

Abstract
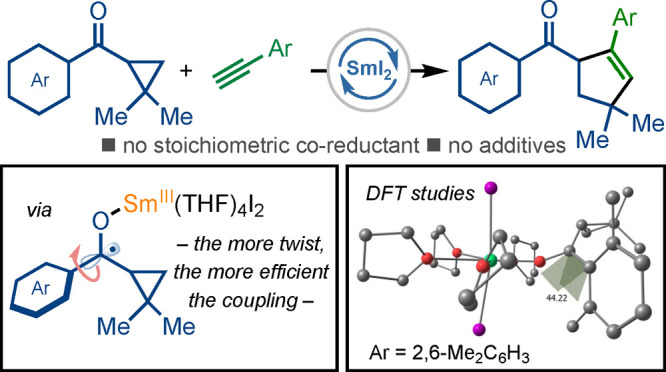
The archetypal single electron transfer reductant, samarium(II) diiodide (SmI2, Kagan’s reagent), remains one of the most important reducing agents and mediators of radical chemistry after four decades of widespread use in synthesis. While the chemistry of SmI2 is very often unique, and thus the reagent is indispensable, it is almost invariably used in superstoichiometric amounts, thus raising issues of cost and waste. Of the few reports of the use of catalytic SmI2, all require the use of superstoichiometric amounts of a metal coreductant to regenerate Sm(II). Here, we describe a SmI2-catalyzed intermolecular radical coupling of aryl cyclopropyl ketones and alkynes. The process shows broad substrate scope and delivers a library of decorated cyclopentenes with loadings of SmI2 as low as 15 mol %. The radical relay strategy negates the need for a superstoichiometric coreductant and additives to regenerate SmI2. Crucially, our study uncovers an intriguing link between ketone conformation and efficient cross-coupling and thus provides an insight into the mechanism of radical relays involving SmI2. The study lays further groundwork for the future use of the classical reagent SmI2 in contemporary radical catalysis.
Introduction
The archetypal single electron transfer (SET)1 reductant, samarium(II) diiodide (SmI2, Kagan’s reagent),2 remains one of the most important reducing agents and mediators of radical chemistry after four decades of widespread use in synthesis.3 Intramolecular processes using the commercially available reagent are particularly popular, and SmI2-mediated radical cyclizations feature in the total synthesis of numerous high profile and complex natural products.4 Intermolecular processes using SmI2 are inherently more challenging as intermolecular radical C–C bond formation must outrun the competing reduction of radicals to carbanions. While the chemistry of SmI2 is very often unique, and thus the reagent is indispensable,2−4 it is almost invariably used in superstoichiometric amounts, thus raising issues of cost and waste. Of the few reports of the use of catalytic SmI2, all require the use of superstoichiometric amounts of a metal coreductant to regenerate Sm(II).5 For example, Corey described one of the very few SmI2-catalyzed intermolecular coupling processes:5b Unfortunately, the catalytic system requires 15 equiv of Zn/Hg amalgam (Scheme 1A).
Scheme 1. SmI2-Catalyzed Intermolecular Radical Couplings.

(A) Using a stoichiometric coreductant to regenerate Sm(II). (B) This work. Using a radical relay to regenerate Sm(II). The crucial link between conformation and the efficiency of the coupling. TMS = trimethylsilyl.
We recently reported a radical-relay approach to catalysis with SmI2 that negates the need for coreductants and additives: cyclopropyl ketones underwent catalytic radical cyclization to give complex bicyclic ketones.6 We envisaged that unprecedented and more-challenging, intermolecular couplings might be possible using catalytic SmI2, as the reduction of radical intermediates A would be less-problematic at lower concentrations of the reagent.
Herein, we disclose an efficient method for the construction of decorated cyclopentenes using an intermolecular radical coupling of aryl cyclopropyl ketones and alkynes catalyzed by SmI2. Prior to this study, the only previous intermolecular radical coupling of cyclopropyl ketones and alkynes was an enantioselective process utilizing a noncommercial, chiral-at-rhodium complex and requiring imidazolyl cyclopropyl ketones capable of two-point binding to the metal.8f Crucially, our studies uncover an intriguing link between ketone conformation and efficient coupling and thus provide an insight into the mechanism of radical relays7,8 involving SmI2 (Scheme 1B).
Results and Discussion
Optimization studies began with the SmI2-mediated coupling of readily available cyclopropyl phenyl ketones 1a–c and phenylacetylene 2a (Table 1). While the use of 25 mol % of SmI2 with phenyl ketone 1a resulted in low conversion and a 35% yield of 3a (entry 1), byproduct 4 was also observed in the product mixture (9%). In an attempt to block competing intramolecular radical addition, the use of 2-methylphenyl ketone 1b was investigated; under identical conditions, 1b gave 3b in 99% isolated yield after 45 min (entry 2). Lowering the reaction temperature (entry 3) or the catalytic loading of SmI2 to 20 mol % (entry 4) and 15 mol % (entry 5) led to lower conversion. However, switching to 2,6-dimethylphenyl ketone 1c and using 15 mol % SmI2 gave 3c in 87% yield (entry 6). The use of the corresponding cyclohexyl cyclopropyl ketone 1d resulted in no product formation, and starting materials were recovered unchanged (entry 7) (vide infra). This is likely due to reversible reduction of the carbonyl and/or reversible fragmentation.
