

Abstract
Understanding the antioxidant activity of flavonoids is important to investigate their biological activities as well as to design novel molecules with low toxicity and high activity. Aromaticity is a chemical property found in cyclic structures that plays an important role in their stability and reactivity, and its investigation can help us to understand the antioxidant activity of some heterocyclic compounds. In the present study, we applied the density functional theory (DFT) to investigate the properties of seven flavonoid structures with well-reported antioxidant activity: flavan, anthocyanidin, flavanone, flavonol, isoflavone, flavone, and flavan-3-ol. Conformational, structural, magnetic, and electronic analyses were performed using nuclear magnetic resonance, ionization potentials, electron affinity, bond dissociation energy, proton affinity, frontier molecular orbitals (highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO)), and aromaticity through nucleus-independent chemical shifts to analyze these seven flavonoid structures. We revised the influence of hydroxyl groups on the properties of flavonoids and also investigated the influence of the aromaticity of these seven flavonoids on the antioxidant activity.
1. Introduction
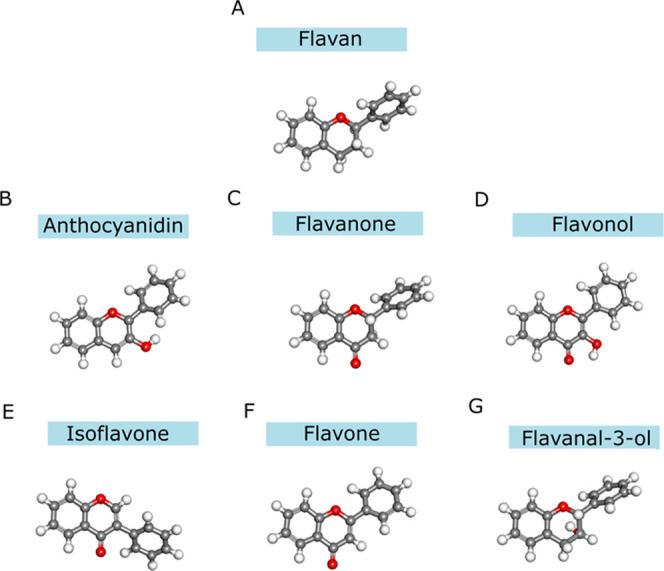
Flavonoids are a class of natural compounds characterized by a backbone containing 15 carbon atoms, two phenyl rings A and B linked by heterocyclic (pyran) ring C (Figure 1).1 Benzo-γ-pyran (chromone) refers to the heterocycle formed by the phenyl ring (A) and the pyran ring (C).2 Flavonoids are divided into six subclasses: anthocyanidins, flavanones, flavonols, isoflavones, flavone, and flavan-3-ol.3−5
Figure 1.

Molecular structures of the investigated flavonoids showing the atoms and ring labels, as well as the studied dihedral angles. (A) Flavan, (B) anthocyanidin, (C) flavanone, (D) flavonol, (E) isoflavone, (F) flavone, and (G) flavan-3-ol.
Flavonoids possess a wide range of biological activities, such as anti-inflammatory,6 antibacterial,7 anticancer,8,9 and antiviral.10,11 Different computational studies have investigated the structural properties of these molecules related to their biological activities, especially their antioxidant activity; however, the influence of the aromaticity for their reactivity remains unknown.12−19 The antioxidant property is related to the ability to neutralize free radicals that are involved with the oxidative stress in the cell.20,21 Free radicals, such as reactive oxygen species, cause serious damages to cell structures, such as lipids, proteins, and nucleic acids, and they are involved in food deterioration and the development of several human diseases.22 Flavonoids are phenolic compounds with proven antioxidant activity that prevent the action of free radicals, thus protecting the cell against their harmful effects.
Investigating the antioxidant activity through electronic properties is important to design novel antioxidant molecules with low toxicity and high activity.23,24 Three major mechanisms have been proposed to explain the antioxidant activity of compounds.25−27 It has been proposed that flavonoids (FlavOH) can neutralize the free radical by donating a hydrogen atom (eq 1).28−30
| 1 |
Another mechanism consists of the electron transfer, where the antioxidant can donate an electron to the free radical (eqs 2 and 3).
| 2 |
| 3 |
The second mechanism (2) is known as hydrogen atom transfer (HAT), which occurs in (3) single-electron transfer followed by proton transfer (SET-PT). The third mechanism is known as sequential proton loss electron transfer (SPLET), which consists of the following mechanism (eqs 4 and5)
| 4 |
| 5 |
where occurs the transference of a proton from the antioxidant (4) followed by electron donation. The parameters applied to evaluate the antioxidant activity in the mechanisms of HAT, SET-PT, and SPLET include the calculation of molecular properties, such as bond dissociation enthalpy (BDE), ionization potential (IP), and proton affinity (PA), respectively.31 Besides these mechanisms, the free radicals that emerge in both reactions (FlavO* and FlavOH*+) must be neutralized to avoid oxidative stress.28
The aromaticity of cyclic structures is a key parameter for their reactivity.32,33 Aromatic rings show diamagnetism and increase the stability of the structures. In contrast, antiaromatic rings show paramagnetism and less stability. The aromaticity can be addressed through chemical shielding in 1H nuclear magnetic resonance (NMR) spectra.34,35 Moreover, the antioxidant activity of compounds can be mapped by the frontier molecular orbitals (highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO)) and ionization potential.36
In the present study, we performed a systematic analysis of seven flavonoid structures—flavan, anthocyanidin, flavanone, flavonol, isoflavone, flavone, and flavan-3-ol—using structural, conformational, and electronic analyses of protonated (neutral) and deprotonated forms. We compared the conformational properties of these structures with previous theoretical studies, analyzing the lower energy conformations, especially the electron density of the benzo-γ-pyran ring and the influence of the hydroxyl and ketone groups and aromatic rings on their antioxidant activity.
2. Results and Discussion
Different computational approaches have been used to investigate flavonoid structures.3,31,37,38 Recently, Zheng et al. performed a comparative study focusing on the interactions of the hydrogens in the solvated systems of 5,7-dihydroxy-2-(3,4-dihydroxyphenyl)-chrome-4-one (luteolin) using DFT (B3LYP method) and concluded that luteolin presents a nonplanar conformation.37 Similarly, Matteini et al. performed a theoretical study with the flavonoid rutin, using molecular mechanics (MM) and semiempirical calculations using Austin Model 1 (AM1), and they found 16 stable conformations with overall minimum energy due to the formation of intermolecular H-bond interactions between the hydroxyl group and C12 attached to ring B.38 Herein, we optimized the geometries of the seven flavonoid structures using the CAM-B3LYP/def2TZV calculations (Figure 2). The atoms were numbered according to the flavonoid scaffold (see Figure 1).
Figure 2.
Optimized conformations of the investigated flavonoid structures. (A) Flavan, (B) anthocyanidin, (C) flavanone, (D) flavonol, (E) isoflavone, (F) flavone, and (G) flavan-3-ol.
The dipole moments (Debye), interatomic distances, and dihedrals angles for the neutrals and deprotonated structures optimized with CAM-B3LYP/def2TZV are shown in Table 1. The largest dipole moments were found for the flavonol and flavone structures, while anthocyanidin showed the smallest dipole moment. Table 1 shows the dihedral angles τ: O1–C2–C1′–C6′, ω: C9–O1–C2–C3, and ϕ: C10–C4–C3–C2.
