

Abstract
The covalent linkage of catalytic units to aptamer sequence-specific nucleic acids exhibiting selective binding affinities for substrates leads to functional scaffolds mimicking native enzymes, nucleoapzymes. The binding of the substrates to the aptamer and their structural orientation with respect to the catalytic units duplicate the functions of the active center of enzymes. The possibility of linking the catalytic sites directly, or through spacer units, to the 5′-end, 3′-end, and middle positions of the aptamers allows the design of nucleoapzyme libraries, revealing structure–functions diversities, and these can be modeled by molecular dynamics simulations. Catalytic sites integrated into nucleoapzymes include DNAzymes, transition metal complexes, and organic ligands. Catalytic transformations driven by nucleoapzymes are exemplified by the oxidation of dopamine or l-arginine, hydroxylation of tyrosine to l-DOPA, hydrolysis of ATP, and cholic acid-modified esters. The covalent linkage of photosensitizers to the tyrosinamide aptamer leads to a photonucleoapzyme scaffold that binds the N-methyl-N′-(3-aminopropane)-4,4′-bipyridinium-functionalized tyrosinamide to the aptamer. By linking the photosensitizer directly, or through a spacer bridge to the 5′-end or 3′-end of the aptamer, we demonstrate a library of supramolecular photosensitizer/electron acceptor photonucleoapzymes mimicking the functions of photosystem I in the photosynthetic apparatus. The photonucleoapzymes catalyze the photoinduced generation of NADPH, in the presence of ferredoxin-NADP+-reductase (FNR), or the photoinduced H2 evolution catalyzed by Pt nanoparticles. The future prospects of nucleoapzymes and photonucleoapzymes are discussed.
The information encoded in the base sequence of nucleic acids has been used, in the past two decades, to apply the biopolymer as a functional material to develop new catalysts.1 The discovery of native nucleic acids (ribozymes) sparked tremendous efforts to evolve synthetically sequence-specific RNA- or DNA-based catalysts (ribozymes or DNAzymes).2,3 Indeed, a wide variety of catalytic nucleic acids catalyzing the nicking or ligation of nucleic acid substrates, using cofactor-dependent sequence-specific oligonucleotides (cofactor as a metal ion or amino acid), were reported.4,5 These catalytic transformations are limited, however, to substrates that form duplex recognition complexes with their substrates. In addition, the hemin cofactor conjugated to different configurations of the G-quadruplex acts as a versatile horseradish peroxidase-mimicking DNAzyme that catalyzes the H2O2-mediated oxidation of organic substrates that yields, similar to the native peroxidase, chromophores (dyes) or fluorescent products6−9 and generates chemiluminescence, in the presence of luminol.10 Also, hemin/G-quadruplex DNAzymes were broadly applied to mimic peroxidase reactions, such as the H2O2-catalyzed oxidation of phenols,11 thiols,12 NADH,13 or aniline.14 During these transformations, no specific binding of the substrates to the catalytically active intermediates occurred, leading to limited turnover rates as compared to those of the native enzymes. A different approach for using nucleic acids as functional materials to develop catalysts has involved the direct linkage of metal ions to the oligonucleotide scaffolds or the binding of metal ions to ligands covalently tethered to the DNA scaffolds. Different catalyzed transformations, such as carbon–carbon bond formation,15 Diels–Alder,16−18 or Michael addition reactions,19 and the phosphorylation of hydroxyl groups,20 were demonstrated by these oligonucleotide/metal complex hybrids. Although impressive chiroselective yields induced by the chiral DNA ligands were demonstrated, the systems revealed low turnover rates due to the lack of engineered binding sites.
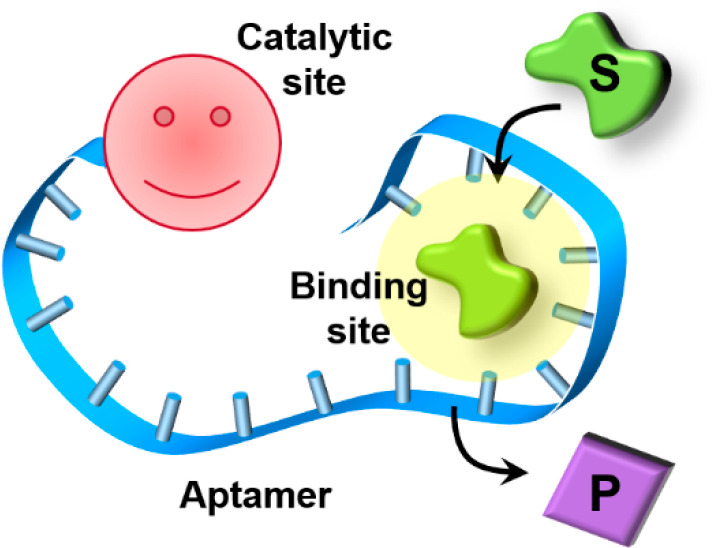
Aptamers are sequence-specific nucleic acids that exhibit selective binding properties with respect to low-molecular weight substrates and macromolecules.21,22 These binding features of aptamers were used to develop sensors,23−26 switches,27−29 and stimuli-responsive drug carriers,30,31 and the aptamers were applied as therapeutic agents.32−34 In addition, it was demonstrated that mutation of the base sequences of aptamers and the modification of aptamers with redox groups35 or photoisomerizable36 units can enhance or switch the binding properties of aptamers. These unique features of aptamers guided the development of a new class of nucleic acid biocatalysts as schematically outlined in Figure 1A. The covalent tethering of a catalytic unit to the chiral aptamer-binding sequence yields a hybrid structure that mimics the functions of native enzymes.37 The aptamer-binding site concentrates the substrate in spatial proximity to the catalytic unit, thereby mimicking the fundamental features of native enzymes, where the concentration of the substrate in the proximity of the active catalytic unit provides nature’s “reactivity secret”. The possibility of tethering the catalytic site at the 3′- or 5′-end of the aptamer or in intrachain positions and the feasibility of tethering the catalyst to the aptamer via flexible bridges or linking the catalyst between split aptamer subunits (Figure 1B) provide a library of possible catalysts, termed by us “nucleoapzymes”, that are expected to reveal variable catalytic functions.37 An attempt to understand the structure–function relationships within this set of nucleoapzymes by means of molecular dynamics simulations could eventually allow, in the future, the in silico design of superior nucleoapzymes. Different catalysts were anchored to aptamers to construct the nucleoapzymes, and these included DNAzymes or transition metal complexes. In this Perspective, we summarize recent advances in the development of nucleoapzymes and broaden the concept to photonucleoapzymes, where photocatalytic–aptamer conjugates mimic the functions of photosynthesis.
Figure 1.
