

Abstract
Mass spectrometry (MS) serves as the centerpiece technology for proteome, lipidome, and metabolome analysis. To have a better understanding of the multi-faceted networks of myriad regulatory layers in complex organisms, an integration on different layers of multi-omics is up-and-coming, including joint extraction methods of diverse biomolecular classes and comprehensive data analyses of different omics. Despite the versatility of MS systems, fractured methodology drives nearly all MS laboratories to specialize in analysis of a single ome at the exclusion of the others. Although liquid chromatography-mass spectrometry (LC-MS) analysis is similar for different biomolecular classes, the integration on the instrument level is lagging behind. The recent advancement on high flow proteomics enables us to take a first step on integration of proteins and lipids analysis. Here, we describe a technology to achieve broad and deep coverage of multiple molecular classes simultaneously through multi-omic single-shot technology (MOST) requiring only one column, one LC-MS instrument and a simplified workflow. MOST achieved great robustness and reproducibility. Its application on a Saccharomyces cerevisiae study consisting of 20 conditions revealed 2,842 protein groups and 325 lipids, and potential molecular relationships.
Graphical Abstract
Myriad regulatory layers involving tens of thousands of biomolecules – including nucleic acids, proteins, lipids, and metabolites – modulate the cellular processes that govern complex organisms. Untangling these multi-faceted networks will require innovative technologies to globally monitor diverse classes of biomolecules. For example, gene expression profiles have failed to fill unresolved gaps in many biosynthesis pathways implicated in many human diseases. Mass spectrometry (MS) has propelled systems biology by offering access to the proteome, lipidome, and metabolome. For each of these classes, remarkably similar MS methods, consisting of chromatographic separation, mass measurement, and tandem MS1,2,3, have been developed and are used widely. Despite this methodological common ground, nearly all MS laboratories specialize in niche applications targeting a single ome.
This arrangement causes two major problems. First, the confinement to a single ome across published data impoverishes the MS community’s understanding of their interplay4,5,6,7; a PubMed search of publications from the last five years reveals that though multi-omic studies have tripled in that time, less than ~ 7.5% of MS-based omic studies incorporated multi-omic analysis in 2019. Second, the majority of existing multi-omic analyses are conducted across laboratories, potentially leading to noise introduced by sample heterogeneity, variable sample handling, and instrument variance that can then obscure otherwise strong biomolecular associations8,9,10,11. We posit that an integrated workflow could both improve data quality and unlock more comprehensive coverage of the biological system.
Thus, in contrast to the fragmented single-ome status quo, we envision an integrated technology for the metabolome, lipidome, and proteome that allows these omes to be jointly extracted and prepared from a single sample; loaded onto and separated by a single LC column; and jointly mass analyzed as they elute on a single MS platform. We term this method multi-omic single-shot technology (MOST). As researchers have been trying to overcome the bottleneck of co-extraction of multiple molecular classes such as metabolites, lipids and proteins12,13,14, here, we demonstrate that an integrated analysis of proteins and lipids is possible. To our knowledge, MOST is the first technology that integrates proteome and lipidome analysis in a single LC-MS run using a single reverse-phase (RP) column and a binary mobile phase system15,16,17 – i.e., without the need to maintain dedicated LC-MS systems or work across multiple laboratories.
EXPERIMENTAL SECTION
Materials and reagents.
Solvents and reagents are listed in the Supporting Information with vendor and catalog number (Supplementary Text S1).
Yeast cultures.
Details of yeast cultures are described in the Supporting Information (Supplementary Text S2).
Sample preparation for mass spectrometry.
The procedure was adapted from previous literature18. Frozen cell pellets were thawed on ice and mixed with 250 μL of methanol, 750 μL of methyl tert-butyl ether (MTBE), and 200 μL of water. The samples were vortexed for 10 s and sonicated for 5 min. Phase separation was completed after centrifugation (12,000 g, 5 min, 4 °C). 200 μL of the upper hydrophobic layer was aliquoted into a glass insert amber autosampler vial, dried, and reconstituted in 100 μL of ACN/IPA/H2O (65:30:5, v/v/v).
To the lower hydrophilic layer, 200 μL of 6M GnHCl and 100 mM tris (pH = 8.0) was added. The samples were boiled at 100 °C for 5 min, rested at room temperature for 5 min, were boiled at 100 °C for 5 min again. Proteins were precipitated by addition with methanol to 90%, vortex for ~ 10 s, and centrifugation at 12,000 g for 5 min. The resulting protein pellet was re-suspended in the lysis buffer containing 8M urea, 10 mM tris(2-carboxyethyl)phosphine, 40 mM chloroacetamide, and 100 mM tris (pH = 8.0). The samples were digested with lysyl endopeptidase (1:50, enzyme/protein) for 4 hrs. The samples were diluted four times by 50 mM tris and incubated with trypsin (1:50, enzyme/protein) for overnight digestion. The samples were acidified and desalted using Strata cartridges. A quantitative colorimetric peptide assay (Thermo Scientific) was performed to measured peptide concentration.
MOST LC-MS.
