) = 1.29, P = 0.25. b n = 6–7 mice; Group: F (3, 88) = 31.13, P < 0.0001; Time: F (3, 88) = 4.49, P = 0.0056; Interaction: F (9, 88) = 1.74, P = 0.093. c n = 7 mice; Group: F (3, 106) = 23.89, P < 0.0001; Time: F (3, 106) = 5.46, P = 0.0016; Interaction: F (9, 106) = 1.67, P = 0.1. d n = 9 mice; Group: F (3, 124) = 34.87, P < 0.0001; Time: F (3, 124) = 3.92, P = 0.01; Interaction: F (9, 124) = 5.3, P < 0.0001. e n = 7 mice; Group: F (1, 62) = 4.24, P = 0.05; Time: F (3, 62) = 1.52, P = 0.22; Interaction: F (3, 62) = 0.35, P = 0.79. f n = 7 mice; Group: F (1, 62) = 3.27, P = 0.075; Time: F (3, 62) = 0.61, P = 0.61; Interaction: F (3, 62) = 0.13, P = 0.94. *P < 0.05, **P < 0.01, compared with baseline. Asterisks in red or green represent comparisons with their own baseline.
Intrathecal injection of L-dopa improves mechanical allodynia by activating D2Rs in parkinsonian mice
Both L-dopa and apomorphine are nonselective dopamine receptor agonists and can activate both D1Rs and D2Rs in spinal cord neurons. To determine which kind of dopamine receptor mediates the antinociceptive effect of L-dopa in the 6-OHDA-lesioned mice, we evaluated the mechanical paw withdrawal threshold in the 6-OHDA-lesioned mice after a single intrathecal injection of L-dopa plus either a D1R selective antagonist, SKF83566 (10 nmol), or a D2R selective antagonist, sulpiride (10 nmol). Similar to L-dopa alone (Fig. 4a), L-dopa (10 nmol) plus SKF83566 significantly attenuated mechanical allodynia on both the contralesional and ipsilesional hindpaws 1 h after administration in the 6-OHDA-lesioned mice (Fig. 5a). Unlike L-dopa alone (Fig. 4a), coadministration of L-dopa with sulpiride did not change mechanical allodynia in the 6-OHDA-lesioned mice (Fig. 5b). However, co-application of L-dopa with either sulpiride or SKF83566 had no effect on the mechanical paw withdrawal threshold in the saline control mice. These data suggest that the antiallodynia effect of L-dopa is likely mediated by dopamine D2Rs in the spinal cord.
Fig. 5. D2 receptors mediate the antinociceptive effects of L-dopa in unilateral 6-OHDA lesioned mice.
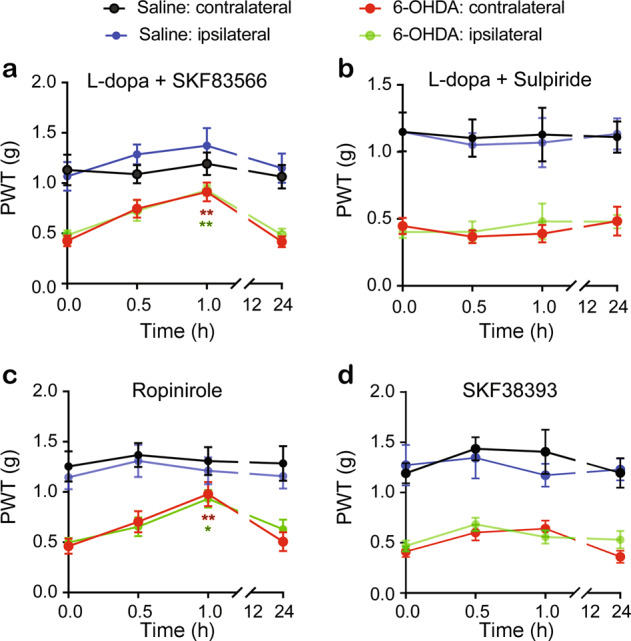
L-dopa plus SKF83566 (a), L-dopa plus sulpiride (b), SKF38393 (c), and ropinirole (d) were intrathecally administered to the saline control or unilateral 6-OHDA-lesioned mice. Two consecutive drug applications were separated by at least 3 days. PWTs in both hindpaws were measured in separate experiments before and 0.5, 1 and 24 h after drug injection. Two-way ANOVA was used to analyze the data in (a–d). a n = 7 in the saline group, n = 9 in the 6-OHDA group; Group: F (3, 119) = 30.71, P < 0.0001; Time: F (3, 119) = 8.16, P < 0.0001; Interaction: F (9, 119) = 1.01, P = 0.43. b n = 6 in the saline group, n = 9 in the 6-OHDA group; Group: F (3, 105) = 51.92, P < 0.0001; Time: F (3, 105) = 0.31, P = 0.82; Interaction: F (9, 105) = 0.12, P = 0.99. c n = 6 in the saline group, n = 9 in the 6-OHDA group; Group: F (3, 105) = 62.05, P < 0.0001; Time: F (3, 105) = 2.71, P = 0.06; Interaction: F (9, 105) = 0.46, P = 0.89. d n = 6 in the saline group, n = 9 in the 6-OHDA group; Group: F (3, 105) = 33.95, P < 0.0001; Time: F (3, 105) = 4.39, P = 0.006; Interaction: F (9, 106) = 1.03, P = 0.42. *P < 0.05, **P < 0.01, compared with baseline. Asterisks in red or green represent comparisons to their own baseline.