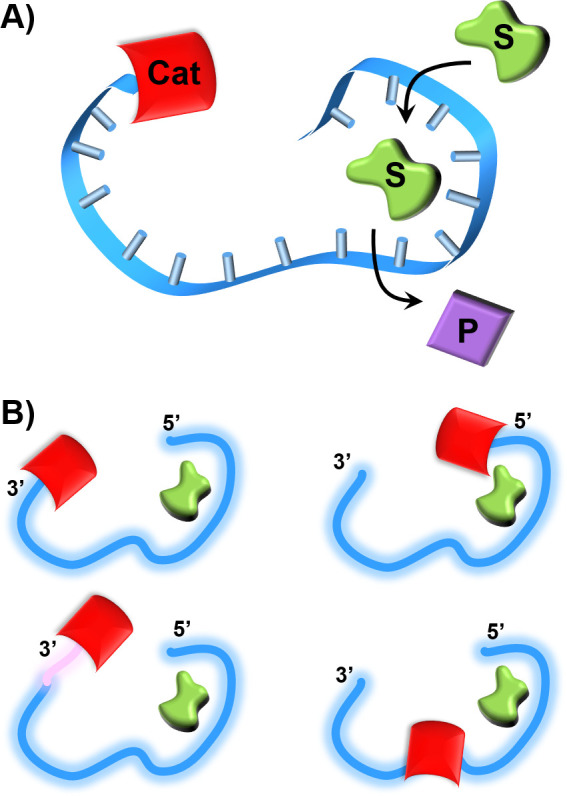
(A) Schematic structure of a nucleoapzyme consisting of a catalyst (Cat) and aptamer conjugate. S = substrate, P = product (B) Schematic library of nucleoapzymes consisting of a catalyst linked directly or through a spacer bridge (pink domain) to the 3′- or 5′-end of the aptamer scaffold or where the catalyst separates the “split” aptamer subunits.
DNAzyme–Aptamer Conjugates as Nucleoapzymes
Realizing that peroxidases catalyze the oxidation of dopamine (1) to aminochrome (2), Golub et al. prepared a series of hemin/G-quadruplex-dopamine aptamer nucleoapzymes37 (Figure 2A). Figure 2B presents the rates of oxidation of the dopamine substrate (1) by H2O2 in the presence of the hemin/G-quadruplex-dopamine aptamer nucleoapzymes, where the catalyst is conjugated to the 5′-end of the aptamer through a TATA spacer (structure I, curve a), to the 5′-end of the aptamer through a single A base (structure II, curve b), or to the 3′-end of the aptamer through a TATA spacer (structure III, curve c). For comparison, the H2O2-catalyzed oxidation of the dopamine substrate (1) by the separated hemin/G-quadruplex and aptamer units (configuration IV) is shown in curve d. The different nucleoapzymes examined in the study demonstrated superior catalytic activities compared to the oxidation of dopamine (1) to aminochrome (2) by the separated units. Nonetheless, the catalytic activities of the nucleoapzymes revealed structure-controlled activities. While the nucleoapzyme in configuration I revealed a 20-fold catalytic enhancement as compared to the separated components, the nucleoapzyme in configuration III showed an only 3-fold catalytic enhancement as compared to the separated components. Also, the flexibility of the tethering chain linking the catalyst to the aptamer scaffold affected the catalytic activity of the resulting nucleoapzyme. The different nucleoapzyme revealed a Michaelis–Menten type kinetics, exhibiting saturation rates upon full occupation of the aptamer-binding sites: Vmax values of 13.5 ± 0.5, 9.6 ± 0.2, and 2.6 ± 0.2 nM s–1 and kcat values of (18.3 ± 0.9), (13.0 ± 0.7), and (3.1 ± 0.1) × 10–3 s–1 for configurations I–III, respectively. As the binding affinities of dopamine (1) for the different nucleoapzymes were similar, the structures of the intermediate substrate complexes have a dominant effect on the catalytic performance of the nucleoapzymes.
Figure 2.

(A) Hemin/G-quadruplex-dopamine aptamer nucleoapzyme structures where in configuration I the catalyst is linked through a TATA tether to the 5′-end of the aptamer. In configuration II, the catalyst is tethered to the 5′-end of the aptamer through a single A bridge. In configuration III, the catalyst is linked to the 3′-end of the aptamer through a TATA tether. Configuration IV represents the control system, where the hemin/G-quadruplex catalyst is separated from the aptamer scaffold. The reaction driven by the systems corresponds to the hemin/G-quadruplex-catalyzed oxidation of dopamine (1) by H2O2 to aminodopachrome (2). (B) Rates of oxidation of dopamine (1) by H2O2 at different concentrations of dopamine (1): (a) nucleoapzyme configuration I, (b) nucleoapzyme configuration II, (c) nucleoapzyme configuration III and (d) control system consisting of the separated components (configuration IV). (C) Molecular dynamics energy-minimized structures of (I) nucleoapzyme in configuration I and (II) nucleoapzyme in configuration III.
Molecular dynamics (MD) simulations were used to rationalize the experimental differences in the catalytic activities of the nucleoapzymes in terms of energetically favored structures of the hemin/G-quadruplex-dopamine aptamer conjugates that define the distances and orientations of the catalytic sites with respect to the aptamer-binding site. These two parameters dictate the frequency and probability of intimate contact between the catalyst and the substrate associated with the binding site. Besides the spatial and dynamic relation between the catalytic units and the substrate-binding site, the accessibility of the catalytic site to the binding site can be similarly visualized by MD simulations. Panel I of Figure 2C depicts the MD-simulated structure of the most active hemin/G-quadruplex-dopamine aptamer nucleoapzyme composed of the catalytic site linked through the TATA spacer to the 5′-end of the aptamer (configuration I). The catalytic site is separated from the aptamer wide rim pocket by a distance corresponding to 3–5 nm, leading to a short reaction distance and accessibility of the catalyst to the dopamine substrate (1) associated with the aptamer-binding site. The MD-stimulated structure of the least effective nucleoapzyme, nucleoapzyme in configuration III, where the catalytic site is linked to the 3′-end of the aptamer through the TATA tether, is shown in panel II of Figure 2C. The distance separating the catalytic site from the binding site is substantially longer (9–15 nm), and the catalyst is oriented toward the narrow rim of the binding site, where the association of the dopamine substrate (1) is perturbed. All of these structural features of the nucleoapzyme in configuration II lead to the lower activity of the hybrid conjugate. This concept of hemin/G-quadruplex-aptamer catalytic conjugates was further applied to design nucleoapzymes for the H2O2-mediated oxidation of l-arginine to citrulline using the l-arginine aptamer as the binding site.37
Metal–Ligand Complex-Functionalized Aptamers as Nucleoapzymes
The available number of DNAzymes that can be conjugated to aptamers limits the broadening of the concept of a nucleoapzyme. The substitution of DNAzymes by metal–ligand complexes, which exhibit catalytic functions, as catalytic units conjugated to aptamer-binding sequences, may provide a versatile path to yield metal–ligand complex-functionalized aptamers as enzyme-mimicking hybrid systems. This concept has been exemplified with the development of a series of Cu(II)–terpyridine-modified dopamine aptamers and a series of Fe(III)–terpyridine-modified dopamine aptamers as nucleoapzymes that catalyze the H2O2-mediated oxidation of dopamine (1) to aminochrome (2).38Figure 3A depicts the Fe(III)–terpyridine-modified dopamine aptamer catalyzed oxidation of dopamine (1) to aminochrome (2) using the nucleoapzyme consisting of the Fe(III)–terpyridine catalyst tethered to the 5′-end of the aptamer through a 4T bridge, configuration I, and the nucleoapzyme composed of the catalyst linked to the 3′-end of the aptamer through the 4T bridge, configuration II. Figure 3B shows that the rates of dopamine oxidation by the nucleoapzymes in configurations I and II are 180- and 35-fold faster, respectively, than the rates of oxidation of the substrate by the separated catalyst and aptamer units (curves a and b vs curve c). Kinetic analysis of the nucleoapzymes activity reveals that the values of kcat and KM are 267 × 10–4 s–1 and 33 ± 12 μM and 200 × 10–4 s–1 and 39 ± 18 μM for configurations I and II, respectively. As the binding affinity of the substrate for the two nucleoapzymes was similar, the enhanced catalytic activity of configuration I was attributed to the higher catalytic performance of the catalytic site in configuration I. Indeed, MD simulations revealed that the Fe(III)–terpyridine catalytic site is closer (30 Å) to the dopamine-binding site in configuration I, panel I of Figure 3C, as compared to the longer spatial separation of the catalytic site from the substrate-binding site in configuration II (43 Å), panel II of Figure 3C, suggesting the increased probability of the flexible catalytic unit reacting with the substrate in nucleoapzyme configuration I (Figure 3C).