LC–MS analysis was performed on a Waters C18 reverse-phase BEH column (150 mm × 1.0 mm × 2.1 μm particle size) at 50 °C and 60 μL/min flow rate. Mobile phase A consisted of 0.2% formic acid in H2O. Mobile phase B consisted of 0.2% formic acid and 5 mM ammonium formate in IPA/ACN (90:10, v/v). 5 μL of lipid extracts were injected followed by the injection of 20 μg of peptides at 0% of mobile phase B. For the gradient, mobile phase B was held at 0% for 1 min, increased to 28% over 52 min, reached to 70% at 60 min, increased to 100% over 20 min. The washing step at 100% mobile phase B was 5 min. and then the re-equilibration step at 0% mobile phase B was 14 min. Eluting molecules were subjected to electrospray ionization using the heated electrospray ionization source (HESI II) on a Q Exactive HF mass spectrometer (Thermo Scientific). Sheath gas and auxiliary gas were set to 30 units and 6 units, respectively. The spray voltage was set to ±4.5 kV. Capillary temperature and aux gas heater temperature were set to 275 °C and 300 °C, respectively. A 34 gauge spray needle was used.
For peptides scanning, only positive mode was used. MS1 data acquisition was from 0–60 min at a resolution of 60,000 with the automatic gain control (AGC) target set to 3 × 106, m/z range to 300–1350, and maximum injection time (IT) to 50 ms. MS2 data were acquired at a resolution of 30,000 with the AGC target set to 1 × 105, maximum injection time to 60 ms, and loop count to 10 (top10). For lipids scanning, polarity switching was used. MS1 data acquisition was from 60–90 min at a resolution of 30,000 with the AGC target set to 1 × 106, m/z range to 200–1600, and maximum IT to 100 ms. MS2 data were acquired at a resolution of 30,000 with the AGC target set to 1 × 105, maximum injection time to 50 ms, and loop count to 2 (top2).
Data processing.
For proteomics, raw data files were processed by MaxQuant19 (Version 1.5.8.3). The database of reviewed yeast proteins plus isoforms was downloaded from Uniprot on July 23, 2019. Searches used target-decoy strategy20 and the Andromeda21 search algorithm. Default parameters in MaxQuant were used. Label-free quantification (LFQ) was calculated with minimum ratio count of 2. The match between runs was on. MS/MS spectra were not required for LFQ comparisons. Imputation was based on normal distribution using mean and standard deviation of the lowest 1% of LFQ values by Perseus (Version 1.6.0.7). Triplicate protein LFQ values were pooled, log2 transformed, averaged, and then normalized against the WT (mean log2[Δgene/WT], n = 3). P values were obtained from two-tailed t-tests.
For lipidomics, raw data files were processed using Compound Discoverer 2.1 (Thermo Scientific) and Lipidex22. The parameters were mainly adapted from previous literature23,24,25,26. Lipid retention time range was set to 60.1–89.0 min (0.5 min tolerance) and the precursor mass range was set to 100–5000 Da (10 ppm mass tolerance) for aggregation of chromatographic peaks into distinct compound groups. Peak detection required a signal-to-noise ratio of 1.5, a minimum peak intensity of 5 × 105, and a maximum peak width of 0.75 min. Peaks were marked as background if they had a < 5-fold intensity increase over blanks. An in-silico generated lipid spectral library (48 lipid classes and 3.5 × 104 molecular compositions) was used for MS/MS spectra searching. Spectral matches were excluded with a < 500 dot product score and a < 700 reverse dot product score. MS/MS spectra were identified at the individual fatty acid substituent level of structural resolution with a minimum spectral purity of 75%. MS/MS spectra were identified at the sum of the fatty acid substituents level of structural resolution when individual fatty acid substituents were unresolved. Triplicate lipid intensities were pooled, log2 transformed, averaged, and then normalized against the WT (mean log2[Δgene/WT], n = 3). P values were obtained from two-tailed t-tests.
Molecule covariance network analysis.
Pearson’s correlation coefficients (r) were obtained by Pearson’s correlation analysis using fold changes for molecules from all strains. P values were obtained by two-tailed t-tests and were corrected for multiple hypothesis testing (Benjamini-Hochberg). Correlations with |r| ≥ 0.8 and P < 0.001 were kept. Gephi Open Graph Visualization Platform (version 0.9.2) was used to visualize the entire correlation network, where the layout was set to Fruchterman–Reingold with area of 10,000 and gravity of 30. Where applicable, nearest neighbor covariance networks for certain molecules are visualized through igraph() function27 in R statistical environment28.