Table 1. Screening of Catalytic Conditionsa.
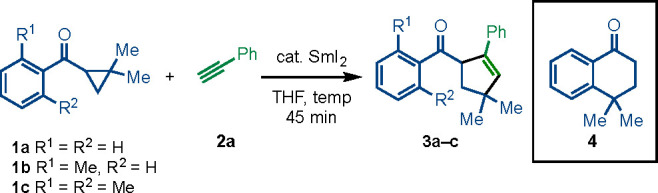
| entry | ketone | temp. (°C) | SmI2 loading | Conversion | Yield of 3a |
|---|---|---|---|---|---|
| 1 | 1a | 55 | 25 mol % | 40% | 35%b |
| 2 | 1b | 55 | 25 mol % | 100% | 99%b |
| 3 | 1b | RT | 25 mol % | 85% | 82%c |
| 4 | 1b | 55 | 20 mol % | 85% | 79% |
| 5 | 1b | 55 | 15 mol % | 63% | 50% |
| 6 | 1c | 55 | 15 mol % | 89% | 87% |
| 7 | 1dd | 55 | 25 mol % | 0% | 0% |
Reaction conditions: 1a–c (1 equiv), 2a (5 equiv), SmI2 (0.1 M in THF), in THF (0.5 mL/0.1 mmol of substrate) under nitrogen. aNMR yield using nitromethane as internal standard.
Isolated yield given.
Reaction time, 16 h.
Cyclohexyl 2,2-dimethylcyclopropylketone 1d was used. THF = tetrahydrofuran.
The scope of the reaction with regard to the aryl alkyne was explored using 2-methylphenyl ketone 1b (Figure 1). The presence of electron-releasing alkyl, alkoxy, amino, and trifluoromethoxy groups on the aryl substituent of the alkyne was tolerated (3e-3q). In line with the intermolecular addition of a nucleophilic radical (cf. A in Scheme 1B) to the alkyne, aryl alkynes bearing electron-withdrawing groups (e.g., bromo, fluoro, trifluoromethyl, phenyl, nitrile, and carbomethoxy) generally gave higher yields of 3 (3r–3ad). Diynes and a triyne underwent monocoupling to give 3ae-3ag in high yield. Naphthyl (3ah), phenanthrenyl (3ai), and pyrenyl (3aj) motifs were tolerated, as was the important heteroaromatic, thiophene (3ak). Crucially, functional groups that are typically reduced by SmI2 (e.g., carbomethoxy, nitrile and bromo) are unreactive under the catalytic conditions. The alkyl substituted alkyne, prop-2-yn-1-ylcyclopentane, was unreactive, as was phenyl propiolate. Attempted coupling with benzofuran was unsuccessful, and starting materials were recovered.6 For ineffective coupling partners, it appears that trapping of the radical formed upon reversible fragmentation of the cyclopropyl ring is inefficient and starting ketone is recovered. See the Supporting Information for further details and a table of unsuccessful coupling partners.
Figure 1.

Scope with respect to the aryl alkyne. Reaction conditions: 1b (1 equiv), 2 (typically 5 equiv), 25 mol % SmI2 (0.1 M in THF), in THF (0.5 mL/0.1 mmol of substrate) under nitrogen. Isolated yields; a using 2.5 equiv of 2; b using 40 mol % SmI2.
We next varied the aryl cyclopropyl ketone partner 1 (Figure 2). As noted during optimization studies, the presence of an ortho-methyl substituent on the aryl ring had a marked, beneficial effect on the efficiency of the catalytic radical coupling. In addition, ethyl (3al), fluoro (3am, 3at), chloro (3ao), phenyl (3as), and iodo (3au) substituents on the aryl ring of the ketone were compatible with the catalytic coupling. Ortho substitution was again seen to have a clear, beneficial impact on the efficiency of coupling; compare the yield of 3c with that of 3ap. The use of conveniently prepared spirocyclic cyclopropylketones gave spirocycles 3av and 3aw in 73% and 52%, respectively.
Figure 2.

Scope with respect to the aryl cyclopropyl ketone. Reaction conditions: 1 (1 equiv), 2 (typically 5 equiv), 25 mol % SmI2 (0.1 M in THF), in THF (0.5 mL/0.1 mmol of substrate) under nitrogen. Isolated yields. a Using 1.01 g of ketone partner. b Using 15 mol % of SmI2. c Using 40 mol % SmI2. d 4-Ethynylbenzonitrile was used as the alkyne partner. e Starting ketone was recovered.