Figure 3.

(A) Schematic structures of Fe3+–terpyridine-modified dopamine aptamer nucleoapzymes for the catalyzed oxidation of dopamine (1) by H2O2 to yield dopachrome (2). (B) Rates of oxidation of dopamine (1) to dopachrome (2) in the presence of H2O2 using variable concentrations of dopamine (1): (a) using the nucleoapzyme in configuration I, (b) using the nucleoapzyme in configuration II, and (c) using the separated Fe3+–terpyridine catalyst and the dopamine aptamer. (C) Molecular dynamics energy-minimized structures of (I) the nucleoapzyme in configuration I and (II) the nucleoapzyme in configuration II.
In addition, the Fe(III)–terpyridine complex linked to the tyrosinamide aptamer was found to act as a nucleoapzyme that catalyzes the oxidation of tyrosinamide (3) to amidodopachrome (5) by H2O2 (Figure 4A).39 This oxygen-insertion process occurred only in the presence of an ascorbic acid/H2O2 mixture. A set of Fe(III)–terpyridine-functionalized tyrosinamide aptamers, in which the catalytic site was attached directly to the 5′- and 3′-ends of the aptamer (configurations I and II, respectively) or through 4T bridging tethers (configurations III and IV), were assembled. In the primary step, the oxidation of tyrosinamide (3) to the amidated l-DOPA (4) proceeded, and this was followed by the oxidation of 4 to amidodopachrome (5). Similarly, the cascaded oxidation of tyrosine (6) to l-DOPA (7) and dopachrome (8) was driven by the Fe(III)–terpyridine-functionalized tyrosinamide aptamer. Figure 4B depicts the oxidation of tyrosinamide (3) by nucleoapzyme configurations III and IV, in the presence of a H2O2/ascorbic acid mixture, in comparison to oxidation of 3 by the separated catalyst and aptamer components (curves a and b vs curve c). A 100- and 80-fold enhanced oxidation of 3 by the nucleoapzyme in configurations III and IV was observed (for configuration III, kcat = 11.8 ± 0 s–1 and KM = 205 μM; for configuration IV, kcat = 9.1 ± 0.2 s–1 and KM = 1.93 μM). Mechanistic insight into the catalytic oxidation of tyrosinamide to the catechol product by the H2O2/ascorbic acid mixture was obtained by electron spin resonance (ESR) measurements (Figure 4C). While the formation of •OH and ascorbate radical by the nucleoapzyme in configuration III is inefficient, in the presence of the H2O2/ascorbic acid mixture effective formation of the two radicals was detected (Figure 4D). The effective formation of the two radicals was attributed to a mutual synergistic cooperative formation of the two radicals, where the small amount of ascorbate radicals reacts with H2O2 to yield peroxo ascorbate and •OH. The peroxo ascorbate dissociates to ascorbate radical and •OH, and the resulting •OH re-forms the ascorbate (Figure 4D). This chain reaction provides a constant supply of the reactive oxygen species, •OH, and ascorbate radical. The two reactive species participate then in the formation of the tyrosine radical that recombines with •OH.
Figure 4.

(A) Schematic stepwise oxidation of tyrosinamide (3) or tyrosine (6) by H2O2/ascorbic acid to amidodopachrome (4) and dopachrome (8) using Fe3+–terpyridine-modified tyrosinamide aptamers as nucleoapzymes. (B) Rates of oxidation of tyrosinamide by a H2O2/ascorbic acid mixture to amidodopachrome (5) at different concentrations in the presence of (a) the Fe3+–terpyridine-functionalized tyrosinamide nucleoapzyme in configuration III, (b) the Fe3+–terpyridine-functionalized tyrosinamide nucleoapzyme in configuration IV, and (c) the separated Fe3+–terpyridine catalyst and the tyrosinamide aptamer. (C) Suggested cooperative feedback radical chain reaction driven by H2O2 and ascorbic acid in the presence of the Fe3+–terpyridine catalyst leading to the insertion of •OH into the tyrosine residue. (D) ESR spectrum corresponding to the •OH and ascorbate radical generated by the nucleoapzyme in configuration III in the presence of the H2O2/ascorbic acid mixture.
Besides oxidative nucleoapzymes, hydrolytic metal–organic complexes linked to aptamer-binding scaffolds were reported. The catalytic hydrolysis of ATP to ADP by a set of nucleoapzymes consisting of bis-Zn2+-pyridyl-salen type complexes linked directly or through 2T spacers to the 5′-end or 3′-end of the ATP aptamer (configurations I–IV) is shown in Figure 5A.40Figure 5B depicts the catalytic performance of the 3′-2T-catalyst-ATP aptamer conjugate (configuration III) and the 5′-2T-catalyst-ATP aptamer conjugate (configuration IV) in comparison to the separated catalyst and aptamer components (curves a–c, respectively). While the separated components do not show catalytic hydrolysis of ATP, the 3′-2T-catalyst-modified aptamer conjugate shows a superior catalytic rate compared to that of the 5′-end catalyst-modified aptamer (for configuration III, kcat = 688 × 10–2 min–1 and KM = 38 ± 7 μM; for configuration IV, kcat = 297 × 10–2 min–1 and KM = 33 ± 6 μM). As the binding affinities of ATP for the different nucleoapzymes were similar (Kd = 19 μM), it was concluded that the difference in the catalytic rates of the nucleoapzyme in configurations III and IV originates from the favored catalytic activity of the bis-Zn2+-pyridyl-salen complex in nucleoapzyme configuration III. MD simulations of the energetically stabilized structures of the nucleoapzyme in configurations III and IV indicated that the catalytic site in the nucleoapzyme in configuration III is positioned in a sterically favored configuration with respect to the hydrolytic reaction site, as compared to the spatial separation of the catalytic site from the reaction site in the nucleoapzyme in configuration IV (Figure 5C). While the distance separating the catalytic site from the reaction site in the nucleoapzyme in configuration III is 18 Å, panel I Figure 5C, the distance separating the catalytic site from the reaction site in nucleoapzyme configuration IV corresponds to 44 Å, panel II of Figure 5C.
Figure 5.

