

Abstract
fms-like tyrosine like kinase 3 internal tandem duplication (FLT3-ITD) is present in acute myeloid leukemia (AML) in 30% of patients and is associated with short disease-free survival. FLT3 inhibitor efficacy is limited and transient but may be enhanced by multi-targeting of FLT3-ITD signaling pathways. FLT3-ITD drives both STAT5-dependent transcription of oncogenic Pim-1 kinase and inactivation of the tumor suppressor protein phosphatase 2A (PP2A), and FLT3-ITD, Pim-1 and PP2A all regulate the c-Myc oncogene. We studied mechanisms of action of co-treatment of FLT3-ITD-expressing cells with FLT3 inhibitors and PP2A-activating drug (PADs), which are in development. PADs, including FTY720 and DT-061, enhanced FLT3 inhibitor growth suppression and apoptosis induction in FLT3-ITD-expressing cell lines and primary AML cells in vitro and MV4-11 growth suppression in vivo. PAD and FLT3 inhibitor co-treatment independently downregulated c-Myc and Pim-1 protein through enhanced proteasomal degradation. c-Myc and Pim-1 downregulation was preceded by AKT inactivation, did not occur in cells expressing myristoylated (constitutively active) AKT1, and could be induced by AKT inhibition. AKT inactivation resulted in activation of GSK-3β, and GSK-3β inhibition blocked downregulation of both c-Myc and Pim-1 by PAD and FLT3 inhibitor co-treatment. GSK-3β activation increased c-Myc proteasomal degradation through c-Myc phosphorylation on T58; infection with c-Myc with T58A substitution, preventing phosphorylation, blocked downregulation of c-Myc by PAD and FLT3 inhibitor co-treatment. GSK-3β also phosphorylated Pim-1L/Pim-1S on S95/S4. Thus, PADs enhance efficacy of FLT3 inhibitors in FLT3-ITD-expressing cells through a novel mechanism involving AKT inhibition-dependent GSK-3β-mediated increased c-Myc and Pim-1 proteasomal degradation.
Keywords: FLT3 inhibitor, PP2A-activating drug, acute myeloid leukemia, FLT3-ITD, c-Myc, Pim kinase, AKT, glycogen synthase kinase-3β
Introduction
Internal tandem duplication of the fms-like tyrosine kinase 3 receptor tyrosine kinase (FLT3-ITD), resulting in constitutive and aberrant FLT3 signaling (1), is present in AML cells of 30% of patients (2), and FLT3-ITD AML patients have short disease-free survival (2). FLT3 tyrosine kinase inhibitors (TKIs) are cytotoxic toward FLT3-ITD-expressing AML cells in vitro and in vivo, but clinical responses are generally limited and transient (3). Efforts have focused on identifying drug combinations to improve responses to FLT3 inhibitors, and thus patient outcomes (3). FLT3-ITD oncogenic tyrosine kinase activity promotes leukemogenesis through both STAT5-dependent (1) overexpression of the oncogenic serine/threonine kinase Pim-1 (4) and inactivation of the tumor suppressor multimeric serine-threonine protein phosphatase 2A (PP2A) (5,6), providing additional therapeutic targets.
FLT3-ITD both constitutively activates FLT3 and causes aberrant signaling through STAT5 and downstream Pim-1, in addition to signaling through PI3 kinase (PI3K)/AKT and mitogen-activated protein kinase (MEK)/extracellular-signal-regulated kinase (ERK) (1). Pim-1 contributes to FLT3-ITD proliferative and anti-apoptotic effects through phosphorylation-dependent stabilization of regulators of cell growth and survival, including the c-Myc oncogene (7). It also phosphorylates and stabilizes FLT3 in a positive feedback loop in FLT3-ITD-expressing cells (8,9). Inhibition of Pim activity enhances FLT3 inhibitor cytotoxicity in FLT3-ITD-expressing AML cells in vitro and in vivo (8–11).
PP2A is a heterotrimeric protein composed of 65 and 36 kDa structural/scaffold A and catalytic C subunits and a diverse repertoire of structurally distinct regulatory B subunits that dictate subcellular localization and substrate specificity (12). PP2A enzymatic activity is negatively regulated through binding of inhibitory proteins including the cancerous inhibitor of PP2A (CIP2A), SET, and the binding protein for SET (SETBP1) (12). CIP2A upregulation has been reported as a mechanism of PP2A inactivation in FLT3-ITD-expressing cells (13). Co-treatment with the PP2A-activating drugs (PADs) FTY720 or OP449 was shown to enhance FLT3 inhibitor cytotoxicity in FLT3-ITD-expressing cells in vitro (5,6).
Pim-1 kinase is a PP2A substrate, and its PP2A-dependent dephosphorylation decreases its expression through decreased protein stability (14). Mechanistically, Pim-1 interacts with the PP2A regulatory B subunit B56β; B56β knockdown decreases Pim-1 ubiquitination and increases Pim-1 protein half-life, increasing expression of both of the Pim-1 isoforms, 44 kDa Pim-1L and 33 kDa Pim-1S (15).
FLT3-ITD, PP2A and Pim-1 all regulate expression of the transcription factor c-Myc (7,16,17). c-Myc is transcriptionally upregulated downstream of FLT3-ITD, and c-Myc or Pim-1 knockdown has anti-proliferative effects in FLT3-ITD-expressing cells (16). In contrast, PP2A and Pim-1 both regulate c-Myc expression post-translationally (7,17,18). Two N-terminus phosphorylation sites, serine 62 (S62) and threonine 58 (T58), are important in regulating c-Myc protein stability (18). Pim-1 increases S62 phosphorylation, stabilizing c-Myc protein, in association with decreased T58 phosphorylation (7), while the PP2A regulatory subunit B56α selectively associates with the c-Myc N-terminus and PP2A dephosphorylates c-Myc at S62, resulting in reduced c-Myc protein stability (17). Thus FLT3 inhibition decreases c-Myc transcription, while both Pim-1 inhibition and PP2A activation decrease c-Myc protein stability.
PADs are in development (19). Here, we studied mechanisms by which they enhance FLT3 inhibitor efficacy in cells with FLT3-ITD.
Materials and Methods
Cell lines
Mouse Ba/F3 cells transfected with human FLT3-ITD (Ba/F3-ITD) and wild-type FLT3 (FLT3-WT) and the FLT3-ITD-expressing MV4-11 human AML cell line were obtained and cultured as previously described (20). The FLT3-WT cell lines OCI-AML2 and THP-1 were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), Braunschweig, GER. Mycoplasma testing was performed every six months with the Mycoplasma Pcr Detection Kit (GeneCopoeia, Rockville, MD).
Retroviral infection of Ba/F3-ITD cells
Ba/F3-ITD cells were infected with a pMX-puro retroviral vector encoding Flag-K67M kinase-dead (KD) Pim-1, from Dr. Tomasz Skorski, Temple University, Philadelphia, PA, or empty vector control, as described (20). Pim-1 overexpression was confirmed by immunoblotting.
The myc-estrogen receptor (ER) expression vector pBABEpuro-myc-ER (plasmid #19128) (21) and pBABE-puro empty vector control were from Addgene (Cambridge, MA). Approximately 80% confluent Phoenix-AMPHO packaging cells (ATCC CRL-3213) were incubated in 25 μM chloroquine for 1 hour, then transfected with 20 μg retroviral plasmid DNA by the calcium phosphate method. Ba/F3-ITD cells were infected with virus-containing medium collected after 24 hours in the presence of polybrene (4 μg/ml). Cells were seeded in 2 ml virus-containing medium, centrifuged at 1800 rpm at 32°C for 45 minutes, then incubated at 32°C for 4 hours, centrifuged at 1800 rpm at 32°C for 45 minutes, then incubated in fresh virus-containing medium at 32°C for 2 hours. The cells were then incubated at 37°C for 24 hours, infected with virus-containing medium and incubated at 32°C for 5 hours. Infected cells were incubated in fresh virus-free medium overnight, then cultured with 1 mg/ml puromycin for 14 days. Myc overexpression was confirmed by immunoblotting. Myc-ER-expressing Ba/F3-ITD cells were treated with 300 nM 4-hydroxytamoxifen (4-OHT) (Sigma-Aldrich, St. Louis, MO) to activate the myc-ER fusion protein via translocation from cytoplasm to nucleus.
Ba/F3-ITD cells were also infected with constitutively active myristoylated AKT (Myr-AKT), pBabe-Puro-Myr-Flag-AKT1 (Addgene plasmid #15294) (22), or pBABE-puro empty vector control, as above.
Finally, Ba/F3-ITD cells were infected with pMSCVpuro-Flag-cMyc-T58A plasmid (Addgene plasmid #20076) (18) containing c-Myc with a T58 mutation changing threonine to alanine, inhibiting phosphorylation, or pMSCVpuro empty vector control (Takara Bio USA, Mountain View, CA).
Patient samples
Blood and bone marrow samples were obtained from FLT3-ITD and FLT3-WT AML patients (Supplementary Table S1) on a University of Maryland Baltimore Institutional Review Board-approved protocol, following written informed consent, in accordance with the Declaration of Helsinki. Mononuclear cells isolated by density centrifugation over Ficoll-Paque (Sigma-Aldrich) were cultured in RPMI 1640 with 20% fetal bovine serum (FBS), without cytokine supplementation, with and without FLT3 inhibitors and/or PADs. Cells were studied fresh, without prior cryopreservation.
Materials
Gilteritinib (4) (ASP2215; Active Biochem, Maplewood, NJ) and quizartinib (4) (AC220; Selleck Chemicals, Houston, TX), type I and II FLT3 inhibitors, respectively, clinically active in FLT3-ITD AML, were used at pharmacologically relevant concentrations (23,24). The SET-sequestering PAD FTY720 (25) (Fingolimod; Cayman Chemical Company, Ann Arbor, MI) was also used at pharmacologically relevant concentrations. DT-061, an orally bioavailable small molecule activator of PP2A (SMAP) developed by reengineering tricyclic neuroleptics and proposed to directly bind the PP2A Aα subunit (26–29), was provided by Dr. Goutham Narla. The pan-Pim inhibitor AZD1208 and GSK-3β inhibitors TC-G 24 and TWS119 were from Tocris Bioscience, Minneapolis, MN, the c-Myc inhibitor 10058-F4 from Sigma-Aldrich, and the pan-AKT inhibitor MK-2206 from Selleck Chemicals, Houston, TX.
Cytotoxicity assay
Cytotoxicity was measured using the WST-1 assay after 48-hour culture (20). IC50 values were determined by non-linear curve fitting to a dose-response curve using Prism V software (GraphPad, La Jolla, CA).
Cell proliferation assay
Cells cultured in RPMI 1640 medium with 10% FBS with and without drug(s) were collected at serial time points and live cells were counted after trypan blue dye exclusion, as described (20).
Measurement of apoptosis
Apoptosis was measured by annexin V/PI staining, as described (20).
