

Abstract
Succinate dehydrogenase (SDH) present in the inner mitochondrial membrane is an important target enzyme for the design of SDH inhibitor-type fungicides. Using SDH as the target enzyme, 22 novel longifolene-derived diacylhydrazine compounds were designed and synthesized using the renewable natural product longifolene as the starting material. Their structures were confirmed by IR, 1H NMR, 13C NMR, electrospray mass spectrometry, and elemental analysis. In vitro antifungal activity of the target compounds was preliminarily evaluated. As a result, some of them showed better or comparable antifungal activity than that of the commercial fungicide chlorothalonil, in which compound 5a had inhibitory rates of 97.5, 80.5, 72.1, and 67.1% against Physalospora piricola, Colletotrichum orbiculare, Alternaria solani, and Gibberella zeae, respectively, presenting excellent and broad-spectrum activity that deserved further study. Besides, a reasonable and effective three-dimensional structure–activity quantitative relationship model has been established. There was a significant positive correlation between the antifungal activity and the docking-based binding energy analyzed using Spearman’s rank correlation algorithm. Also, the simulative binding pattern of the target compounds with SDH was investigated by molecular docking study. Furthermore, the diacylhydrazine and phenol groups of the target compounds were proposed to be the potential pharmacophores by frontier molecular orbital analysis.
Introduction
The number of commercial succinate dehydrogenase (or succinate-ubiquinone oxidoreductase, EC 1.3.5.1) inhibitor (SDHI) fungicides has been increasing in recent years because of their highly effective and broad-spectrum fungicidal performance, which was owing to their unique action mode with the target enzyme.1 At present, about 20 SDHI fungicides have been registered according to the statistics from the Fungicide Resistance Action Committee including carboxin, fluxapyroxad, boscalid, benzovindiflupyr, bixafen, penflufen, and so forth.2 All of these compounds are carboxamides. Meanwhile, fruitful research studies for discovering novel succinate dehydrogenase (SDH) inhibitor molecules have been performed incessantly,3−8 and some other pharmacophore-type compounds for SDHI fungicides have been found such as carbohydrazides9 and sulfonamides.10
SDH is the only enzyme involved in both the respiratory chain and the tricarboxylic acid cycle. It catalyzes the oxidation of succinate to fumarate, coupled with the reduction of ubiquinone to ubiquinol in the inner mitochondrial membrane. It has been identified as a significant target for the design of agrochemical fungicides.11,12 The kinetics research of SDH inhibition through 10 commercial carboxamide fungicides provided the useful reference for the design of new SDHI fungicides. The result showed that the carboxamide molecule competed with ubiquinone rather than succinate, and the carbonyl oxygen atom of the carboxamide formed hydrogen bonds with two key amino acid residues TRP and TYR, the acid moiety interacted with the residues ARG, SER, IIE, and PRO, and the amine moiety interacted with the residues TRP, IIE, and IIE.13
We also noted that diacylhydrazine compounds containing double carboxamide groups exhibited diverse pharmacological properties, such as insecticidal,14 herbicidal,15 antifungal,16 antitumor,17,18 and antimalarial19 activities. Meanwhile, compounds bearing a phenol pharmacophore comparable to ubiquinol showed a wide range of biological activities, especially antifungal activity.20−22
On the other hand, longifolene (4,8,8-trimethyl-9-methylenedecahydro-1,4-methanoazulene), a naturally occurring tricyclic sesquiterpene, is the main constituent of heavy turpentine, which is a byproduct in the production of rosin and turpentine from living pine trees, but just used as a less expensive boiler fuel.23 By the isomerization–aromatization reaction, longifolene can be converted into 7-isopropyl-1, 1-dimethyltetralin (longifolene-derived tetraline),24 whose structural modification was explored to be used in perfume25 or medicine26 and so forth. In light of these clues above and as the continuation of our interest in the work of high value-added application of forest resources for the sustainable development of the resin industy,26−30 a series of novel longifolene-derived diacylhydrazine compounds incorporating phenol and diacylhydrazine groups were designed by the strategy of molecular docking-based virtual screening based on the crystal structure of SDH (UniProtKB AC P32420, homology modeling on SWISS-MODEL web) and synthesized and characterized. In vitro antifungal activity of all the target compounds was preliminarily evaluated against eight fungi. Furthermore, a three-dimensional structure–activity quantitative relationship (3D-QSAR) model was built by the comparative molecular field analysis (CoMFA) method, molecular docking was conducted to explore the binding mode for these molecules with SDH, and the frontier molecular orbital was calculated to analyze the potential pharmacophore for the target molecules.
Results and Discussion
Synthesis and Characterization of Compounds
The target compounds were synthesized according to the route shown in Scheme 1. Longifolene-derived tetralin 2 was prepared by the isomerization–aromatization reaction using nanocrystalline sulfated zirconia as a catalyst and further oxidized by TBHP oxidant to give longifolene-derived tetralone 3. Compound 2 and 3 were prepared according to our previous report.25 Compound 4 was prepared by the Baeyer–Villager oxidation reaction using m-CPBA as the oxidant. 22 longifolene-derived diacylhydrazine compounds 5a–5v were then synthesized by the hydrazinolysis reaction of compound 4 with different substituted acylhydrazine compounds in 75.0–80.0% yields.