RESULTS AND DISCUSSION
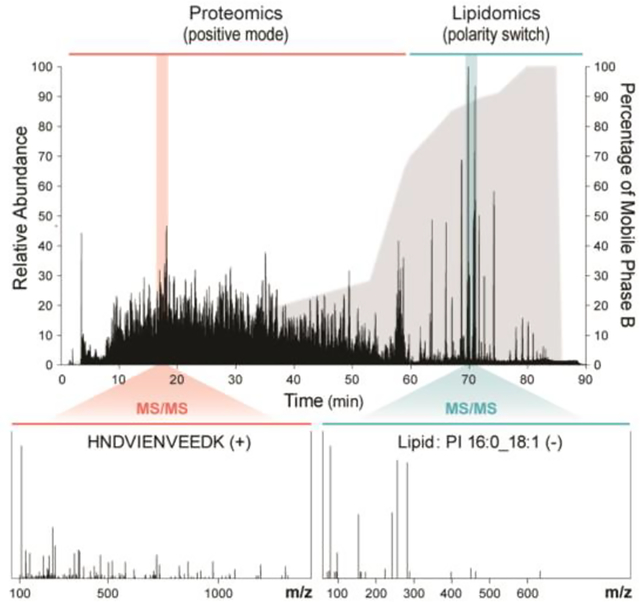
To test our hypothesis that peptides and lipids can be co-analyzed in a single LC-MS methodology, we first sequentially loaded a complex mixture of lipids and then peptides from a yeast cell lysate onto an RP LC column. Note for highest compatibility with developed lipid separation methods, we elected to use a higher flow separation setup – a one-millimeter inner diameter column with microliter flow rates. It is also robust, because typically the column can separate > 800 samples while maintaining good peak shapes and without substantial increase in backpressure. We observed no clogging or other LC-related issues developing the method and running samples. Recent work by Kuster et al. and others have demonstrated such high flow conditions can work well for peptides separations, though they do come at the cost of reduced sensitivity and sampling depth29,30,31,32. In our case, high flow led to 33.7% less unique peptides and 30.7% less protein groups compared to nano flow loaded with 10-fold less samples. Using a modified gradient lasting 90 minutes, we eluted first the peptides (0–60 mins) and then the lipids (60–90 mins) into a quadrupole Orbitrap hybrid MS (Figure 1, Supplementary Figure S1a). Given the hydrophobicity differences of peptide and lipids, these two molecule classes are easily separated temporally for durations we control using our MOST gradient. During peptide elution, the MS was operated in positive-ion mode and peptides were selected for tandem MS using a data-dependent top-ten method. Then, when mostly lipids begin to elute at approximately 60 minutes, the MS is instructed to begin collecting in polarity-switching mode, now collecting with a data-dependent top-two method. Note best performance was achieved with a gradient comprising 90% isopropanol and 10% acetonitrile, a gradient commonly used for lipid separations3,33.
Figure 1. Diagram showing comparison between traditional workflow and MOST workflow.
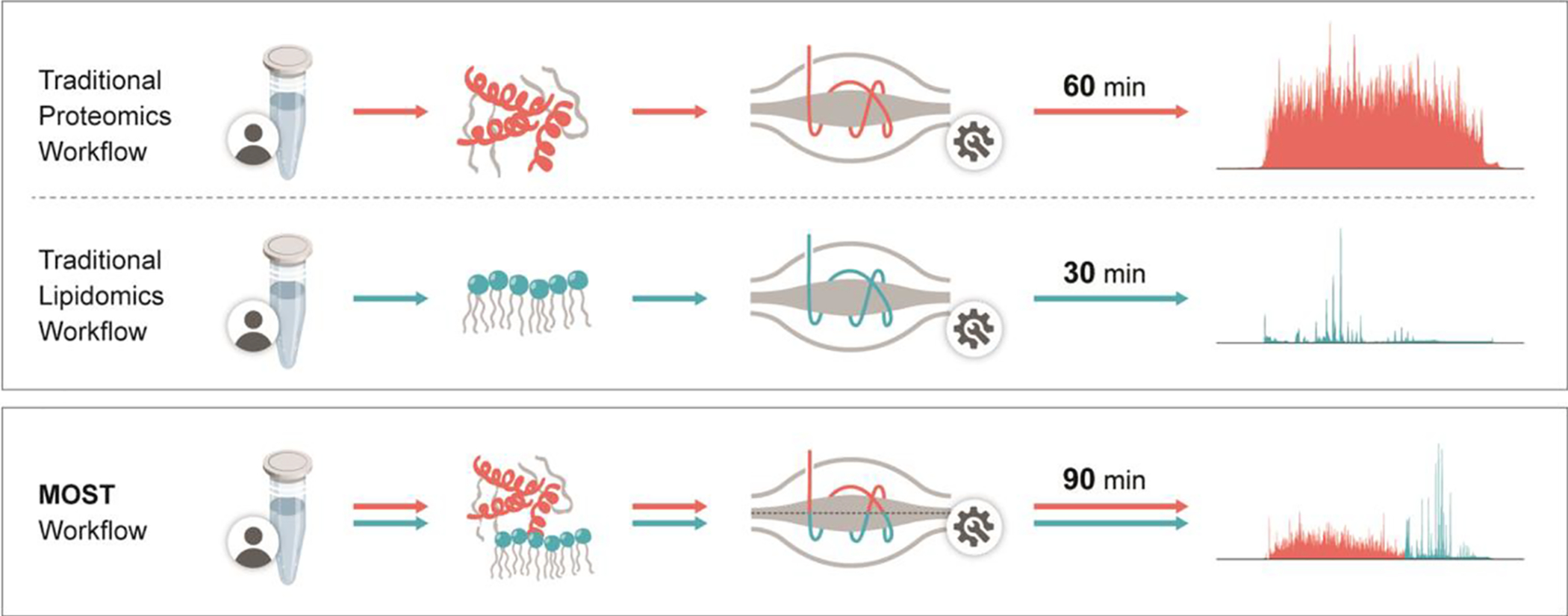
For traditional proteomics, a 60 min nLC-MS/MS was applied, in which gradient was from water to acetonitrile/water (80:20,v/v). For traditional lipidomics, a 30 min HPLC-MS/MS was applied, in which gradient was from acetonitrile/water (70:30,v/v). to isopropanol/acetonitrile (90:10,v/v). For MOST, a special 90 min HPLC-MS/MS was applied, in which gradient was from water to isopropanol/acetonitrile (90:10,v/v). Mobile phase additives were detailed in Experiment Section.
