

Abstract
The pathogenic mechanism of neuropathy-associated aminoacyl-tRNA synthetase (ARS) gene variants is poorly defined. Mullen et al. generate new models of pathogenic, dominant HARS1 mutations and show that they increase EIF2α phosphorylation and decrease protein translation in neurons. These results are consistent with a dominant-negative mechanism of ARS-mediated peripheral neuropathy.
Keywords: aminoacyl-tRNA synthetase, HARS1, peripheral neuropathy, protein translation, dominant-negative
To synthesize proteins, cells require a pool of tRNAs charged with the corresponding amino acid. This reaction is catalyzed by the aminoacyl-tRNA synthetase (ARS) family of enzymes, which can function in either the cytoplasm or the mitochondria to supply the protein translation machinery in each compartment1. Mutations in ARS-encoding genes can cause both recessive and dominant human phenotypes2,3. Although recessive ARS-mediated disease usually severely affects the development and function of broad array of tissues, dominant ARS mutations primarily cause a later-onset peripheral neuropathy, with clinical heterogeneity including variable severity and age of onset. The mechanism of ARS-associated peripheral neuropathy is poorly understood and the primary mechanism is frequently debated in the literature2. One possible explanation is that heterozygosity for neuropathy-associated ARS mutations, which are primarily loss-of-function mutations but do not significantly compromise protein stability, results in a dominant-negative effect where the mutant protein represses the ability of the remaining wild-type ARS protein to function. This argument is strengthened by the fact that all five ARS enzymes implicated in peripheral neuropathy function as homodimers2. An alternate argument is that pathogenic mutations cause conformational changes that allow the mutant ARS enzyme to aberrantly interact with other cellular proteins, which then affects other neuronal pathways that are required for neuron development or health4. In this issue of The FEBS Journal, Mullen and colleagues report a detailed study of pathogenic histidyl-tRNA synthetase (HARS1) mutations and find molecular and cellular phenotypes that are consistent with a dominant-negative mechanism of disease5.
Mullen et. al. studied three independent HARS1 variants that have been linked to peripheral neuropathy. Two (V155G and Y330C) were previously found to segregate with disease in small pedigrees, displayed loss-of-function characteristics in yeast complementation assays, and were functionally impaired in enzymatic assays6. The third (R137Q) was found in a single patient with a late-onset dominant peripheral neuropathy, caused reduced yeast growth, and caused aberrant axon morphologies when over-expressed in C. elegans7. Although these variants were interpreted as loss-of-function mutations, it was unclear how they impacted protein synthesis and how they might damage neurons.
To address the above questions, the authors paired two over-expression models: PC12 cells (which can be differentiated to generate axon-like projections) and zebrafish embryos. PC12 cells expressing the mutant HARS1 proteins showed an increase of phosphorylated EIF2α, a marker of the integrated stress response and an indication of accumulating uncharged tRNA8, consistent with significantly reduced HARS1 function. This was accompanied by an approximate 20% reduction in global protein synthesis, as measured by OP-Puro incorporation. This downregulation in protein synthesis did not dramatically affect PC12 morphology, viability, or number of neurites formed; however, it did modestly decrease the length of the longest neurite in each cell. These findings suggest that the ability to form or maintain long neuronal processes, such as the long axons of the peripheral nerve, is dependent on protein synthesis.
To complement this approach with an in vivo model, the authors injected wild-type zebrafish embryos with V155G or Y330C human HARS1 mRNA. Similar to what was observed when over-expressing R137Q HARS1 in worm7, by 48 hours post fertilization zebrafish neurons showed improper guidance. Neuronal processes in mutant-expressing fish were also shorter than those of fish expressing wild-type human HARS1 protein. Unsurprisingly for such severe morphological defects, the fish also displayed motor deficits in behavioral assays. This latter phenotype indicates that, at least in the context of over-expression, loss-of-function HARS1 mutations are sufficient to impair proper neuronal development in fish.
Although the three HARS1 mutations studied were previously shown to decrease enzyme activity, it is still possible that they also gain aberrant neomorphic properties that contribute to pathogenicity. Therefore, it is critical to determine if inhibiting HARS1 activity or protein synthesis is sufficient to reproduce the cellular and morphological phenotypes caused by the HARS1 mutations. When Mullen and colleagues treated differentiating PC12 cells with histidinol—a small molecule inhibitor of HARS1—the neurites showed increased EIF2α phosphorylation, reduced global protein translation, and a reduced length of the longest neurite in each cell; all of these findings are comparable to the effects observed upon over-expression of each HARS1 mutant protein. Similarly, treatment with cycloheximide—a protein synthesis inhibitor—reduced the length of the longest neurite in each cell. Notably, cycloheximide also phenocopied the HARS1 mutations in zebrafish, shortening the length of the neuronal processes in the dorsal root ganglia. These data demonstrate that chemically inhibiting protein synthesis will mimic the phenotype of HARS1 mutations, supporting the hypothesis that reduced protein synthesis is part of the ARS-associated neuropathy disease mechanism.