Table 1. Dipole Moments (Debye), Interatomic Distances between the Carbons (Å), and Torsion Angles (Degrees) of the Optimized Structures of Flavonoids at the CAM-B3LYP/def2TZV Level.
| dipole | C1′–C2′ | C1′–C6′ | C1′–C2 | C2–C3 | O1–HC6′ | τ | ω | ϕ | |
|---|---|---|---|---|---|---|---|---|---|
| luteolin37 | 1.41 | 1.40 | 1.47 | 1.35 | –160.7 | –0.05 | –1.50 | ||
| experimental | 1.38 | 1.42 | 1.46 | 1.36c | 19.00 | 55.30c | 36.30c | ||
| 1.40b | 1.40b | 1.47b | 1.36b | –164.6c | |||||
| –178.4d | 0.00d | 0.00d | |||||||
| flavan | 1.83 | 1.40 | 1.40 | 1.51 | 1.53 | 2.46 | –147.17 | –44.03 | –47.84 |
| anthocyanidin | 3.44 | 1.41 | 1.41 | 1.45 | 1.39 | 2.46 | 171.21 | 0.21 | 1.36 |
| anthocyanidind | 5.44 | –178.79 | –0.01 | –0.57 | |||||
| flavanone | 2.61 | 1.40 | 1.40 | 1.50 | 1.53 | 2.50 | 144.44 | 51.21 | 33.11 |
| flavonol | 3.58 | 1.40 | 1.40 | 1.48 | 1.34 | 2.32 | 169.08 | 0.11 | 1.56 |
| flavonola | 8.52 | 179.00 | –0.19 | –0.42 | |||||
| isoflavone | 3.03 | 1.40 | 1.40 | 1.34 | –177.93 | –0.23 | –2.46 | ||
| flavone | 4.42 | 1.40 | 1.40 | 1.47 | 1.35 | 2.40 | 157.75 | 0.28 | 1.6 |
| flavan-3-ol | 1.92 | 1.39 | 1.39 | 1.50 | 1.53 | 2.50 | 147.13 | 52.43 | 46.12 |
| flavan-3-ola | 116.44 | 33.96 | 57.11 |
The interatomic distances obtained in the present study were compared with the luteolin structure calculated at the B3LYP level.37 We noted that there is no significant difference between the calculated interatomic distances of the investigated structures of flavonoids (Table 1) with those previously investigated in the literature.37
Lau et al., using B3LYP/6-31G(d) calculations, suggested that the planarities of 5,7-dihydroxyflavone and 7,8-dihydroxyflavone were associated with the induced dipole interaction between O1 and the hydrogen of C6 (O1–HC6) with variations between 2.32 and 2.46 Å.43 Similarly, Machado et al., investigated flavonoids with a hydroxyl group in rings A and C using B3LYP/6-31G(d,p), and they found the values of the torsion angle τ between 0 and 20° and the distance values of O1–HC′ between 2.31 and 2.39 Å.44 Aparicio investigated 16 different flavonoid structures using B3LYP/6-311++G(d,p) and associated the planarity of the molecules with the hydroxyl group bonded to C3 that forms a hydrogen bond with C6′ in ring B.3 Our analyses showed that the protonated structures of anthocyanidin, flavonol, and flavone have a planar conformation with distances of 2.31, 2.31, and 2.40 Å between the hydrogen bonded to C6′ and O1, respectively. The distance between O3H and C4 is 2.54 Å in anthocyanidin with a torsion angle τ of 171.21°. Flavan-3-ol does show a great conformational change in its structure with a distance of 2.00 Å between O3H and C3 and a torsion angle τ of 147.13°. Previous theoretical and experimental studies obtained a torsion angle τ close to 180°.38,39 However, the CAM-B3LYP calculations showed a planar conformation only for the deprotonated anthocyanidin and flavonol; the deprotonated flavan-3-ol showed no planarity between rings B and C. Our computational analysis indicated that the anthocyanidin structure did not show an interaction between hydroxyl and the hydrogen bond to C6′ of ring B as confirmed previously.3,44
The protonated forms of flavonol and anthocyanidin showed planar conformation with a C6′–O1 distance of 2.30 Å and a O1–C6′–H angle of 60°. In contrast, flavan and flavan-3-ol showed conformational changes with atomic distances of C6′–O1 equal to 2.46 and 2.50 Å, respectively, and angles of O1–C6′–H equal to 62.52 and 61.45°, respectively. These results indicate that the stabilization of these molecules is not due to the formation of a hydrogen bond between C6′ and O1.
The deprotonation of anthocyanidin and flavonol did not change the planarity of their structures, which suggests that the hydroxyl group has no influence on the stabilization of planar structures. Machado et al.44 indicated an interaction between the oxygen of O3H and the ketone group of ring C that stabilizes the planar conformation by the electronic redistribution through the molecule. Comparing the results of anthocyanidin, flavonol, and flavan-3-ol (Figure 2), we found an H-bond interaction between the hydroxyl group with ring C. Similar to Machado et al., we also identified an interaction between the hydroxyl group and the oxygen of the ketone group.44Figure 3 shows the relationships between the energy and conformational variations related to the torsion angles τ of the protonated molecules. All conformational analyzes were performed using the CAM-B3LYP/def2TZV protocol.
Figure 3.

Conformational analysis of the torsion angle τ of the investigated flavonoids (in degrees). (A) Flavan, (B) flavanone, (C) isoflavone, and (D) flavone.
Flavan and flavanone showed almost the same pattern, with the maximum energy found for torsion angles τ between 80 and 140°, while isoflavone and flavone have the maximum energy at almost 150 and 130°. Table 2 shows the bond distance values obtained using the CAM-B3LYP/def2TZV protocol of the deprotonated flavonoid molecules.
Table 2. Interatomic Distances (Å) and Angles (Degrees) of the Hydrogen Bond were Obtained from the Conformational Analysis of the Flavonoid Structures.
| anthocyanidin | flavonol | flavan-3-ol | |
|---|---|---|---|
| O3H···O | 2.06 | ||
| O2–H–O3 | 115 | ||
| HO···HC4 | 2.66 | 2.58 | |
| HO···H (ring B) | 2.09 | 2.15 | 2.99 |
| C4–O2–H | 86.57 | 97.69 |
Cappelli et al. performed a conformational analysis of flavonoids analyzing the hydroxyl groups of all rings using molecular mechanics and DFT in the gas phase and the methanol solution.45 The simulations showed nine different conformations for the investigated structures. In contrast, Machado et al.44 did not find interactions between the hydroxyl group and the carbons of ring C in the investigated conformations. However, it is important to highlight that detailed analyses of the electronic properties of the flavonoids will be necessary to corroborate with the aforementioned suggestions.44,45 We noted that when the hydroxyl group interacts with the oxygen of the ketone group, it leads to a decrease in the overall energy of flavonol with a dihedral angle τ equal to 169.08°. Additionally, flavonol and anthocyanidin tend to be planar (Table 2).
Figure 4 shows the conformational analysis of anthocyanidin (Figure 4A), flavonol (Figure 4B), and flavan-3-ol (Figure 4C). We noted a significant reduction in the energy values of the flavonol conformation due to the alterations of O3H orientation. The structures of the lowest-energy conformations of anthocyanidin, flavonol, and flavan-3-ol are related to the positions of the hydroxyl group interacting with the hydrogen of C4, as well as the ketone group. The rotational barrier of flavanol is the highest compared to anthocyanidin and flavan-3-ol, which corroborates the previous findings.3 Additionally, these structures form a hydrogen bond interaction between the hydroxyl groups of C3 (ring C) and O4 (ring C), as well as the hydroxyl group of C3 (ring C) and the hydrogen of C6′ (ring B).
Figure 4.

Conformational analysis of flavonoid structures on torsion angle τ (in degrees) and their respective high-energy and low-energy conformational states. (A, B) Flavonol, (C, D) flavan-3-ol, and (E, F) anthocyanidin.