Determination of synergy
Cells plated in triplicate on 96-well plates were treated with drugs at various concentrations alone and in combinations. Assays were terminated after 48 hours and combination indexes were determined according to the Chou-Talalay method using CompuSyn software, as previously described (11). Values less than 1, equal to 1 and greater than 1 are synergistic, additive and antagonistic, respectively.
in vivo study
NOD-scid IL2Rgnull (NSG) mice age 6–8 weeks were purchased from the Jackson Laboratory (Bar Harbor, ME). Exponentially growing MV4-11-luc cells (1×106), from Dr. Sharyn Baker, The Ohio State University, Columbus, OH, were injected intravenously into the lateral tail veins of restrained mice (11). Engraftment was assessed seven days later using the Xenogen IVIS Spectrum Imaging System (Caliper Life Sciences, Hopkinton, MA) after intraperitoneal injection of 150 mg/kg D-luciferin, and mice were sorted into four treatment groups (n=5 each) with equal mean signal intensity. Gilteritinib (Chemietek, Indianapolis, IN) and DT-061 (MedChemExpress USA, Monmouth Junction, NJ) were formulated via loading into preformed liposomes by transmembrane ammonium sulfate gradient or passive equilibration techniques, respectively. Gilteritinib (2 mg/kg) and/or DT-061 (5 mg/kg) were administered intravenously to MV4-11-luc-bearing mice every other day beginning on Day 7 after inoculation. Leukemia burden was measured weekly by bioluminescence imaging, as above. Bioluminescent image data were analyzed with Living Image software (PerkinElmer, Waltham, MA). Total bioluminescent signal was obtained as photons/second. Endpoints were 20% body weight loss, hind limb paralysis or lack of mobility to eat/drink. The University of Maryland Institutional Animal Care and Use Committee approved the study.
Immunoblotting
Cells were lysed in 150 mM NaCl lysis buffer with protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentration was measured using the Dye Reagent Concentrate: Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA) and 20 μg from each sample was immunoblotted (11,20). Immunoblots were incubated with antibodies (catalog numbers in parentheses) including polyclonal antibodies to c-Myc (9402), Pim-1 (2907), BAD (9292), p-BAD (S112) (9291), p-p44/42 MAPK (ERK1/2) (T202/Y204) (9101) and ubiquitin (3933) and monoclonal antibodies to p44/42 MAPK (ERK1/2) (9107), GSK-3α/β (5676), p-GSK-3α/β (S9/S21) (8566), AKT (9272), p-AKT (S473) (4060), p-AKT (T308) (2965) and p44/42MAPK (ERK1/2) (9107) (Cell Signaling Technology, Danvers, MA), β-actin (clone C-11, sc-1615 HRP) (Santa Cruz Biotechnology, Dallas, TX) vinculin (V9264) (Sigma-Aldrich), p-Myc (S62) (ab78318) and p-Myc (T58) (ab185655) (Abcam, Cambridge, MA) overnight at 4°C, then with horseradish peroxidase-conjugated secondary antibodies for one hour at room temperature. Band intensities measured by densitometry (VisionWorks®LS, UVP, Upland, CA) at serial time points were compared to pre-treatment intensities, defined as 100%.
Protein turnover and proteasomal degradation
To study protein turnover, cells were treated with 100 μg/mL cycloheximide (CHX) (Sigma-Aldrich) for 60 minutes to block new protein translation before addition of FLT3 inhibitor and/or PAD or DMSO control. Protein expression was studied at serial time points by immunoblotting. Band intensities measured by densitometry at serial time points were compared to pre-treatment intensity, defined as 100%. Protein half-life was calculated using the algorithm t1/2=0.693tn/ln(C0/Cn), where n represents the last time point (30).
To study the effect of proteasomal degradation, cells were treated with CHX as above, with and without addition of the proteasome inhibitor carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG-132; Calbiochem, San Diego, CA) (20 μM) 30 minutes after adding CHX and 30 minutes before adding FLT3 inhibitor and/or PAD.
Co-immunoprecipitation
Ba/F3-ITD cells were treated with 15 nM gilteritinib and 2 μM FTY720 or DMSO control for 4 hours. Cell lysates were pulled down with c-Myc antibody (Cell Signaling Technology) and immunoprecipitated protein was probed with ubiquitin antibody (Cell Signaling Technology). Ba/F3-ITD cells were also treated with 1 nM quizartinib and 2 μM FTY720, or DMSO control, for 1 hour. Cell lysates were pulled down with Pim-1 antibody (Santa Cruz Biotechnology) and immunoprecipitated protein was probed with ubiquitin antibody.
Quantitative real time polymerase chain reaction (qRT-PCR)
RNA was isolated using Trizol Reagents (Thermo Fisher Scientific, Waltham, MA) and cDNA was created using Superscript IV Reverse Transcriptase (Thermo Fisher Scientific). RT-qPCR was performed using SYBER® Green (MilliporeSigma, St. Louis, MO). All reactions were performed in triplicate. Primers are shown in Supplementary Table S2. The ΔCt method for relative quantification of gene expression was used to determine mRNA expression levels.
GSK-3β phosphorylation of Pim-1
The Pim-1 amino acid sequence (https://www.uniprot.org/uniprot/P11309#P11309-1) was searched for potential GSK-3β phosphorylation sites using the Group-based Prediction System algorithm (GPS 5.0, http://gps.biocuckoo.cn), with cutoff set to a medium threshold with a false prediction rate below 6% for serine/threonine kinases. Sites identified included S95/S4 on Pim-1L/Pim-1S, which is present in a sequence (MLLSKINSL) highly conserved among mammalian species. Wild-type peptide with S95/S4 (EVGMLLSKINSL) and synthetic peptide with S95A/S4A mutation (EVGMLLAKINSL) preventing phosphorylation at this site were obtained from LifeTein (Somerset, NJ). GSK-3β phosphorylation was measured using the non-radioactive ADP-Glo™ GSK-3β kinase assay (Promega, Madison, WI), according to the manufacturer’s instructions. Wild-type and mutant peptides (10 μg) were added to 0.2 ng GSK-3β kinase enzyme, 1μg GSK-3β kinase substrate as a positive control and 50μM ATP and the reaction was incubated at room temperature for 60 minutes in the dark. ADP-Glo reagent (5μl) was then added to each well, followed by incubation for 30 minutes in the dark (ATP depletion phase). Kinase Glo detection reagent (10 μl) was then added, followed by incubation for 30 minutes at room temperature in the dark (ADP detection phase). The luminescent signal was detected using the FlexStation 3 multi-mode microplate reader (Molecular Devices, San Jose, CA) and data were analyzed by SoftMax Pro 5.4.6. software (Molecular Devices). Luminescent signal for mutant peptide was normalized to luminescent signal for wild-type peptide.
Statistical analysis
All in vitro data were derived from at least three independent experiments, with error bars representing standard error of the mean (SEM). Statistical analysis was performed by two-way ANOVA with post hoc Bonferroni testing, using Prism 5 (GraphPad, San Diego, CA). In the in vivo model, photon intensity in mice treated with DT-061 and gilteritinib combination versus gilteritinib alone was compared by 2-way ANOVA with Sidak’s multiple comparison test.
Results
PP2A-activating drugs enhance efficacy of FLT3 inhibitors in cells with FLT3-ITD
To demonstrate the effect of co-treatment on cell growth in vitro, MV4-11 and Ba/F3-ITD cells, with FLT3-ITD, were grown in RPMI 1640 medium with 10% FBS with FLT3 inhibitors (gilteritinib or quizartinib) and/or PADs (FTY720 or DT-061) at their IC50 concentrations for these cell lines (Supplementary Table S3), or DMSO control, and viable cells were counted at serial time points. While FLT3 inhibitors and PADs as single agents decreased growth of both cell lines relative to DMSO control, concurrent treatment with PAD and FLT3 inhibitor markedly decreased growth relative to single-drug treatments in MV4-11 (Figure 1A) and Ba/F3-ITD (Supplementary Figure S1A) cells.
Figure 1. Concurrent PP2A-activating drug treatment increases cytotoxicity of FLT3 inhibitors in MV4-11 cells and AML patient blasts with FLT3-ITD and in an MV4-11 orthotopic mouse model.

A. Combination treatment decreases MV4-11 growth. MV4-11 human FLT3-ITD AML cells were cultured in triplicate with 1 nM quizartinib or 15 nM gilteritinib and/or 2 μM FTY720 or 10 μM DT-061, or DMSO control, and viable cells were counted after 24, 48 and 72 hours. B. Combination treatment increases MV4-11 apoptosis. MV4-11 cells were cultured in triplicate with 1 nM quizartinib or 15 nM gilteritinib and/or FTY720 or DT-061 at the concentrations shown, or DMSO control, and apoptosis was measured after 48 hours by annexin V/PI staining. Statistical analysis was performed by two-way ANOVA with post hoc Bonferroni testing (*p<0.05, **p<0.005, ***p<0.001). C. Combination treatment is synergistic in MV4-11 cells. MV4-11 cells cultured in triplicate on 96-well plates were treated with gilteritinib and FTY720 alone and in combination at the concentrations shown. Combination indexes were determined by Chou-Talalay analysis. D. DT-061 enhances efficacy of gilteritinib in vivo. NSG mice inoculated with MV411-luc cells on Day 0 were treated with liposomal gilteritinib and/or DT-061, or vehicle control, beginning on Day 7 (D7). Left panel shows change in photon intensity, measured by bioluminescence imaging, over time, with ***p=0.0004, comparing DT-061 and gilteritinib combination vs. gilteritinib alone on Day 49 by 2-way ANOVA with Sidak’s multiple comparison test. Right panel shows two representative mice from each treatment group on Days 7, 28, 35 and 49. E. Combination treatment increases FLT3-ITD AML blast apoptosis. FLT3-ITD AML patient blasts were plated in triplicate in RPMI 1640 medium with 1 nM quizartinib or 15 nM gilteritinib and/or FTY720 or DT-061 at the concentrations shown, or DMSO control, and apoptosis was measured after 48 hours by annexin V/PI staining. Statistical analysis was performed by two-way ANOVA with post hoc Bonferroni testing (*p<0.05, **p<0.005, ***p<0.001).
To investigate the cytotoxic effects of co-treatment, MV4-11 and Ba/F3-ITD cells were cultured for 48 hours with the FLT3 inhibitors gilteritinib or quizartinib at their IC50 concentrations for these cell lines (Supplementary Table S3) with FTY720 or DT-061 at increasing concentrations, and apoptosis was measured by annexin V/propidium iodide (PI) labeling, detected by flow cytometry. PAD and FLT3 inhibitor combinations significantly increased apoptosis, relative to single-drug treatments in MV4-11 (Figure 1B) and Ba/F3-ITD (Supplementary Figure S1B) cells.