Scheme 1. Synthetic Route of the Longifolene-Derived Diacylhydrazine Compounds 5a–5v (Table 1).

Reagents and conditions: (a) ZnCl2, 140.0 °C, reflux 8 h; (b) TBHP, CH3CN, CuCl2, 40.0 °C; (c) m-CPBA, Cl2Cl2, rt; and (d) EtOH, a series of different acylhydrazine compounds, 78.0 °C.
The structures of the target compounds were confirmed by IR, NMR, electrospray mass spectrometry (ESI-MS), and elemental analysis. In the IR spectra, the characteristic absorption bands at about 3438–3318 and 3289–3187 cm–1 were attributed to the stretching vibrations of the O–H and N–H, respectively. The characteristic absorption bands at about 1706–1661 cm–1 were assigned to the stretching vibrations of C=O. The 1H NMR spectra exhibited characteristic signals at δ 6.93–6.69 ppm, which were assigned to the protons of the benzene ring, and the characteristic signals at about δ 10.21–8.88 ppm were assigned to the amino protons of the diacylhydrazine moiety. The 13C NMR spectra of the target compounds showed peaks for two carbon atoms of C=O at δ 173.94–163.23 ppm and carbon atoms of the benzene ring at δ 146.11–116.38 ppm. The other saturated carbons displayed signals in the region of δ 37.60–24.81 ppm. Their molecular weights and the C, H, and N elemental ratios agreed with the results of ESI-MS and elemental analysis, respectively.
In Vitro Antifungal Activity
The antifungal activities of the target compounds 5a–5v were evaluated by the agar dilution method31 against eight plant pathogens at 50 mg/L, including apple root spot (Physalospora piricola), wheat scab (Gibberella zeae), speckle on peanut (Cercospora arachidicola), tomato early blight (Alternaria solani), fusarium wilt on cucumber (Fusarium oxysporumf. sp.Cucumerinum), watermelon anthracnose (Colletotrichum orbiculare), corn southern leaf blight (Bipolaris maydis), and rice sheath blight (Rhizoctonia solani). The commercial fungicide chlorothalonil was used as a positive control. The results are listed in Table 2. It was found that most of the tested compounds displayed certain antifungal activity against the tested fungi. Compared with that of the commercial fungicide (chlorothalonil), some compounds exhibited excellent antifungal activity. For instance, compound 5a (R = o-I Ph) had inhibitory rates of 97.5, 80.5, 72.1, and 67.1% against P. piricola, C. orbiculare, A. solani, and G. zeae, respectively, presenting better antifungal effect than that of the positive control with inhibitory rates of 92.9, 75.0, 45.0, and 58.3%, respectively. Besides, compounds 5s (R = o-Cl Ph), 5j (R = o-NO2 Ph), 5d (R = p-OCH3 Ph), and 5g (R = p-F Ph) had inhibitory rates of 97.5, 87.1, 80.8, and 80.8%, respectively, against P. piricola, 5h (R = o-F Ph) had an inhibitory rate of 70.8% against G. zeae, 5k (R = o-Cl Ph) had an inhibitory rate of 72.1% against A. solani, and 5b (R = 2,4-OH Ph) had an inhibitory rate of 73.5% against R. solani, showing excellent to moderate antifungal activity. Overall, compound 5a (R = o-I Ph) exhibited excellent and broad-spectrum antifungal activity against most of the tested fungi. It was also found that the R groups had a noticeable influence on activity, so a 3D-QSAR study was performed subsequently.
Table 2. In Vitro Antifungal Activity of the Target Compounds 5a–5v.