To further explore the role of D1Rs and D2Rs in mechanical allodynia in unilateral 6-OHDA-lesioned mice, a D2R agonist, ropinirole (20 nmol), was intrathecally applied to the 6-OHDA-lesioned mice and the saline control mice. As illustrated in Fig. 5c, mechanical allodynia was improved in both the ipsilesional and contralesional hindpaws of the 6-OHDA-lesioned mice, but not in those of the saline control mice. However, a single intrathecal injection of the D1R agonist SKF38393 (10 nmol) had no effect on mechanical allodynia in the 6-OHDA-lesioned mice (Fig. 5d). Either ropinirole or SKF38393 had no effect on the mechanical paw withdrawal threshold in the saline control mice.
These data further confirmed that activation of D2Rs in the spinal cord mimics the antinociceptive effect of L-dopa in the 6-OHDA-lesioned mice.
Modulation of glutamatergic synaptic transmission by D2Rs is enhanced in the SDH neurons of the mice with parkinsonism
As intrathecal administration of L-dopa attenuated pain hypersensitivity in the unilateral 6-OHDA-lesioned mice through activation of D2Rs, we wondered whether a D2R agonist reversed hyperexcitability in the SDH neurons in these mice. As illustrated in Fig. 6a, b, incubation with the D2R agonist ropinirole (1 μM) reduced the frequency of depolarizing current-evoked firing in the 6-OHDA-lesioned mice but not in the saline control mice. Furthermore, ropinirole (1 μM) also significantly hyperpolarized the SDH neurons in the 6-OHDA-lesioned mice to restore the membrane potentials of these neurons to levels similar to those in the saline control mice (Fig. 6c). These results indicate that the responses of D2Rs in the SDH neurons to its agonist may be enhanced in the 6-OHDA-lesioned mice, compared with those in the saline control mice.
Fig. 6. D2Rs are enhanced in spinal cord dorsal horn in the unilateral 6-OHDA-lesioned mice.

a Representative current-clamp recordings showing firing evoked by 80 pA current injection from an SDH neuron of a saline control mouse (left panels) and that from an SDH neuron of a 6-OHDA-lesioned mouse (right panels) in the absence (upper panels) and presence (middle panels) of ropinirole (10 µM). b Frequencies of firing evoked by depolarizing current steps in the SDH neurons in the saline control and 6-OHDA-lesioned mice in the absence and presence of ropinirole. Two-way ANOVA was used to analyze the data. n = 8 in each group; Group: F (3, 232) = 34.47, P < 0.0001; Current injection: F (13, 232) = 21.3, P < 0.0001; Interaction: F (39, 232) = 1.29, P = 0.13. c Summarized data showing the mean resting membrane potential of the SDH neurons in the saline control mice and the 6-OHDA-lesioned mice with and without ropinirole. One-way ANOVA: F (3, 28) = 6.71, P = 0.0015, n = 8 neurons in each group; Sal vs Sal+Rop, P = 0.94; 6-OHDA vs 6-OHDA + Rop, P = 0.0028; Sal vs 6-OHDA, P = 0.018; Sal vs 6-OHDA + Rop, P = 0.88. d Time course of the sEPSC frequency before, during, and after 5 min ropinirole (10 µM) in an SDH neuron in a unilateral 6-OHDA-lesioned mouse. e Summarized data showing the inhibition of the sEPSC frequency by ropinirole in the saline control mice and the unilateral 6-OHDA-lesioned mice. Saline (n = 15) vs 6-OHDA (n = 18), t = 3.88, P = 0.0005, unpaired t-test. f Summarized data showing the changes in the sEPSC amplitude by ropinirole in the SDH neurons in the saline control mice and the 6-OHDA-lesioned mice. Saline (n = 15) vs 6-OHDA (n = 18), t = 0.52, P = 0.61, unpaired t-test.