It is expected that neutral structures of anthocyanidin, flavonol, and flavone present high aromaticity when compared with the other analyzed flavonoids. Therefore, the aromaticity of the structures can contribute to understand the antioxidant mechanism. The number and position of hydroxyl groups have been widely studied regarding the antioxidant activity.14,46−51 For example, for luteolin, it was demonstrated that by removing the O3H group in ring C, the antioxidant activity consequently decreases.46 Moreover, the importance of ring C unsaturation was pointed out, which allows electron delocalization for stabilization of the aryloxyl radical.46 We have found that isotropic NICS(1) values of anthocyanidin ring C are larger than the values of other studied flavonoid structures, suggesting that the ketone group decreases the aromaticity, while the hydroxyl group increases the aromaticity jointly to the hydrogen bond, as expected from the substituent effects. Besides, high dipole moments were found for the deprotonated molecules; however, in neutral molecules, the highest values were found in flavone, flavonol, and anthocyanidin, which are also the planar structures (Table 1). Additionally, flavonol and anthocyanidin tend to be planar (Table 2). In contrast to flavan-3-ol, the molecular planarity of anthocyanidin and flavanol is characterized by the presence of the O3H group through a weak hydrogen bond with HC6′, which is in agreement with the literature.3
Figure 5 shows the NICS plots (ppm per r distance in Å), which are representative of the isotropic, anisotropic, and ZZ (NICSzz) fields. Negative values of absolute shielding computed at the center of the molecule indicate the presence of induced diatropic current (aromaticity), whereas positive values indicate paratropic currents (antiaromaticity).52 The r distances were obtained from the neutral structures of the molecules: flavan, anthocyanidin, flavanone, flavonol, isoflavone, flavone, and flavan-3-ol. Our NICS results showed that the ring C of the analyzed flavonoids exhibited a significant reduction in their aromaticity.
Figure 5.

NICS values as a function of the distance r of the neutral and isotropic NICS(1) displayed numerically for ring C. The isotropic NICS(1) value (in ppm) was shown for all analyzed flavonoids.
The NICS values demonstrated that only anthocyanidin has an aromatic ring C (Figure 5). The formation of the interaction between the hydroxyl group and C4 could improve the planarity of the structure and also increase the π electrons in ring C. However, it is important to highlight that we should not neglect the influence of ketone oxygen for aromaticity due to the presence of the π electrons associated with this atom. The antioxidant activity of flavonoids was shown to be dependent on the stabilization of the O3H group through unsaturation of ring C.14,46−51 Despite the presence of planarity in the molecules, the ZZ term (NICSzz) is well aligned with the anisotropic (NICSanisotropic) term of ring B (Figure 5) and the aromaticity only occurs in the ring C of anthocyanidin (NICSzz and NICSisotropic terms). This result corroborates the high antioxidant activity previously reported for an anthocyanidin analogue.46 The chemical shifts (δ) extracted from the NMR of carbon (13C NMR) and oxygen (17O NMR) are shown in Tables 3 and 4. Similar results were found for the calculated 13C NMR concerning previous experimental studies.49,53,54
Table 3. Chemical Shifts (δ) in ppm for 13C Obtained from CAM-B3LYP/def2TZV and HSEh1PBE/cc-pVDZ Calculations Using Tetramethylsilane (TMS) as a Reference.
| CAM-B3LYP/def2TZV |
HSEh1PBE/cc-pVDZ |
|||||||
|---|---|---|---|---|---|---|---|---|
| C3 | C4 | C8 | C10 | C3 | C4 | C8 | C10 | |
| Theoretical | ||||||||
| flavan | 37.2 | 29.0 | 127.6 | 132.2 | 33.7 | 27.4 | 114.7 | 120.5 |
| anthocyanidin | 156.2 | 143.6 | 127.8 | 130.3 | 146.3 | 132.1 | 116.0 | 121.9 |
| flavanone | 53.0 | 213.9 | 127.6 | 131.6 | 47.1 | 194.0 | 115.3 | 120.2 |
| flavanol | 148.9 | 190.1 | 126.8 | 130.5 | 139.9 | 174.7 | 115.0 | 121.1 |
| isoflavone | 137.8 | 193.2 | 126.8 | 134.9 | 128.8 | 176.5 | 114.7 | 125.3 |
| flavone | 117.7 | 194.7 | 126.5 | 134.8 | 108.4 | 177.6 | 194.7 | 124.7 |
| flavan-3-ol | 140.6 | 37.3 | 128.2 | 132.5 | 125.3 | 34.6 | 115.3 | 121.6 |
| Experimental | ||||||||
| anthocyanidina | 143.9 | 130.9 | 94.0 | 111.1 | ||||
| flavonolb | 136.2 | 176.4 | 93.8 | 98.7 | ||||
| flavonolc | 135.5 | 175.7 | 93.4 | 102.9 | ||||
| flavonec | 103.3 | 182.2 | 94.2 | 104.2 | ||||
Table 4. Chemical Shifts (δ) in ppm of Oxygen (17O NMR) Obtained from CAM-B3LYP/def2TZV and HSEh1PBE/cc-pVDZ (in Parenthesis) Calculations.
| compound/atoms | O1 | O3 | O4 |
|---|---|---|---|
| flavan | 197.3 (209.3) | ||
| anthocyanidin | 20.5 (59.1) | 215.4 (224.9) | |
| anthocyanidina | 14.5 (54.1) | –41.6 (−28.1) | |
| flavanone | 187.6 (201.7) | –348.7 (−274.6) | |
| flavonol | 133.6 (153.4) | 247.9 (252.8) | –160.2 (−112.8) |
| flavonola | 136.1 (149.6) | –20.27 (−15.0) | –253.4 (−190.3) |
| isoflavone | 118.9 (132.2) | –249.3 (−181.9) | |
| flavone | 120.6 (137.9) | –258.3 (−202.7) | |
| flavan-3-ol | 181.3 (189.2) | 156.8 (181.0) | |
| flavan-3-ola | 178.9 (189.9) | 223.7 (207.4) |
Deprotonated structures.
The analysis of 17O NMR using CAM-B3LYP/def2TZV and HSEh1PBE/cc-pVDZ showed that: (1) the results with CAM-B3LYP/def2TZV are underestimated concerning the results obtained with HSEh1PBE/cc-pVDZ and (2) in the neutral flavonol structure, the H-bond interaction formed by O3 is stronger than that by O1, and O1 is stronger than O4. Similarly, in the structure of anthocyanidin, the H-bond interaction formed by O3 is stronger than O1.
The 17O NMR analysis of flavonol and anthocyanidin oxygen indicated the formation of an interaction between O3H and the oxygen of ketone and C4 of ring C, respectively, thus stabilizing the molecule in a planar conformation. Moreover, the change in the shielding (ppm) of the oxygen atoms of flavonol due to deprotonation occurs along with an increase of 80.09° in the torsion angle τ. Aromatic rings tend to planarity, but a similar conformational change does not occur with ring C of flavan-3-ol that differently from flavonol does not have ketone oxygen. The change in the shielding (ppm) of the oxygen due to deprotonation occurs in anthocyanidin similar to flavonol, but without a change in aromaticity. The energy density at the critical point divided by the electron density distribution (H(rc)/ρc) was obtained from AIM calculations, which suggest for the O3H···HC6′ interaction of anthocyanidin and flavonol structures, a hydrogen bond with a partial electrostatic character, showing 0.15 and 0.18 Hartree/electron, respectively.
To better understand the relevance of the structural differences of the investigated flavonoids for their antioxidant activity, we also analyzed the energy of the HOMO/LUMO molecular orbitals using CAM-B3LYP/def2TZV and HSEh1PBE/cc-pvDZ protocols. Both quantum chemistry calculation protocols showed the same profile. Therefore, Figure 6 shows the HOMO and LUMO molecular orbitals of the investigated flavonoid structures, calculated at the CAM-B3LYP/def2TZV level. The HOMO topology of a phenolic compound may indicate its active site of the free-radical elimination due to hydrogen abstraction after electron transfer.55
Figure 6.

Frontier molecular orbitals obtained using CAM-B3LYP/def2TZV calculations. Negative regions are represented in red color and positive in green. (A) Flavan, (B) anthocyanidin, (C) flavanone, (D) flavonol, (E) isoflavone, (F) flavone, and (G) flavan-3-ol.