Finally, the importance of gem-dialkyl substitution on the cyclopropane ring in 1 was probed; monomethyl substrate 1q gave 3ax in moderate yield and as a 2:1 mixture of diastereoisomers, while the use of the simple, unsubstituted cyclopropyl ketone failed to deliver 3ay. Cyclopropyl ketone 1s, bearing a phenyl substituent on the cyclopropane ring, failed to deliver 3az and starting ketone was recovered; although the radical anion intermediate derived from 1s is likely to undergo facile ring-opening,9a the benzylic radical from cyclopropane fragmentation appears to be insufficiently reactive to be trapped by the alkyne. Pleasingly, 3c was prepared on a 1 g scale in 86% while 3ar and 3c were prepared on a 1 mmol scale in 90% yield and 82% yield, respectively, with a reduced 15 mol % loading of SmI2.
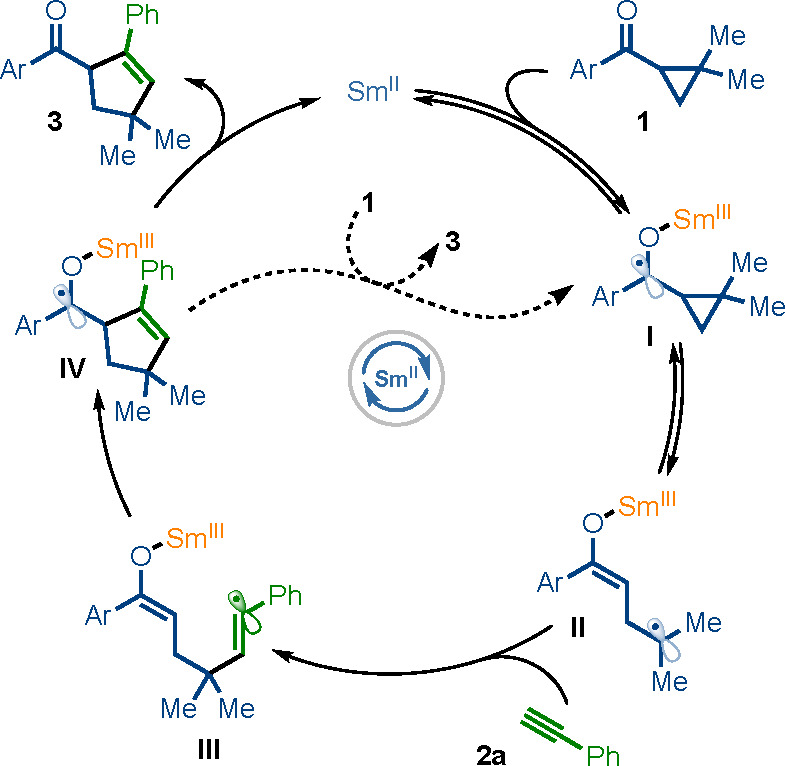
In line with our previous mechanistic studies,6 we propose a radical-relay mechanism for the SmI2-catalyzed, intermolecular radical coupling (Scheme 2): Note that exposure of 1b and 2a to various Lewis acids (e.g., SmI3, Yb(OTf)3, La(OTf)3) gave no trace of 3b, thus ruling out a Lewis acid-mediated coupling.10 Reversible SET from SmI2 to ketone 1 gives ketyl radical I(11) which fragments9b to give enolate/radical II. Intermolecular coupling with 2a then generates radical III which rebounds by addition to the Sm(III)-enolate moiety, generating new ketyl radical IV. Back electron transfer to Sm(III) regenerates the SmI2 catalyst and liberates product 3. It is also possible that ketyl radical IV directly reduces starting ketone 1.12 Calculations (vide infra) suggest that product ketyl radical IV has a similar reducing ability to the starting ketyl radical I. Thus, rather than a case of “reductant upconversion”,12b in which a more reducing radical is formed from a less-reducing radical and an electron-transfer chain process results, we believe it is the instability of the starting ketyl radical I and the formation of a more stable product ketyl radical IV that is key to the catalytic process.
Scheme 2. Proposed Radical Relay for the Intermolecular Radical Coupling.
Computational studies (PBE0/Def2-TZVP/PCM(THF)/D3(B-J)//Def2-SVP) have been used to probe the mechanism of the catalytic, intermolecular, radical coupling and, in particular, the crucial impact that ortho-substitution in the aryl ketones 1 has on the efficiency of coupling (Figure 3). First, we examined the conformation of the samarium ketyl radicals derived from ketones 1a–c (cf. I in Scheme 2) and the distribution of spin density in the radicals (Figure 3A). The main impact of the ortho-methyl substituents in ketones 1b and 1c is that the aryl rings are twisted out of the plane of the ketone carbonyl. This can be seen for ketyl radicals I-1b and I-1c, in which the aryl rings are 13° and 44°, respectively, out of plane. This renders the ketyl radicals I-1b and I-1c less stable, with less spin density in the aromatic ring and more of the spin density localized on what was the ketone carbonyl carbon (31% spin density on the aryl ring I-1a, 28% in I-1b and 18% in I-1c) (Figure 3A).
Figure 3.