We next examined whether D2Rs in presynaptic glutamatergic terminals synapsing onto SDH neurons are also modified in the 6-OHDA-lesioned mice. We recorded EPSCs from the SDH neurons in the presence of a GABAA receptor antagonist, bicuculline (10 μM), and after a 5–10 min stable baseline was obtained, we added a D2R agonist, ropinirole (1 μM), in perfusate for 6–8 min (Fig. 6d). We observed that most of the recorded SDH neurons responded to ropinirole (15 out of 17 neurons in the saline control mice, 18 out of 20 neurons in the 6-OHDA-lesioned mice). Ropinirole significantly reduced the sEPSC frequency by 36% and 21% in the SDH neurons of the 6-OHDA-lesioned mice and the saline control mice, respectively (Fig. 6e). The inhibition of the sEPSC frequency by ropinirole in the 6-OHDA-lesioned mice was substantially stronger than that in the saline control mice. No significant change in EPSC amplitude in the SDH neurons was observed during the application of ropinirole in either the 6-OHDA-lesioned mice or the saline control mice (Fig. 6f). Therefore, in the 6-OHDA-lesioned mice, D2Rs in the presynaptic glutamatergic terminals synapsing onto SDH neurons were also enhanced.
Thus, the hyperexcitability of the SDH neurons and the function of D2Rs on both glutamatergic terminals and SDH neurons may be enhanced in the 6-OHDA-lesioned mice.
Tonic activation of dopamine D2 receptors in the spinal cord dorsal horn is impaired in the mice with unilateral parkinsonism
We then assessed whether the tonic activation of D2Rs was also affected in the 6-OHDA-lesioned mice. We tested the effect of sulpiride, a D2R antagonist, on the sEPSCs in the SDH neurons. The sEPSCs were recorded before, during, and after a 5 min bath application of sulpiride (10 µM) (Fig. 7a–d). We observed that bath application of sulpiride increased the frequency of the EPSCs in the SDH neurons more strongly in the saline control mice (~48%) than in the 6-OHDA-lesioned mice (~17%) (Fig. 7e). However, the sEPSC amplitude remained unchanged in the presence of sulpiride in the SDH neurons in either the unilateral 6-OHDA-lesioned mice or the saline control mice (Fig. 7f). These results suggest that tonic activation of D2Rs on glutamatergic terminals synapsing onto SDH neurons may be impaired, probably due to deficits in dopamine release in the spinal cord dorsal horn.
Fig. 7. Tonic activation of D2Rs in spinal cord dorsal horn is impaired in the unilateral 6-OHDA-lesioned mice.

Typical traces of sEPSCs recorded from an SDH neuron of a saline control mouse (a) and from a neuron of a 6-OHDA-lesioned mouse (c). Time course of the sEPSC frequency before, during, and after 6 min of sulpiride (10 µM) in an SDH neuron from a saline control mouse (b) and in an SDH neuron from a 6-OHDA-lesioned mouse (d). e Summarized data showing potentiation of the sEPSC frequency by sulpiride in SDH neurons from the saline control mice and the 6-OHDA-lesioned mice. Saline (n = 15) vs 6-OHDA (n = 13), t = 3.17, P = 0.004, unpaired t-test. f Summarized data showing the changes in the sEPSC amplitude in SDH neurons from the saline control mice and the 6-OHDA-lesioned mice. Saline (n = 15) vs 6-OHDA (n = 15), t = 1.78, P = 0.086, unpaired t-test.
Discussion
In the present study, we used behavioral tests and electrophysiological techniques to examine the antinociceptive effects of dopamine receptor agonists in the spinal cord dorsal horn following unilateral partial lesion of SNc DA neurons with microinjection of 6-OHDA in the right MFB. We demonstrated that (1) the 6-OHDA-lesioned mice displayed lower mechanical and thermal nociceptive thresholds on both hindpaws, which were not associated with motor dysfunction; (2) intrathecal administration of L-dopa or apomorphine increased the mechanical and thermal nociceptive thresholds. This effect was blocked by a D2R antagonist, but not by a D1R antagonist, while the effect was mimicked by the D2R agonist, but not by the D1R agonist; (3) the 6-OHDA-lesioned mice showed higher neuronal excitability in the lumber SDH neurons, which was reversed by the D2R agonist, than the control mice; (4) a D2R agonist inhibited the sEPSCs in the SDH neurons more strongly in the 6-OHDA-lesioned mice than in the control mice. These findings suggest that the hyperexcitability of the SDH neurons could be a key pathophysiology underlying parkinsonian pain hypersensitivity, and both presynaptic and postsynaptic D2Rs in the SDH of the spinal cord may be potentiated following SNc DA neuron lesions and may be rational targets for the treatment of pain symptoms in PD.