As previous work has shown that haploinsufficiency is not a possible explanation for these peripheral neuropathies2, reduced protein synthesis would have to be a result of a dominant-negative mechanism. This requires mutant ARS subunits to dimerize with, and repress, the function of the wild-type subunit. Previously, V155G and Y330C were shown to have sedimentation velocities almost identical to that of wild-type HARS16—here, Mullen et al. show that R137Q behaves similarly, consistent with all three mutations retaining their ability to dimerize. Of note, R137Q affects a highly conserved residue at the dimer interface, underscoring that the effects of pathogenic mutations on dimerization cannot be predicted from structural assessments alone.
The severity of the phenotypes observed by Mullen et al. may be a result of exogenous HARS1 expression; it is unclear if endogenous levels of mutant HARS1 would have the same effect. Endogenous levels of mutant HARS1 could have a milder dominant-negative effect, which may not suppress protein synthesis to the extent that impairs neuronal health. Moving forward, it will be important to confirm the effects of endogenous dominant ARS mutations on neuronal protein translation. Additionally, although Mullen et al. demonstrate that pathogenic HARS1 mutations maintain their ability to dimerize with wild-type HARS1, this does not directly test whether dimerization is required for pathogenicity. This will be a critical experiment for fully demonstrating a dominant negative mechanism of ARS-mediated peripheral neuropathy.
As a whole, this study provides strong evidence for dominant, loss-of-function HARS1 mutations function impairing protein translation and neuronal health, consistent with a dominant-negative effect (see Figure). Moving forward, it will be important to test if reducing dimerization of the mutant allele can improve these phenotypes. Additionally, by phenocopying these mutations with chemical inhibitors of HARS1 and protein synthesis, Mullen et al. weaken the argument that ARS mutations act through neomorphic gain-of-function interactions, unrelated to protein translation. These results also indicate that therapeutic efforts for ARS-mediated peripheral neuropathy should be aimed at restoring ARS function and improving protein translation.
Figure.
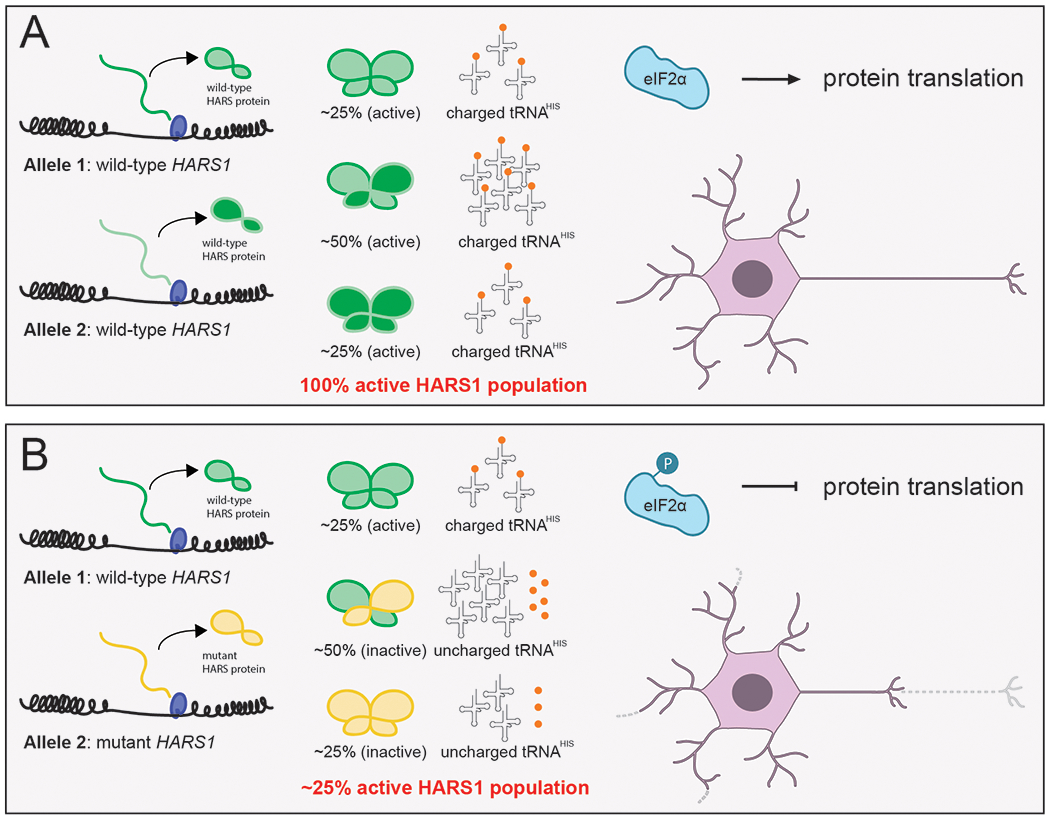
Proposed mechanism for HARS1-mediated peripheral neuropathy. (A) In an unaffected individual, two wild-type HARS1 alleles produce a fully active population of HARS1 enzymes and a sufficiently charged population of tRNAHIS for global protein translation to proceed. Long neuronal processes can develop properly and can be maintained for full neuronal function. (B) In a patient with dominant neuropathy, heterozygosity for a dominant-negative HARS1 mutation leads to a heterogeneous HARS1 population. The majority of HARS1 homodimers comprise either two mutant subunits or one mutant and one wild-type subunit, all of which have reduced activity. There is an abundance of uncharged tRNAHIS in the neuron, which leads to EIF2α phosphorylation, a global reduction in protein synthesis, and degeneration of the long neuronal processes required for a healthy peripheral nervous system.