| relative inhibitory rate (%) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compounds | R | P. piricola | G. zeae | C. arachidicola | A. solani | F. oxysporum f. sp. Cucumerinum | C. orbiculare | B. maydis | R. solani |
| 5a | o-I Ph | 97.5 | 67.1 | 58.1 | 72.1 | 76.0 | 80.5 | 65.0 | 44.1 |
| 5b | 2,4-OH Ph | 70.4 | 52.9 | 69.3 | 68.6 | 44.0 | 41.8 | 40.0 | 73.5 |
| 5c | o-OH Ph | 42.9 | 70.7 | 69.7 | 54.8 | 65.3 | 46.4 | 57.5 | 14.7 |
| 5d | p-OCH3 Ph | 80.8 | 45.7 | 69.3 | 51.4 | 56.8 | 62.3 | 57.5 | 18.2 |
| 5e | o-Br Ph | 74.6 | 56.4 | 61.9 | 58.3 | 67.4 | 55.5 | 60.0 | 0 |
| 5f | p-Br Ph | 51.7 | 52.9 | 54.4 | 51.4 | 48.3 | 48.6 | 45.0 | 41.8 |
| 5g | p-F Ph | 80.8 | 52.9 | 58.1 | 47.9 | 50.4 | 53.2 | 60.0 | 26.5 |
| 5h | o-F Ph | 53.8 | 70.8 | 54.4 | 51.4 | 48.3 | 50.9 | 42.5 | 0 |
| 5i | o-CF3 Ph | 51.3 | 70.7 | 61.9 | 54.8 | 65.3 | 41.8 | 60.0 | 22.9 |
| 5j | o-NO2 Ph | 87.1 | 52.9 | 54.4 | 54.8 | 56.8 | 55.5 | 55.0 | 0 |
| 5k | o-Cl Ph | 47.4 | 70.7 | 65.6 | 72.1 | 39.8 | 62.3 | 52.5 | 0 |
| 5l | m-Cl Ph | 55.8 | 74.3 | 50.7 | 54.8 | 58.9 | 57.7 | 55.0 | 0 |
| 5m | p-Cl Ph | 47.5 | 70.7 | 58.1 | 44.5 | 58.9 | 48.6 | 60.0 | 0 |
| 5n | p-CH3 Ph | 76.3 | 38.6 | 39.6 | 47.9 | 56.8 | 50.9 | 50.0 | 31.2 |
| 5o | o-CH3 Ph | 62.1 | 56.4 | 50.7 | 54.8 | 52.6 | 53.2 | 50.0 | 0 |
| 5p | p-CH2OH Ph | 56.4 | 56.4 | 69.3 | 54.8 | 37.7 | 46.4 | 35.0 | 20.6 |
| 5q | p-C(CH3)3 Ph | 32.9 | 70.7 | 61.9 | 51.4 | 35.5 | 30.5 | 32.5 | 0 |
| 5r | Ph | 45.4 | 74.3 | 50.7 | 51.4 | 35.5 | 37.3 | 27.5 | 0 |
| 5s | CH3 | 97.5 | 60.0 | 58.1 | 61.7 | 44.0 | 46.4 | 32.5 | 0 |
| 5t | CH2 Ph | 70.0 | 70.7 | 65.6 | 44.5 | 50.4 | 53.2 | 45.0 | 0 |
| 5u | CH2CN | 46.7 | 60.0 | 50.7 | 61.7 | 50.4 | 44.1 | 40.0 | 0 |
| 5v | α-furyl | 45.4 | 35.0 | 58.1 | 51.4 | 50.4 | 46.4 | 40.0 | 14.7 |
| chlorothalonil | 92.9 | 58.3 | 94.4 | 45.0 | 91.7 | 75.0 | 81.8 | 96.3 | |
Table 1. R Groups of the Compounds 5a–5v.
| compounds | R | compounds | R | compounds | R |
|---|---|---|---|---|---|
| 5a | o-I Ph | 5i | o-CF3 Ph | 5q | p-C(CH3)3 Ph |
| 5b | 2,4-OH Ph | 5j | o-NO2 Ph | 5r | Ph |
| 5c | o-OH Ph | 5k | o-Cl Ph | 5s | CH3 |
| 5d | p-OCH3 Ph | 5l | m-Cl Ph | 5t | CH2 Ph |
| 5e | o-Br Ph | 5m | p-Cl Ph | 5u | CH2CN |
| 5f | p-Br Ph | 5n | p-CH3 Ph | 5v | α-furyl |
| 5g | p-F Ph | 5o | o-CH3 Ph | ||
| 5h | o-F Ph | 5p | p-CH2OH Ph |
3D-QSAR Analysis
3D-QSAR analysis of the experimental and predicted antifungal activity against P. piricola for the target compounds was carried out by the CoMFA method according to our previous report,32 and the 16 target compounds in the training set and the 2 target compounds in the test set are presented in Table 3. The result is shown in Table 4. A 3D-QSAR model was established with the conventional correlation r2 = 0.997 and the cross-validated coefficient q2 = 0.574. Referring to the report,33 the inhibitory rate against P. piricola was converted to an active factor (AF). The scatter plot of the predicted AF values versus experimental AF values is presented in Figure 1, where all data were concentrated near the X = Y line, illustrating that the 3D-QSAR model was reliable and had a good predictive ability.
Table 3. AF Values of Experimental and Predicted Activities for the Target Compounds 5a–5ra.