We observed that 6-OHDA was administered unilaterally in the MFB to cause a rapid but partial lesion of DA neurons in the ipsilateral SNc. In these parkinsonian mice, motor function was moderately impaired 1 week after the 6-OHDA lesion, but gradually improved 3 weeks after the lesion. This finding suggests the occurrence of compensatory changes in motor circuits, which was consistent with previous studies [22]. In contrast, the nociceptive thresholds were significantly reduced 1 week after the 6-OHDA lesion and persisted for at least 4 weeks. The difference in the time courses between the development of motor deficit and pain hypersensitivity suggests that pain hypersensitivity after the 6-OHDA lesion may not be associated with motor dysfunction. Similar to previous studies [23–25], we observed reduced mechanical and thermal pain thresholds in the bilateral hindpaws of the unilateral 6-OHDA-lesioned mice. However, lesions of unilateral SNc DA neurons impair motor function on the contralateral side [19, 26]. This finding might be additional evidence to support the notion that 6-OHDA lesions may cause pain hypersensitivity and motor deficits through different pathophysiological mechanisms. It also explains why the treatment for motor deficits does not produce comparable benefits for pain symptoms in PD.
Systemically administered dopamine receptor agonists have been reported to influence various nociceptive behaviors in animals [13]. Using intrathecal injection, we found that activation of D2Rs in the spinal cord with L-dopa and apomorphine is sufficient to convey antinociceptive effects in parkinsonian mice. Notably, the onset of the L-dopa effect (60 min after administration) lagged behind that of apomorphine (30 min after administration). This finding may be because L-dopa is a precursor of dopamine, and it needs to be converted into dopamine in DA terminals and released to act on dopamine receptors, while apomorphine is a dopamine receptor agonist and activates dopamine receptors after entering the spinal cord. These results suggest that pain hypersensitivity in parkinsonian mice may be related to dysfunction of the DA system in the spinal cord, and compensation for dopamine is a valid strategy to mitigate the hypersensitivity.
Five dopamine receptor subtypes have been classified into D1-like (D1 and D5) or D2-like (D2, D3, and D4) receptors (D1Rs or D2Rs). Dopamine receptor agonists may modulate nociceptive behaviors by activating either D1Rs or D2Rs. Classically, D1Rs, when activated, are coupled with the mobilization of stimulatory G protein (Gs), followed by activation of adenylyl cyclase and subsequent elevation of cAMP levels and activation of protein kinase A, whereas activated D2-like receptors inhibit adenylyl cyclase through coupling to Gi/o, attenuating cAMP production and protein phosphorylation [27]. D1Rs and D2Rs modulate multiple proteins and physiological functions through these second messenger cascades. In terms of regulation of neuronal activity and neurotransmitter release in the spinal cord, D2R activation results in inhibition of high voltage-gated calcium channels and EPSCs [28–30] and potentiation of inward rectification potassium channels [31, 32]. D1R activation inhibits tetrodotoxin-resistant sodium channels [33] and transient receptor potential vanilloid type 1 (TRPV1) receptors [34], but potentiates C-fiber stimulation-evoked EPSCs [35].
It has been demonstrated that both D1Rs and D2Rs are densely located in the superficial spinal cord dorsal horn [36, 37] and on primary afferent fibers from the dorsal root ganglion neurons [33, 38]. However, there is no information available about the levels of D1Rs and D2Rs in specific neuron types in the spinal cord dorsal horn. We demonstrated that dopamine receptor agonist-induced reduction in nociceptive responses was mediated by D2Rs, because the D2R agonist, but not the D1R agonist, consistently and significantly increased mechanical nociceptive thresholds in both hindpaws of the 6-OHDA-lesioned mice. Moreover, the D2R antagonist blocked the antinociceptive effect of L-dopa. These data are consistent with previous studies showing that spinal D2Rs are the major mediators of the antinociceptive effect of systemically administered dopamine receptor agonists [13, 14]. The attenuation of nociception by D2R agonists has also been observed in inflammatory pain [39, 40] and peripheral nerve injury-induced neuropathic pain [41, 42]. Here, we showed that activation of D2Rs in the spinal cord relieved pain hypersensitivity in mouse models of parkinsonism. Thus, D2Rs within the spinal cord dorsal horn may play a significant regulatory role in a variety of pain sensations.