Synergy of PAD and FLT3 inhibitor combinations was also demonstrated cells by Chou-Talalay analysis in the FLT3-ITD cell lines MV4-11 (Figure 1C) and Ba/F3-ITD (Supplementary Figure S1C), while synergy was not seen in the FLT3-WT AML cell lines THP-1 and OCI-AML2 (Supplementary Figure S2).
To study PAD and/or FLT3 inhibitor effects in vivo, NSG mice with MV4-11-luc cells injected intravenously and allowed to engraft were treated with gilteritinib and/or DT-061 or vehicle control, five mice per treatment group, as described in Materials and Methods, and leukemia burden was measured by luciferin imaging. By Day 35, leukemia burden was significantly lower in mice treated with DT-061 and gilteritnib combination, compared to gilteritinib alone (p= 0.0018 by 2-way ANOVA with Sidak’s multiple comparison test) (Figure 1D), and also compared to DT-061 alone (p<0.0001). Images of all mice are shown in Supplementary Figure S3.
Finally, FLT3-ITD AML patient blasts were also cultured for 48 hours with gilteritinib (15 nM) or quizartinib (1 nM) with and without 4 μM FTY720 or 10 μM DT-061. PAD treatment significantly increased apoptosis induction by FLT3 inhibitors (Figure 1E). In contrast, PAD and FLT3 inhibitor combinations at these concentrations were not cytotoxic in blasts from patients with AML with FLT3-WT or marrow cells from AML patients in complete remission (CR) or CR with incomplete platelet recovery (Supplementary Figure S4).
PAD and FLT3 inhibitor treatment downregulates c-Myc and Pim-1 expression in FLT3-ITD-expressing cells
To study effects of PAD and FLT3 inhibitor treatment on c-Myc and Pim-1, expression of these proteins was measured by immunoblotting at serial time points in Ba/F3-ITD and MV4-11 cells and FLT3-ITD AML patient blasts treated with gilteritinib and/or FTY720 or DT-061, or DMSO control. Co-treatment with gilteritinib and FTY720 or DT-061 markedly downregulated both c-Myc and Pim-1 protein expression, relative to treatment with single drugs or DMSO control (Figure 2, A-D). Expression of p-T58- and p-S62-c-Myc relative to total c-Myc was also studied, demonstrating that p-T58-c-Myc expression was increased and more sustained than p-S62- and total c-Myc expression in both Ba/F3-ITD and MV4-11 cells treated with gilteritinib and FTY720 combination, though expression was more sustained in Ba/F3-ITD (Supplementary Figure S6).
Figure 2. Concurrent treatment with PP2A activator and FLT3 inhibitor downregulates c-Myc and Pim-1 protein expression.
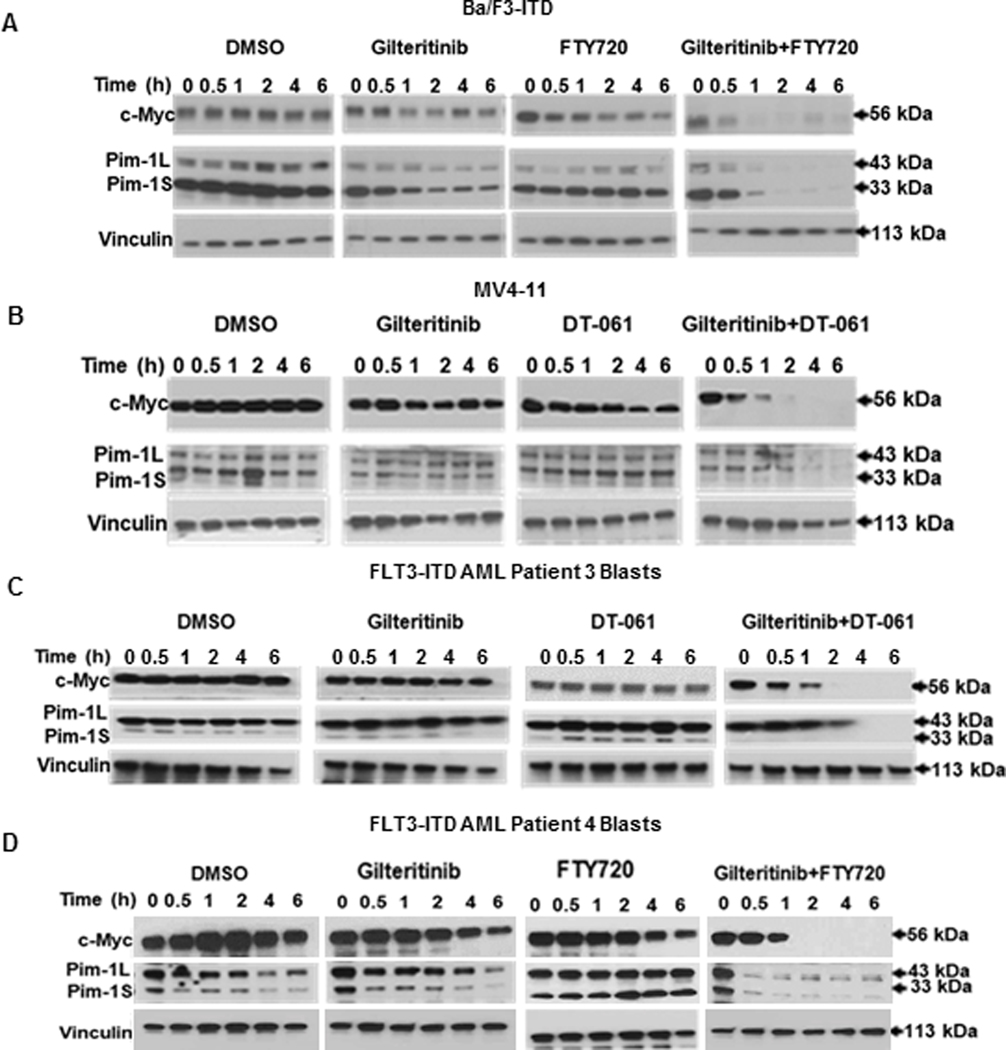
Ba/F3-ITD (A) and MV4-11 (B) cells and FLT3-ITD AML patient blasts (C,D) cultured with 15 nM gilteritinib and/or 2 μM (cell lines) or 4 μM (patient blasts) FTY720 or 10 μM DT-061, or DMSO control, were harvested at the time points shown, and c-Myc and Pim-1 protein expression was measured by immunoblotting, with vinculin protein expression as a loading control. Results are also shown graphically in Supplementary Figure S4.
mRNA expression was also studied (Supplementary Figure S7). c-Myc mRNA expression was stable in Ba/F3-ITD cells treated with gilteritinib and FTY720, but decreased and then stabilized in similarly treated MV4-11 cells, as did Pim-1 mRNA expression in both cell lines. Progressive mRNA downregulation, as seen for protein expression, was not observed.
Finally, effects of PAD and FLT3 inhibitor treatment on c-Myc and Pim-1 in blasts from Patient 7, with FLT3-WT AML, are shown in Supplementary Figure S8. c-Myc and Pim-1 were not downregulated by combination treatment.
PAD and FLT3 inhibitor treatment increases c-Myc and Pim-1 protein turnover in FLT3-ITD-expressing cells through enhanced proteasomal degradation
As an initial approach to testing whether c-Myc and Pim-1 downregulation by PAD and FLT3 inhibitor co-treatment is post-translational, c-Myc and Pim-1 protein expression was measured by immunoblotting at serial time points in Ba/F3-ITD cells treated with quizartinib and/or FTY720, or DMSO control, with and without pre-treatment with the proteasome inhibitor MG-132. c-Myc and Pim-1 protein expression was downregulated in Ba/F3-ITD cells co-treated with quizartinib and FTY720, and pre-treatment with MG-132 inhibited this downregulation (Supplementary Figure S9). This suggested increased proteasomal degradation as a mechanism for the observed downregulation of c-Myc and Pim-1 protein expression in FLT3-ITD-expressing cells co-treated with PAD and FLT3 inhibitor.
To confirm that c-Myc and Pim-1 protein downregulation was a result of increased proteasomal degradation, Ba/F3-ITD (Figure 3A) and MV4-11 (Figure 3B) cells were pretreated with CHX for 60 minutes to block new protein translation, with and without addition of MG-132 after 30 minutes, then treated with gilteritinib and FTY720, or DMSO control. c-Myc and Pim-1 protein turnover increased with gilteritinib and FTY720 co-treatment, compared to DMSO control, with a marked reduction in the half-lives of c-Myc and Pim-1 proteins (Supplementary Figure S10). MG-132 treatment decreased both c-Myc and Pim-1 protein turnover (Figure 3A,B; Supplementary Figure S10), consistent with increased proteasomal degradation as the mechanism for the c-Myc and Pim-1 protein downregulation in FLT3-ITD-expressing cells co-treated with PAD and FLT3 inhibitor.
Figure 3. Concurrent PP2A-activating drug and FLT3 inhibitor treatment increases c-Myc and Pim-1 ubiquitination and proteasomal degradation.
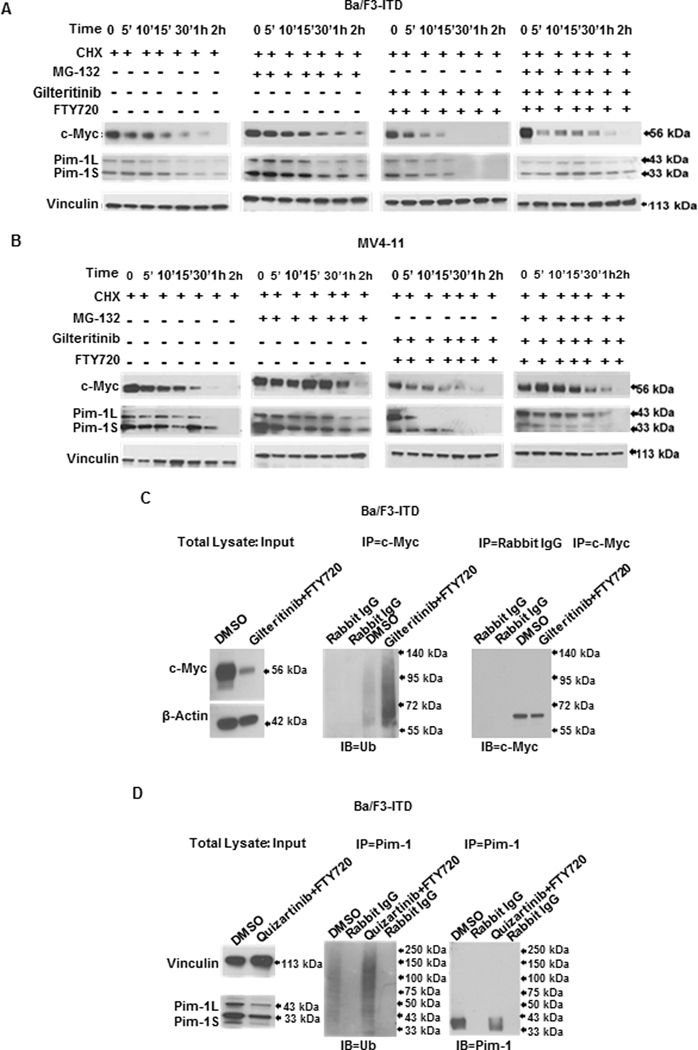
Ba/F3-ITD (A) and MV4-11 (B) cells pre-treated with 100 μg/ml cycloheximide (CHX) to inhibit new protein translation, with or without addition of the proteasome inhibitor MG-132 (20 μM), were treated with 1 nM quizartinib and/or 2 μM FTY720, or DMSO control, and c-Myc and Pim-1 protein expression was measured by immunoblotting at the time points shown. Results are also shown graphically in Supplementary Figure S4. C. Ba/F3-ITD cell lysates prepared after treatment with 15 nM gilteritinib and 2 μM FTY720, or DMSO control, for 4 hours were pulled down with c-Myc antibody and the immunoprecipitated protein complex was immunoblotted and probed with ubiquitin antibody. D. Ba/F3-ITD cell lysates prepared after treatment with 1nM quizartinib and 2 μM FTY720, or DMSO control, for one hour were pulled down with Pim-1 antibody and the immunoprecipitated protein complex was immunoblotted and probed with ubiquitin antibody.