Encouraged by these initial proof-of-concept results, we next sought to optimize the balance between peptide and lipid detection and instrumentation demands. First, to achieve an optimal peptide injection amount under high flow conditions, we assessed serial dilutions (Supplementary Figure S1b) and determined that an optimal load of 20 μg of peptides produced > 18,000 unique peptide identifications corresponding to > 2,200 protein groups. In that same experiment, we typically detect ~ 150 lipids, a number comparable to the amount observed when we perform lipidomics separately on yeast (Supplementary Figure S1c) as well as published data for triplicate wildtype34. For both omes, we analyzed approximately equivalent amounts of starting material. Using MOST for mammalian cell line analysis, we identified and quantified > 2,600 protein groups and > 500 lipids from a HAP1 cell line. These results were achieved without the addition of dimethyl sulfoxide (DMSO)31,32,35. Although DMSO enhances ionization and increases identification, in our case those benefits were short-term, as it rapidly caused instrument fouling. To restore the performance, cleaning of the front part of the mass spectrometer (i.e., S-lens and ion transfer tube) was needed, usually followed by ~ one day of vacuum regeneration, bakeout, and calibration procedure. Considering the instrument downtime and the frequency of cleaning, we decide against using DMSO. Additionally, in contrast to other high flow proteomic methods31,32, we incorporated ammonium formate to the mobile phase. Ammonium salt is a common additive to lipidomics analyses to improve neutral lipid detection. Here, its use led to a slight reduction in peptide identifications but a substantial increase in lipid identifications compared to no-ammonium analyses: 5.8% fewer unique peptides and 3.3% fewer protein groups as compared to an 11.6% gain in lipid identifications. It also led to the detection of key lipid classes, including cardiolipins (CLs) and triglycerides (TGs) (Supplementary Figure S1d–e). Finally, we tested two types of RP columns, C18 BEH and C18 CSH. The BEH column led to more identifications in both proteomic and lipidomic analyses: 1.3% more unique peptides and protein groups, 14.9% more lipids (Supplementary Figure S1f–g).
MOST showed excellent reproducibility as measured by quantification and retention time (RT) stability (Figure 2a; Supplementary Figure S2a). Importantly, protein quantification (R2 = 0.995) and peptide RT (R2 = 1) were not affected by the presence of lipids on the column. Similarly, lipid quantification (R2 = 0.994) and lipid RT (R2 = 0.996) were identical (Figure 2b; Supplementary Figure S2b). In addition, comparable results were obtained for mass error (0.40 vs. 0.41 absolute median ppm for peptides and 2.73 vs. 2.72 absolute median ppm for lipids) and identifications (< 10 identified protein groups or lipids differed between single-ome high flow platforms and our newly developed MOST platform) (Supplementary Figure S2c–e).