Similar to mice suffering from neuropathic pain [16, 21], the 6-OHDA-lesioned mice displayed hyperexcitability in the SDH neurons (Fig. 3). We observed that in the 6-OHDA-lesioned mice, the membrane properties of the SDH neurons were similar on both the contralesional and ipsilesional sides and displayed hyperexcitability, relative to those in the saline control mice. The SDH neurons in the 6-OHDA-lesioned mice exhibited more depolarized resting membrane potentials and more robust responses to depolarizing current stimulation than those in the saline control mice.
Pain hypersensitivity may be associated with the hyperexcitability of the SDH neurons in the 6-OHDA-lesioned mice because these two events have similar features. Both occurred in the contralesional and ipsilesional sides; both were reversed by activation of D2Rs in the spinal cord; and in the saline-treated mice, the D2R agonist altered neither the excitability of the SDH neurons nor the pain thresholds. Therefore, we postulate that the hyperexcitability in the SDH neurons may be a central sensitization mechanism underlying pain hypersensitivity in the 6-OHDA-lesioned mice.
The SDH receives glutamatergic inputs from C fibers and Aδ fibers of the dorsal root ganglia, spinal cord glutamatergic interneurons [43], and brain stem reticular formation [30], and a D2R agonist inhibits these inputs [30, 44]. Similar to these studies, we observed that D2R agonists significantly inhibited the sEPSC frequency in the SDH neurons, and the effect was much stronger in the 6-OHDA-lesioned mice than in the saline control mice. This finding suggests that the function of D2Rs on glutamatergic terminals synapsing onto SDH neurons was enhanced in the 6-OHDA-lesioned mice. If tonic dopamine release occurs in the spinal cord dorsal horn, it may cause tonic inhibition of sEPSCs, and inhibition of D2Rs may facilitate sEPSCs. Indeed, consistent with previous studies [45, 46], we observed that the D2R antagonist inhibited the sEPSCs in the SDH neurons, but the effect was stronger in the control mice than in the 6-OHDA-lesioned mice. This result seems to contradict the enhancement of D2R function in the 6-OHDA-lesioned mice. As the physiological outcomes of a D2R antagonist depend on the tonic activation of D2Rs by endogenous dopamine, the lack of effect of a D2R antagonist on the sEPSCs in the 6-OHDA-lesioned mice may suggest dopamine depletion in the spinal cord. A previous study reported that the reduction in dopamine content in the spinal cord was not statistically significant in a parkinsonian rat model that received bilateral 6-OHDA lesions in the dorsal striatum [47]. As 6-OHDA injection in the striatum causes a slower appearance of lesions and fewer lesions of SNc DA neurons than 6-OHDA injection in the MFB [48], it may not exclude the possibility that the parkinsonian mice in the present study exhibit dopamine depletion in the spinal cord. Further neurochemical investigations are warranted to clarify this possibility.
Notably, the DA fibers in the spinal cord dorsal horn originate from hypothalamic A11 periventricular DA neurons, but not from SNc DA neurons [13]. The A11 periventricular DA neurons may be affected following the lesion of SNc DA neurons. Further investigations are required to elucidate the alterations in the physiological properties of A11 DA neurons and in the intracellular signaling pathways that mediate D2R modulation of SDH neurons and glutamatergic terminals in the spinal cord dorsal horn.
In summary, the present study identified the neuronal and synaptic mechanisms underlying pain hypersensitivity and the antinociceptive effects of dopamine agonists in parkinsonian mice. Although SNc DA neurons do not project to the spinal cord dorsal horn, lesions of these neurons in one hemisphere caused hyperexcitability in the SDH neurons, reduced tonic activity of D2Rs, and potentiated D2R function on both the ipsilesional and contralesional sides. Dopamine agonists may activate D2Rs in both SDH neurons and their glutamatergic inputs to restore normal excitability in the SDH neurons and relieve pain hypersensitivity in parkinsonian mice. Therefore, spinal D2Rs can be promising therapeutic targets for the treatment of chronic pain in PD.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81701100 (CYZ), 81870891 (CX), 81971038 (CYZ)), the Fund for Jiangsu Province Specially Appointed Professor (CX, CYZ), the Natural Science Foundation of Jiangsu Province (BK20171160, CYZ), The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (17KJA320007 (CX), 18KJA320009 (CYZ)), the Jiangsu Province Fund for Dominant Discipline (Anesthesiology), and Startup Funds from Xuzhou Medical University (D2017009, D2017010).