The analysis of the natural bond orbital (NBO) of ring C showed π bonds (1) in C9–C10 of flavan; (2) in C3–C4 and C9–C10 of anthocyanidin; (3) in O2–C4 and C9–C10 of flavanone; (4) in O3–C4, C9–C10, and C2–C3 of flavonol; (5) in C2–C3 and C9–C10 of isoflavone; (6) in O2–C4, C2–C3, and C9–C10 of flavone; and (7) in C9–C10 of flavan-3-ol. Mendes et al. studied the antioxidant activities of flavonoids using B3LYP/6-31G(d) calculations and they found ΔEL–H ranging from 1.72 to 5.88 eV.36 Payán-Gómez et al. investigated rutin using DFT with M05-2X/6-31+G(d,p) calculations, and they showed a HOMO–LUMO transition of 4.28 eV, which is in the region of ultraviolet electronic transition.56 However, it is noteworthy that a transition of 3.88 eV was also previously found for this molecule.56 According to Payán-Gómez et al., the HOMO–LUMO transitions were characterized by charge transfers along with the entire flavonoid structure.56Table 5 shows the energies (eV) of the HOMO–LUMO molecular orbitals, the energy values of ΔEL–H, IP (kcal·mol–1), and EA (kcal·mol–1); the charges of ring B and benzo-γ-pyran were obtained using CAM-B3LYP/def2TZV and HSEh1PBE/cc-pVDZ calculations.
Table 5. ΔEL–H (ELUMO – EHOMO) Values (eV), IP (kcal·mol–1), EA (kcal·mol–1), and NBO Atomic Charges of the Benzo-γ-pyran (Chromone) and Ring B for the Neutral and Deprotonated (d) Structures of the Analyzed Flavonoids.
| flavan | anthocyanidin | anthocyanidind | flavanone | flavonol | flavonold | isoflavone | flavone | flavan-3-ol | flavan-3-old | |
|---|---|---|---|---|---|---|---|---|---|---|
| CAM-B3LYP/def2TZV | ||||||||||
| HOMO | –7.55 | –11.66 | –6.68 | –8.14 | –7.64 | –2.30 | –7.88 | –8.13 | –7.90 | –1.94 |
| ΔEL–H | 8.46 | 5.50 | 4.47 | 7.35 | 6.34 | 4.84 | 7.04 | 7.09 | 8.62 | 6.19 |
| IP | 174.00 | 268.94 | 153.94 | 188.19 | 176.18 | 52.95 | 181.71 | 187.43 | 182.13 | 44.68 |
| EA | 21.27 | 142.20 | 50.73 | 18.10 | 30.11 | –58.97 | 19.28 | 24.08 | –16.77 | –98.68 |
| NBOchromone | 0.36 | 0.75 | –0.83 | –0.03 | –0.06 | –0,86 | –0.04 | –0.05 | –0.02 | –0.99 |
| NBOring B | 0.01 | 0.25 | –0.17 | 0.03 | 0.06 | –0,14 | 0.04 | 0.05 | 0.02 | –0.05 |
| HSEh1PBE/cc-pVTZ | ||||||||||
| HOMO | –5.81 | –10.12 | –5.14 | –6.40 | –5.87 | –0.64 | –6.20 | –8.39 | –6.08 | 0.17 |
| ΔEL–H | 5.32 | 2.83 | 1.87 | 4.40 | 3.50 | 2.20 | 4.19 | 6.17 | 5.51 | 2.8 |
| IP | 134.12 | 233.42 | 118.41 | 147.27 | 135.35 | 17.79 | 142.83 | 147.43 | 140.17 | –4.02 |
| EA | 11.30 | 168.03 | 74.92 | 45.79 | 54.56 | –35.90 | 46.14 | 51.29 | 13.07 | –68.57 |
| NBOchromone | 0.048 | 0.72 | –0.07 | 0.03 | –0.02 | –0.75 | –0.14 | –0.04 | –0.06 | –0.74 |
| NBOring B | –0.05 | 0.28 | 0.15 | –0.02 | 0.02 | –0.25 | 0.00 | 0.04 | –0.02 | –0.34 |
The energies of the frontier molecular orbitals (HOMO–LUMO) are associated with electron donation and acceptance through ionization potential and electron affinity, respectively.57,58 Although anthocyanidin has a +1 charge, due to the pyrylium ion, the EA value showed to be high when compared with other molecules, showing the known values previously reported for this molecule. Anthocyanidin also showed aromaticity in ring C, but the low values of BDE and PA suggest that the high antioxidant activity is explained by the hydroxyl group. This result is consistent with the sequential proton loss electron transfer mechanism (SPLET).25−27 It is important to note that the values with HSEh1PBE/cc-pVTZ are underestimated concerning the CAM-B3LYP/def2TZV protocol.59
According to Matteini et al., in the rutin structure, the HOMO–LUMO transitions are characterized by charge transfers in ring B showing a ΔEL–H value of 7.78 eV.38 Ninh The et al. studied isoflavones using B3LYP/6-311(d) and found a ΔEL–H between 4 and 5 eV.60 We found a similar ΔEL–H value for isoflavone using the HSEh1PBE/cc-pVTZ protocol.51 Regarding the ΔEL–H values obtained using the CAM-B3LYP/def2TZV calculations, the investigated flavonoid structures showed values consistent with those obtained in previous studies.38Table 4 shows the atomic charges of benzo-γ-pyran and ring B for the investigated structures. Using the CAM-B3LYP/def2TZV calculations, we demonstrated that the charge is shifted from benzo-γ-pyran to ring B (Table 4). The flavan, protonated anthocyanidin, and flavanone structures are consistent with the results obtained using the HSEh1PBE/cc-pVTZ calculations.3 The long-range functional CAM-B3LYP/def2TZV showed different results for the charges when compared with the functional hybrid HSEh1PBE/cc-pVDZ.61 We noted that the results obtained with the CAM-B3LYP/def2TZV protocol are more accurate due to the additional polarization functions. Table 5 shows bond dissociation energy values and proton affinity calculated at 298.15 K and 1 atm. The calculations were carried out using CAM-B3LYP/def2TZV.
The BDE of O–H bonds is a key concept to analyze the antioxidant activity, while PA is a conceptual parameter to evaluate the deprotonation. It is expected that weaker O–H bonds favor the reaction to inactivate the free radical, as this low value of BDE favors the transfer of hydrogen from the O3H group to the free radical.57,58,62,63
| 6 |
| 7 |
Table 6 presents the BDE and PA values calculated at the CAM-B3LYP/def2TZV level. Our BDE values showed to be larger than the values from previous DFT studies of about 85.2 kcal·mol–1 for a flavonol analogue, and 80.9 kcal·mol–1 for quercetin.64,65 The present study investigated flavonoid structures with fewer hydroxyls than these cited studies, and it could explain the differences between the obtained BDE values. Anthocyanidin has the lowest BDE value, which is in agreement with the highest antioxidant activity found for this molecule when compared with the flavonol and flavan-3-ol.46
Table 6. Bond Dissociation Energy Values (BDE, kcal·mol–1) and Proton Affinity (PA, kcal·mol–1) of Anthocyanidin, Flavonol, and Flavan-3-ol.
| anthocyanidin | flavonol | flavan-3-ol | |
|---|---|---|---|
| BDE | 63.32 | 91.36 | 110.55 |
| PA | 239.16 | 337.77 | 394.32 |
It is well known that SPLET is determined by proton affinity.46,66 Our PA values of the flavonoid structures corroborate those investigated by Ninh The et al.51 The low BDE and PA values of anthocyanidin demonstrate that its structure possesses a strong antioxidant activity that could be explained by the dissociation of the hydrogen bond.
The values presented in Table 7 are smaller (absolute value) than those obtained at B3LYP/6-311++G**.3 The values of hydrogens of O3H and the oxygen of ketone (OC4) of the flavonol structure showed differences due to the bond formation. Similar results were obtained for the oxygen of the O3H and hydrogen (HC6′) of the flavonol structure. The OC4 and O3H′ hydrogen charges also suggest the formation of a hydrogen bond in the flavonol structure.