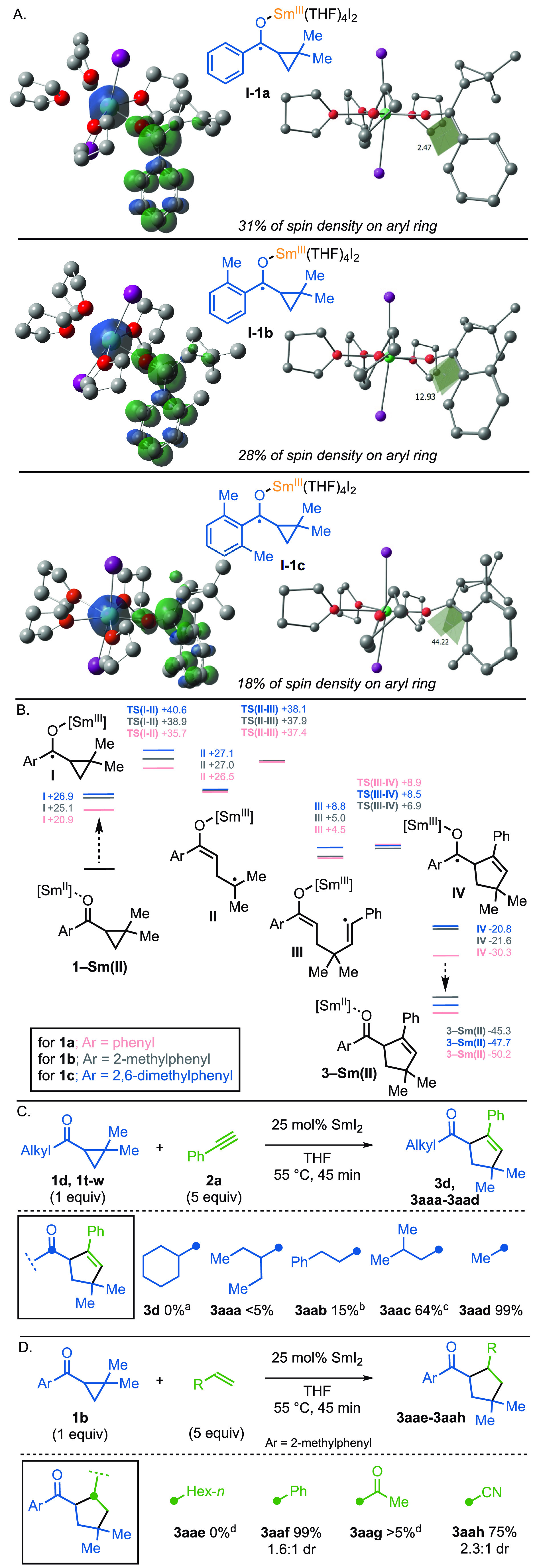
Computational studies. (A) Probing the importance of conformation on ketyl radical stability. (B) Mechanism of the catalytic radical coupling and the influence of the aryl substituent. Level of theory; PBE0/Def2-TZVP/PCM(THF)/D3(B-J)//Def2-SVP. (C) Scope with respect to alkyl cyclopropyl ketones. Reaction conditions: 1 (1 equiv), 2a (5 equiv), 25 mol % SmI2 (0.1 M in THF), in THF (0.5 mL/0.1 mmol of substrate) under nitrogen. Isolated yields. a Starting ketone was recovered. b Starting ketone was recovered in 81% yield. c Starting ketone was recovered in 22% yield. (D) Survey of alkene partners. Reaction conditions: 1 (1 equiv), alkene (5 equiv), 25 mol % SmI2 (0.1 M in THF), in THF (0.5 mL/0.1 mmol of substrate) under nitrogen. Isolated yields. d Starting ketone was recovered.
Crucially, the destabilization of the ketyl radical I appears to lower the barrier for ring-opening of the cyclopropyl ring to give radicals II; the computed barrier for ring opening of I-1a is 14.8 kcal mol–1, while those for I-1b and I-1c are 13.8 and 13.7 kcal mol–1, respectively (Figure 3B). It is interesting to note that the TS(I–II) is also destabilized, as ortho substituents are introduced to the aryl ring, but to a slighter lesser extent, while the stability of the radical product of ring-opening II is largely unaffected by the nature of the aryl ring as the spin density is now remote from the aryl substituent. It is important to note that destabilization of ketyl radical I makes SET from Sm(II) to the ketone a higher energy process (20.9 kcal mol–1 for 1a; 25.1 kcal mol–1 for 1b, and; 26.9 kcal mol–1 for 1c); however, this energy cost is repaid in full at the end of the relay during the favorable reduction of Sm(III) by the ketyl radical.
Building on our proposal that aryl cyclopropyl ketones are more effective substrates when their aryl rings are twisted out of the plane, thus rendering the ketyl radicals formed upon reduction less stable, we revisited the use of alkyl cyclopropyl ketones in the reaction. As discussed previously, cyclohexyl cyclopropyl ketone 1d was unreactive and the use of other bulky alkyl cyclopropyl ketones resulted in low yields or the return of only starting material. However, i-butyl and methyl cyclopropyl ketones underwent smooth coupling, to give 3aac and 3aad, respectively (Figure 3C). Attempts to switch alkyne partners for alkenes showed that the use of simple alkenes was not effective (e.g., oct-1-ene), although coupling was seen with the activated acceptors, styrene and acrylonitrile, to give 3aaf and 3aah, respectively (Figure 3D).