To further confirm this mechanism, ubiquitinated c-Myc and Pim-1 was measured in Ba/F3-ITD cells treated with gilteritinib and FTY720, or DMSO control, for 4 hours and 1 hour, respectively. A marked increase in ubiquitinated c-Myc (Figure 3C) and Pim-1 (Figure 3D) was seen with gilteritinib and FTY720 treatment, compared to control.
Decreases in c-Myc and Pim-1 expression by PAD and FLT3 inhibitor co-treatment occur independently
To determine whether c-Myc downregulation by PAD and FLT3 inhibitor co-treatment occurs via effects on Pim-1, Ba/F3-ITD cells treated with the pan-Pim kinase inhibitor AZD1208 and Ba/F3-ITD cells expressing kinase-dead Pim-1 were studied. Ba/F3-ITD cells were treated with FTY720 and gilteritinib with and without pre-treatment with the pan-Pim kinase inhibitor AZD1208, and c-Myc expression was measured at serial time points. c-Myc was similarly downregulated in cells with and without AZD1208 pre-treatment (Figure 4A). c-Myc expression was also studied at serial time points in parental Ba/F3-ITD cells and Ba/F3-ITD cells expressing kinase-dead Pim-1 or empty vector control treated with gilteritinib and FTY720. c-Myc was similarly downregulated in all three (Figure 4B). Decreased p-BAD (S112) demonstrates inhibition of Pim-1 activity in Figures 4, A and B.
Figure 4. Effects of PP2A-activating drug and FLT3 inhibitor combination treatment on c-Myc and Pim-1 expression are independent.
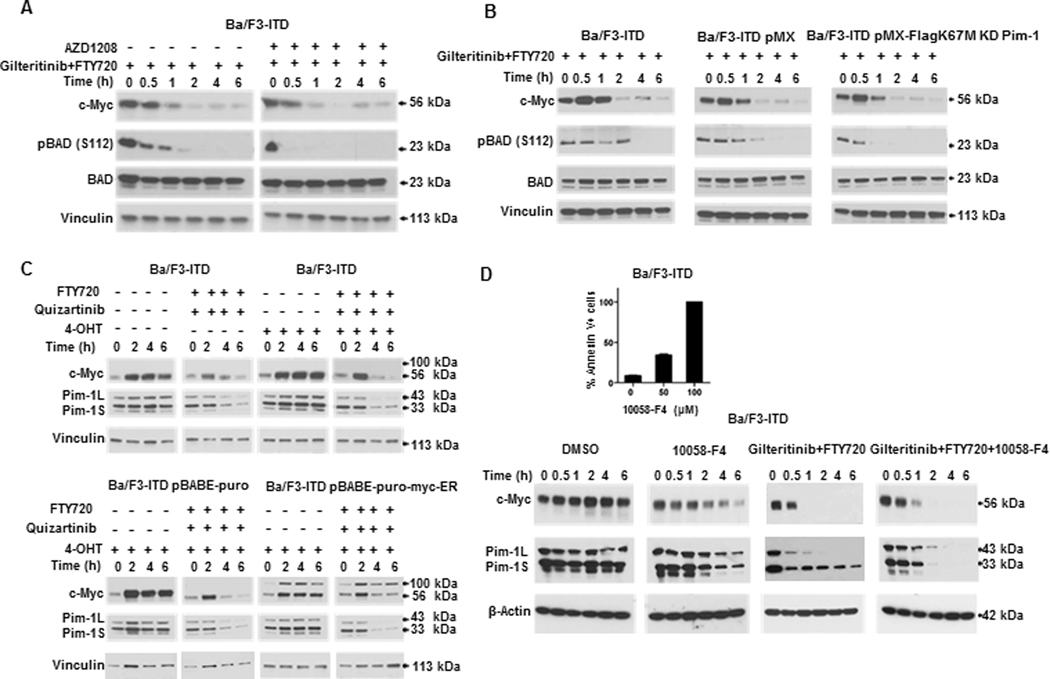
A. Pre-treatment of Ba/F3-ITD cells with the pan-Pim kinase inhibitor AZD1208 does not alter c-Myc downregulation by combination treatment. Ba/F3-ITD cells were treated with 15 nM gilteritinib and 2 μM FTY720 with or without 30-minute pre-treatment with 1 μM AZD1208, and c-Myc protein expression was measured by immunoblotting at the time points shown. Decreased p-BAD (S112) demonstrates inhibition of Pim-1 activity. B. c-Myc is similarly downregulated by combination treatment in parental Ba/F3-ITD and Ba/F3-ITD infected with kinase-dead Pim-1 kinase or empty vector control. Parental Ba/F3-ITD cells and Ba/F3-ITD cells infected with kinase-dead Pim-1 kinase (Ba/F3-ITD pMX-FlagK67M) or with empty vector control (Ba/F3-ITD pMX) were treated with 15 nM gilteritinib and 2 μM FTY720 and c-Myc expression was measured by immunoblotting at the time points shown. Decreased p-BAD (S112) demonstrates inhibition of Pim-1 activity. C. Pim-1 kinase is similarly downregulated by combination treatment in parental Ba/F3-ITD and Ba/F3-ITD overexpressing c-Myc or empty vector control. Parental Ba/F3-ITD and Ba/F3-ITD expressing ER-Myc or empty vector control were treated with 1 nM quizartinib and 2 μM FTY720, or DMSO control. Cells expressing ER-Myc were cultured with 300 nM 4-hydroxytamoxifen (4-OHT) to activate the myc-ER fusion protein via translocation from cytoplasm to nucleus, and parental cells and empty vector cells were similarly cultured with 4-OHT. c-Myc and Pim-1 protein was measured by immunoblotting at the time points shown. D. Myc inhibition does not downregulate Pim-1. Ba/F3-ITD cells were treated with the Myc inhibitor 10058-F4 (100 μM) and Pim-1 protein expression was measured by immunoblotting at the time points shown. Immunoblots of Ba/F3-ITD cells treated with gilteritinib and FTY720 and/or 10058-F4 are shown. The inset demonstrates apoptosis induction in Ba/F3-ITD cells by 48-hour 100 μM 10058-F4 treatment.
To determine whether Pim-1 downregulation by FTY720 and quizartinib co-treatment is dependent on c-Myc downregulation, Pim-1 expression was measured at serial time points in Ba/F3-ITD cells and Ba/F3-ITD cells infected with ER-Myc plasmid or empty vector control. Pim-1 was similarly downregulated in all three (Figure 4C). Additionally, treatment of Ba/F3-ITD cells with the c-Myc inhibitor 10058-F4 at 100 μM, a concentration at which it induced apoptosis (Figure 4D inset), did not downregulate Pim-1 expression (Figure 4D).
PAD and FLT3 inhibitor treatment inactivates AKT
Because CIP2A overexpression has been associated with significantly higher c-Myc, STAT5, and p-AKT (S473) (active) levels in FLT3-ITD-expressing cells (13) and p-AKT (S473) regulates c-Myc stability in prostate cancer cells (31), we hypothesized that p-AKT (S473) might regulate c-Myc protein turnover in FLT3-ITD-expressing cells. To test this hypothesis, we studied p-AKT (S473) expression in FLT3-ITD-expressing cells treated with PAD and/or FLT3 inhibitor.
p-AKT (S473) decreased rapidly in Ba/F3-ITD and MV4-11 cells and FLT3-ITD AML patient blasts co-treated with quizartinib or gilteritinib and FTY720 or DT-061, but not in cells treated with single drugs or with DMSO control (Figure 5A-C). p-AKT (S473) decrease preceded Pim-1 and c-Myc downregulation (Figure 5A-C). p-AKT (T308) also decreased with drug co-treatment (Figure 5B).
Figure 5. PP2A-activating drug and FLT3 inhibitor combination rapidly inactivates AKT,
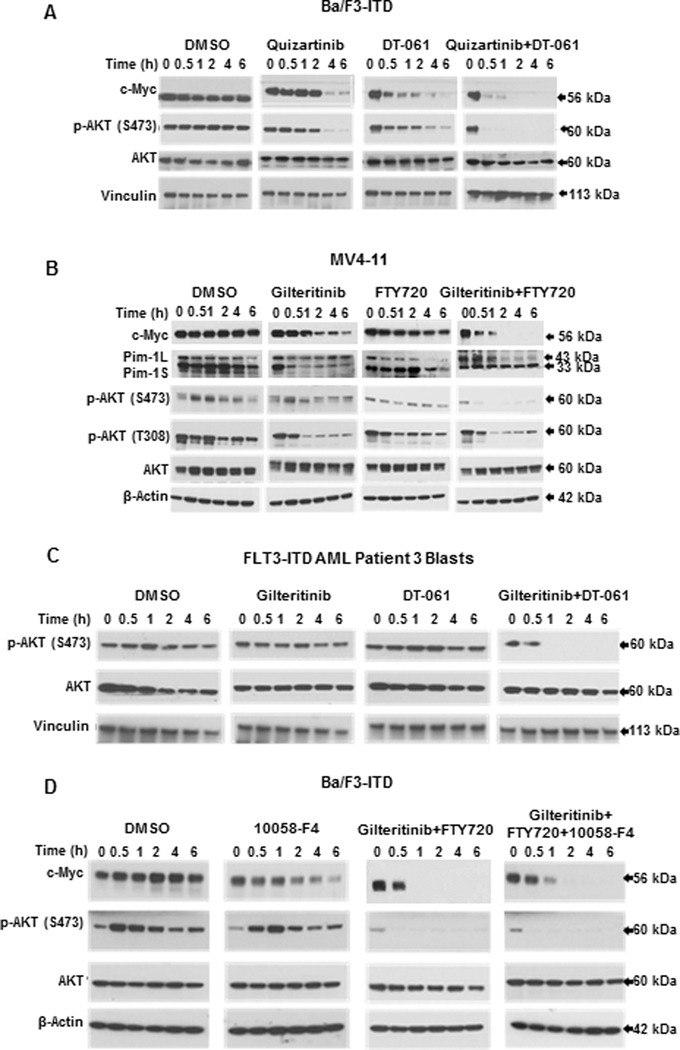
A. Treatment of Ba/F3-ITD cells with DT-061 and quizartinib combination causes rapid decrease in p-AKT (S473). Ba/F3-ITD cells were treated with 1 nM quizartinib and/or 10 μM DT-061, or DMSO control, and protein expression was measured by immunoblotting at the time points shown. B. Treatment of MV4-11 cells with gilteritinib and FTY720 combination rapidly decreases p-AKT (S473) and (T308). MV4-11 cells were treated with 15 nM gilteritinib and/or 2 μM FTY720, or DMSO control, and protein expression was measured by immunoblotting at the time points shown. This blot is also shown in Figure 2B. C. Treatment of FLT3-ITD AML patient blasts with gilteritinib and DT-061 causes rapid decrease in p-AKT (S473). FLT3-ITD AML patient blasts were treated with 15 nM gilteritinib and/or 10 μM DT-061, or DMSO control, and protein expression was measured by immunoblotting at the time points shown. This blot is also shown in Figure 2C. D. c-Myc inhibition does not inactivate AKT. Ba/F3/ITD cells were treated with the c-Myc inhibitor 10058-F4 (100 μM), and p-AKT (S473) and AKT protein expression was measured at serial time points. c-Myc inhibition did not inactivate AKT, consistent with c-Myc being downstream of AKT. Immunoblots of Ba/F3/ITD cells treated with gilteritinib and FTY720 without and with 10058-F4 are shown as controls.