Table 7. Calculated Atomic Charge (NBO) of Oxygen (O3H), Hydrogen (HC6′), Hydrogen (O3H), and Carbon-Bound Oxygen (OC4) of the Flavonoids Using the CAM-B3LYP/def2TZV Calculations.
| atoms | flavan | anthocyanidin | flavanone | flavonol | isoflavone | flavone | flavan-3-ol |
|---|---|---|---|---|---|---|---|
| O3H(O) | –0.633 | –0.655 | –0.696 | ||||
| HC6′ | 0.261 | 0.213 | 0.256 | 0.234 | 0.221 | 0.209 | |
| O3H(H) | 0.503 | 0.505 | 0.463 | ||||
| OC4 | –0.512 | –0.583 | –0.541 | –0.542 |
3. Conclusions
In the present study, we revised conformational, electronic, and structural properties related to the antioxidant activity of seven flavonoid structures and presented novel information regarding their magnetic properties. The conformational analyzes of anthocyanidin, flavonol, and flavan-3-ol indicate the stabilization of the molecules with increased planarity due to the formation of the H-bond interaction between O1 and HC6′. The analysis of flavonol also indicates the existence of a hydrogen bond between the hydroxyl group (O3H) and the oxygen of ketone that stabilizes its molecular structure, improving the planar conformation. Comparing the aromaticity results of flavonol, flavone, and anthocyanidin, we found that the planarity and the interaction between O3H and HC6′ contributed to the aromaticity of ring C. Similar to the expected behavior of the substituent effect, the analysis of the aromaticity of ring C showed that the hydroxyl group improves the aromaticity and the ketone group decreases the aromaticity.67 The importance of the ketone group is also seen in the oxygen NMR chemical shift of flavonol, as the δ in O2 shows that it is more strongly bonded than O1. NMR also indicates an H bond between O3H and the oxygen of ketone.
Regarding anthocyanidin, our results demonstrated that its lower PA and BDE values, when compared with flavonol and flavan-3-ol, indicate that it has the highest antioxidant activity. Furthermore, our results indicate that the anthocyanidin aromaticity is not related to electron transfer.
Finally, the NBO atomic charge values are displaced from ring B to benzo-γ-pyran in flavan, neutral anthocyanidin, flavanone, and neutral flavan-3-ol. The values obtained for the charges are related to the interactions between O3H and the oxygen of ketone and between O3H and HC6′.
4. Methodology
4.1. Flavonoid Structures
Initially, the flavonoid structures were obtained in the PubChem database in the MOL2 format.68 The following flavonoid structures from the natural origin were selected due to their reported antioxidant activities: flavan (CID 94156, C15H14O, Figure 2A), anthocyanidin (CID 414159, (C15H11O2)+; Figure 2B), flavanone (CID 10251, C15H12O2; Figure 2C), flavonol (CID 11349, C15H10O3; Figure 2D), isoflavone (CID 72304, C15H10O2; Figure 2E), flavone (CID 10680, C15H10O2; Figure 2F), and flavan-3-ol (CID 3707243, C15H14O2; Figure 2G).
4.2. Electronic and Structural Analysis
The self-consistent field calculations of flavonoid structures were performed using DFT.69 DFT has been a useful method to study the structural, electronic, and magnetic molecular properties of compounds.16,37,58,70−72 The CAM-B3LYP functional was used with the split valence def2TZV basis set of Ahlrichs et al. (CAM-B3LYP/def2TZV).73−76 We selected the CAM-B3LYP functional due to its well-established and useful application to investigate chemical structures.77 The correlation consistent cc-pVDZ basis set was also applied to study the aromaticity and the oxidation of the aromatic ring in the flavonoid structures.78 We used CAM-B3LYP/def2TZV to performs the conformational, electronic, and magnetic analyses. We performed a relaxed scanning for all structural optimizations of the flavonoids. The balanced polarized triple-zeta def2TZV basis set has presented satisfactory structural results in the literature.79 Recently, we have used CAM-B3LYP and def2TZV basis sets for structural analyses and the results showed to agree with the experimental data.71,80 These computational methods are efficient and less time-consuming to analyze conformations. Besides, we compared the conformational analyses obtained with CAM-B3LYP/def2TZV calculations with the hybrid functional HSEh1PBE/cc-pVDZ. The HSEh1PBE level of density functional theory has been widely used to study the frontier molecular orbitals and NMR spectra.81
The calculations were carried out in the Gaussian09 package82,83 in the gas phase (vacuum). Conformational analyses were performed varying the dihedral angles τ present in the atoms O1–C2–C1′–C6′ with steps containing 10° totalizing 18 minimization cycles using CAM-B3LYP/def2TZV. Frontier molecular orbitals HOMO and LUMO were analyzed using the CAM-B3LYP/def2TZV and HSEh1PBE/cc-pVDZ levels. Atoms in molecules (AIM) analysis was carried out using the AIMAll package.84
4.3. Magnetic Properties Analysis
The investigation of the aromaticity was carried out through the nucleus-independent chemical shift (NICS) method.35 This method consists of the calculation of 1H NMR, employing ghost atoms defined preferably in the center of the molecule ring (NICS 0) or at a point outside the plane and perpendicular to the molecule at 1 Å (NICS 1). The NICS values were calculated with the Gauge-independent atomic orbital (GIAO),85 using the cc-pVDZ basis set with the hybrid functional HSEh1PBE (HSEh1PBE/cc-pVDZ) and compared with the results obtained from CAM-B3LYP/def2TZV calculations.
The NICS method has been extensively tested on planar and cyclic molecules, but aromaticity can also occur on nonplanar molecules.86,87 An NICSisotropic(1) value more negative than NICSisotropic(0) is expected in planar aromatic rings. All calculated NICS values are on the z-axis of the ring. The NICSzz procedure has been used to investigate the aromaticity of cyclic structures.88 We applied the NICS(0) plus NICS(1) procedure to explore the aromaticity of the rings of the seven selected flavonoids.
Acknowledgments
The authors are grateful to the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 310071/2018-6) and the Foundation for Research Support of the Federal District/Brazil (FAPDF 0193.001545/2017) for the financial support. K.S.d.C. is also grateful for the scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Grant Number 88882.466102/2019-01).
The authors declare no competing financial interest.