(A) Schematic configurations of bis-Zn2+-pyridyl-salen-modified ATP aptamers acting as nucleoapzymes for the catalyzed hydrolysis of ATP to ADP. (B) Rates of hydrolysis of ATP to ADP by representative nucleoapzymes: (a) nucleoapzyme in configuration III, (b) nucleoapzyme in configuration IV, and (c) control experiment using the separated bis-Zn2+-pyridyl-salen catalyst and the ATP aptamer. (C) Molecular dynamics energy-minimized structures of the nucleoapzyme in (I) configuration III and (II) configuration IV.
Ligand-Functionalized Nucleoapzymes
Specific amino acids in proteins such as histidine, lysine, or glutamate, in the proximity of the active site of enzymes, often participate in the catalytic process within the catalytic center by providing Lewis acids or bases, or nucleophilic functionalities for the activation of the active site-bound reaction substrates. In the past, these functionalities were tethered to artificial receptors such as crown ethers41,42 or cyclodextrins43 as part of efforts to mimic native enzymes by supramolecular structures. Similarly, low-molecular weight ligands were tethered to aptamers as part of efforts to develop nucleoapzymes. For example, imidazole was tethered to the sequence-specific aptamer recognizing cholic acid. The imidazole-functionalized aptamer (Dcat1) acted as a nucleoapzyme catalyzing the hydrolysis of coumarin-modified cholic acid ester (9) to cholic acid (10) (Figure 6). The catalyzed hydrolysis of 9 was 100-fold faster than the hydrolysis of 9 by the separated aptamer and imidazole units. Kinetic characterization of the nucleoapzyme revealed Michaelis–Menten kinetics (kcat = 0.8 ± 0.1 h–1, and KM = 26 ± 6 μM). A set of related nucleoapzymes that included the imidazole ligand at other positions of the aptamer was prepared. All nucleoapzymes revealed enhanced activity with respect to the hydrolysis of 9, as compared to the separated components, yet DCat1 exhibited superior hydrolytic activity among the nucleoapzymes, demonstrating structure–function relationships of the nucleoapzyme library.44
Figure 6.
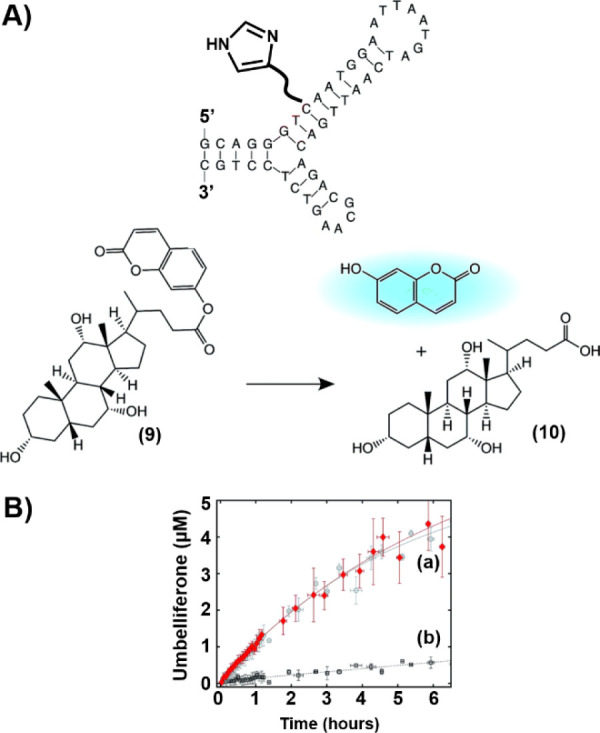
(A) Catalyzed hydrolysis of coumarin-modified cholic acid ester (9) by the imidazole-functionalized cholic acid aptamer. (B) Time-dependent hydrolysis of 9 by (a) the imidazole-modified aptamer and (b) the separated imidazole and aptamer units.
Photonucleoapzymes as Photosynthetic Model Systems
Mimicking photosynthesis by artificial means is one of the “holey-grail” challenges in science.45,46 These efforts include attempts to mimic the primary electron transfer events of the native photosystems of the photosynthetic apparatus (Z-scheme) by means of ingenious supramolecular systems47−50 and organized microenvironments50,51 that control the forward electron transfer (ET)/back reaction processes and involve efforts to utilize the photogenerated redox species to drive chemical transformations such as H2 evolution,52−54 CO2 reduction,55,56 or biocatalytic processes.57 Different heterogeneous catalysts, such as Pt,58,59 Pd,56 or Ru55 nanoparticles, were applied to catalyze H2 evolution or CO2 fixation processes, and homogeneous catalysts catalyzing H2 evolution60,61 and enzymes catalyzing the generation of NAD(P)H,57 CO2 fixation, or the synthesis of amino acids62 were coupled to the photoinduced ET transfer reactions to induce secondary fuel-generating processes or valuable chemical-forming transformations.
The successful introduction of the nucleoapzyme concept that organizes the catalytic units and reaction substrate by means of an aptamer scaffold was adapted to organize supramolecular photonucleoapzyme structures, where the photosensitizer unit and electron acceptor unit are forced into spatial proximity by means of an aptamer scaffold. That is, the noncovalent supramolecular association of an acceptor with a photosensitizer-functionalized aptamer yields the primary intimate structure between the light-harnessing component and the primary electron acceptor unit, where effective photoinduced ET proceeds, similar to the primary events in the photosynthetic reaction center. Figure 7A introduces the concept of a photonucleoapzyme by the functionalization of the tyrosinamide aptamer with a Ru(bppy)32+ photosensitizer.63 The association of the tyrosinamide ligand modified with the N-methyl-N′-(3-aminopropane)-4,4′-bipyridinium electron acceptor (11) with the aptamer scaffold generated the supramolecular photonucleoapzyme assembly in which effective intracomplex electron transfer occurs. The photogenerated redox products were subsequently coupled to photosynthetic transformations involving the synthesis of NADPH and the evolution of hydrogen. Four different photonucleoapzymes were prepared, where the Ru(bppy)32+ photosensitizer was covalently linked directly to the 5′- and 3′-ends of the aptamer (configurations I and II, respectively) or bridged to the 5′- and 3′-ends through 4T spacers (configurations III and IV, respectively). Effective, nonlinear, intramolecular ET quenching within all photonucleoapzyme structures was demonstrated (Figure 7B, curves a–d). The quenching efficiencies revealed structure dependence and decreased in the following order: IV > III > II > I. Nonetheless, the quenching efficiencies for all photosensitizer-aptamer conjugates were substantially higher than those of the diffusional ET quenching process of the separated Ru(bppy)32+ photosensitizer and the tyrosinamide aptamer in the presence of the tyrosinamide/bipyridinium quencher (Figure 7B, curve e). The structure-controlled electron transfer quenching efficiencies within the set of photonucleoapzymes dictated the yields of the resulting tyrosinamide/bipyridinium radical cation, TA-MV+•, formed under steady-state irradiation in the presence of Na2EDTA as a sacrificial electron donor.
Figure 7.