To confirm that AKT inactivation occurs upstream of c-Myc downregulation, Ba/F3-ITD cells were treated with the Myc inhibitor 10058-F4. Myc inhibition did not decrease p-AKT (S473) nor inhibit the decrease in p-AKT induced by gilteritinib and FTY720 co-treatment (Figure 5D).
AKT inactivation is necessary and sufficient for post-translational c-Myc and Pim-1 downregulation and apoptosis induction by PAD and FLT3 inhibitor co-treatment
To determine whether AKT inactivation is necessary and sufficient for c-Myc and Pim-1 downregulation and apoptosis induction by PAD and FLT3 inhibitor co-treatment, Ba/F3-ITD cells were infected with myristoylated AKT (Myr-AKT1), causing constitutive AKT activation, and were also treated with the pan-AKT inhibitor MK-2206.
In Ba/F3-ITD cells infected with pBabe-Puro-Myr-Flag-AKT1, FTY720 and quizartinib co-treatment did not downregulate c-Myc or Pim-1 expression, in contrast to findings in parental Ba/F3-ITD and Ba/F3-ITD infected with empty vector control (Figure 6A). pBabe-Puro-Myr-Flag-AKT1 expression also inhibited induction of increased c-Myc and Pim-1 protein turnover by FTY720 and quizartinib co-treatment (Figure 6B). Moreover, FTY720 and quizartinib co-treatment did not induce apoptosis in Ba/F3-ITD cells infected with pBabe-Puro-Myr-Flag-AKT1, in contrast to findings in parental Ba/F3-ITD and Ba/F3-ITD infected with empty vector control (Figure 6C). Therefore, AKT inactivation is necessary for downregulation of c-Myc and Pim via increased protein turnover and induction of apoptosis by PAD and FLT3 inhibitor co-treatment.
Figure 6. AKT inactivation is necessary and sufficient for c-Myc and Pim-1 downregulation and induction of apoptosis by PP2A-activating drug and FLT3 inhibitor co-treatment.
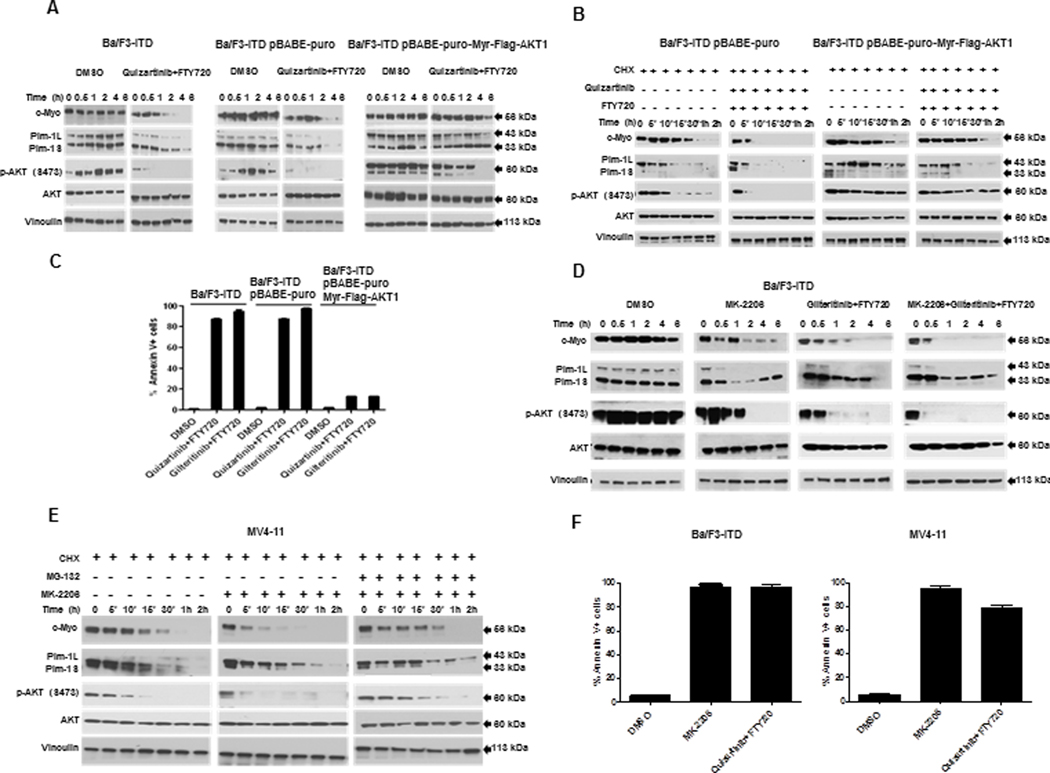
A. Constitutive AKT activation inhibits c-Myc and Pim-1 downregulation by combination treatment. c-Myc, Pim-1, p-AKT (S473) and AKT protein expression was measured by immunoblotting in parental Ba/F3-ITD cells and Ba/F3-ITD cells expressing myristoylated AKT (pBABE-Puro-Myr-FLAG-AKT1) or empty vector control (pBABE-Puro) treated with 1 nM quizartinib and 2 μM FTY720. B. Constitutive AKT activation inhibits increase in c-Myc and Pim-1 turnover in co-treated cells. c-Myc, Pim-1, p-AKT (S473) and AKT protein expression was measured by immunoblotting in Ba/F3-ITD cells expressing myristoylated AKT or empty vector control treated with 100 μg/ml cycloheximide (CHX) to block new protein translation, then with 1 nM quizartinib and 2 μM FTY720, or DMSO control. C. Constitutive AKT activation prevents induction of apoptosis by FLT3 inhibitor and PP2A activator combination. Apoptosis was measured by annexin V/PI labeling in parental Ba/F3-ITD cells and Ba/F3-ITD cells expressing myristoylated AKT or empty vector control treated with 1 nM quizartinib or 15 nM gilteritinib and 2 μM FTY720 for 48 hours. Constitutive AKT activation prevented induction of apoptosis (P<0.0005). D. AKT inhibition downregulates Pim-1 and c-Myc expression. c-Myc, Pim-1, p-AKT (S473) and AKT protein expression was measured by immunoblotting in Ba/F3-ITD cells treated with the AKT inhibitor MK-2206 (5 μM) and/or 15 nM gilteritinib and 2 μM FTY720. E AKT inhibition increases Pim-1 and c-Myc proteasomal degradation. c-Myc and Pim-1 expression was measured by immunoblotting in MV4-11 cells treated with 100 μg/ml cycloheximide (CHX), with and without the proteasome inhibitor MG-132 (20 μM), then with the AKT inhibitor MK-2206 (5 μM). F. AKT inhibition induces apoptosis of cells with FLT3-ITD. Apoptosis was measured in Ba/F3-ITD and MV4-11 cells treated with the AKT inhibitor MK-2206 (5 μM) or 1 nM quizartinib and 2 μM FTY720 or DMSO control for 48 hours. AKT inhibition induced apoptosis (P<0.0005).
Additionally, treatment with the AKT inhibitor MK-2206 downregulated c-Myc and Pim-1 protein expression similarly to FTY720 and gilteritinib co-treatment (Figure 6D). Moreover, MK-2206 increased c-Myc and Pim-1 protein turnover, and this effect was inhibited by pre-treatment with MG-132 (Figure 6E). Finally, MK-2206 induced apoptosis of Ba/F3-ITD and MV4-11 cells similarly to FTY720 and gilteritinib co-treatment (Figure 6F). Thus, AKT inactivation is sufficient for c-Myc and Pim-1 downregulation via increased protein turnover and for induction of apoptosis in FLT3-ITD-expressing cells.
We also studied p-ERK1/2 expression in Ba/F3-ITD and MV4-11 cells treated with gilteritinib and/or FTY720. p-ERK1/2 (T202/Y204) was downregulated in cells treated with drug combination, but later than, and to a lesser extent than, p-AKT (S473) (Supplementary Figure S11).
Based on the fact that c-Myc inhibition potently induces apoptosis of cells with FLT3-ITD (Figure 4D), c-Myc is likely is likely responsible for the apoptosis observed in Figure 6F, but it cannot be stated with certainty that it is solely responsible.
AKT inactivation regulates c-Myc and Pim-1 proteasomal degradation through GSK-3β activation
Since GSK-3β phosphorylates c-Myc at T58 to enhance its proteasomal degradation (18) and GSK-3β is an AKT substrate (32), we hypothesized that AKT inactivation by PAD and FLT3 inhibitor co-treatment decreases GSK-3β phosphorylation, thereby activating it, resulting in c-Myc T58 phosphorylation and thus enhanced c-Myc proteasomal degradation.
Indeed, gilteritinib and DT-061 co-treatment caused rapid GSK-3α/β dephosphorylation (activation) in MV4-11 cells (Figure 7A). Moreover, co-treatment with the GSK3-β inhibitor TC-G 24 prevented c-Myc and Pim-1 downregulation in Ba/F3-ITD cells (Figure 7B) and FLT3-ITD AML patient blasts (Figure 7C) treated with gilteritinib and DT-061. Treatment with TC-G 24 also inhibited apoptosis induction by PAD and FLT3 inhibitor combinations (Figure 7D). A second GSK3-β inhibitor, TWS119, also inhibited c-Myc and Pim-1 downregulation in Ba/F3-ITD cells (Supplementary Figure S12A) and inhibited apoptosis induction (Supplementary Figure S12B) by PAD and FLT3 inhibitor combinations.