Figure 2. Performance characteristics of MOST.
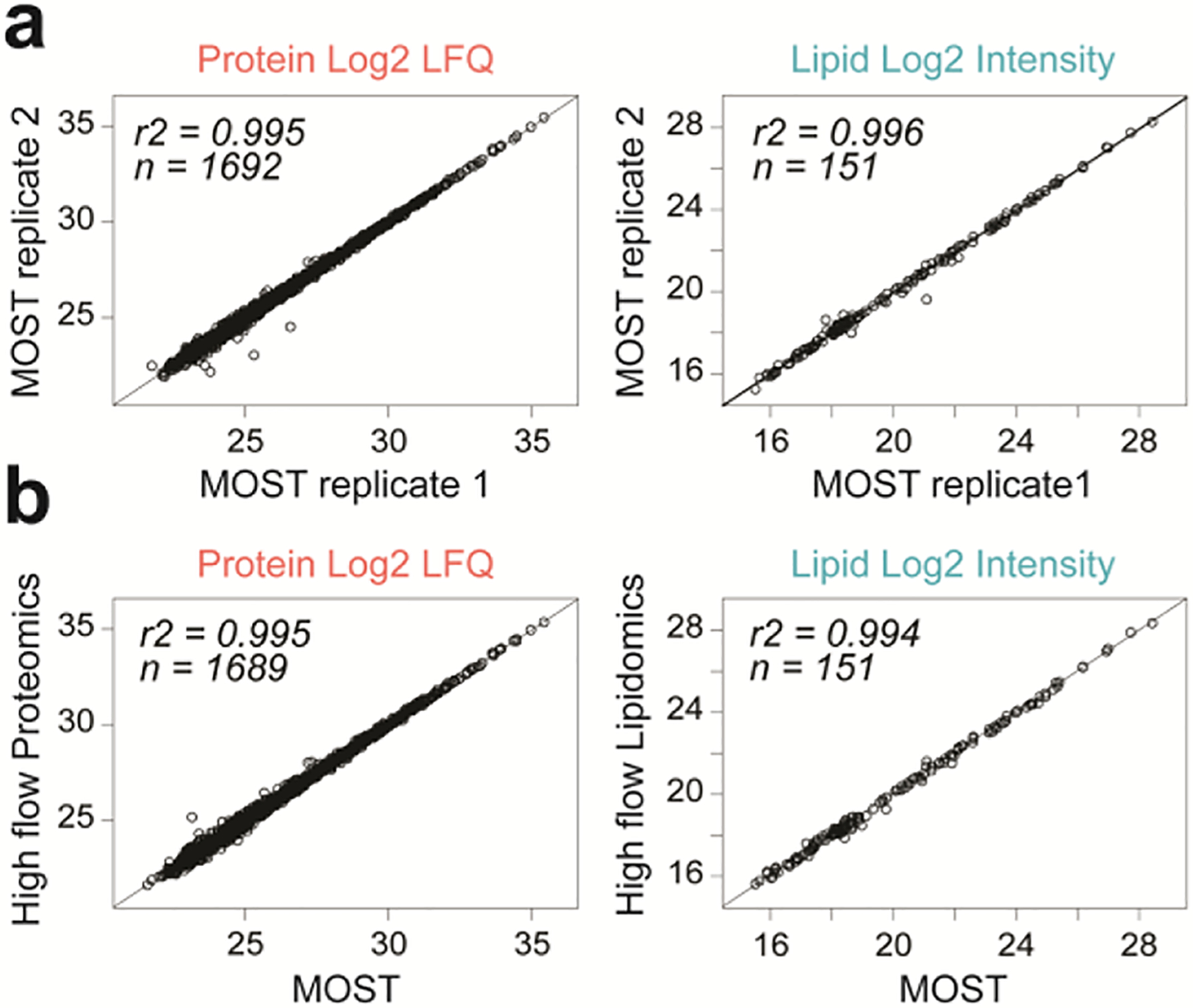
(a) Scatter plot of two MOST runs showing good reproducibility of quantification of proteins and lipids. (b) Scatter plot of a MOST run versus a high flow proteomics/lipidomics run showing no interference on protein/lipid quantification from each other.
As one objective of MOST is to deliver high throughput multi-omic measurements, we next applied our system to study the yeast lipid metabolism – an enterprise that benefits from quantitative measurement of both proteins and lipids. We examined 20 Saccharomyces cerevisiae strains with mitochondria- and lipid-related gene deletions (Δgene) in biological triplicate. Using MOST, we identified and quantified a total of 2,842 protein groups and 325 lipids among 20 S. cerevisiae strains from a single LC-MS method. Again, capability of measuring fold change (up to three orders of magnitude) and CV (15–16% median CV) were comparable. Highlighting the high level of agreement between these datasets, the distribution of gene strains in principal component analysis (PCA) followed the same pattern in both platforms. Finally, by every one of these metrics in this expanded comparison, there is no difference in performance of MOST when both lipids and peptides are present on the column as compared to when they are loaded individually.
Having validated the quality of multi-omic data generated by MOST, we next aimed to assess what it can reveal about the biological system of interest, particularly perturbations to lipid biosynthesis pathways. The Δgene strains studied here can be divided into three categories based on their target pathway: coenzyme Q6 (CoQ6) biosynthesis, cardiolipin (CL) biosynthesis, and those of uncharacterized function. We discovered that on average 647 molecules (575 protein groups and 72 lipids, 20.4% of total measured) were perturbed with each gene deletion as compared to wild-type. For example, the gene products of tam41 and taz1 function in the early and late stage of CL biosynthesis, respectively. Accordingly, we observed significant changes in both protein and lipid abundances in Δtam41 and Δtaz1, which results an ultimate reduction of CL levels (Figure 3a). Similarly, for CoQ6 biosynthesis-related Δgene strains, we observed expected dramatic changes in CoQ6 biosynthesis proteins, the lipid CoQ6 itself, and its upstream intermediates (Figure 3b). To our knowledge, this is the first time such multi-omic data has been acquired simultaneously.
Figure 3. Biological study by MOST.