References
- Panche A. N.; Diwan A. D.; Chandra S. R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, e47 10.1017/jns.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo L. Polyphenols: Chemistry, Dietary Sources, Metabolism, and Nutritional Significance. Nutr. Rev. 1998, 56, 317–333. 10.1111/j.1753-4887.1998.tb01670.x. [DOI] [PubMed] [Google Scholar]
- Aparicio S. A Systematic Computational Study on Flavonoids. Int. J. Mol. Sci. 2010, 11, 2017–2038. 10.3390/ijms11052017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsha S. N.; Anilakumar K. R. In Vitro Free Radical Scavenging and DNA Damage Protective Property of Coriandrum sativum L. Leaves Extract. J. Food Sci. Technol. 2014, 51, 1533–1539. 10.1007/s13197-012-0648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucá M. M.; Cysne Filho F. M. S.; de Almeida J. C.; Mesquita D.; da S.; Barriga J. R.; de M.; Dias K. C. F.; Barbosa T. M.; Vasconcelos L. C.; Leal L. K. A. M.; Ribeiro J. E.; et al. Flavonoids: Biological Activities and Therapeutic Potential. Nat. Prod. Res. 2020, 34, 692–705. 10.1080/14786419.2018.1493588. [DOI] [PubMed] [Google Scholar]
- Wang D.; Wang X.; Zhang C.; Ma Y.; Zhao X. Calf Thymus DNA-Binding Ability Study of Anthocyanins from Purple Sweet Potatoes (Ipomoea batatas L.). J. Agric. Food Chem. 2011, 59, 7405–7409. 10.1021/jf200851h. [DOI] [PubMed] [Google Scholar]
- Farhadi F.; Khameneh B.; Iranshahi M.; Iranshahy M. Antibacterial Activity of Flavonoids and Their Structure-Activity Relationship: An Update Review. Phytother. Res. 2019, 33, 13–40. 10.1002/ptr.6208. [DOI] [PubMed] [Google Scholar]
- De Souza L. A.; Soeiro M. M.; De Almeida W. B. A DFT Study of Molecular Structure and 1H NMR, IR, and UV-Vis Spectrum of Zn(II)-Kaempferol Complexes: A Metal-Flavonoid Complex Showing Enhanced Anticancer Activity. Int. J. Quantum Chem. 2018, 118, e25773 10.1002/qua.25773. [DOI] [Google Scholar]
- Jiang C. H.; Sun T. L.; Xiang D. X.; Wei S. S.; Li W. Q. Anticancer Activity and Mechanism of Xanthohumol: A Prenylated Flavonoid from Hops (Humulus lupulus L.). Front. Pharmacol. 2018, 9, 530 10.3389/fphar.2018.00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani S.; Poh C. L. Flavonoids as Antiviral Agents for Enterovirus A71 (EV-A71). Viruses 2020, 12, 184 10.3390/v12020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anusuya S.; Gromiha M. M. Structural Basis of Flavonoids as Dengue Polymerase Inhibitors: Insights from QSAR and Docking Studies. J. Biomol. Struct. Dyn. 2019, 37, 104–115. 10.1080/07391102.2017.1419146. [DOI] [PubMed] [Google Scholar]
- Galúcio J. M.; Monteiro E. F.; de Jesus D. A.; Costa C. H.; Siqueira R. C.; dos Santos G. B.; Lameira J.; da Costa K. S. In Silico Identification of Natural Products with Anticancer Activity Using a Chemo-Structural Database of Brazilian Biodiversity. Comput. Biol. Chem. 2019, 83, 107102 10.1016/j.compbiolchem.2019.107102. [DOI] [PubMed] [Google Scholar]
- De Souza L. A.; Da Silva H. C.; De Almeida W. B. Structural Determination of Antioxidant and Anticancer Flavonoid Rutin in Solution through DFT Calculations of 1H NMR Chemical Shifts. ChemistryOpen 2018, 7, 902–913. 10.1002/open.201800209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza L. A.; Tavares W. M. G.; Lopes A. P. M.; Soeiro M. M.; De Almeida W. B. Structural Analysis of Flavonoids in Solution through DFT 1H NMR Chemical Shift Calculations: Epigallocatechin, Kaempferol and Quercetin. Chem. Phys. Lett. 2017, 676, 46–52. 10.1016/j.cplett.2017.03.038. [DOI] [Google Scholar]
- Xiaowei H.; Xiaobo Z.; Jiewen Z.; Jiyong S.; Xiaolei Z.; Holmes M. Measurement of Total Anthocyanins Content in Flowering Tea Using near Infrared Spectroscopy Combined with Ant Colony Optimization Models. Food Chem. 2014, 164, 536–543. 10.1016/j.foodchem.2014.05.072. [DOI] [PubMed] [Google Scholar]
- Todorova T. Z.; Traykov M. G.; Tadjer A. V.; Velkov Z. A. Structure of Flavones and Flavonols. Part I: Role of Substituents on the Planarity of the System. Comput. Theor. Chem. 2013, 1017, 85–90. 10.1016/j.comptc.2013.05.005. [DOI] [Google Scholar]
- van Acker S. A. B. E.; de Groot M. J.; van den Berg D. J.; Tromp M. N. J. L.; den Kelder G. D.-O.; van Der Vijgh W. J. F.; Bast A. A Quantum Chemical Explanation of the Antioxidant Activity of Flavonoids. Chem. Res. Toxicol. 1996, 9, 1305–1312. 10.1021/tx9600964. [DOI] [PubMed] [Google Scholar]
- Lameira J.; Alves C. N.; Santos L. S.; Santos A. S.; de Almeida Santos R. H.; Souza J.; Silva C. C.; da Silva A. B. F. A Combined X-Ray and Theoretical Study of Flavonoid Compounds with Anti-Inflammatory Activity. J. Mol. Struct.: THEOCHEM 2008, 862, 16–20. 10.1016/j.theochem.2008.04.029. [DOI] [Google Scholar]
- Rasouli H.; Hosseini Ghazvini S. M. B.; Yarani R.; Altıntaş A.; Jooneghani S. G. N.; Ramalho T. C. Deciphering Inhibitory Activity of Flavonoids against Tau Protein Kinases: A Coupled Molecular Docking and Quantum Chemical Study. J. Biomol. Struct. Dyn. 2020, 1–14. 10.1080/07391102.2020.1814868. [DOI] [PubMed] [Google Scholar]
- Martins H. F. P.; Leal J. P.; Fernandez M. T.; Lopes V. H. C.; Cordeiro M. N. D. S. Toward the Prediction of the Activity of Antioxidants: Experimental and Theoretical Study of the Gas-Phase Acidities of Flavonoids. J. Am. Soc. Mass Spectrom. 2004, 15, 848–861. 10.1016/j.jasms.2004.02.007. [DOI] [PubMed] [Google Scholar]
- do Nascimento L. D.; de Moraes A. A. B.; da Costa K. S.; Galúcio J. M. P.; Taube P. S.; Costa C. M. L.; Cruz J. N.; de Aguiar Andrade E. H.; de Faria L. J. G. Bioactive Natural Compounds and Antioxidant Activity of Essential Oils from Spice Plants: New Findings and Potential Applications. Biomolecules 2020, 10, 988 10.3390/biom10070988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkadi H. A Review on Free Radicals and Antioxidants. Infect. Disord. Drug Targets 2020, 20, 16–26. 10.2174/1871526518666180628124323. [DOI] [PubMed] [Google Scholar]
- Kabanda M. M.; Mammino L.; Murulana L. C.; Mwangi H. M.; Mabusela W. T. Antioxidant Radical Scavenging Properties of Phenolic Pent-4-En-1-Yne Derivatives Isolated From Hypoxis rooperi. A DFT Study in Vacuo and in Solution. Int. J. Food Prop. 2015, 18, 149–164. 10.1080/10942912.2013.825842. [DOI] [Google Scholar]
- Zhang H. Y.; Sun Y. M.; Wang X. L. Substituent Effects on O-H Bond Dissociation Enthalpies and Ionization Potentials of Catechols: A DFT Study and Its Implications in the Rational Design of Phenolic Antioxidants and Elucidation of Structure-Activity Relationships for Flavonoid Antioxidan. Chem. – Eur. J. 2003, 9, 502–508. 10.1002/chem.200390052. [DOI] [PubMed] [Google Scholar]