The products of the SmI2-catalyzed cross-coupling are versatile building blocks for synthesis. For example, cyclopentene 3c can be selectively oxidized and reduced to give epoxide 5 and ketone 6, respectively (Figure 4). Furthermore, the product of cross-coupling 3ar, bearing the pentamethylphenyl ketone motif, can be efficiently converted to the corresponding alcohol 7, acid 8, and ester 9.13
Figure 4.
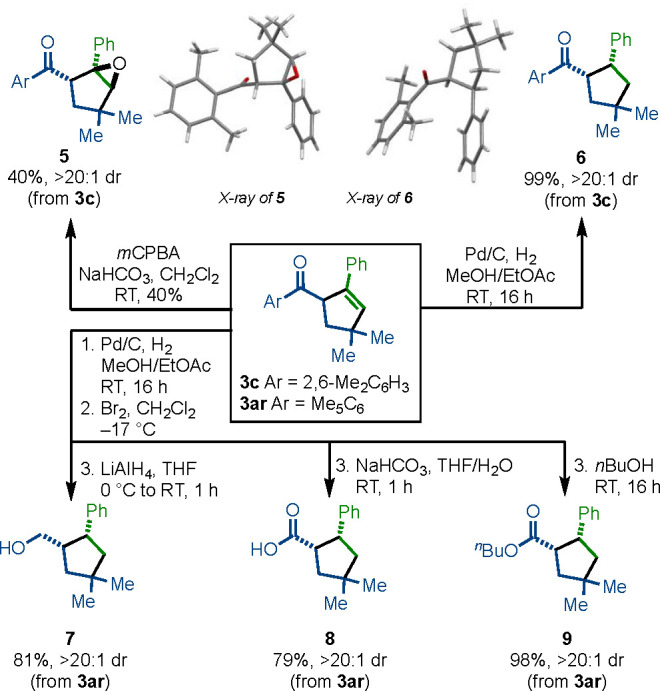
Selective manipulation of the products of SmI2-catalyzed intermolecular coupling.
Conclusion
In summary, SmI2 catalyzes the radical cross-coupling of aryl cyclopropyl ketones and alkynes. The process shows broad substrate scope and delivers a library of decorated cyclopentenes with loadings down to 15 mol % of SmI2. We invoke the operation of a radical relay mechanism that negates the need for a superstoichiometric coreductant and additives to regenerate Sm(II). Crucially, our study uncovers an intriguing link between ketone conformation and efficient cross-coupling and thus provides an insight into the mechanism of radical relays involving SmI2. The study lays further groundwork for the future use of the classical SET reagent SmI2 in contemporary radical catalysis.
Experimental Section
Preparation of SmI2
An oven-dried round-bottom flask, equipped with a stirrer bar, was flushed with a strong flow of N2 for 30 min. The flask was then loaded with samarium metal (∼40 mesh, 1.4 equiv), washed diiodoethane (1 equiv), and the flask was flushed for another 30 min with N2. Freshly distilled and degassed THF (0.1 M) was added, and the mixture was stirred overnight at room temperature. Finally, the mixture was allowed to settle for at least 1 h and titrated prior to use.14
General Procedure for the Catalytic Intermolecular Coupling
To an oven–dried microwave reaction vial containing a stirrer bar, was added ketone 1 (0.1 mmol, 1 equiv), and the vial was flushed with N2. After 15 min, THF (0.5 mL) and alkyne 2 (0.5 mmol, 5 equiv) were introduced by syringe. The vial was placed in a preheated oil bath at 55 °C, followed by the addition of freshly prepared SmI2 (typically 25 mol %, 0.1 M, 0.250 mL). The reaction was stirred vigorously (400 rpm) for 45 min. The reaction mixture was cooled to room temperature and filtered through a silica gel pad (100–200 mesh size), washing with CH2Cl2 (15 mL). Solvent was removed in vacuo, and the desired compound 3 was obtained without further purification. In a few cases, the product 3 was purified by column chromatography on silica gel (100- 200 mesh size) with hexane/ethyl acetate as eluent.
Acknowledgments
We thank the EPSRC (PDRA to S.A. and PDRA to N.A.B.; EP/R029938/1), the European Union Horizon 2020 (Fellowship to S.A.; Marie Sklodowska-Curie IF, EU project 891623 - SmART), and the University of Manchester.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications Web site. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c01356.