(A) Schematic configurations of photonucleoapzymes as model systems mimicking photosynthesis. The artificial photosynthetic systems drive the light-induced synthesis of NADPH or the evolution of hydrogen. The photonucleoapzymes consist of a photosensitizer S [S = Ru(II)-tris-pyridine electron acceptor] linked to the tyrosinamide aptamer that binds the N-methyl-N′-(3-aminopropane)-4,4′-bipyridinium-tyrosinamide electron acceptor. The resulting supramolecular photosensitizer/electron acceptor complex mimics the primary electron transfer cascade of native photosystem I. The scheme outlines four different configurations, I–IV, of photonucleoapzymes. (B) Fluorescence quenching curves revealed by the photonucleoapzymes in (a) configuration I, (b) configuration II, (c) configuration III, and (d) configuration IV. (e) Diffusional electron transfer quenching of the separated photosensitizer and the N-methyl-N′-(3-aminopropane)-4,4′-bipyridinium-tyrosinamide electron acceptor bound to the aptamer.
The yields of TA-MV+· guided the yields of the subsequent catalyzed chemical transformations (Figure 8A). For example, Figure 8B depicts the time-dependent TA-MV+• driven reduction of NADP+ to NADPH catalyzed by ferredoxin-NADP+-reductase (FNR). The quantum yields for the formation of NADPH by the different photonucleoapzymes followed the primary ET quenching efficiencies: IV (4.1%) > III (3.6%) > II (2.9%) > I (2.6%). These were substantially higher than the quantum yield for the generation of NADPH by the separated Ru(bppy)32+ and aptamer in the presence of TA-MV2+ (<0.2%) (cf. Figure 8B, curve e). Also, the photosensitized generation of TA-MV+• was coupled to the Pt nanoparticle-catalyzed evolution of the hydrogen fuel (Figure 8A). The time-dependent yields of H2 produced by the different photonucleoapzymes are presented in Figure 8C. The H2 evolution yields by all photonucleoapzyme nanostructures (configurations I–IV) are substantially higher than the H2 evolution yield of the separated Ru(bppy)32+ and aptamer system in the presence of TA-MV2+ (curves a–d vs curve e in Figure 8C). The quantum yields of H2 evolution followed the primary ET quenching efficiencies in the supramolecular photonucleoapzyme structures [IV (3.0%) > III (3.2%) > II (2.2%) > I (1.8%) versus the quantum yield of H2 evolution by the separated components (0.31%)]. The concept of photonucleoapzymes was further expanded by designing a library of Zn(II)-protoporphyrin IX/G-quadruplex chromophore-tyrosinamide aptamer conjugates operating as photonucleoapzymes, thus demonstrating the versatility of the concept for the development of new photocatalytic systems.64
Figure 8.
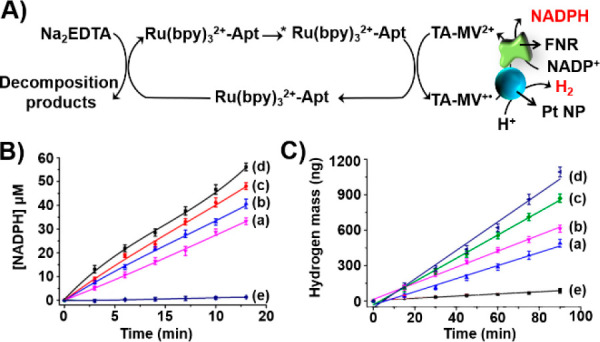
(A) Schematic photosensitized FNR-catalyzed reduction of NADP+ to NADPH and Pt nanoparticle-catalyzed H2 evolution driven by the series of photonucleoapzymes in the presence of the N,N′-bipyridinium-tyrosinamide electron acceptor and Na2EDTA as a sacrificial electron donor. (B) Time-dependent light-induced FNR-catalyzed formation of NADPH by (a) photonucleoapzyme configuration I, (b) photonucleoapzyme configuration II, (c) photonucleoapzyme configuration III, (d) photonucleoapayme configuration IV, and (e) a reference system consisting of the separated Ru(II)-bipyridine photosensitizer and the N-methyl-N′-(3-aminopropane)-4,4′-bipyridinium-tyrosinamide electron acceptor bound to the tyrosinamide aptamer. (C) Time-dependent light-induced Pt nanoparticle-catalyzed H2 evolution by (a) photonucleoapzyme configuration I, (b) photonucleoapzyme configuration II, (c) photonucleoapzyme configuration III, (d) photonucleoapayme configuration IV, and (e) a reference system consisting of the separated Ru(II)-bipyridine photosensitizer and the N-methyl-N′-(3-aminopropane)-4,4′-bipyridinium-tyrosinamide electron acceptor bound to the tyrosinamide aptamer.
Conclusions and Perspectives
The structural modification of aptamers with catalytic and photocatalytic units to yield nucleoapzymes and photonucleoapzymes proved to be a viable and versatile approach for mimicking native enzymes and photosynthetic reaction centers. Although substantial advances were demonstrated, important challenges are ahead of us. (i) At present, the catalytic performance of nucleoapzymes is substantially lower than that of native enzymes. For example, the kcat value of the ATPase-mimicking nucleoapzyme, configuration III, is ∼103-fold lower than that of the native F1-ATPase.65 Nonetheless, the advantages of the nucleoapzyme as compared to native enzymes should be mentioned. These include the stability of the nucleoapzymes as compared to the native enzyme and, specifically, the diversity and programmability of the nucleoapzyme with respect to diverse substrates dictated by the elicited aptamers. This programmability is nonexistent in native enzymes. Although substantial advances in the engineering of nucleoapzymes were demonstrated, several pathways to further improve the catalytic performance of nucleoapzymes may be suggested. The catalytic rates of the nucleoapzymes should be improved. This could be accomplished by enhancing the binding affinity of the substrates for the aptamer-binding site (increase in the local molarity). This may be achieved by the mutation of the aptamer-binding sites66 and by chemical modules that allosterically stabilize substrate-aptamer complexes.35 In addition, the programmed positioning of the catalytic sites with respect to the substrate-binding site by means of the DNA scaffold could be a general means of controlling the distance between the catalytic unit and the substrate-binding site. Indeed, such a programmable hemin/G-quadruplex/dopamine aptamer supramolecular “ruler” demonstrated distance-controlled catalytic functions.67 Furthermore, the design of new catalytic modules, e.g., other ligand–metal ion complexes, or cofactor-dependent DNAzymes, e.g., metal ion, or amino acid (histidine)-dependent DNAzymes to the aptamer would provide a versatile means of localizing active sites in the proximity of the substrates. (ii) Molecular dynamics simulations provide a useful computational tool for evaluating the structure–function relationships of nucleoapzymes. Our results suggest that adapting this tool for the in silico design of new nucleoapzymes provides insights into new catalytic scaffolds. (iii) The nucleic acid scaffolds being a part of the nucleoapzymes or photonucleoapzymes allow, in principle, the assembly of bifunctional or trifunctional nucleoapzymes or photonucleoapzymes. For example, by the conjugation of two (or three) aptamers by duplex or Y-shaped bridges, each modified by a different catalyst, the operation of catalytic cascades may be envisaged. Similarly, by the conjugation of one aptamer (or more) to a photosensitizer-functionalized aptamer scaffold, photoinduced electron transfer cascades leading to effective charge separation could be realized. In addition, the conjugation of the photosensitizer-aptamer scaffolds would allow the programmed biphotonic operation of photocatalytic assemblies mimicking the Z-scheme of the native photosynthetic apparatus. (iv) The integration of nucleoapzymes and photonucleoapzymes in cell-like containers, such as vesicles,68,69 microcapsules,70,71 or polymersomes,72,73 could lead to synthetic cell systems, thus providing new concepts in the field of systems chemistry. All of these challenges provide a broad interdisciplinary area for chemists, physicists, material scientists, and researchers active in nanoscience and nanobiotechnology.