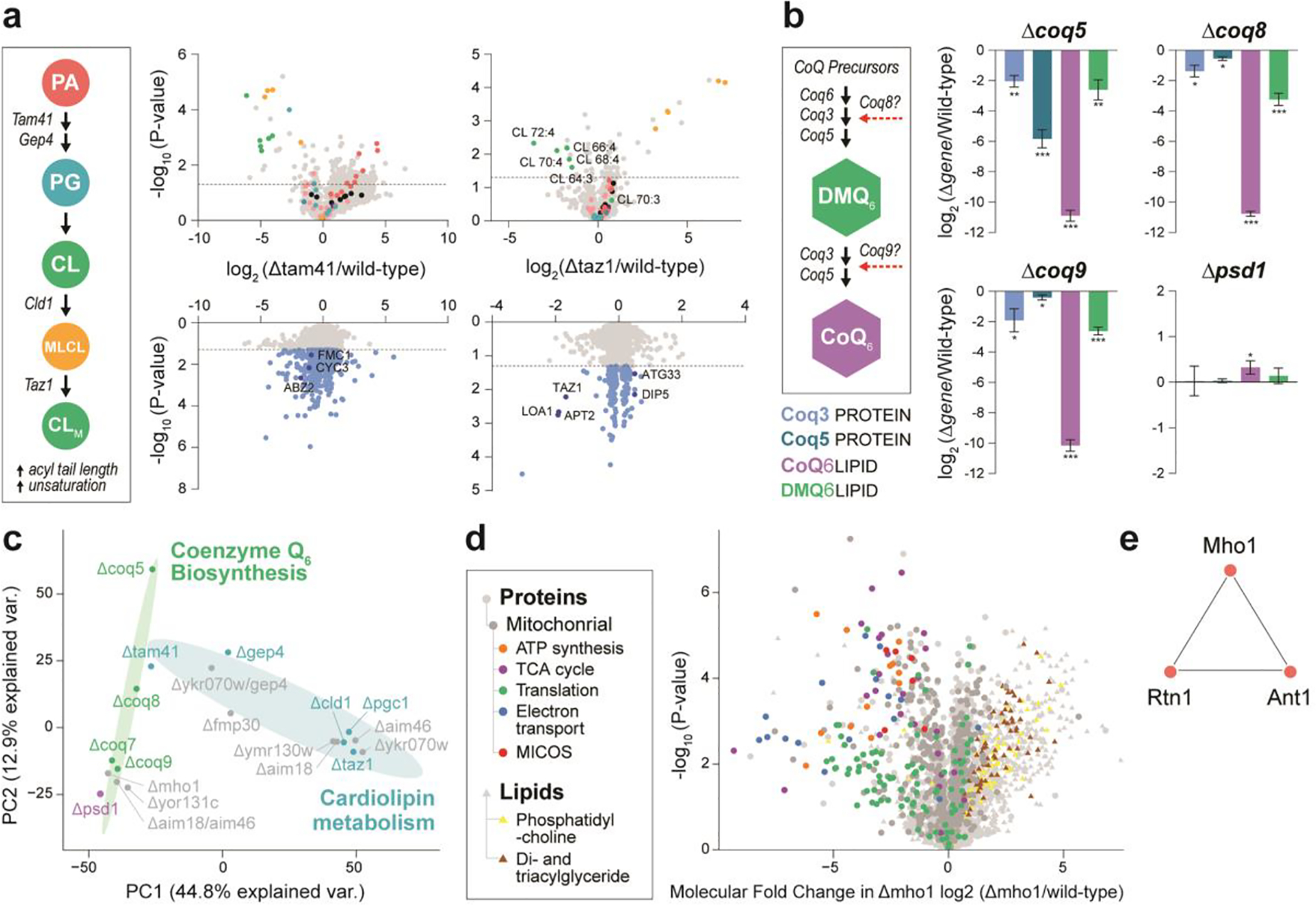
(a) Schematic pathway of cardiolipin biosynthsis and volcano plots across related gene knockout strains. PA, phosphatidic acid. PG, phosphoglycerol. CL, cardiolipin. MLCL, monolysocardiolipin. (b) Schematic pathway of Co-enzyme Q6 biosynthsis and molecule abundance across related gene knockout strains. *, p-value < 0.05. **, p-value < 0.01. ***, p-value < 0.001. (c) Principal component analysis (PCA) of Δgene strains. (d) Volcano plot showing average fold-change in molecule abundances (mean log2[Δmho1/wild-type]) versus statistical significance, showing select functional groups (GO terms and lipid class) significantly (Benjamini-Hochberg adjusted p-value < 0.05) enriched in either upregulated or downregulated molecules. (e) Nearest neighbor covariance network for Mho1.
Serving the multi-omic nature of our MS profiles, we determined pairwise covariance between proteins and lipids to infer gene function (Supplementary Figure S4). Examination of correlations across all proteins and lipids revealed numerous molecular relationships of this multi-omic covariance network, visualized as nodes for molecules and edges for correlations. After applying strict correlation thresholds (Pearson |r| ≥ 0. 8, Benjamini-Hochberg adjusted p-value < 0.001), a striking 477,090 edges between 2,618 nodes remained. For example, loss of Mho1 leads to a severe respiratory deficiency, decreased abundance of prominent mitochondrial proteins, and increased abundance of di- and triacylglycerides (Figure 3c–d). We observed that the poorly characterized yeast protein Mho1 correlates with Rtn1 and Ant1 (Figure 3e), which are important mediators of peroxisomal biogenesis and metabolic activity36,37,38. The mammalian homolog of Mho1, MEMO1, is a copper-binding enzyme that produces O2− and regulates microtubule stability during cell migration39,40. One hypothesis drawn from our covariance analysis is that Mho1 exerts a similar enzymatic activity in yeast and produces ROS that signals to and controls peroxisome homeostasis. While further experimentation is required to verify whether a functional connection exists between Mho1 and peroxisomal organization, we present MOST as an MS approach that can deliver multi-omic data expeditiously and thereby accelerate biochemical discovery.