Figure 7. PP2A-activating drug and FLT3 inhibitor combination downregulates c-Myc and Pim-1 expression through GSK-3β activation and AKT inactivation is necessary and sufficient for GSK-3β activation by PP2A-activating drug and FLT3 inhibitor combination.
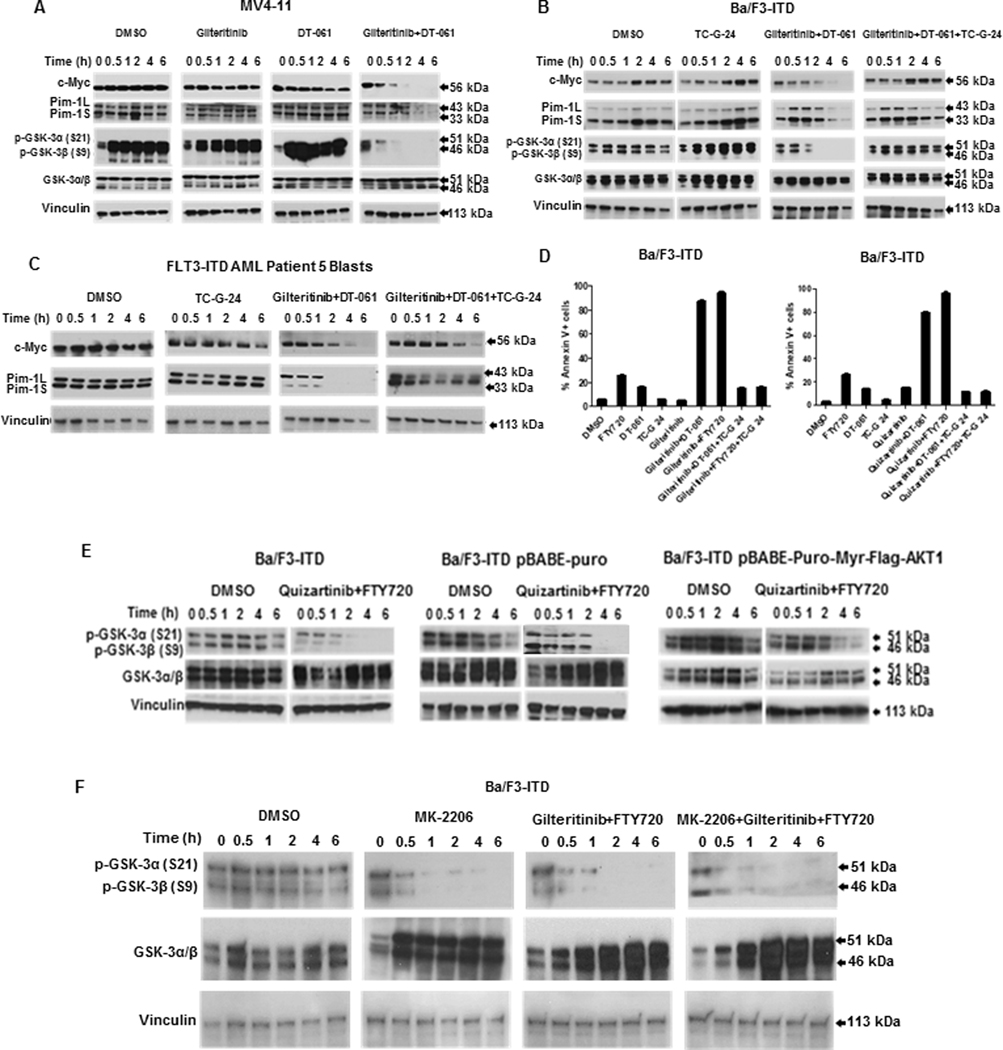
A. Combination treatment activates GSK-3α/β. c-Myc, Pim-1, p-GSK-3α/β (S21/S9) and GSK-3α/β protein expression was measured by immunoblotting in MV4-11 cells treated with 15 nM gilteritinib and/or 10 μM DT-061, or DMSO control, showing rapid decrease in p-GSK-3α/β (S21/S9) (GSK-3α/β activation) with combination treatment. This blot is also shown in Figure 2B. B. GSK-3β inhibitor prevents c-Myc and Pim-1 downregulation by combination treatment. c-Myc, Pim-1, p-GSKα/β (S21/S9) and GSKα/β protein expression was measured by immunoblotting in Ba/F3-ITD cells treated with the GSK-3β inhibitor TC-G 24 (17 nM) and/or 15 nM gilteritinib and 10 μM DT-061. TC-G 24 increased p-GSKα/β, demonstrating GSKα/β inactivation. C. GSK-3β inhibitor prevents c-Myc and Pim-1 downregulation by combination treatment in FLT3-ITD AML patient blasts. c-Myc and Pim-1 expression was measured by immunoblotting in blasts treated with the GSK-3β inhibitor TC-G 24 (17 nM) and/or 15 nM gilteritinib and 10 μM DT-061. D. GSK-3β inhibitor prevents apoptosis induction by PP2A-activating drug and FLT3 inhibitor combination. Apoptosis was measured by annexin V/PI staining in Ba/F3-ITD cells cultured for 48 hours with DMSO control, FTY720 or DT-061 and and/or gilteritinib (left) or quizartinib (right), GSK-3β inhibitor TC-G 24 or combinations. GSK-3β inhibitor prevented apoptosis induction (P<0.0005). E. Constitutive AKT activation prevents GSK-3β activation. p-GSK-3α/β and GSK-3α/β protein expression was measured in parental Ba/F3-ITD and Ba/F3-ITD cells infected with empty vector or myristoylated AKT1 treated with 1 nM quizartinib and 2 μM FTY720. F. AKT inhibitor activates p-GSK-3α/β. p-GSK-3α/β and GSK-3α/β expression was measured in Ba/F3-ITD cells treated with the AKT inhibitor MK-2206 (5 μM) and/or 15 nM gilteritinib and 2 μM FTY720.
We next demonstrated that AKT inactivation is necessary and sufficient for GSK-3β activation by PAD and FLT3 inhibitor co-treatment. FTY720 and quizartinib co-treatment caused GSK-3α and -β dephosphorylation (activation) in parental Ba/F3-ITD cells and Ba/F3-ITD cells infected with empty vector, but not with myristoylated (constitutively activated) AKT (Figure 7E). Additionally, treatment with the AKT inhibitor MK-2206 resulted in GSK-3α and -β dephosphorylation, similarly to FTY720 and gilteritinib co-treatment (Figure 7F).
To demonstrate that AKT-mediated c-Myc proteasomal degradation occurs through c-Myc phosphorylation at T58, Ba/F3-ITD cells infected with pMSCVpuro-Flag-cMyc-T58A plasmid with c-Myc mutation at T58, inhibiting phosphorylation, or empty vector control were co-treated with FTY720 and gilteritinib. FTY720 and gilteritinib co-treatment did not increase c-Myc protein turnover in cells expressing c-Myc with the T58A mutation (Figure 8A), demonstrating that FLT3 inhibitor and PAD co-treatment increases c-Myc proteasomal degradation through c-Myc T58 phosphorylation. pMCVpuro-FLAG-cMYC-WT is unavailable as a control, but it should be noted that Pim-1 kinase is similarly downregulated by combination treatment in parental Ba/F3-ITD and Ba/F3-ITD overexpressing c-Myc or empty vector control (Figure 4C). Additionally, expression of c-Myc with the T58A mutation protected against apoptosis induction by quizartinib and FTY720 combination (Figure 8B).
Figure 8. GSK-3β phosphorylates c-Myc on T58 and Pim-1L/Pim-1S on S95/S4C.
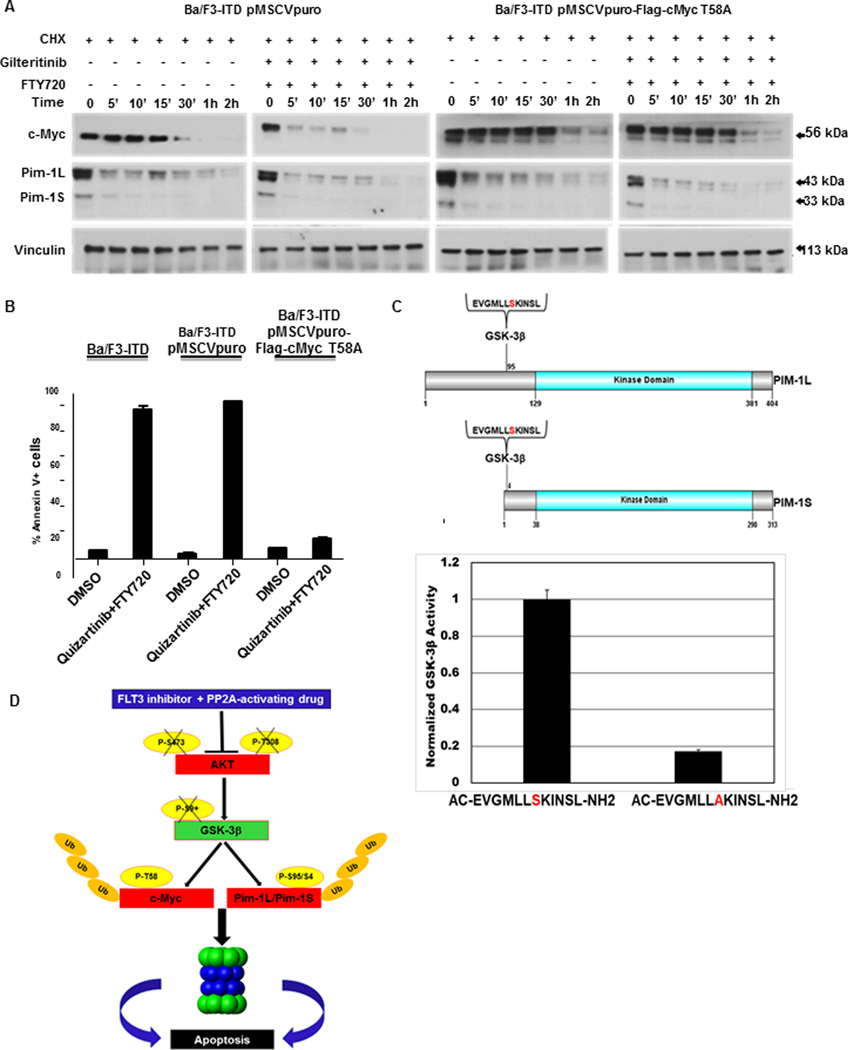
A. GSK-3β phosphorylates c-Myc on T58. c-Myc and Pim-1 protein expression was measured by immunoblotting in Ba/F3-ITD cells infected with pMSCVpuro-Flag-cMyc T58A plasmid, with a mutation inhibiting c-Myc T58 phosphorylation, or pMSCVpuro empty vector control, treated with 100 μg/ml cycloheximide (CHX) to block new protein translation, then gilteritinib 15 nM and 2 μM FTY720, or DMSO control. Expression of T58A inhibited increased turnover of c-Myc, but not Pim-1. B. Expression of c-Myc with the T58A mutation protects against apoptosis induction by quizartinib and FTY720 combination. Parental Ba/F3-ITD cells and Ba/F3-ITD cells Ba/F3-ITD cells infected with pMSCVpuro-Flag-cMyc T58A plasmid or pMSCVpuro empty vector control were treated with 1 nM quizartinib and 2 μM FTY720, or DMSO control, for 48 hours and apoptosis was measured by Annexin V/PI labeling. C.GSK-3β phosphorylates Pim-1L/Pim-1S on S95/S4. The wild-type Pim-1 peptide EVGMLLSKINSL, including S95/S4 on Pim-1L/Pim-1S, and EVGMLLAKINSL, the corresponding peptide with S95A/S4A mutation preventing serine phosphorylation, are shown schematically in the upper panel. Marked decreased in GSK-3β phosphorylation of EVGMLLAKINSL, compared to EVGMLLSKIN, in a GSK-3β kinase assay is shown in the lower panel (P<0.0005). D. Schematic summarizing the pathway affected by PP2A-activating drug and FLT3 inhibitor combination treatment.