Our research on nucleoapzymes and photonucleoapzymes is supported by the Volkswagen Foundation, Germany.
The authors declare no competing financial interest.
References
- Wang F.; Lu C.-H.; Willner I. (2014) From Cascaded Catalytic Nucleic Acids to Enzyme–DNA Nanostructures: Controlling Reactivity, Sensing, Logic Operations, and Assembly of Complex Structures. Chem. Rev. 114, 2881–2941. 10.1021/cr400354z. [DOI] [PubMed] [Google Scholar]
- Breaker R. R.; Joyce G. F. (1994) A DNA enzyme that cleaves RNA. Chem. Biol. 1, 223–229. 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Silverman S. K. (2016) Catalytic DNA: Scope, Applications, and Biochemistry of Deoxyribozymes. Trends Biochem. Sci. 41, 595–609. 10.1016/j.tibs.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S.-M.; Jeong S.; Kim J.-M.; Chong B.-O.; Park Y.-K.; Park H.; Yu J. (1999) Cholesterol Esterase Activity by in Vitro Selection of RNA against a Phosphate Transition-State Analogue. J. Am. Chem. Soc. 121, 10844–10845. 10.1021/ja991848u. [DOI] [Google Scholar]
- Famulok M.; Hartig J. S.; Mayer G. (2007) Functional Aptamers and Aptazymes in Biotechnology, Diagnostics, and Therapy. Chem. Rev. 107, 3715–3743. 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- Sen D.; Poon L. C. H. (2011) RNA and DNA complexes with hemin [Fe(III) heme] are efficient peroxidases and peroxygenases: how do they do it and what does it mean?. Crit. Rev. Biochem. Mol. Biol. 46, 478–492. 10.3109/10409238.2011.618220. [DOI] [PubMed] [Google Scholar]
- Travascio P.; Bennet A. J.; Wang D. Y.; Sen D. (1999) A ribozyme and a catalytic DNA with peroxidase activity: active sites versus cofactor-binding sites. Chem. Biol. 6, 779–787. 10.1016/S1074-5521(99)80125-2. [DOI] [PubMed] [Google Scholar]
- Nakayama S.; Wang J.; Sintim H. O. (2011) DNA-Based Peroxidation Catalyst-What Is the Exact Role of Topology on Catalysis and Is There a Special Binding Site for Catalysis?. Chem. - Eur. J. 17, 5691–5698. 10.1002/chem.201002349. [DOI] [PubMed] [Google Scholar]
- Georgiades S. N.; Abd Karim N. H.; Suntharalingam K.; Vilar R. (2010) Interaction of Metal Complexes with G-Quadruplex DNA. Angew. Chem., Int. Ed. 49, 4020–4034. 10.1002/anie.200906363. [DOI] [PubMed] [Google Scholar]
- Pavlov V.; Xiao Y.; Gill R.; Dishon A.; Kotler M.; Willner I. (2004) Amplified Chemiluminescence Surface Detection of DNA and Telomerase Activity Using Catalytic Nucleic Acid Labels. Anal. Chem. 76, 2152–2156. 10.1021/ac035219l. [DOI] [PubMed] [Google Scholar]
- Nakayama S.; Sintim H. O. (2012) Investigating the interactions between cations, peroxidation substrates and G-quadruplex topology in DNAzyme peroxidation reactions using statistical testing. Anal. Chim. Acta 747, 1–6. 10.1016/j.aca.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Golub E.; Freeman R.; Willner I. (2013) Hemin/G-Quadruplex-Catalyzed Aerobic Oxidation of Thiols to Disulfides: Application of the Process for the Development of Sensors and Aptasensors and for Probing Acetylcholine Esterase Activity. Anal. Chem. 85, 12126–12133. 10.1021/ac403305k. [DOI] [PubMed] [Google Scholar]
- Golub E.; Freeman R.; Willner I. (2011) A Hemin/G-Quadruplex Acts as an NADH Oxidase and NADH Peroxidase Mimicking DNAzyme. Angew. Chem., Int. Ed. 50, 11710–11714. 10.1002/anie.201103853. [DOI] [PubMed] [Google Scholar]
- Wang Z.-G.; Zhan P.; Ding B. (2013) Self-Assembled Catalytic DNA Nanostructures for Synthesis of Para-directed Polyaniline. ACS Nano 7, 1591–1598. 10.1021/nn305424e. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Megens R. P.; Feringa B. L.; Roelfes G. (2010) DNA-based asymmetric catalysis. Chem. Soc. Rev. 39, 2083–2092. 10.1039/b811349c. [DOI] [PubMed] [Google Scholar]
- Caprioara M.; Fiammengo R.; Engeser M.; Jäschke A. (2007) DNA-Based Phosphane Ligands. Chem. - Eur. J. 13, 2089–2095. 10.1002/chem.200601058. [DOI] [PubMed] [Google Scholar]
- Ropartz L.; Meeuwenoord N. J.; van der Marel G. A.; van Leeuwen P. W. N. M.; Slawin A. M. Z.; Kamer P. C. J. (2007) Phosphine containing oligonucleotides for the development of metallodeoxyribozymes. Chem. Commun. 1556–1558. 10.1039/b617871e. [DOI] [PubMed] [Google Scholar]
- Roelfes G.; Feringa B. L. (2005) DNA-Based Asymmetric Catalysis. Angew. Chem., Int. Ed. 44, 3230–3232. 10.1002/anie.200500298. [DOI] [PubMed] [Google Scholar]
- Coquière D.; Feringa B. L.; Roelfes G. (2007) DNA-Based Catalytic Enantioselective Michael Reactions in Water. Angew. Chem., Int. Ed. 46, 9308–9311. 10.1002/anie.200703459. [DOI] [PubMed] [Google Scholar]
- Walsh S. M.; Sachdeva A.; Silverman S. K. (2013) DNA Catalysts with Tyrosine Kinase Activity. J. Am. Chem. Soc. 135, 14928–14931. 10.1021/ja407586u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Osborne S. E.; Ellington A. D. (1997) Nucleic Acid Selection and the Challenge of Combinatorial Chemistry. Chem. Rev. 97, 349–370. 10.1021/cr960009c. [DOI] [PubMed] [Google Scholar]
- Willner I.; Zayats M. (2007) Electronic Aptamer-Based Sensors. Angew. Chem., Int. Ed. 46, 6408–6418. 10.1002/anie.200604524. [DOI] [PubMed] [Google Scholar]
- Du Y.; Li B.; Wang E. (2013) Fitting” Makes “Sensing” Simple: Label-Free Detection Strategies Based on Nucleic Acid Aptamers. Acc. Chem. Res. 46, 203–213. 10.1021/ar300011g. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510. 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Wilking M.; Hennecke U. (2013) The influence of G-quadruplex structure on DNA-based asymmetric catalysis using the G-quadruplex-bound cationic porphyrin TMPyP4·Cu. Org. Biomol. Chem. 11, 6940–6945. 10.1039/c3ob41366g. [DOI] [PubMed] [Google Scholar]