| compounds | R | MW | AF | AF′ | residual |
|---|---|---|---|---|---|
| 5a | o-I Ph | 494.11 | –1.103 | –1.148 | 0.045 |
| 5b | 2,4-OH Ph | 400.20 | –2.226 | –2.228 | 0.002 |
| 5c | o-OH Ph | 384.20 | –2.709 | –2.725 | 0.016 |
| 5d | p-OCH3 Ph | 398.22 | –1.976 | –1.987 | 0.011 |
| 5e | o-Br Ph | 446.12 | –2.182 | –2.066 | –0.116 |
| 5f | p-Br Ph | 446.12 | –2.620 | –2.655 | 0.035 |
| 5g | p-F Ph | 386.20 | –1.963 | –1.956 | –0.007 |
| 5h | o-F Ph | 386.20 | –2.521 | –2.509 | –0.012 |
| 5i | o-CF3 Ph | 436.20 | –2.617 | –2.615 | –0.002 |
| 5j | o-NO2 Ph | 413.20 | –1.787 | –1.800 | 0.013 |
| 5k | o-Cl Ph | 402.17 | –2.650 | –2.709 | 0.059 |
| 5l | m-Cl Ph | 402.17 | –2.503 | –2.505 | 0.002 |
| 5m | p-Cl Ph | 402.17 | –2.648 | –2.626 | –0.022 |
| 5n | p-CH3 Ph | 382.23 | –2.075 | –2.076 | –0.001 |
| 5o | o-CH3 Ph | 382.23 | –2.368 | –2.358 | –0.010 |
| 5p | p-CH2OH Ph | 398.22 | –2.488 | –2.496 | 0.008 |
| 5q* | p-C(CH3)3 Ph | 424.27 | –2.937 | –2.930 | –0.007 |
| 5r* | Ph | 368.21 | –2.647 | –2.622 | –0.025 |
AF: experimental value; AF′: predicted value; *: test set compounds.
Table 4. Summary of CoMFA.
| contribution (%) |
||||||
|---|---|---|---|---|---|---|
| q2 | r2 | S | F | steric | electrostatic | |
| CoMFA | 0.574 | 0.997 | 0.045 | 105.314 | 0.682 | 0.318 |
Figure 1.

Scatter plot of predicted AF values vs experimental AF values.
The contribution rates for steric and electrostatic fields were 68.2 and 31.8% (Table 4), respectively, showing that the steric field was the major contributor to the increase in activity. The steric and electrostatic field contour maps for the R groups at the benzene ring are presented in Figure 2. The field contours were represented with different colors: In the steric contour map, the green enclosed volume represented that the R group embedding this area will favor the increase in activity, while yellow defines the opposite. In the electrostatic contour map, the blue enclosed volume represented that the R group with an electropositive surface embedding in this area will favor the increase in activity, while red defines the opposite. As shown in Figure 2, a green block is suspended above the 2-position of the benzene ring and a blue block is around the 4-position. Namely, the introduction of R groups embedding the green enclosed volume will favor the increase of activity, but cannot in the opposite. For example, as shown in Figure 2a, compound 5a (R = o-I Ph), of which the iodine atom can embed in the green block (Figure 2b), showed higher activity than compound 5k (R = o-Cl Ph), of which the chlorine atom cannot embed in the green block (Figure 2c). Likewise, as shown in Figure 2d, compound 5d (R = p-OCH3 Ph), which had a methoxy group with an electropositive surface located at the 4-position of the benzene ring (Figure 2e), showed better antifungal activity than compound 5n (R = p-CH3 Ph, Figure 2f). Other compounds shared a similar case.
Figure 2.
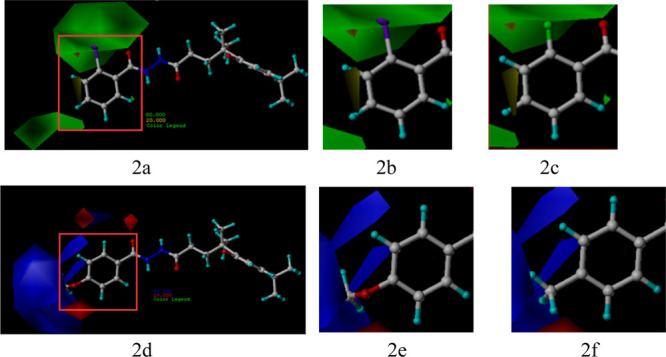
Contours of CoMFA: (a) contour of the steric contribution represented in yellow and green, (b) expanded contour of compound 5a, (c) expanded contour of compound 5k, (d) contour of electrostatic contribution represented in red and blue, (e) expanded contour of compound 5d, and (f) expanded contour of compound 5n.
Molecular Docking and Frontier Molecular Orbital
The molecular docking study for the target compounds was performed using AutoDock 4.2.6 software34 according to our previous work.26 The simulation of the binding pattern for the best antifungal activity of compound 5a and commercial SDHI carboxin with SDH is shown in Figure 3. The oxygen atoms of carbonyl and phenol moieties interacted with the residues TRP173 and HIS60 via the H bond (Figure 3a), which are similar to the commercial SDHI carboxin (Figure 3b). Besides, compound 5a was surrounded by the hydrophobic residues HIS104, VAL59, and TYR58.
Figure 3.

Binding mode and the interaction of (a) compound 5a with SDH and (b) commercial SDHI carboxin with SDH.
In fact, some diacylhydrazine compounds were used as commercial pesticides such as tebufenozide, chromafenozide, and methoxyfenozide, which were ecdysone receptor agonists. Therefore, molecular docking investigation for the comparison between the target compound 5a and the commercial pesticide tebufenozide was carried out using the heterodimer ecdysteroid receptor/ultraspiracle (EcR/USP) as the target protein (PDB ID 1R20). The result is shown in Figure 4. As a whole, compound 5a embeds incompletely in the ligand (chromafenozide)-binding domain (Figure 4a), while the tebufenozide showed the opposite (Figure 4b), implying that compound 5a showed a potentially weak action with EcR/USP.