We next sought to determine the mechanism by which GSK-3β regulates Pim-1 protein turnover. To investigate potential GSK-3β phosphorylation sites on Pim-1L and Pim-1S, bioinformatic analysis of their protein sequences (GPS 5.0, http://gps.biocuckoo.cn) was performed. Putative phosphorylation sites identified included S65, S69, S74 and S95 on Pim-1L and S4 on Pim-1S. The S95 sequence in Pim-1L (MLLAKINSL) is homologous to the S4 sequence in Pim-1S (Figure 8C, upper panel) and is also conserved in multiple mammalian species, supporting essential biological functions of these residues. We therefore tested the peptide sequence EVGMLLSKINSL, which includes the putative phosphorylation sites S95 in Pim-1L and S4 in Pim-1S, as potential substrates for GSK-3β kinase activity in an in vitro kinase assay. Commercially available GSK-3β active kinase was incubated with this peptide or its corresponding mutant version in which the predicted phosphorylation serine (S) was substituted by alanine (A), which cannot be phosphorylated. GSK-3β phosphorylated the wild-type peptide, but not the mutant peptide with A substituted for S (Figure 8C, lower panel). We therefore tested this site, S95/S4 on Pim-1/Pim-1L, with peptides EVGMLLSKINSL and EVGMLLAKINSL, the latter with a mutated putative GSK-3β phosphorylation site, in a GSK-3β kinase assay, described in Materials and Methods. GSK-3β kinase activity was markedly decreased when EVGMLLAKINSL, rather than EVGMLLSKINSL, was present in the phosphorylation reaction (Figure 8C, lower panel), consistent with GSK-3β phosphorylation of S95/S4 on Pim-1L/Pim-1S.
A schematic summarizing the pathway affected by the PAD and FLT3 inhibitor combination treatment is shown in Figure 8D.
Discussion
FLT3-ITD is present in AML cells of 30% of patients (2), and results in constitutive and aberrant FLT3 signaling (1). FLT3-ITD AML patients have short disease-free survival (2). FLT3 inhibitors are cytotoxic toward FLT3-ITD-expressing AML cells and produce clinical benefit, with the FLT3/multikinase inhibitor midostaurin approved for use after chemotherapy for newly diagnosed AML with FLT3 mutations (33), and the more potent and specific FLT3 inhibitor gilteritinib approved for single-agent treatment of refractory or relapsed AML with FLT3 mutations (34). Nevertheless, clinical responses to FLT3 inhibitors are generally limited and transient, with rapid development of resistance (3). Combinations with drugs targeting other molecules in FLT3-ITD signaling pathways may improve responses to FLT3 inhibitors (3). Drugs that have been shown to enhance efficacy of FLT3 inhibitors when given in combination have included Pim kinase inhibitors (8–11,35), AKT inhibitors (36,37), the Bcl-2 inhibitor venetoclax (38) and PADs (5,6).
PADs and FLT3 inhibitors have been previously shown to produce synergistic cytotoxicity in FLT3-ITD-expressing AML cells in vitro (5,6). The PAD OP449, which activates PP2A by antagonizing its inhibition by SET, is cytotoxic toward the FLT3-ITD expressing cell line MOLM-14 and results in decreased STAT5, AKT, and S6 ribosomal protein phosphorylation, and OP449 and quizartinib co-treatment synergistically reduces MOLM-14 cell growth (5). Treatment with the PAD FTY720 and its analogue AAL(S) decreases ERK and AKT phosphorylation, induces death of FLT3-ITD-expressing cells and inhibits colony formation, and FTY720 and AAL(S) had synergistic cytotoxicity with FLT3 inhibitors, including in the presence of bone marrow stroma, and synergistic effects on colony formation by FLT3-ITD-expressing cells, but not normal CD34+ bone marrow cells (6). It was hypothesized that since PP2A induces degradation and inactivation of Pim-1 (14), FTY720 or AAL(S) might inhibit Pim-1 as the mechanism for synergy with FLT3 inhibitors (6). Here we demonstrate that PAD and FLT3 inhibitor co-treatment of FLT3-ITD-expressing cells results in marked downregulation of both c-Myc and Pim-1 protein expression through increased proteasomal degradation of both proteins, resulting from AKT inactivation, through a GSK-3β-dependent mechanism. Our work adds to the growing literature on effectiveness of the dual therapeutic strategy of activating phosphatases while inhibiting kinases to enhance efficacy of kinase inhibitors (39), and elucidates for the first time the mechanism of action of this dual therapeutic strategy in FLT3-ITD AML.
Combined PAD and kinase inhibitor treatment also has therapeutic efficacy in other malignant cell types. Co-treatment with OP449 and dovitinib, an orally active multi-receptor TKI, synergistically reduced T-acute lymphoblastic leukemia cell viability, via c-Myc, p-c-Myc (S62), p-ERK1/2, p-AKT and p70S6 kinase downregulation (40). DT-061 co-treatment sensitized KRAS-mutant lung cancer cells to MEK inhibitor via p-AKT and c-Myc downregulation (41). Correspondingly, PP2A inactivation by recurrent mutation in the scaffolding subunit resulted in resistance to MEK inhibitors (42). In another study, combination of PADs and the TKI afatinib was synergistic in TKI-sensitive and -resistant lung adenocarcinoma cells, with TKI re-sensitization of resistant cells thought to be due to AKT and MAP kinase (MAPK) pathway inactivation, as acquired TKI resistance commonly occurs through reactivation of PI3K and MAPK pathways downstream of EGFR (43). Finally, PAD and mTOR inhibitor co-treatment decreased pancreatic ductal adenocarcinoma growth through suppression of AKT/mTOR signaling and post-translational c-Myc protein downregulation (44).
AKT is activated downstream of FLT3-ITD by phosphorylation on S473 and T308 (1). We found that combined PAD and FLT3 inhibitor treatment rapidly inactivates AKT in FLT3-ITD-expressing cells through dephosphorylation of the S473 and T308 residues. The dual PI3K/PDK-1 inhibitor BAG956 was previously shown to be cytotoxic toward cell lines and patient-derived FL3-ITD-expressing AML cells, including Ba/F3-ITD cells resistant to the FLT3 inhibitor midostaurin (PKC412) (36). In a subsequent study using a combinatorial high-throughput drug screen, selective AKT inhibitors, including AT7867, GSK690693 and MK-2206, were found to synergize with FLT3 inhibitors in FLT3-ITD-expressing AML cells in either the absence or presence of stroma (37). Additionally, in two cell lines with induced resistance to sorafenib without acquired FLT3 mutations, and in sorafenib-resistant FLT3-ITD AML patient blasts, FLT3 signaling was inhibited by sorafenib, but the PI3K/mTOR pathway remained activated, and cells were sensitive to the selective PI3K/mTOR inhibitor gedatolisib (45). In other work, STAT5 activation by FLT3-ITD was found to protect cells treated with PI3K/Akt pathway inhibitors from apoptosis by maintaining Mcl-1 expression through the mTORC1/4EBP1/eIF4E pathway (46), potentially supporting efficacy of co-inhibition of FLT3 and the PI3K/Akt pathway in FLT3-ITD-expressing cells.
We further found that AKT inactivation by PAD and FLT3 inhibitor co-treatment resulted in post-translational downregulation of Pim-1 and c-Myc protein expression. We found that post-translational c-Myc downregulation occurred through T58 phosphorylation, in turn caused by GSK-3β activation, and that treatment with GSK-3β inhibitors replicated the effects of PAD and FLT3 inhibitor co-treatment. Additionally, whereas GSK-3β has been found to be a Pim-1 substrate (47), we found here that Pim-1 is a potential GSK-3β substrate, elucidating a potential negative feedback loop, in which Pim-1 inhibits GSK-3β by phosphorylating it on S9 (47), and GSK-3β phosphorylates Pim-1, resulting in its post-translational upregulation, shown here.
In addition to elucidating a novel regulatory pathway, our findings have potential clinical applicability, supporting the use of PAD and FLT3 inhibitor combinations to treat FLT3-ITD AML. PADs are in development (19). The PAD FTY720 (Fingolimod) is approved by the United States Food and Drug Administration for treatment of multiple sclerosis (MS) and is thought to have an acceptable risk to benefit ratio for treatment of AML, given its lack of bone marrow toxicity and low rate of serious toxicities in healthy subjects (48) and in MS patients (49). Novel FTY720 analogues are also in development (50,51).The orally bioavailable phenothiazine derivative DT-061 is in preclinical development (19). Our findings support potential therapeutic efficacy of PAD and FLT3 inhibitor combinations in AML with FLT3-ITD. They also support potential efficacy of AKT inhibitors and GSK-3β inhibitors in this disease subtype.
FLT3 inhibitor resistance mechanisms include FLT3 tyrosine kinase domain mutations, RAS mutations, and Pim overexpression (3). Dual targeting of FLT3-ITD signaling pathways may overcome FLT3 inhibitor resistance (3). Future work will address PAD co-treatment efficacy in preventing and overcoming FLT3 inhibitor resistance in FT3-ITD AML. Notably, a genome-wide CRISPR screen identified GSK-3 as a gene critical for resistance to quizartinib (52). Expression of GSK-3α was markedly reduced in quizartinib-resistant FLT3-ITD cells, and GSK-3 knockout resulted in quizartinib resistance, through reactivation of FLT3-ITD downstream signaling pathways (52). Additionally, GSK-3 inhibits Wnt signaling, and GSK-3 deletion restores it (52). Co-treatment with PAD and FLT3 inhibitor inactivates AKT, which activates GSK-3, and might overcome this mechanism of quizartinib resistance. PAD co-treatment warrants further study as strategy for preventing or reversing FLT3 inhibitor resistance.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Nicole Glynn-Cunningham, M.S., University of Maryland Greenebaum Comprehensive Cancer Center, for tissue procurement, Pedram Azimzadeh, M.S. and Nariman Balenga, Ph.D., Department of Surgery, and Rossana Trotta, Ph.D., Department of Microbiology and Immunology, University of Maryland School of Medicine and Greenebaum Comprehensive Cancer Center, for technical advice.