Experiments, experimental procedures, characterization data, CCDC numbers for X-ray crystal structures, details of computational studies, and spectra for all new compounds (PDF)
Accession Codes
CCDC 2039352–2039355 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews on SET and radical chemistry, see:; a Gansäuer A.; Bluhm H. Reagent-Controlled Transition-Metal-Catalyzed Radical Reactions. Chem. Rev. 2000, 100, 2771–2788. 10.1021/cr9902648. [DOI] [PubMed] [Google Scholar]; b Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]; c Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Plesniak M. P.; Huang H.-M.; Procter D. J. Radical cascade reactions triggered by single electron transfer. Nature Rev. Chem. 2017, 1, 0077–0092. 10.1038/s41570-017-0077. [DOI] [Google Scholar]
- a Girard P.; Namy J.-L.; Kagan H. B. Divalent lanthanide derivatives in organic synthesis. 1. Mild preparation of SmI2 and YbI2 and their use as reducing or coupling agents. J. Am. Chem. Soc. 1980, 102, 2693–2698. 10.1021/ja00528a029. [DOI] [Google Scholar]; For selected reviews of SmI2 chemistry, see:; b Szostak M.; Fazakerley N. J.; Parmar D.; Procter D. J. Cross-Coupling Reactions Using Samarium(II) Iodide. Chem. Rev. 2014, 114, 5959–6039. 10.1021/cr400685r. [DOI] [PubMed] [Google Scholar]; c Molander G. A.; Harris C. R. Sequencing Reactions with Samarium(II) Iodide. Chem. Rev. 1996, 96, 307–338. 10.1021/cr950019y. [DOI] [PubMed] [Google Scholar]; d Flowers R. A. Mechanistic Studies on the Roles of Cosolvents and Additives in Samarium(II)-Based Reductions. Synlett 2008, 10, 1427–1439. 10.1055/s-2008-1078414. [DOI] [Google Scholar]
- Selected applications of SmI2. Nitrogen fixation;; a Ashida Y.; Arashiba K.; Nakajima K.; Nishibayashi Y. Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 2019, 568, 536–540. 10.1038/s41586-019-1134-2. [DOI] [PubMed] [Google Scholar]; Polymer chemistry;; b Conticello V. P.; Gin D. L.; Grubbs R. H. Ring-opening metathesis polymerization of substituted bicyclo[2.2.2]octadienes: a new precursor route to poly(1,4-phenylenevinylene). J. Am. Chem. Soc. 1992, 114, 9708–9710. 10.1021/ja00050a088. [DOI] [Google Scholar]; Industrial medicinal chemistry;; c Austad B. C.; Calkins T. L.; Chase C. E.; Fang F. G.; Horstmann T. E.; Hu Y.; Lewis B. M.; Niu X.; Noland T. A.; Orr J. D.; Schnaderbeck M. J.; Zhang H.; Asakawa N.; Asai N.; Chiba H.; Hasebe T.; Hoshino Y.; Ishizuka H.; Kajima T.; Kayano A.; Komatsu Y.; Kubota M.; Kuroda H.; Miyazawa M.; Tagami K.; Watanabe T. Commercial Manufacture of Halaven®: Chemoselective Transformations En Route to Structurally Complex Macrocyclic Ketones. Synlett 2013, 24, 333–337. 10.1055/s-0032-1318026. [DOI] [Google Scholar]
- For reviews of SmI2 in natural product synthesis, see:; a Edmonds D. J.; Johnston D.; Procter D. J. Samarium(II)-iodide-mediated cyclizations in natural product synthesis. Chem. Rev. 2004, 104, 3371–3403. 10.1021/cr030017a. [DOI] [PubMed] [Google Scholar]; b Nicolaou K. C.; Ellery S. P.; Chen J. S. Samarium diiodide mediated reactions in total synthesis. Angew. Chem., Int. Ed. 2009, 48, 7140–7165. 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected examples of SmI2 in natural product synthesis, see:; c Mukaiyama T.; Shiina I.; Iwadare H.; Saitoh M.; Nishimura T.; Ohkawa N.; Sakoh H.; Nishimura K.; Tani Y-i.; Hasegawa M.; Yamada K.; Saitoh K. Asymmetric Total Synthesis of Taxol®. Chem. - Eur. J. 1999, 5, 121–161. 10.1002/(SICI)1521-3765(19990104)5:1<121::AID-CHEM121>3.0.CO;2-O. [DOI] [Google Scholar]; d Beemelmanns C.; Reissig H.-U. A Short Formal Total Synthesis of Strychnine with a Samarium Diiodide Induced Cascade Reaction as the Key Step. Angew. Chem., Int. Ed. 2010, 49, 8021–8025. 10.1002/anie.201003320. [DOI] [PubMed] [Google Scholar]; e Cha J. Y.; Yeoman J. T. S.; Reisman S. E. A Concise Total Synthesis of (−)-Maoecrystal Z. J. Am. Chem. Soc. 2011, 133, 14964–14967. 10.1021/ja2073356. [DOI] [PubMed] [Google Scholar]; f Fazakerley N. J.; Helm M. D.; Procter D. J. Total Synthesis of (+)-Pleuromutilin. Chem. - Eur. J. 2013, 19, 6718–6723. 