Figure 4.

Binding mode and the interaction of (a) compound 5a with EcR/USP and (b) tebufenozide with EcR/USP.
Meanwhile, the correlation between the binding energy and antifungal activity was evaluated. There were 20 conformations docked for each target compound (Figure 5), and the lowest binding energy in the maximum cluster for the docked conformations was chosen as the representative binding energy for the corresponding compound. The scatter plot of AF values versus binding energies for the title compounds is shown in Figure 6. All of the data were concentrated near the line Y = 0.3066X – 0.8523, illustrating that there was a clear positive monotonic association between AF values and binding energies. In addition, Spearman’s rank correlation coefficient analytical approach was carried out using IBM SPSS STATITICS 22 software to investigate the correlation between AF values and binding energies. The result is listed in Table 5. It was found that the correlation was significant at 0.001 (at 0.01 level). The Spearman correlation coefficient was 0.762, indicating that there was a significant positive correlation, namely, the activity gradually increased with the increase in binding energies.
Figure 5.

Cluster of 20 docked conformations for compound 5a.
Figure 6.

Monotonicity of AF values vs binding energies for the target compounds.
Table 5. Spearman Rank Correlation Coefficient of AF Values Versus Binding Energies for the Target Compounds.
| binding energy | AF values | ||
|---|---|---|---|
| binding energy | correlation coefficient | 1.000 | 0.762 |
| Sig. (1-tailed) | 0.001 | ||
| AF values | correlation coefficient | 0.762 | 1.000 |
| Sig. (1-tailed) | 0.001 |
According to frontier molecular orbital theory, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are the two important factors affecting the bioactivity of compounds, because the HOMO has the priority to provide electrons, while the LUMO easily accepts electrons. The group with frontier molecular orbitals was a potential pharmacophore.35 Therefore, the frontier molecular orbitals of compound 5s with good inhibitory activity were calculated using the mean of the DFT/B3LYP method in the Gaussian 09 package.36 The total energy of compound 5s was −998.65190379 a.u., and the energy gap between the HOMO and LUMO was 0.217 a.u. The DFT-derived graphic results are presented using GaussView 5 software37 in Figure 7. The HOMO is located on the phenol moiety and the LUMO is located on the diacylhydrazine moiety, implying that these moieties were potential pharmacophores for the contribution of bioactivity.
Figure 7.

Optimized conformer and the maps of HOMO and LUMO for compound 5s.
Conclusions
In summary, 22 novel longifolene-derived diacylhydrazine compounds were designed by molecular docking-based virtual screening based on the crystal structure of SDH, synthesized using the renewable natural forest product longifolene as the starting material, and confirmed by IR, 1H NMR, 13C NMR, ESI-MS, and elemental analysis. The evaluation of the in vitro antifungal activity for the target compounds showed that some of them exhibited excellent inhibitory activity against the tested fungi. Meanwhile, a reasonable and effective 3D-QSAR model has been established for the further study of these types of compounds. There was a significant positive Spearman’s rank correlation between the antifungal activity and the docking-based binding energy. Molecular docking study revealed that there were H bonds and hydrophobic interactions between diacylhydrazine compounds and SDH. The distributed situation of the frontier molecular orbital showed that the phenol and diacylhydrazine moieties were potential pharmacophores for the contribution of bioactivity. Overall, compound 5a with excellent and broad-spectrum activity was the potential SDHI leading compound worthy of further study.
Materials and Methods
Synthesis and Characterization of Compounds
The synthesized compounds were characterized by IR (Nicolet IS 50 FT-IR spectrometer using KBr tableting), 1H NMR and 13C NMR (Bruker AVANCE III HD 600 MHz spectrometer using CDCl3 or dimethyl sulfoxide as the solvent and TMS as an internal standard), ESI-MS (TSQ Quantum Access MAX HPLC-MS apparatus), and elemental analysis (PerkinElmer 2400II elemental analyzer). The melting points were measured using a Hanon MP420 automatic melting point apparatus. All the characterization data above can be found in the Supporting Information. Longifolene 1 was provided by Wuzhou Pine Chemicals Co., Ltd., Wuzhou, China (65.0%, GC analyses). All other reagents were purchased from commercial suppliers and used as received. Using longifolene as the starting material, the longifolene-derived diacylhydrazine target compounds were synthesized. The longifolene-derived tetraline 2 and the longifolene-derived tetralone 3 were prepared according to our previously reported method.
General Procedure for the Synthesis of Compound 4
Compound 3 (1.00 g, 9.25 mmol) and m-CPBA (3.19 g, 18.50 mmol) were mixed in CH2Cl2 (5 mL). The mixture was continuously stirred at room temperature. After the completion of the reaction, monitored by the thin layer chromatography method, the resulting mixture was washed three times with 5.0% NaHCO3 aqueous solution. The organic layer was dried over anhydrous Na2SO4 and evaporated in a vacuum. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 80:1, v/v) to give the colorless liquid compound 4.