Financial support: Supported by Merit Review Award BX002184 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Service (M.R.B.), NIH-NCI grants RO1 CA163800 (D.P.), R01 CA181654 (G.N.), R01 CA240993 (G.N.) and P30 CA134274, NIH-NHLBI grant HL144741R01 (G.N.), DOD grant W81XWH-19-BCRP-BTA12 (G.N.), University of Maryland, Baltimore UMMG Cancer Research Grant #CH 649 CRF issued by the State of Maryland Department of Health and Mental Hygiene (DHMH) under the Cigarette Restitution Fund Program, Rogel Cancer Gift Funds (G.N.), the Valanda Wilson Leukemia Research Fund and Mary Ellen’s Angelic Fund for Leukemia Research.
Footnotes
Conflict of Interest Statement: The Icahn School of Medicine at Mount Sinai has filed patents covering composition of matter on the small molecules disclosed herein for the treatment of human cancer and other diseases (International Application Numbers: PCT/US15/19770, PCT/US15/19764; and US Patent: US 9,540,358 B2). Mount Sinai is actively seeking commercial partners for the further development of the technology. G.N. has a financial interest in the commercialization of the technology. D.P. has ownership interest in US Patents 8,633,161; 9,220,706 and 8,318,812.
References
- 1.Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin Cancer Res 2003;9:2140–50. [PubMed] [Google Scholar]
- 2.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: Current status and future directions. Mol Cancer Ther 2017;16;991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim KT, Baird K, Ahn JY, Meltzer P, Lilly M, Levis M, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood 2005;105:1759–67. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res 2014;20:2092–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith AM, Dun MD, Lee EM, Harrison C, Kahl R, Flanagan H, et al. Activation of protein phosphatase 2A in FLT3+ acute myeloid leukemia cells enhances the cytotoxicity of FLT3 tyrosine kinase inhibitors. Oncotarget 2016;7:47465–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008;27:4809–19. [DOI] [PubMed] [Google Scholar]
- 8.Natarajan K, Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase phosphorylates and stabilizes 130 kDa FLT3 and promotes aberrant STAT5 signaling in acute myeloid leukemia with FLT3 internal tandem duplication. PLoS One 2013;8:e74653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green AS, Maciel TT, Hospital M-A, Yin C, Mazed F, Townsend EC, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv 2015;1:e1500221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fathi AT, Arowojolu O, Swinnen I, Sato T, Rajkhowa T, Small D, et al. A potential therapeutic target for FLT3-ITD AML: PIM1 kinase. Leuk Res 2012;36:224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapoor S, Natarajan K, Baldwin PR, Doshi KA, Lapidus RG, Mathias TJ, et al. Concurrent inhibition of Pim and FLT3 kinases enhances apoptosis of FLT3-ITD acute myeloid leukemia cells through increased Mcl-1 proteasomal degradation. Clin Cancer Res 2018;24:234–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol 2013;14:e229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucas CM, Scott LJ, Carmell N, Holcroft AK, Hills RK, Burnett AK, et al. CIP2A- and SETBP1-mediated PP2A inhibition reveals AKT S473 phosphorylation to be a new biomarker in AML. Blood Adv 2018;2:964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Losman JA, Chen XP, Vuong BQ, Fay S, Rothman PB. Protein phosphatase 2A regulates the stability of Pim protein kinases. J Biol Chem 2003;278:4800–5. [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Arnold HK, Lilly MB, Sears RC, Kraft AS. Negative regulation of Pim-1 protein kinase levels by the B56beta subunit of PP2A. Oncogene 2007;26: 5145–53. [DOI] [PubMed] [Google Scholar]
- 16.Kim KT, Baird K, Davis S, Piloto O, Levis M, Li L, et al. Constitutive Fms-like tyrosine kinase 3 activation results in specific changes in gene expression in myeloid leukaemic cells. Br J Haematol 2007;138:603–15. [DOI] [PubMed] [Google Scholar]
- 17.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-Myc and negatively regulates c-Myc accumulation. Mol Cell Biol 2006;26:2832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signaling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004;6:308–18. [DOI] [PubMed] [Google Scholar]
- 19.O’Connor CM, Perl A, Leonard D, Sangodkar J, Narla G. Therapeutic targeting of PP2A. Int J Biochem Cell Biol 2018;96:182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doshi KA, Trotta R, Natarajan K, Rassool FV, Tron AE, Huszar D, et al. Pim kinase inhibition sensitizes FLT3-ITD acute myeloid leukemia cells to topoisomerase 2 inhibitors through increased DNA damage and oxidative stress. Oncotarget 2016;7:48280–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricci MS, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, et al. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol 2004;24:8541–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007;129:1065–79. [DOI] [PubMed] [Google Scholar]
- 23.Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol 2013;31:3681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, et al. Pre-clinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017;129:257–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujita T, Hirose R, Yoneta M, Sasaki S, Inoue K, Kiuchi M, et al. Potent immunosuppressants, 2-alkyl-2-aminopropane-1,3-diols. J Med Chem 1996;39:4451–9. [DOI] [PubMed] [Google Scholar]
- 26.Sangodkar J, Perl A, Tohmé R, Kiselar J, Kastrinsky DB, Zaware N, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 2017;127:2081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McClinch K, Avelar RA, Callejas D, Izadmehr S, Wiredja D, Perl A, et al. Small-molecule activators of protein phosphatase 2A for the treatment of castration-resistant prostate cancer. Cancer Res 2018;78:2065–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrington C, Yuan E, Mazhar S, Izadmehr S, Hurst L, Allen-Petersen B, et al. Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J Biol Chem 2020;295:757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonard D, Huang W, Izadmehr S, O’Connor CM, Wiredja DD, Wang Z, et al. Selective PP2A enhancement through biased heterotrimer stabilization. Cell 2020;181:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lüscher B, Eisenman RN. c-myc and c-myb protein degradation: effect of metabolic inhibitors and heat shock. Mol Cell Biol 1988;8:2504–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quan Y, Wang N, Chen Q, Xu J, Cheng W, Di M, et al. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 2015;6:26494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 1998;273:19929–32. [DOI] [PubMed] [Google Scholar]
- 33.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med 2019;381:1728–40. [DOI] [PubMed] [Google Scholar]
- 35.Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res 2006;66:3828–35. [DOI] [PubMed] [Google Scholar]
- 36.Weisberg E, Banerji L, Wright RD, Barrett R, Ray A, Moreno D, et al. Potentiation of antileukemic therapies by the dual PI3K/PDK-1 inhibitor, BAG956: effects on BCR-ABL- and mutant FLT3-expressing cells. Blood 2008;111:3723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weisberg E, Liu Q, Zhang X, Nelson E, Sattler M, Liu F, et al. Selective Akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PLoS One 2013;8:e56473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma J, Zhao S, Qiao X, Knight T, Edwards H, Polin L, et al. Inhibition of Bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin Cancer Res 2019;25:6815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westermarck J. Targeted therapies don’t work for a reason; the neglected tumor suppressor phosphatase PP2A strikes back. FEBS J 2018;285:4139–45. [DOI] [PubMed] [Google Scholar]
- 40.Richard NP, Pippa R, Cleary MM, Puri A, Tibbitts D, Mahmood S, et al. Combined targeting of SET and tyrosine kinases provides an effective therapeutic approach in human T-cell acute lymphoblastic leukemia. Oncotarget 2016;7:84214–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kauko O, O’Connor CM, Kulesskiy E, Sangodkar J, Aakula A, Izadmehr S, et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci Transl Med 2018;10:eaaq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Connor CM, Leonard D, Wiredja D, Avelar RA, Wang Z, Schlatzer D, et al. Inactivation of PP2A by a recurrent mutation drives resistance to MEK inhibitors. Oncogene 2020;39:703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tohmé R, Izadmehr S, Gandhe S, Tabaro G, Vallabhaneni S, Thomas A, et al. Direct activation of PP2A for the treatment of tyrosine kinase inhibitor-resistant lung adenocarcinoma. JCI Insight 2019;4:e125693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen-Petersen BL, Risom T, Feng Z, Wang Z, Jenny ZP, Thoma MC, et al. Activation of PP2A and inhibition of mTOR synergistically reduce MYC signaling and decrease tumor growth in pancreatic ductal adenocarcinoma. Cancer Res 2019;79:209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, Kabir NN, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016;35:5119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nogami A, Oshikawa G, Okada K, Fukutake S, Umezawa Y, Nagao T, et al. FLT3-ITD confers resistance to the PI3K/Akt pathway inhibitors by protecting the mTOR/4EBP1/Mcl-1 pathway through STAT5 activation in acute myeloid leukemia. Oncotarget 2015;6:9189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santio NM, Salmela M, Arola H, Eerola SK, Heino J, Rainio EM, et al. The PIM1 kinase promotes prostate cancer cell migration and adhesion via multiple signalling pathways. Exp Cell Res 2016;342:113–24. [DOI] [PubMed] [Google Scholar]
- 48.Kovarik JM, Schmouder R, Barilla D, Riviere GJ, Wang Y, Hunt T. Multiple-dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol 2004;44:532–7. [DOI] [PubMed] [Google Scholar]
- 49.Enjeti AK, D’Crus A, Melville K, Verrills NM, Rowlings P. A systematic evaluation of the safety and toxicity of fingolimod for its potential use in the treatment of acute myeloid leukaemia. Anticancer Drugs 2016;27:560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vicente C, Arriazu E, Martínez-Balsalobre E, Peris I, Marcotegui N, García-Ramírez P, et al. A novel FTY720 analogue targets SET-PP2A interaction and inhibits growth of acute myeloid leukemia cells without inducing cardiac toxicity. Cancer Lett 2020;468:1–13. [DOI] [PubMed] [Google Scholar]
- 51.Toop HD, Dun MD, Ross BK, Flanagan HM, Verrills NM, Morris JC. Development of novel PP2A activators for use in the treatment of acute myeloid leukaemia. Org Biomol Chem 2016;14:4605–16. [DOI] [PubMed] [Google Scholar]
- 52.Hou P, Wu C, Wang Y, Qi R, Bhavanasi D, Zuo Z, et al. A genome-wide CRISPR screen identifies genes critical for resistance to FLT3 inhibitor AC220. Cancer Res 2017;77:4402–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.