- Wu N.; Willner I. (2017) Programmed dissociation of dimer and trimer origami structures by aptamer–ligand complexes. Nanoscale 9, 1416–1422. 10.1039/C6NR08209B. [DOI] [PubMed] [Google Scholar]
- Liu J.; Cao Z.; Lu Y. (2009) Functional Nucleic Acid Sensors. Chem. Rev. 109, 1948–1998. 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz J.; Moshe M.; Willner I. (2009) Coherent Activation of DNA Tweezers: A “SET-RESET” Logic System. Angew. Chem., Int. Ed. 48, 3834–3837. 10.1002/anie.200805819. [DOI] [PubMed] [Google Scholar]
- Chen W.-H.; Yu X.; Liao W.-C.; Sohn Y. S.; Cecconello A.; Kozell A.; Nechushtai R.; Willner I. (2017) ATP-Responsive Aptamer-Based Metal-Organic Framework Nanoparticles (NMOFs) for the Controlled Release of Loads and Drugs. Adv. Funct. Mater. 27, 1702102. 10.1002/adfm.201702102. [DOI] [Google Scholar]
- Zhang Z.; Balogh D.; Wang F.; Willner I. (2013) Smart Mesoporous SiO2 Nanoparticles for the DNAzyme-Induced Multiplexed Release of Substrates. J. Am. Chem. Soc. 135, 1934–1940. 10.1021/ja311385y. [DOI] [PubMed] [Google Scholar]
- Rohloff J. C.; Gelinas A. D.; Jarvis T. C.; Ochsner U. A.; Schneider D. J.; Gold L.; Janjic N. (2014) Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol. Ther.--Nucleic Acids 3, e201 10.1038/mtna.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan D.; Li Y.; Tang Z.; Cao Z. C.; Chen H. W.; Mallikaratchy P.; Sefah K.; Yang C. J.; Tan W. (2006) Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. U. S. A. 103, 11838–11843. 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. (2010) Aptamers as therapeutics. Nat. Rev. Drug Discovery 9, 537–550. 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biniuri Y.; Luo G.-F.; Fadeev M.; Wulf V.; Willner I. (2019) Redox-Switchable Binding Properties of the ATP–Aptamer. J. Am. Chem. Soc. 141, 15567–15576. 10.1021/jacs.9b06256. [DOI] [PubMed] [Google Scholar]
- Phillips J. A.; Liu H.; O’Donoghue M. B.; Xiong X.; Wang R.; You M.; Sefah K.; Tan W. (2011) Using Azobenzene Incorporated DNA Aptamers to Probe Molecular Binding Interactions. Bioconjugate Chem. 22, 282–288. 10.1021/bc100402p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub E.; Albada H. B.; Liao W.-C.; Biniuri Y.; Willner I. (2016) Nucleoapzymes: Hemin/G-Quadruplex DNAzyme–Aptamer Binding Site Conjugates with Superior Enzyme-like Catalytic Functions. J. Am. Chem. Soc. 138, 164–172. 10.1021/jacs.5b09457. [DOI] [PubMed] [Google Scholar]
- Biniuri Y.; Albada B.; Wolff M.; Golub E.; Gelman D.; Willner I. (2018) Cu2+ or Fe3+ Terpyridine/Aptamer Conjugates: Nucleoapzymes Catalyzing the Oxidation of Dopamine to Aminochrome. ACS Catal. 8, 1802–1809. 10.1021/acscatal.7b03454. [DOI] [Google Scholar]
- Luo G. F.; Biniuri Y.; Vázquez-González M.; Wulf V.; Fadeev M.; Lavi R.; Willner I. (2019) Metal Ion-Terpyridine-Functionalized L-Tyrosinamide Aptamers: Nucleoapzymes for Oxygen Insertion into C-H Bonds and the Transformation of L-Tyrosinamide into Amidodopachrome. Adv. Funct. Mater. 29, 1901484. 10.1002/adfm.201901484. [DOI] [Google Scholar]
- Biniuri Y.; Shpilt Z.; Albada B.; Vázquez-González M.; Wolff M.; Hazan C.; Golub E.; Gelman D.; Willner I. (2020) A Bis-Zn2+-Pyridyl-Salen-Type Complex Conjugated to the ATP Aptamer: An ATPase-Mimicking Nucleoapzyme. ChemBioChem 21, 53–58. 10.1002/cbic.201900182. [DOI] [PubMed] [Google Scholar]
- Kirby A. J. (1996) Enzyme Mechanisms, Models, and Mimics. Angew. Chem., Int. Ed. Engl. 35, 706–724. 10.1002/anie.199607061. [DOI] [Google Scholar]
- Kralj M.; Tušek-Božić L.; Frkanec L. (2008) Biomedical Potentials of Crown Ethers: Prospective Antitumor Agents. ChemMedChem 3, 1478–1492. 10.1002/cmdc.200800118. [DOI] [PubMed] [Google Scholar]
- Dong Z.-Y.; Huang X.; Mao S.-Z.; Liang K.; Liu J.-Q.; Luo G.-M.; Shen J.-C. (2006) Cyclodextrin-Derived Mimic of Glutathione Peroxidase Exhibiting Enzymatic Specificity and High Catalytic Efficiency. Chem. - Eur. J. 12, 3575–3579. 10.1002/chem.200501098. [DOI] [PubMed] [Google Scholar]
- Flanagan M. L.; Arguello A. E.; Colman D. E.; Kim J.; Krejci J. N.; Liu S.; Yao Y.; Zhang Y.; Gorin D. J. (2018) A DNA-conjugated small molecule catalyst enzyme mimic for site-selective ester hydrolysis. Chem. Sci. 9, 2105–2112. 10.1039/C7SC04554A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imahori H. (2004) Porphyrin–fullerene linked systems as artificial photosynthetic mimics. Org. Biomol. Chem. 2, 1425–1433. 10.1039/B403024A. [DOI] [PubMed] [Google Scholar]
- Wasielewski M. R. (1992) Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 92, 435–461. 10.1021/cr00011a005. [DOI] [Google Scholar]
- Samuel A. P. S.; Co D. T.; Stern C. L.; Wasielewski M. R. (2010) Ultrafast Photodriven Intramolecular Electron Transfer from a Zinc Porphyrin to a Readily Reduced Diiron Hydrogenase Model Complex. J. Am. Chem. Soc. 132, 8813–8815. 10.1021/ja100016v. [DOI] [PubMed] [Google Scholar]
- Ward M. D. (1997) Photo-induced electron and energy transfer in non-covalently bonded supramolecular assemblies. Chem. Soc. Rev. 26, 365–375. 10.1039/cs9972600365. [DOI] [Google Scholar]
- Yamamoto M.; Föhlinger J.; Petersson J.; Hammarström L.; Imahori H. (2017) A Ruthenium Complex-Porphyrin-Fullerene-Linked Molecular Pentad as an Integrative Photosynthetic Model. Angew. Chem., Int. Ed. 56, 3329–3333. 10.1002/anie.201612456. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Lin W. (2014) Metal–organic frameworks for artificial photosynthesis and photocatalysis. Chem. Soc. Rev. 43, 5982–5993. 10.1039/C4CS00103F. [DOI] [PubMed] [Google Scholar]