General Procedure for the Synthesis of Longifolene-Derived Diacylhydrazine Compounds 5a–5v
Under stirring, a solution of compound 4 (1.00 g, 4.31 mmol) in ethanol (5 mL) was added dropwise into the solution (5 mL) of substituted acylhydrazine compounds (12.93 mmol) in ethanol at 78.0 °C. After the completion of the reaction, monitored by the thin layer chromatography method, the reaction mixture was washed three times with 5.0% HCl aqueous solution (10 mL each time). The aqueous phase was extracted with Et2O. The combined organic phase was concentrated under reduced pressure to obtain the crude product, which was purified by silica gel column chromatography (petroleum ether/EtOAc = 5:1, v/v) to afford the target compounds 5a–5v.
In Vitro Antifungal Activity Test by the Agar Dilution Method
This test procedure was carried out according to the reported method and is described briefly as follows. Three copies of a culture plate containing 50 μg/mL tested compound and plant pathogenic fungi were cultured at 24.0 ± 1.0 °C for 48 h. Meanwhile, aseptic distilled water was used as a blank control. There were three replicates for each tested compound. The inhibition rate was calculated by comparing the mycelium diameter of the fungi treated with the emulsion to the blank control.
3D-QSAR Analysis
The 3D-QSAR model was built using the CoMFA method of Sybyl-X 2.1.1 software. The structures of compounds 5a–5v were optimized based on the Tripos force field and Gasteiger-Hückel charges. Compound 5a with the best activity was used as the template molecule and the common skeleton atoms are marked with an asterisk, as shown in Figure 8. The 18 target compounds, of which the R group contains the benzene ring, were superimposed and the result is displayed in Figure 9. The inhibition rate against P. piricola was converted to the AF using the formula: AF = log{[relative inhibitory rate/(100 – relative inhibitory rate)] × molecular weight}.
Figure 8.

Common skeleton atoms marked with an asterisk.
Figure 9.
Superposition mode for the 18 target compounds containing the benzene ring on the R group.
The built 3D-QSAR model was checked by the partial least-squares method. Its predictive capability was judged by a cross-validated value squared (q2), a correlation coefficient squared (r2), a standard deviation (S), and a Fisher validation value (F).
Molecular Docking and Frontier Molecular Orbital
The molecular docking was carried out using AutoDock 4.2.6 software. The PDB file of the target enzyme SDH–carboxin complex (UniProtKB AC P32420, homology modeling) was downloaded from the SWISS-MODEL web. The small molecules in the SDH–carboxin complex were removed. A 60 × 60 × 60 point grid box around the Q-site was set as the docking area. The docking parameters were searched using the Lamarckian genetic algorithm (GA) and the number of GA runs was set to 20 (namely, there were 20 conformations for each compound). The lowest binding energy in the maximum cluster for the docked conformations was chosen as the representative binding energy for the corresponding compound.
Moreover, the frontier molecular orbital for the target compound 5s was calculated by the DFTB3LYP/6-31G (d, p) method in the Gaussian 09 package on the Computer Supercomputing Platform at Guangxi University, and the result was viewed using GaussView 5 software.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (no. 31870556) and the Open Fund of Guangxi Key Laboratory of Chemistry and Engineering of Forest Products (no. GXFK2001). The authors are grateful to the State Key Laboratory of Element-Organic Chemistry, Nankai University, China, for the bioassay test.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00217.
Structure of compounds 5a–5v; IR, 1H NMR, 13C NMR, and ESI-MS analysis and spectra; model quality estimate of homology modeling on SWISS-MODEL web; and partial result of molecular docking-based virtual screening for the designed compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gu L. L.; Bai Y. L. Application and development of 10 kinds of new pesticides with potential. Mod. Agrochem. 2018, 17, 1–7. [Google Scholar]
- Qiu S. S.; Bai Y. L. Progress on research and development of succinate dehydrogenase inhibitor fungicides (I). Mod. Agrochem. 2014, 13, 1–7. [Google Scholar]