Author contributions
CX and CYZ designed the experiments. CX and CYZ analyzed and illustrated the data. CX and CYZ collected the electrophysiological data. DLT and YWL managed the mouse colony. DLT, YWL, and CYZ performed the behavioral tests. DLT and YWL performed the mouse survival surgery. CYZ, CX, and YWL collected image data. CX and CYZ wrote the manuscript. Every author read and approved the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Chun-yi Zhou, Email: Chunyi.zhou@xzhmu.edu.cn.
Cheng Xiao, Email: xchengxj@xzhmu.edu.cn.
References
- 1.Santos-Garcia D, Abella-Corral J, Aneiros-Diaz A, Santos-Canelles H, Llaneza-Gonzalez MA, Macias-Arribi M. [Pain in Parkinson’s disease: prevalence, characteristics, associated factors, and relation with other non motor symptoms, quality of life, autonomy, and caregiver burden] Rev Neurol. 2011;52:385–93. [PubMed] [Google Scholar]
- 2.Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464–74. doi: 10.1016/S1474-4422(09)70068-7. [DOI] [PubMed] [Google Scholar]
- 3.Ha AD, Jankovic J. Pain in Parkinson’s disease. Mov Disord. 2012;27:485–91. doi: 10.1002/mds.23959. [DOI] [PubMed] [Google Scholar]
- 4.Wasner G, Deuschl G. Pains in Parkinson disease–many syndromes under one umbrella. Nat Rev Neurol. 2012;8:284–94. doi: 10.1038/nrneurol.2012.54. [DOI] [PubMed] [Google Scholar]
- 5.Brefel-Courbon C, Payoux P, Thalamas C, Ory F, Quelven I, Chollet F, et al. Effect of levodopa on pain threshold in Parkinson’s disease: a clinical and positron emission tomography study. Mov Disord. 2005;20:1557–63. doi: 10.1002/mds.20629. [DOI] [PubMed] [Google Scholar]
- 6.Schestatsky P, Kumru H, Valls-Sole J, Valldeoriola F, Marti MJ, Tolosa E, et al. Neurophysiologic study of central pain in patients with Parkinson disease. Neurology. 2007;69:2162–9. doi: 10.1212/01.wnl.0000295669.12443.d3. [DOI] [PubMed] [Google Scholar]
- 7.Gerdelat-Mas A, Simonetta-Moreau M, Thalamas C, Ory-Magne F, Slaoui T, Rascol O, et al. Levodopa raises objective pain threshold in Parkinson’s disease: a RIII reflex study. J Neurol Neurosurg Psychiatry. 2007;78:1140–2. doi: 10.1136/jnnp.2007.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J, Lim CS, Seo H, Park CA, Zhuo M, Kaang BK, et al. Pain perception in acute model mice of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Mol Pain. 2015;11:28. doi: 10.1186/s12990-015-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domenici RA, Campos ACP, Maciel ST, Berzuino MB, Hernandes MS, Fonoff ET, et al. Parkinson's disease and pain: modulation of nociceptive circuitry in a rat model of nigrostriatal lesion. Exp Neurol. 2019;315:72–81. doi: 10.1016/j.expneurol.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Rukavina K, Leta V, Sportelli C, Buhidma Y, Duty S, Malcangio M, et al. Pain in Parkinson’s disease: new concepts in pathogenesis and treatment. Curr Opin Neurol. 2019;32:579–88. doi: 10.1097/WCO.0000000000000711. [DOI] [PubMed] [Google Scholar]
- 11.Perrotta A, Sandrini G, Serrao M, Buscone S, Tassorelli C, Tinazzi M, et al. Facilitated temporal summation of pain at spinal level in Parkinson’s disease. Mov Disord. 2011;26:442–8. doi: 10.1002/mds.23458. [DOI] [PubMed] [Google Scholar]
- 12.Pertovaara A, Wei H. Dual influence of the striatum on neuropathic hypersensitivity. Pain. 2008;137:50–9. doi: 10.1016/j.pain.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Puopolo M. The hypothalamic-spinal dopaminergic system: a target for pain modulation. Neural Regen Res. 2019;14:925–30. doi: 10.4103/1673-5374.250567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Liu S, Lu X, Tao F. Role of descending dopaminergic pathways in pain modulation. Curr Neuropharmacol. 2019;17:1176–82. doi: 10.2174/1570159X17666190430102531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishioka K, Nakajima M. Beneficial therapeutic effects of spinal cord stimulation in advanced cases of Parkinson’s disease with intractable chronic pain: a case series. Neuromodulation. 2015;18:751–3. doi: 10.1111/ner.12315. [DOI] [PubMed] [Google Scholar]