ACKNOWLEDGEMENTS
R.M. is supported by the Michigan Pre-doctoral Training in Genetics Program (GM007544) and an individual National Research Service Award (NRSA) from the National Institute of Neurological Diseases and Stroke (NS108510). A.A. is supported by a grant from the National Institute of General Medical Sciences (GM118647).
ABBREVIATIONS
- ARS
aminoacyl-tRNA synthetases
- HARS1
cytoplasmic histidyl-tRNA synthetase
- tRNA
transfer ribonucleic acid
- EIF2α
eukaryotic initiation factor 2 subunit α
- PC12
rat pheochromocytoma cells
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
REFERENCES
- (1).Antonellis A; Green ED The Role of Aminoacyl-TRNA Synthetases in Genetic Diseases. Annual review of genomics and human genetics 2008, 9 (1), 87–107. [DOI] [PubMed] [Google Scholar]
- (2).Meyer-Schuman R; Antonellis A Emerging Mechanisms of Aminoacyl-TRNA Synthetase Mutations in Recessive and Dominant Human Disease. Human molecular genetics 2017, 26 (R2), R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kuo ME; Antonellis A Ubiquitously Expressed Proteins and Restricted Phenotypes: Exploring Cell-Specific Sensitivities to Impaired TRNA Charging. Trends in Genetics 2020, 36 (2), 105–117. 10.1016/j.tig.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wei N; Zhang Q; Yang X-L Neurodegenerative Charcot–Marie–Tooth Disease as a Case Study to Decipher Novel Functions of Aminoacyl-TRNA Synthetases. J Biol Chem 2019, 294 (14), 5321–5339. 10.1074/jbc.REV118.002955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mullen P; Abbott JA; Wellman T; Aktar M; Fjeld C; Demeler B; Ebert AM; Francklyn CS Neuropathy-Associated Histidyl-TRNA Synthetase Variants Attenuate Protein Synthesis in Vitro and Disrupt Axon Outgrowth in Developing Zebrafish. FEBS J. 2020. 10.1111/febs.15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Abbott JA; Meyer-Schuman R; Lupo V; Feely S; Mademan I; Oprescu SN; Griffin LB; Alberti MA; Casasnovas C; Aharoni S; Basel-Vanagaite L; Züchner S; De Jonghe P; Baets J; Shy ME; Espinós C; Demeler B; Antonellis A; Francklyn C Substrate Interaction Defects in Histidyl-TRNA Synthetase Linked to Dominant Axonal Peripheral Neuropathy. Hum. Mutat 2018, 39 (3), 415–432. 10.1002/humu.23380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Vester A; Velez Ruiz G; McLaughlin HM; Lupski JR; Talbot K; Vance JM; Züchner S; Roda RH; Fischbeck KH; Biesecker LG; Nicholson G; Beg AA; Antonellis A A Loss-of-Function Variant in the Human Histidyl-TRNA Synthetase (HARS) Gene Is Neurotoxic In Vivo. Human mutation 2013, 34 (1), 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bond S; Lopez-Lloreda C; Gannon PJ; Akay-Espinoza C; Jordan-Sciutto KL The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2α in Neurodegeneration. Journal of Neuropathology & Experimental Neurology 2020, 79 (2), 123–143. 10.1093/jnen/nlz129. [DOI] [PMC free article] [PubMed] [Google Scholar]