- Liu X. H.; Qiao L.; Zhai Z. W.; Cai P. P.; Cantrell C. L.; Tan C. X.; Weng J. Q.; Han L.; Wu H. K. Novel 4-pyrazole carboxamide derivatives containing flexible chain motif: design, synthesis and antifungal activity. Pest Manage. Sci. 2019, 75, 2892–2900. 10.1002/ps.5463. [DOI] [PubMed] [Google Scholar]
- Yang D.; Zhao B.; Fan Z.; Yu B.; Zhang N.; Li Z.; Zhu Y.; Zhou J.; Kalinina T. A.; Glukhareva T. V. Synthesis and biological activity of novel succinate dehydrogenase inhibitor derivatives as potent fungicide candidates. J. Agric. Food Chem. 2019, 67, 13185–13194. 10.1021/acs.jafc.9b05751. [DOI] [PubMed] [Google Scholar]
- Ren Z.-L.; Liu H.; Jiao D.; Hu H.-T.; Wang W.; Gong J.-X.; Wang A.-L.; Cao H.-Q.; Lv X.-H. Design, synthesis, and antifungal activity of novel cinnamon-pyrazole carboxamide derivatives. Drug Dev. Res. 2018, 79, 307–312. 10.1002/ddr.21469. [DOI] [PubMed] [Google Scholar]
- Du S.; Li Z. H.; Tian Z. M.; Xu L. Synthesis, antifungal activity and QSAR of novel pyrazole amides as succinate dehydrogenase inhibitors. Heterocycles 2018, 96, 74–85. 10.3987/COM-17-13826. [DOI] [Google Scholar]
- Xiong L.; Li H.; Jiang L.-N.; Ge J.-M.; Yang W.-C.; Zhu X. L.; Yang G.-F. Structure-based discovery of potential fungicides as succinate ubiquinone oxidoreductase inhibitors. J. Agric. Food Chem. 2017, 65, 1021–1029. 10.1021/acs.jafc.6b05134. [DOI] [PubMed] [Google Scholar]
- Yao T.-T.; Xiao D.-X.; Li Z.-S.; Cheng J.-L.; Fang S.-W.; Du Y.-J.; Zhao J.-H.; Dong X.-W.; Zhu G.-N. Design, synthesis, and fungicidal evaluation of novel pyrazole-furan and pyrazole-pyrrole carboxamide as succinate dehydrogenase inhibitors. J. Agric. Food Chem. 2017, 65, 5397–5403. 10.1021/acs.jafc.7b01251. [DOI] [PubMed] [Google Scholar]
- Wu Y.-Y.; Shao W.-B.; Zhu J.-J.; Long Z.-Q.; Liu L.-W.; Wang P.-Y.; Li Z.; Yang S. Novel 1,3,4-oxadiazole-2-carbohydrazides as prospective agricultural antifungal agents potentially targeting succinate dehydrogenase. J. Agric. Food Chem. 2019, 67, 13892–13903. 10.1021/acs.jafc.9b05942. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Shen Y.-Q.; Pan X.-Y.; Hou Y.-P.; Wu Q.-Y.; Yang G.-F. Discovery of 1,2,4-triazole-1,3- disulfonamides as dual inhibitors of mitochondrial complex II and complex III. New J. Chem. 2015, 39, 7281–7292. 10.1039/c5nj00215j. [DOI] [Google Scholar]
- Saraste M. Oxidative phosphorylation at the fin de siècle. Science 1999, 283, 1488–1493. 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- Sun F.; Huo X.; Zhai Y.; Wang A.; Xu J.; Su D.; Bartlam M.; Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Zhu X.-L.; Xiong L.; Li H.; Song X.-Y.; Liu J.-J.; Yang G.-F. Computational and experimental insight into the molecular mechanism of carboxamide inhibitors of succinate-ubquinone oxidoreductase. ChemMedChem 2014, 9, 1512–1521. 10.1002/cmdc.201300456. [DOI] [PubMed] [Google Scholar]
- Shang J.; Sun R. F.; Li Y. Q.; Huang R. Q.; Bi F. C.; Wang Q. M. Synthesis and insecticidal evaluation of N - tert - Butyl - N′ - thio [1- (6 - chloro - 3 - pyridylmethyl)-2-nitroiminoimidazolidine]-N, N′-diacylhydrazines. J. Agric. Food Chem. 2010, 58, 1834–1837. 10.1021/jf903642s. [DOI] [PubMed] [Google Scholar]
- Tan Y.; He H. Q.; Liu X. H.; Weng J. Q.; Tan C. X. Synthesis and herbicidal activities of novel pyridine substituted pyrazole diacylhydrazine derivatives. Chin. J. Synth. Chem. 2018, 26, 727–732. [Google Scholar]
- Wu W. N.; Wang R.; Du B.; Ouyang G. P. Synthesis and fungicidal activity evaluation of diacylhydrazine derivatives containing pyrimidine moiety. Chem. Res. Appl. 2017, 29, 810–815. [Google Scholar]
- Seow H. A.; Penketh P. G.; Shyam K.; Rockwell S.; Sartorelli A. C. 1,2-Bis (methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl] hydrazine: an anticancer agent targeting hypoxic cells. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 9282–9287. 10.1073/pnas.0409013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajani O. O.; Obafemi C. A.; Nwinyi O. C.; Akinpelu D. A. Microwave assisted synthesis and antimicrobial activity of 2-quinoxalinone-3-hydrazone derivatives. Bioorg. Med. Chem. 2010, 18, 214–221. 10.1016/j.bmc.2009.10.064. [DOI] [PubMed] [Google Scholar]