- 16.Zhou C, Luo ZD. Nerve injury-induced calcium channel alpha-2-delta-1 protein dysregulation leads to increased pre-synaptic excitatory input into deep dorsal horn neurons and neuropathic allodynia. Eur J Pain. 2015;19:1267–76. doi: 10.1002/ejp.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T, Zhu H, Hou Y, Gu W, Wu H, Luan Y, et al. Galantamine reversed early postoperative cognitive deficit via alleviating inflammation and enhancing synaptic transmission in mouse hippocampus. Eur J Pharmacol. 2019;846:63–72. doi: 10.1016/j.ejphar.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 18.Xiao C, Cho JR, Zhou C, Treweek JB, Chan K, McKinney SL, et al. Cholinergic mesopontine signals govern locomotion and reward through dissociable midbrain pathways. Neuron. 2016;90:333–47. doi: 10.1016/j.neuron.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou C, Gu W, Wu H, Yan X, Deshpande P, Xiao C, et al. Bidirectional dopamine modulation of excitatory and inhibitory synaptic inputs to subthalamic neuron subsets containing alpha4beta2 or alpha7 nAChRs. Neuropharmacology. 2019;148:220–8. doi: 10.1016/j.neuropharm.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 20.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou C, Luo ZD. Electrophysiological characterization of spinal neuron sensitization by elevated calcium channel alpha-2-delta-1 subunit protein. Eur J Pain. 2014;18:649–58. doi: 10.1002/j.1532-2149.2013.00416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuter K, Olech L, Glowacka U. Prolonged dysfunction of astrocytes and activation of microglia accelerate degeneration of dopaminergic neurons in the rat substantia nigra and block compensation of early motor dysfunction induced by 6-OHDA. Mol Neurobiol. 2018;55:3049–66. doi: 10.1007/s12035-017-0529-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gee LE, Chen N, Ramirez-Zamora A, Shin DS, Pilitsis JG. The effects of subthalamic deep brain stimulation on mechanical and thermal thresholds in 6-OHDA-lesioned rats. Eur J Neurosci. 2015;42:2061–9. doi: 10.1111/ejn.12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez-Paz A, Drucker-Colin R, Milan-Aldaco D, Palomero-Rivero M, Ambriz-Tututi M. Intrastriatal chromospheres’ transplant reduces nociception in hemiparkinsonian rats. Neuroscience. 2018;387:123–34. doi: 10.1016/j.neuroscience.2017.08.052. [DOI] [PubMed] [Google Scholar]
- 25.Chudler EH, Lu Y. Nociceptive behavioral responses to chemical, thermal and mechanical stimulation after unilateral, intrastriatal administration of 6-hydroxydopamine. Brain Res. 2008;1213:41–7. doi: 10.1016/j.brainres.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon HH, Park JH, Kim YH, Min J, Hwang E, Lee CJ, et al. Optogenetic inactivation of the subthalamic nucleus improves forelimb akinesia in a rat model of Parkinson disease. Neurosurgery. 2014;74:533–40. doi: 10.1227/NEU.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 27.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 28.Almanza A, Segura-Chama P, Leon-Olea M, Luis E, Garduno-Gutierrez R, Mercado-Reyes J, et al. Cellular mechanism for specific mechanical antinociception by D2-like receptor at the spinal cord level. Neuroscience. 2019;417:81–94. doi: 10.1016/j.neuroscience.2019.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Wang D, Grillner S, Wallen P. 5-HT and dopamine modulates CaV1.3 calcium channels involved in postinhibitory rebound in the spinal network for locomotion in lamprey. J Neurophysiol. 2011;105:1212–24. doi: 10.1152/jn.00324.2009. [DOI] [PubMed] [Google Scholar]
- 30.Svensson E, Wikstrom MA, Hill RH, Grillner S. Endogenous and exogenous dopamine presynaptically inhibits glutamatergic reticulospinal transmission via an action of D2-receptors on N-type Ca2+ channels. Eur J Neurosci. 2003;17:447–54. doi: 10.1046/j.1460-9568.2003.02466.x. [DOI] [PubMed] [Google Scholar]