- Willner I.; Yang J.-M.; Laane C.; Otvos J. W.; Calvin M. (1981) The function of silicon dioxide colloids in photoinduced redox reactions. Interfacial effects on the quenching, charge separation, and quantum yields. J. Phys. Chem. 85, 3277–3282. 10.1021/j150622a014. [DOI] [Google Scholar]
- Tachibana Y.; Vayssieres L.; Durrant J. R. (2012) Artificial photosynthesis for solar water-splitting. Nat. Photonics 6, 511–518. 10.1038/nphoton.2012.175. [DOI] [Google Scholar]
- Turro N. J.; Grätzel M.; Braun A. M. (1980) Photophysical and Photochemical Processes in Micellar Systems. Angew. Chem., Int. Ed. Engl. 19, 675–696. 10.1002/anie.198006751. [DOI] [Google Scholar]
- Amao Y.; Maki Y.; Fuchino Y. (2009) Photoinduced Hydrogen Production with Artificial Photosynthesis System Based on Carotenoid-Chlorophyll Conjugated Micelles. J. Phys. Chem. C 113, 16811–16815. 10.1021/jp900063r. [DOI] [Google Scholar]
- Willner I.; Maidan R.; Mandler D.; Duerr H.; Doerr G.; Zengerle K. (1987) Photosensitized reduction of carbon dioxide to methane and hydrogen evolution in the presence of ruthenium and osmium colloids: strategies to design selectivity of products distribution. J. Am. Chem. Soc. 109, 6080–6086. 10.1021/ja00254a029. [DOI] [Google Scholar]
- Willner I.; Mandler D. (1989) Characterization of palladium-.beta.-cyclodextrin colloids as catalysts in the photosensitized reduction of bicarbonate to formate. J. Am. Chem. Soc. 111, 1330–1336. 10.1021/ja00186a028. [DOI] [Google Scholar]
- Mandler D.; Willner I. (1984) Solar Light-Induced Formation of Chiral 2-Butanol in an Enzyme-Catalyzed Chemical-System. J. Am. Chem. Soc. 106, 5352–5353. 10.1021/ja00330a053. [DOI] [Google Scholar]
- Graetzel M. (1981) Artificial photosynthesis: water cleavage into hydrogen and oxygen by visible light. Acc. Chem. Res. 14, 376–384. 10.1021/ar00072a003. [DOI] [Google Scholar]
- Wang C.; deKrafft K. E.; Lin W. (2012) Pt Nanoparticles@Photoactive Metal–Organic Frameworks: Efficient Hydrogen Evolution via Synergistic Photoexcitation and Electron Injection. J. Am. Chem. Soc. 134, 7211–7214. 10.1021/ja300539p. [DOI] [PubMed] [Google Scholar]
- Khnayzer R. S.; Thoi V. S.; Nippe M.; King A. E.; Jurss J. W.; El Roz K. A.; Long J. R.; Chang C. J.; Castellano F. N. (2014) Towards a comprehensive understanding of visible-light photogeneration of hydrogen from water using cobalt(ii) polypyridyl catalysts. Energy Environ. Sci. 7, 1477–1488. 10.1039/C3EE43982H. [DOI] [Google Scholar]
- Fukuzumi S.; Kobayashi T.; Suenobu T. (2011) Photocatalytic Production of Hydrogen by Disproportionation of One-Electron-Reduced Rhodium and Iridium-Ruthenium Complexes in Water. Angew. Chem., Int. Ed. 50, 728–731. 10.1002/anie.201004876. [DOI] [PubMed] [Google Scholar]
- Mandler D.; Willner I. (1986) Photoinduced enzyme-catalysed synthesis of amino acids by visible light. J. Chem. Soc., Chem. Commun. 851–853. 10.1039/c39860000851. [DOI] [Google Scholar]
- Luo G.-F.; Biniuri Y.; Chen W.-H.; Neumann E.; Fadeev M.; Marjault H.-B.; Bedi A.; Gidron O.; Nechushtai R.; Stone D.; Happe T.; Willner I. (2019) Artificial Photosynthesis with Electron Acceptor/Photosensitizer-Aptamer Conjugates. Nano Lett. 19, 6621–6628. 10.1021/acs.nanolett.9b02880. [DOI] [PubMed] [Google Scholar]
- Luo G.-F.; Biniuri Y.; Chen W.-H.; Wang J.; Neumann E.; Marjault H.-B.; Nechushtai R.; Winkler M.; Happe T.; Willner I. (2020) Modelling Photosynthesis with ZnII-Protoporphyrin All-DNA G-Quadruplex/Aptamer Scaffolds. Angew. Chem., Int. Ed. 59, 9163–9170. 10.1002/anie.202002915. [DOI] [PubMed] [Google Scholar]
- Ariga T.; Masaike T.; Noji H.; Yoshida M. (2002) Stepping Rotation of F1-ATPase with One, Two, or Three Altered Catalytic Sites That Bind ATP Only Slowly. J. Biol. Chem. 277, 24870–24874. 10.1074/jbc.M202582200. [DOI] [PubMed] [Google Scholar]
- Biniuri Y.; Albada B.; Willner I. (2018) Probing ATP/ATP-Aptamer or ATP-Aptamer Mutant Complexes by Microscale Thermophoresis and Molecular Dynamics Simulations: Discovery of an ATP-Aptamer Sequence of Superior Binding Properties. J. Phys. Chem. B 122, 9102–9109. 10.1021/acs.jpcb.8b06802. [DOI] [PubMed] [Google Scholar]
- Albada H. B.; Golub E.; Willner I. (2016) Rational design of supramolecular hemin/G-quadruplex–dopamine aptamer nucleoapzyme systems with superior catalytic performance. Chem. Sci. 7, 3092–3101. 10.1039/C5SC04832J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher D. E. (2002) Polymer Vesicles. Science 297, 967–973. 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- Gardner P. M.; Winzer K.; Davis B. G. (2009) Sugar synthesis in a protocellular model leads to a cell signalling response in bacteria. Nat. Chem. 1, 377–383. 10.1038/nchem.296. [DOI] [PubMed] [Google Scholar]
- Chandrawati R.; Caruso F. (2012) Biomimetic Liposome- and Polymersome-Based Multicompartmentalized Assemblies. Langmuir 28, 13798–13807. 10.1021/la301958v. [DOI] [PubMed] [Google Scholar]
- Angelatos A. S.; Radt B.; Caruso F. (2005) Light-Responsive Polyelectrolyte/Gold Nanoparticle Microcapsules. J. Phys. Chem. B 109, 3071–3076. 10.1021/jp045070x. [DOI] [PubMed] [Google Scholar]
- Wu J.; Eisenberg A. (2006) Proton Diffusion across Membranes of Vesicles of Poly(styrene-b-acrylic Acid) Diblock Copolymers. J. Am. Chem. Soc. 128, 2880–2884. 10.1021/ja056064x. [DOI] [PubMed] [Google Scholar]
- Ghoroghchian P. P.; Li G.; Levine D. H.; Davis K. P.; Bates F. S.; Hammer D. A.; Therien M. J. (2006) Bioresorbable Vesicles Formed through Spontaneous Self-Assembly of Amphiphilic Poly(ethylene oxide)-block-polycaprolactone. Macromolecules 39, 1673–1675. 10.1021/ma0519009. [DOI] [PMC free article] [PubMed] [Google Scholar]