- Di Meo F.; Lemaur V.; Cornil J.; Lazzaroni R.; Duroux J. L.; Olivier Y.; Trouillas P. Free Radical Scavenging by Natural Polyphenols: Atom versus Electron Transfer. J. Phys. Chem. A 2013, 117, 2082–2092. 10.1021/jp3116319. [DOI] [PubMed] [Google Scholar]
- Santos-Sánchez N. F.; Salas-Coronado R.; Villanueva-Cañongo C.; Hernández-Carlos B.. Antioxidant Compounds and Their Antioxidant Mechanism. In Antioxidants; IntechOpen, 2019. [Google Scholar]
- Es-Safi N. E.; Ghidouche S.; Ducrot P. H. Flavonoids: Hemisynthesis, Reactivity, Characterization and Free Radical Scavenging Activity. Molecules 2007, 12, 2228 10.3390/12092228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcastro M.; Marino T.; Russo N.; Toscano M. Structural and Electronic Characterization of Antioxidants from Marine Organisms. Theor. Chem. Acc. 2006, 115, 361–369. 10.1007/s00214-006-0077-5. [DOI] [Google Scholar]
- Capaldo L.; Ravelli D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader E. A.; Davidson E. R.; Mayer J. M. Large Ground-State Entropy Changes for Hydrogen Atom Transfer Reactions of Iron Complexes. J. Am. Chem. Soc. 2007, 129, 5153–5166. 10.1021/ja0686918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. Z.; Deng G.; Guo R.; Chen D. F.; Fu Z. M. DFT Studies on the Antioxidant Activity of Naringenin and Its Derivatives: Effects of the Substituents at C3. Int. J. Mol. Sci. 2019, 20, 1450. 10.3390/ijms20061450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatylowicz H.; Marek P. H.; Stasyuk O. A.; Krygowski T. M.; Solà M. Substituted Adenine Quartets: Interplay between Substituent Effect, Hydrogen Bonding, and Aromaticity. RSC Adv. 2020, 10, 23350–23358. 10.1039/D0RA04585C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranac-Stojanović M. Substituent Effect on Triplet State Aromaticity of Benzene. J. Org. Chem. 2020, 85, 4289–4297. 10.1021/acs.joc.9b03472. [DOI] [PubMed] [Google Scholar]
- Baryshnikov G. V.; Valiev R. R.; Karaush N. N.; Sundholm D.; Minaev B. F. Aromaticity of the Doubly Charged [8]Circulenes. Phys. Chem. Chem. Phys. 2016, 18, 8980–8992. 10.1039/c6cp00365f. [DOI] [PubMed] [Google Scholar]
- Stanger A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. 10.1021/jo051746o. [DOI] [PubMed] [Google Scholar]
- Mendes A. P. S.; Borges R. S.; Neto A. M. J. C.; De Macedo L. G. M.; Da Silva A. B. F. The Basic Antioxidant Structure for Flavonoid Derivatives. J. Mol. Model. 2012, 18, 4073–4080. 10.1007/s00894-012-1397-0. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Z.; Zhou Y.; Liang Q.; Chen D. F.; Guo R. Theoretical Studies on the Hydrogen-Bonding Interactions between Luteolin and Water: A DFT Approach. J. Mol. Model. 2016, 22, 257 10.1007/s00894-016-3128-4. [DOI] [PubMed] [Google Scholar]
- Matteini P.; Goti A.; Agati G. Theoretical Conformational Analysis of Rutin. Monatsh. Chem. 2010, 141, 793–800. 10.1007/s00706-010-0330-4. [DOI] [Google Scholar]
- Cox P. J.; Kumarasamy Y.; Nahar L.; Sarker S. D.; Shoeb M. Luteolin. Acta Crystallogr., Sect. E 2003, 59, o975–o977. 10.1107/S1600536803013229. [DOI] [PubMed] [Google Scholar]
- Rossi M.; Rickles L. F.; Halpin W. A. The Crystal and Molecular Structure of Quercetin: A Biologically Active and Naturally Occurring Flavonoid. Bioorg. Chem. 1986, 14, 55–69. 10.1016/0045-2068(86)90018-0. [DOI] [Google Scholar]
- Dacunha-Marinho B.; Martínez A.; Estévez R. J. 7-Hydr-Oxy-5-Meth-Oxy-6,8-Dimethyl-Flavanone: A Natural Flavonoid. Acta Crystallogr., Sect. C 2008, 64, o353–o356. 10.1107/S0108270108014959. [DOI] [PubMed] [Google Scholar]
- Smith G.; Bartley J. P.; Wang E.; Bolt R. C. Two Crystal Polymorphs of a Flavonoid from Melicope Ellyrana. Acta Crystallogr., Sect. C 2001, 57, 1336–1337. 10.1107/S0108270101013695. [DOI] [PubMed] [Google Scholar]
- Lau K. S.; Mantas A.; Chass G. A.; Ferretti F. H.; Estrada M.; Zamarbide G.; Csizmadia I. G. Ab Initio and DFT Conformational Analysis of Selected Flavones: 5,7-Dihydroxyflavone (Chrysin) and 7,8-Dihydroxyflavone. Can. J. Chem. 2002, 80, 845–855. 10.1139/v02-113. [DOI] [Google Scholar]
- Machado N. F. L.; Batista De Carvalho L. A. E.; Otero J. C.; Marques M. P. M. A Conformational Study of Hydroxyflavones by Vibrational Spectroscopy Coupled to DFT Calculations. Spectrochim. Acta, Part A 2013, 109, 116–124. 10.1016/j.saa.2013.01.038. [DOI] [PubMed] [Google Scholar]
- Cappelli C.; Bronco S.; Monti S. Computational Study of Conformational and Chiroptical Properties of 2R,3S,4R-(+)-3,3′,4,4′,7-Flavanpentol. Chirality 2005, 17, 577–589. 10.1002/chir.20210. [DOI] [PubMed] [Google Scholar]
- Rice-Evans C. A.; Miller N. J.; Paganga G. Structure-Antioxidant Activity Relationships of Flavonoids and Phenolic Acids. Free Radicals Biol. Med. 1996, 20, 933–956. 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- Gil E. S.; Couto R. O. Flavonoid Electrochemistry: A Review on the Electroanalytical Applications. Braz. J. Pharmacogn. 2013, 23, 542–558. 10.1590/S0102-695X2013005000031. [DOI] [Google Scholar]
- Amic D.; Davidovic-Amic D.; Beslo D.; Rastija V.; Lucic B.; Trinajstic N. SAR and QSAR of the Antioxidant Activity of Flavonoids. Curr. Med. Chem. 2007, 14, 827–845. 10.2174/092986707780090954. [DOI] [PubMed] [Google Scholar]
- Scotti L.; Fernandes M. B.; Muramatsu E.; de Paula Emereciano V.; Tavares J. F.; da Silva M. S.; Scotti M. T. 13C NMR Spectral Data and Molecular Descriptors to Predict the Antioxidant Activity of Flavonoids. Braz. J. Pharm. Sci. 2011, 47, 241–249. 10.1590/S1984-82502011000200005. [DOI] [Google Scholar]
- Chen Z. Y.; Chan P. T.; Ho K. Y.; Fung K. P.; Wang J. Antioxidant Activity of Natural Flavonoids Is Governed by Number and Location of Their Aromatic Hydroxyl Groups. Chem. Phys. Lipids 1996, 79, 157–163. 10.1016/0009-3084(96)02523-6. [DOI] [PubMed] [Google Scholar]
- Ninh The S.; Do Minh T.; Nguyen Van T. Isoflavones and Isoflavone Glycosides: Structural-Electronic Properties and Antioxidant Relations-A Case of DFT Study. J. Chem. 2019, 2019, 4360175 10.1155/2019/4360175. [DOI] [Google Scholar]
- Mills N. S.; Llagostera K. B. Summation of Nucleus Independent Chemical Shifts as a Measure of Aromaticity. J. Org. Chem. 2007, 72, 9163–9169. 10.1021/jo7013609. [DOI] [PubMed] [Google Scholar]
- Wolniak M.; Wawer I. 13C CPMAS NMR and DFT Calculations of Anthocyanidins. Solid State Nucl. Magn. Reson. 2008, 34, 44–51. 10.1016/j.ssnmr.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Sinha R.; Gadhwal N. K.; Joshi U. J.; Srivastava S.; Govil G. Modifying Effect Og Quercetin on Model Biomembranes: Studied by Molecular Dynamic Simulation, DSC and NMR. Int. J. Curr. Pharm. Res. 2012, 4, 70–79. [Google Scholar]