- 31.Tamae A, Nakatsuka T, Koga K, Kato G, Furue H, Katafuchi T, et al. Direct inhibition of substantia gelatinosa neurones in the rat spinal cord by activation of dopamine D2-like receptors. J Physiol. 2005;568:243–53. doi: 10.1113/jphysiol.2005.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taniguchi W, Nakatsuka T, Miyazaki N, Yamada H, Takeda D, Fujita T, et al. In vivo patch-clamp analysis of dopaminergic antinociceptive actions on substantia gelatinosa neurons in the spinal cord. Pain. 2011;152:95–105. doi: 10.1016/j.pain.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 33.Galbavy W, Safaie E, Rebecchi MJ, Puopolo M. Inhibition of tetrodotoxin-resistant sodium current in dorsal root ganglia neurons mediated by D1/D5 dopamine receptors. Mol Pain. 2013;9:60. doi: 10.1186/1744-8069-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakraborty S, Rebecchi M, Kaczocha M, Puopolo M. Dopamine modulation of transient receptor potential vanilloid type 1 (TRPV1) receptor in dorsal root ganglia neurons. J Physiol. 2016;594:1627–42. doi: 10.1113/JP271198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang HW, Zhou LJ, Hu NW, Xin WJ, Liu XG. Activation of spinal d1/d5 receptors induces late-phase LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2005;94:961–7. doi: 10.1152/jn.01324.2004. [DOI] [PubMed] [Google Scholar]
- 36.Kaur J, Khararjian A, Coleman RA, Constantinescu CC, Pan ML, Mukherjee J. Spinal cord dopamine D2/D3 receptors: in vivo and ex vivo imaging in the rat using 18F/11C-fallypride. Nucl Med Biol. 2014;41:841–7. doi: 10.1016/j.nucmedbio.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levant B, McCarson KE. D(3) dopamine receptors in rat spinal cord: implications for sensory and motor function. Neurosci Lett. 2001;303:9–12. doi: 10.1016/s0304-3940(01)01692-5. [DOI] [PubMed] [Google Scholar]
- 38.Xie GX, Jones K, Peroutka SJ, Palmer PP. Detection of mRNAs and alternatively spliced transcripts of dopamine receptors in rat peripheral sensory and sympathetic ganglia. Brain Res. 1998;785:129–35. doi: 10.1016/s0006-8993(97)01394-2. [DOI] [PubMed] [Google Scholar]
- 39.Gao X, Zhang Y, Wu G. Effects of dopaminergic agents on carrageenan hyperalgesia after intrathecal administration to rats. Eur J Pharmacol. 2001;418:73–7. doi: 10.1016/s0014-2999(01)00930-x. [DOI] [PubMed] [Google Scholar]
- 40.Lapirot O, Melin C, Modolo A, Nicolas C, Messaoudi Y, Monconduit L, et al. Tonic and phasic descending dopaminergic controls of nociceptive transmission in the medullary dorsal horn. Pain. 2011;152:1821–31. doi: 10.1016/j.pain.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 41.Cobacho N, de la Calle JL, Paino CL. Dopaminergic modulation of neuropathic pain: analgesia in rats by a D2-type receptor agonist. Brain Res Bull. 2014;106:62–71. doi: 10.1016/j.brainresbull.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, Tang Y, Shu H, Tatum D, Bai Q, Crawford J, et al. Dopamine receptor D2, but not D1, mediates descending dopaminergic pathway-produced analgesic effect in a trigeminal neuropathic pain mouse model. Pain. 2019;160:334–44. doi: 10.1097/j.pain.0000000000001414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sengul G. Primary afferent projections to the spinal cord. In: Paxinos G, editor. The rat nervous system. 4th ed. Boston: Academic Press; 2015. p. 78–85.
- 44.Lu Y, Doroshenko M, Lauzadis J, Kanjiya MP, Rebecchi MJ, Kaczocha M, et al. Presynaptic inhibition of primary nociceptive signals to dorsal horn lamina I neurons by dopamine. J Neurosci. 2018;38:8809–21. doi: 10.1523/JNEUROSCI.0323-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, Lester HA. Chronic nicotine selectively enhances alpha4beta2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci. 2009;29:12428–39. doi: 10.1523/JNEUROSCI.2939-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kline DD, Takacs KN, Ficker E, Kunze DL. Dopamine modulates synaptic transmission in the nucleus of the solitary tract. J Neurophysiol. 2002;88:2736–44. doi: 10.1152/jn.00224.2002. [DOI] [PubMed] [Google Scholar]
- 47.Cao LF, Peng XY, Huang Y, Wang B, Zhou FM, Cheng RX, et al. Restoring spinal noradrenergic inhibitory tone attenuates pain hypersensitivity in a rat model of Parkinson’s disease. Neural Plast. 2016;2016:6383240. doi: 10.1155/2016/6383240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog Brain Res. 2010;184:17–33. doi: 10.1016/S0079-6123(10)84002-6. [DOI] [PubMed] [Google Scholar]