10.1002/chem.201300968. [DOI] [PubMed] [Google Scholar]; g Breitler S.; Carreira E. M. Total Synthesis of (+)-Crotogoudin. Angew. Chem., Int. Ed. 2013, 52, 11168–11171. 10.1002/anie.201305822. [DOI] [PubMed] [Google Scholar]; h Cai L.; Zhang K.; Kwon O. Catalytic Asymmetric Total Synthesis of (−)-Actinophyllic Acid. J. Am. Chem. Soc. 2016, 138, 3298–3301. 10.1021/jacs.6b00567. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Gong J.; Chen H.; Liu X.-Y.; Wang Z.-X.; Nie W.; Qin Y. Total Synthesis of Atropurpuran. Nat. Commun. 2016, 7, 12183. 10.1038/ncomms12183. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Farney E. P.; Feng S. S.; Schäfers F.; Reisman S. E. Total Synthesis of (+)-Pleuromutilin. J. Am. Chem. Soc. 2018, 140, 1267. 10.1021/jacs.7b13260. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Leung J. C.; Bedermann A. A.; Njardarson J. T.; Spiegel D. A.; Murphy G. K.; Hama N.; Twenter B. M.; Dong P.; Shirahata T.; McDonald I. M.; Inoue M.; Taniguchi N.; McMahon T. C.; Schneider C. M.; Tao N.; Stoltz B. M.; Wood J. L. Total Synthesis of (±)-Phomoidride D. Angew. Chem., Int. Ed. 2018, 57, 1991–1994. 10.1002/anie.201712369. [DOI] [PubMed] [Google Scholar]
- SmI2 catalysis with stoichiometric reductants.; a Nomura R.; Matsuno T.; Endo T. Samarium Iodide-Catalyzed Pinacol Coupling of Carbonyl Compounds. J. Am. Chem. Soc. 1996, 118, 11666–11667. 10.1021/ja962331a. [DOI] [Google Scholar]; b Corey E. J.; Zheng G. Z. Catalytic reactions of samarium (II) iodide. Tetrahedron Lett. 1997, 38, 2045–2048. 10.1016/S0040-4039(97)00263-3. [DOI] [Google Scholar]; c Hélion F.; Namy J.-L. Mischmetall: An Efficient and Low Cost Coreductant for Catalytic Reactions of Samarium Diiodide. J. Org. Chem. 1999, 64, 2944–2946. 10.1021/jo9820667. [DOI] [PubMed] [Google Scholar]; d Aspinall H. C.; Greeves N.; Valla C. Samarium Diiodide Catalyzed Diastereoselective Pinacol Couplings. Org. Lett. 2005, 7, 1919–1922. 10.1021/ol050256f. [DOI] [PubMed] [Google Scholar]; e Maity S.; Flowers R. A. II. Mechanistic Study and Development of Catalytic Reactions of Sm(II). J. Am. Chem. Soc. 2019, 141, 3207–3216. 10.1021/jacs.8b13119. [DOI] [PubMed] [Google Scholar]; Electrocatalytic processes with Sm(II); f Sun L.; Sahloul K.; Mellah M. Use of Electrochemistry to Provide Efficient SmI2 Catalytic System for Coupling Reactions. ACS Catal. 2013, 3, 2568–2573. 10.1021/cs400587s. [DOI] [Google Scholar]; g Zhang Y. F.; Mellah M. Convenient Electrocatalytic Synthesis of Azobenzenes from Nitroaromatic Derivatives Using SmI2. ACS Catal. 2017, 7, 8480–8486. 10.1021/acscatal.7b02940. [DOI] [Google Scholar]; h Bazzi S.; Schulz E.; Mellah M. Electrogenerated Sm(II)-Catalyzed CO2 Activation for Carboxylation of Benzyl Halides. Org. Lett. 2019, 21, 10033–10037. 10.1021/acs.orglett.9b03927. [DOI] [PubMed] [Google Scholar]
- Huang H.-M.; McDouall J. J. W.; Procter D. J. SmI2-catalyzed cyclization cascades by radical relay. Nat. Catal. 2019, 2, 211–218. 10.1038/s41929-018-0219-x. [DOI] [Google Scholar]
- Huang H. M.; Garduño-Castro M. H.; Morrill C.; Procter D. J. Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev. 2019, 48, 4626–4638. 10.1039/C8CS00947C. [DOI] [PubMed] [Google Scholar]
- For examples of related radical relay processes, see.; a Lu Z.; Shen M.; Yoon T. P. [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones by Visible Light Photocatalysis. J. Am. Chem. Soc. 2011, 133, 1162–1164. 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Amador A. G.; Sherbrook E. M.; Yoon T. P. Enantioselective Photocatalytic [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones. J. Am. Chem. Soc. 2016, 138, 4722–4725. 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Amador A.; Sherbrook E.; Lu Z.; Yoon T. A General Protocol for Radical Anion [3 + 2] Cycloaddition Enabled by Tandem Lewis Acid Photoredox Catalysis. Synthesis 2018, 50, 539–547. 10.1055/s-0036-1591500. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hao W.; Wu X.; Sun J. Z.; Siu J. C.; MacMillan S. N.; Lin S. Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc. 2017, 139, 12141–12144. 10.1021/jacs.7b06723. [DOI] [PubMed] [Google Scholar]; e Hao W.; Harenberg J. H.; Wu X.; MacMillan S. N.; Lin S. Diastereo- and Enantioselective Formal [3 + 2] Cycloaddition of Cyclopropyl Ketones and Alkenes via Ti-Catalyzed Radical Redox Relay. J. Am. Chem. Soc. 2018, 140, 3514–3517. 10.1021/jacs.7b13710. [DOI] [PubMed] [Google Scholar]; f Huang X.; Lin J.; Shen T.; Harms K.; Marchini M.; Ceroni P.; Meggers E. Asymmetric [3 + 2] Photocycloadditions of Cyclopropanes with Alkenes or Alkynes through Visible-Light Excitation of Catalyst-Bound Substrates. Angew. Chem., Int. Ed. 2018, 57, 5454–5458. 10.1002/anie.201802316. [DOI] [PubMed] [Google Scholar]; g Robinson S. G.; Wu X.; Jiang B.; Sigman M. S.; Lin S. Mechanistic Studies Inform Design of Improved Ti(salen) Catalysts for Enantioselective [3 + 2] Cycloaddition. J. Am. Chem. Soc. 2020, 142, 18471–18482. 10.1021/jacs.0c07128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stevenson J. P.; Jackson W. F.; Tanko J. M. Cyclopropylcarbinyl-Type Ring Openings. Reconciling the Chemistry of Neutral Radicals and Radical Anions. J. Am. Chem. Soc. 2002, 124, 4271–4281. 10.1021/ja0041831. [DOI] [PubMed] [Google Scholar]; b Molander G. A.; Alonso-Alija C. Opening of cyclopropyl ketones with SmI2. Synthesis of spirocyclic and bicyclic ketones by intramolecular trapping of an electrophile. Tetrahedron 1997, 53, 8067–8084. 10.1016/S0040-4020(97)00499-7. [DOI] [Google Scholar]
- Reissig H.-U.; Zimmer R. Donor–Acceptor-Substituted Cyclopropane Derivatives and Their Application in Organic Synthesis. Chem. Rev. 2003, 103, 1151–1196. 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]
- For seminal mechanistic studies on ketone reduction with SmI2, see:; a Miller R. S.; Sealy J. M.; Shabangi M.; Kuhlman M. L.; Fuchs J. R.; Flowers R. A. II Reactions of SmI2 with Alkyl Halides and Ketones: Inner-Sphere vs Outer-Sphere Electron Transfer in Reactions of Sm(II) Reductants. J. Am. Chem. Soc. 2000, 122, 7718–7722. 10.1021/ja001260j. [DOI] [Google Scholar]; b Chopade P. R.; Prasad E.; Flowers R. A. II. The Role of Proton Donors in SmI2-Mediated Ketone Reduction: New Mechanistic Insights. J. Am. Chem. Soc. 2004, 126, 44–45. 10.1021/ja038363x. [DOI] [PubMed] [Google Scholar]; c Farran H.; Hoz S. Quantifying the Electrostatic Driving Force behind SmI2 Reductions. Org. Lett. 2008, 10, 4875–4877. 10.1021/ol8019692. [DOI] [PubMed] [Google Scholar]
- a Studer A.; Curran D. P. The electron is a catalyst. Nat. Chem. 2014, 6, 765–773. 10.1038/nchem.2031. [DOI] [PubMed] [Google Scholar]; b Syroeshkin M. A.; Kuriakose F.; Saverina E. A.; Timofeeva V. A.; Egorov M. P.; Alabugin I. V. Upconversion of Reductants. Angew. Chem., Int. Ed. 2019, 58, 5532–5550. 10.1002/anie.201807247. [DOI] [PubMed] [Google Scholar]
- a Frost J. R.; Cheong C. B.; Akhtar W. M.; Caputo D. F. J.; Stevenson N. G.; Donohoe T. J. Strategic Application and Transformation of ortho-Disubstituted Phenyl and Cyclopropyl Ketones to Expand the Scope of Hydrogen Borrowing Catalysis. J. Am. Chem. Soc. 2015, 137, 15664–15667. 10.1021/jacs.5b11196. [DOI] [PubMed] [Google Scholar]; b Schubert W. M.; Latourette H. K. The Aromatic Elimination Reaction. II. The Mechanism of the Acid-catalyzed Deacylation of Aromatic Ketones. J. Am. Chem. Soc. 1952, 74, 1829–1834. 10.1021/ja01127a061. [DOI] [Google Scholar]; c Bender M. L.; Chen M. C. Acylium Ion Formation in the Reactions of Carboxylic Acid Derivatives. IV. The Acid catalyzed Hydrolysis of Methyl 4-Substituted-2,6-dimethylbenzoates. J. Am. Chem. Soc. 1963, 85, 37–40. 10.1021/ja00884a007. [DOI] [Google Scholar]
- Szostak M.; Spain M.; Procter D. J. Preparation of Samarium(II) Iodide: Quantitative Evaluation of the Effect of Water, Oxygen, and Peroxide Content, Preparative Methods, and the Activation of Samarium Metal. J. Org. Chem. 2012, 77, 3049–3059. 10.1021/jo300135v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.