We have described a method for integrated multi-omics that enables the co-analysis of lipids and peptides. Leveraging this method to analyze a yeast gene deletion collection, we have demonstrated its ability to detect coordinated multi-omic perturbations. Next, we wondered whether co-processing and co-analyzing proteins and lipids might afford increased ability to detect co-regulation across omes. To do this, we compared correlations generated by the different LC-MS methods: single-ome LC and MOST. In doing so we obtained a comparable density of protein-lipid correlation for MOST compared to single-ome LC (Supplementary Figure S4f). In certain cases, the simultaneous analysis of peptides and lipids afforded by MOST improved the ability to detect coregulated proteins and lipids, here exemplified by the well-known coenzyme-Q biosynthetic pathway (as shown in the nearest neighbor molecule covariance networks in Supplementary Figure S4g–h).
CONCLUSIONS
In conclusion, the MOST method we describe here provides a framework to offer integrated multi-omics data acquisition in a simple and robust manner. As compared to traditional fractured methodology, this method has the potential to increase analysis efficiency and simplicity. Further, we demonstrate that the method generates data of equivalent or higher quality to separate approaches in one analysis. That is, with a single LC-MS system, one can create proteomic and lipidomic data simultaneously. In its future iteration, we envision expanding the platform to nano-flow LC41, applying ultrahigh pressure separation developed and improved by Jorgenson group and Kennedy group42,43, which will greatly reduce the required sample amounts while boosting proteomic and lipidomic depth, as well as integrating metabolite co-analysis for a more comprehensive biological survey. Here we envision incorporating the use of ion mobility and intelligent mass spectrometer data acquisition to quickly direct MS data acquisition between co-eluting metabolites and peptides. Finally, we anticipate that future optimization of sample preparation will allow researchers to comprehensively extract and process all three omes from a single sample in a single vial12,13,14,44. Such a platform could represent an ideal technology for studies constricted by limited sample amount, namely clinical research and, ultimately, single cell analyses.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for support from NIH P41 GM108538 (J.J.C.) and NIH R35GM131795 (D.J.P.) and Robert Kennedy for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary methods and results. Table S1 (lipid classes and corresponding abbreviations used in this study); Text S1 (materials and reagents); Text S2 (methods for yeast cultures); Figure S1 (key parameters for method development); Figure S2 (additional performance characteristics of MOST); Figure S3 (MS analysis metrics and quality assessment for biological study); Figure S4 (features of MOST multi-omic molecule covariance networks) (PDF).
The authors have no competing interests.
REFERENCES
- (1).Aebersold R; Mann M Mass-spectrometric exploration of proteome structure and function. Nature. 2016, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- (2).Karpievitch YV; Polpitiya AD; Anderson GA; Smith RD; Dabney AR Liquid chromatography mass spectrometry-based proteomics: biological and technological aspects. Ann. Appl. Stat 2010, 4(4), 1797–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cajka T; Fiehn O Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Analyt Chem. 2014, 61, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hasin Y; Seldin M; Lusis A Multi-omics approaches to disease. Genome. Biol 2017, 18, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Subramanian I; Verma S; Kumar S; Jere A; Anamika K Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yan J; Risacher SL, Shen L; Saykin AJ Network approaches to systems biology analysis of complex disease: integrative methods for multi-omics data. Brief Bioinform. 2018, 19(6):1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pinu FR; Beale DJ; Paten AM; Kouremenos K; Swarup S; Schirra HJ; Wishart D Systems biology and multi-omics integration: viewpoints from the metabolomics research community. Metabolites. 2019, 9(4), 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kerr K; McAneney H; Smyth LJ et al. A scoping review and proposed workflow for multi-omic rare disease research. Orphanet. J. Rare. Dis 2020, 15, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu Y; Mi Y; Mueller T et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol 2019, 37, 314–322. [DOI] [PubMed] [Google Scholar]
- (10).Blum BC et al. Single-platform ‘multi-omic’ profiling: unified mass spectrometry and computational workflows for integrative proteomics–metabolomics analysis. Mol. Omics 2018, 14, 307–319. [DOI] [PubMed] [Google Scholar]
- (11).Misra B et al. Integrated omics: tools, advances and future approaches. Journal of Molecular Endocrinology. 2019, 62(1), R21–R45. [DOI] [PubMed] [Google Scholar]
- (12).Mubeen U; Giraldi LA; Jüppner J; Giavalisco PA Multi-omics extraction method for the in-depth analysis of synchronized cultures of the green alga Chlamydomonas reinhardtii. J. Vis. Exp 2019, 150, e59547. [DOI] [PubMed] [Google Scholar]
- (13).Burnum-Johnson KE; Kyle JE; Eisfeld AJ; Casey CP; Stratton KG; Gonzalez JF; Habyarimana F; Negretti NM; Sims AC; Chauhan S; Thackray LB; Halfmann PJ; Walters KB; Kim YM; Zink EM; Nicora CD; Weitz KK; Webb-Robertson BM; Nakayasu ES; Ahmer B; Konkel ME; Motin V; Baric RS; Diamond MS; Kawaoka Y; Waters KM; Smith RD; Metz TO MPLEx: a method for simultaneous pathogen inactivation and extraction of samples for multi-omics profiling. The Analyst. 2017, 142(3), 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Coman C; Solari FA; Hentschel A; Sickmann A; Zahedi RP; Ahrends R Simultaneous metabolite, protein, lipid extraction (SIMPLEX): a combinatorial multimolecular omics approach for systems biology. Mol Cell. Proteomics 2016, 15(4), 1453–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li Y et al. A novel approach to the simultaneous extraction and non-targeted analysis of the small molecules metabolome and lipidome using 96-Well solid phase extraction plates with column-switching technology. J. Chromatogr. A 2015, 1409, 277–281. [DOI] [PubMed] [Google Scholar]