- Queiroz A. N.; Gomes B. A. Q. Q.; Moraes W. M. Jr.; Borges R. S. A Theoretical Antioxidant Pharmacophore for Resveratrol. Eur. J. Med. Chem. 2009, 44, 1644–1649. 10.1016/j.ejmech.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Payán-Gómez S. A.; Flores-Holguín N.; Pérez-Hernández A.; Piñón-Miramontes M.; Glossman-Mitnik D. Computational Molecular Characterization of the Flavonoid Rutin. Chem. Cent. J. 2010, 4, 12 10.1186/1752-153X-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saqib M.; Mahmood A.; Akram R.; Khalid B.; Afzal S.; Kamal G. M. Density Functional Theory for Exploring the Structural Characteristics and Their Effects on the Antioxidant Properties. J. Pharm. Appl. Chem. 2015, 71, 65–71. [Google Scholar]
- Tsuneda T. Chemical Reaction Analyses Based on Orbitals and Orbital Energies. Int. J. Quantum Chem. 2015, 115, 270–282. 10.1002/qua.24805. [DOI] [Google Scholar]
- Saqib M.; Iqbal S.; Mahmood A.; Akram R. Theoretical Investigation for Exploring the Antioxidant Potential of Chlorogenic Acid: A Density Functional Theory Study. Int. J. Food Prop. 2016, 19, 745–751. 10.1080/10942912.2015.1042588. [DOI] [Google Scholar]
- Son N. T.; Mai Thanh D. T.; Van Trang N. Flavone Norartocarpetin and Isoflavone 2′-Hydroxygenistein: A Spectroscopic Study for Structure, Electronic Property and Antioxidant Potential Using DFT (Density Functional Theory). J. Mol. Struct. 2019, 1193, 76–88. 10.1016/j.molstruc.2019.05.016. [DOI] [Google Scholar]
- Woon D. E.; Dunning T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for Boron through Neon. J. Chem. Phys. 1995, 103, 4572–4585. 10.1063/1.470645. [DOI] [Google Scholar]
- Ahmadi S. M.; Farhoosh R.; Sharif A.; Rezaie M. Structure-Antioxidant Activity Relationships of Luteolin and Catechin. J. Food Sci. 2020, 85, 298–305. 10.1111/1750-3841.14994. [DOI] [PubMed] [Google Scholar]
- Leopoldini M.; Pitarch I. P.; Russo N.; Toscano M. Structure, Conformation, and Electronic Properties of Apigenin, Luteolin, and Taxifolin Antioxidants. A First Principle Theoretical Study. J. Phys. Chem. A 2004, 108, 92–96. 10.1021/jp035901j. [DOI] [Google Scholar]
- Vo Q. V.; Nam P. C.; Thong N. M.; Trung N. T.; Phan C. T. D.; Mechler A. Antioxidant Motifs in Flavonoids: O–H versus C–H Bond Dissociation. ACS Omega 2019, 4, 8935–8942. 10.1021/acsomega.9b00677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvareda E.; Denis P. A.; Iribarne F.; Paulino M. Bond Dissociation Energies and Enthalpies of Formation of Flavonoids: A G4 and M06-2X Investigation. Comput. Theor. Chem. 2016, 1091, 18–23. 10.1016/j.comptc.2016.06.021. [DOI] [Google Scholar]
- Litwinienko G.; Ingold K. U. Abnormal Solvent Effects on Hydrogen Atom Abstractions. 1. The Reactions of Phenols with 2,2-Diphenyl-1-Picrylhydrazyl (Dpph•) in Alcohols. J. Org. Chem. 2003, 68, 3433–3438. 10.1021/jo026917t. [DOI] [PubMed] [Google Scholar]
- Krygowski T. M.; Ejsmont K.; Stepień B. T.; Cyrański M. K.; Poater J.; Solà M. Relation between the Substituent Effect and Aromaticity. J. Org. Chem. 2004, 69, 6634–6640. 10.1021/jo0492113. [DOI] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C. H.; Leung C. W. The Convolution Equation of Choquet and Deny on [IN]-Groups. Integr. Equations Oper. Theory 2001, 40, 391–402. 10.1007/BF01198136. [DOI] [Google Scholar]
- de Castro E. A. S.; de Oliveira D. A. B.; Farias S. A. S.; Gargano R.; Martins J. B. L. Structure and Electronic Properties of Azadirachtin. J. Mol. Model. 2014, 20, 2084 10.1007/s00894-014-2084-0. [DOI] [PubMed] [Google Scholar]
- Graef E. L.; Martins J. B. L. Analysis of Lowest Energy Transitions at TD-DFT of Pyrene in Vacuum and Solvent. J. Mol. Model. 2019, 25, 183 10.1007/s00894-019-4065-9. [DOI] [PubMed] [Google Scholar]
- Honarparvar B.; Pawar S. A.; Alves C. N.; Lameira J.; Maguire G. E.; Silva J. R. A.; Govender T.; Kruger H. G. Pentacycloundecane Lactam vs Lactone Norstatine Type Protease HIV Inhibitors: Binding Energy Calculations and DFT Study. J. Biomed. Sci. 2015, 22, 15 10.1186/s12929-015-0115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharley J. N.Established DFT Methods Calculation of Conjugation Disturbed in the Presence of Torsional Hyperconjugation. arXiv, September 30, 2016, 1512.04171, ver. 4. https://arxiv.org/abs/1512.04171v4 (accessed March 1, 2021).
- Peintinger M. F.; Oliveira D. V.; Bredow T. Consistent Gaussian Basis Sets of Triple-Zeta Valence with Polarization Quality for Solid-State Calculations. J. Comput. Chem. 2013, 34, 451–459. 10.1002/jcc.23153. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Yanai T.; Tew D. P.; Handy N. C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- Greve D. R. Homoaromaticity in Aza- and Phosphasemibullvalenes. A Computational Study. J. Phys. Org. Chem. 2011, 24, 222–228. 10.1002/poc.1731. [DOI] [Google Scholar]
- Ivanov S. M.; Traven V. F.; Minyaev M. E. Structural Studies of 3-Tert-Butyl-8-(Methylchalcogenyl)Pyrazolo[5,1-c][1,2,4]Triazin-4(1H)-Ones. Struct. Chem. 2020, 31, 1457–1470. 10.1007/s11224-020-01533-9. [DOI] [Google Scholar]
- Santiago P. H. O.; Tiago F. S.; Castro M. S.; Souza P. E. N.; Martins J. B. L.; Gatto C. C. DFT Analysis, Spectroscopic Study and Biological Activity of a Newly Synthesized Benzoylhydrazone Binuclear Cu(II) Complex. J. Inorg. Biochem. 2020, 204, 110949 10.1016/j.jinorgbio.2019.110949. [DOI] [PubMed] [Google Scholar]
- Öner N.; Tamer Ö.; Avci D.; Atalay Y. Quantum Mechanical Calculations on Cis-2,6-Bis(2-Chlorophenyl)-3,3-Dimethylpiperidin-4-One. AIP Conf. Proc. 2016, 1722, 200012 10.1063/1.4944227. [DOI] [Google Scholar]
- Pople J. A.; Head-Gordon M.; Fox D. J.; Raghavachari K.; Curtiss L. A. Gaussian-1 Theory: A General Procedure for Prediction of Molecular Energies. J. Chem. Phys. 1989, 90, 5622–5629. 10.1063/1.456415. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenber D. J.. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford, CT, 2010.
- Keith T. A.AIMAll; TK Gristmill Software: Overland Park, KS, 2019.
- Cheeseman J. R.; et al. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. 10.1063/1.471789. [DOI] [Google Scholar]
- Reisi-Vanani A.; Rezaei A. A. Evaluation of the Aromaticity of Non-Planar and Bowl-Shaped Molecules by NICS Criterion. J. Mol. Graphics Model. 2015, 61, 85–88. 10.1016/j.jmgm.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Park S. S. Structural and Bonding Trends among the B7C11–, B6C2, and B5C31+. Bull. Korean Chem. Soc. 2005, 26, 63–71. 10.5012/bkcs.2005.26.1.063. [DOI] [Google Scholar]
- Matito E.; Duran M.; Solà M. The Aromatic Fluctuation Index (FLU): A New Aromaticity Index Based on Electron Delocalization. J. Chem. Phys. 2005, 122, 014109 10.1063/1.1824895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Sharley J. N.Established DFT Methods Calculation of Conjugation Disturbed in the Presence of Torsional Hyperconjugation. arXiv, September 30, 2016, 1512.04171, ver. 4. https://arxiv.org/abs/1512.04171v4 (accessed March 1, 2021).