- (16).Wang S et al. Simultaneous metabolomics and lipidomics analysis based on novel heart-cutting two-dimensional liquid chromatography-mass spectrometry, Anal. Chim. Acta 2017, 966, 34–40. [DOI] [PubMed] [Google Scholar]
- (17).Schwaiger M et al. Merging metabolomics and lipidomics into one analytical run. Analyst. 2019, 144, 220. [DOI] [PubMed] [Google Scholar]
- (18).Stefely JA et al. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol 2016, 34, 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- (20).Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- (21).Cox J et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 2011, 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- (22).Hutchins PJ, Russell JD, Coon JJ. LipiDex: An integrated software package for high-confidence lipid identification. Cell Systems. 2018, 6, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lee Woojong, Kingstad-Bakke Brock, Paulson Brett, Larsen Autumn et al. Carbomer-based adjuvant elicits CD8 T-cell immunity by inducing a distinct metabolic state in cross-presenting dendritic cells. PLoS pathogens. 2021, 17(1), e1009168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Overmyer KA et al. Large-scale multi-omic analysis of COVID-19 severity. Cell systems. 2021, 12(1), 23–40.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Linke V et al. A large-scale genome-lipid association map guides lipid identification. Nature metabolism. 2020, 2(10), 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu Y et al. Orosomucoid-like 3 supports rhinovirus replication in human epithelial cells. American journal of respiratory cell and molecular biology. 2020, 62(6), 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Csardi G & Nepusz T The igraph software package for complex network research. InterJournal, Complex Systems. 2006, 1695. [Google Scholar]
- (28).R Core Team. R: A language and environment for statistical computing. (2017). https://www.R-project.org/
- (29).Fernández-Niño SMG et al. Standard flow liquid chromatography for shotgun proteomics in bioenergy research. Front. Bioeng. Biotechnol 2015, 3(44), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yin X et al. Plasma proteomics for epidemiology increasing throughput with standard-flow rates. Circ Cardiovasc Genet. 2017, 10, e001808. [DOI] [PubMed] [Google Scholar]
- (31).Lenčo J et al. Conventional-flow liquid chromatography −mass spectrometry for exploratory bottom-up proteomic Analyses. Anal. Chem 2018, 90, 5381–5389. [DOI] [PubMed] [Google Scholar]
- (32).Bian Y, Zheng R, Bayer FP et al. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC–MS/MS. Nat Commun. 2020, 11, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ulmer CZ et al. A robust lipidomics workflow for mammalian cells, plasma, and tissue using liquid-chromatography high-resolution tandem mass spectrometry. Methods Mol Biol. 2017, 1609:91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ejsing CS; Sampaio JL; Surendranath V; Duchoslav E; Ekroos K; Klemm RW; Simons K; Shevchenko A Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. U. S. A 2009, 106(7), 2136–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Distler U et al. Enhancing sensitivity of microflow-based bottom-up proteomics through postcolumn solvent addition. Anal. Chem 2019, 91, 7510–7515. [DOI] [PubMed] [Google Scholar]
- (36).De Craene JO et al. Rtn1p is involved in structuring the cortical endoplasmic reticulum. Mol Biol Cell. 2006, 17(7):3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mast FD; Jamakhandi A; Saleem RA et al. Peroxins Pex30 and Pex29 dynamically associate with reticulons to regulate peroxi-some biogenesis from the endoplasmic reticulum. J. Biol. Chem 2016, 291(30):15408–15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).David C; Koch J; Oeljeklaus S et al. A combined approach of quantitative interaction proteomics and live-cell imaging reveals a regulatory role for endoplasmic reticulum (ER) reticulon homology proteins in peroxisome biogenesis. Mol. Cell. Proteomics 2013, 12(9):2408–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Marone R, Hess D, Dankort D, Muller WJ, Hynes NE, & Badache A Memo mediates ErbB2-driven cell motility. Nature cell biology. 2004, 6(6), 515–522. [DOI] [PubMed] [Google Scholar]
- (40).MacDonald G et al. Memo is a copper-dependent redox protein with an essential role in migration and metastasis. Science signaling. 2014, 7(329), ra56. [DOI] [PubMed] [Google Scholar]
- (41).Danne-Rasche N et al. Nano-LC/NSI MS refines lipidomics by enhancing lipid coverage, measurement sensitivity, and linear dynamic range. Anal. Chem 2018, 90, 809. [DOI] [PubMed] [Google Scholar]
- (42).Grinias KM; Godinho JM; Franklin EG; Stobaugh JT; Jorgenson JW Development of a 45kpsi ultrahigh pressure liquid chromatography instrument for gradient separations of peptides using long microcapillary columns and sub-2μm particles. J. Chromatogr. A 2016, 1469, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sorensen MJ; Miller KE; Jorgenson JW; Kennedy RT Ultrahigh-Performance capillary liquid chromatography-mass spectrometry at 35 kpsi for separation of lipids, J. Chromatogr. A 2020, 1611, 460575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol 2014, 10(10), 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.