- Boss C.; Corminboeuf O.; Grisostomi C.; Weller T. Inhibitors of aspartic proteases-potential antimalarial agents. Expert Opin. Ther. Pat. 2006, 16, 295–317. 10.1517/13543776.16.3.295. [DOI] [Google Scholar]
- Efenberger-Szmechtyk M.; Nowak A.; Czyzowska A.; Kucharska A. Z.; Fecka I. Composition and antibacterial activity of Aronia melanocarpa (Michx.) Elliot, Cornus mas L. and Chaenomeles superba Lindl. leaf extracts. Molecules 2020, 25, 2011. 10.3390/molecules25092011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spréa R. M.; Fernandes Â.; Calhelha R. C.; Pereira C.; Pires T. C. S. P.; Alves M. J.; Canan C.; Barros L.; Amaral J. S.; Ferreira I. C. F. R. Chemical and bioactive characterization of the aromatic plant Levisticum officinale W.D.J. Koch: a comprehensive study. Food Funct. 2020, 11, 1292–1303. 10.1039/c9fo02841b. [DOI] [PubMed] [Google Scholar]
- Han Y. P.; Gong P.; Li K. Y. Study on antibacterial activity of phenol hydroxyl from N-aryl Schiff COS derivatives. Food Mach. 2015, 31, 127–131. [Google Scholar]
- Min D.; Zhao J.; Chen Y.; Zhao Z. D. Research progress of main sesquiterpenes from heavy turpentine. Biomass Chem. Eng. 2017, 51, 54–60. [Google Scholar]
- Tyagi B.; Mishra M. K.; Jasra R. V. Solvent free synthesis of 7-isopropyl-1, 1-dimethyltetralin by the rearrangement of longifolene using nano-crystalline sulfated zirconia catalyst. J. Mol. Catal. A: Chem. 2009, 301, 67–78. 10.1016/j.molcata.2008.11.011. [DOI] [Google Scholar]
- Liu Z.; Jin S.; Wang C.; Zhu L.; Cheng Z.; An X.; Wang S.; Cai Z. Synthesis of new tetralin-type musky compounds. Chem. Ind. For. Prod. 1995, 15, 7–12. [Google Scholar]
- Zhu X.-P.; Lin G.-S.; Duan W.-G.; Li Q.-M.; Li F.-Y.; Lu S.-Z. Synthesis and antiproliferative evaluation of novel longifolene-derived tetralone derivatives bearing 1,2,4-triazole moiety. Molecules 2020, 25, 986–998. 10.3390/molecules25040986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Bai X.; Duan W.; Cen B.; Huang M.; Lu S. High value-added application of sustainable natural forest product α-pinene: synthesis of myrtenal oxime esters as potential KARI inhibitors. ACS Sustain. Chem. Eng. 2019, 7, 7862–7868. 10.1021/acssuschemeng.9b00254. [DOI] [Google Scholar]
- Kang G.-Q.; Duan W.-G.; Lin G.-S.; Yu Y.-P.; Wang X.-Y.; Lu S.-Z. Synthesis of bioactive compounds from 3-carene (II): synthesis, antifungal activity and 3D-QSAR study of (Z)- and (E)-3-caren-5-one oxime sulfonates. Molecules 2019, 24, 477–491. 10.3390/molecules24030477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Chen Z.; Duan W.; Wang X.; Lei F. Synthesis and biological activity of novel myrtenal-derived 2-acyl-1,2,4-triazole-3-thione compounds. Chin. J. Org. Chem. 2018, 38, 2085–2092. 10.6023/cjoc201801043. [DOI] [Google Scholar]
- Lin G.-S.; Duan W.-G.; Yang L.-X.; Huang M.; Lei F.-H. Synthesis and antifungal activity of novel myrtenal-based 4-methyl-1,2,4-triazole-thioethers. Molecules 2017, 22, 193–203. 10.3390/molecules22020193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N.-N.; Li Y.; Yu S.-J.; Zhang X.; Liu X.-H.; Zhao W.-G. Microwave-assisted synthesis of some novel 1, 2, 3-triazoles by click chemistry, and their biological activity. Res. Chem. Intermed. 2013, 39, 759–766. 10.1007/s11164-012-0595-9. [DOI] [Google Scholar]
- Liu X.-H.; Shi Y.-X.; Ma Y.; Zhang C.-Y.; Dong W.-L.; Pan L.; Wang B.-L.; Li B.-J.; Li Z.-M. Synthesis, antifungal activities and 3D-QSAR study of N-(5-substituted-1,3,4-thiadiazol-2-yl)cyclopropanecarboxamides. Eur. J. Med. Chem. 2009, 44, 2782–2786. 10.1016/j.ejmech.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Wang B.-L.; Shi Y.-X.; Zhang S.-J.; Ma Y.; Wang H.-X.; Zhang L.-Y.; Wei W.; Liu X.-H.; Li Y.-H.; Li Z.-M.; Li B.-J. Syntheses, biological activities and SAR studies of novel carboxamide compounds containing piperazine and arylsulfonyl moieties. Eur. J. Med. Chem. 2016, 117, 167–178. 10.1016/j.ejmech.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Sanner M. F. Python: a programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [PubMed] [Google Scholar]
- Sun N.-B.; Fu J.-Q.; Weng J.-Q.; Jin J.-Z.; Tan C.-X.; Liu X.-H. Microwave assisted synthesis, antifungal activity and DFT theoretical study of some novel 1,2,4-triazole derivatives containing the 1,2,3-thiadiazole moiety. Molecules 2013, 18, 12725–12739. 10.3390/molecules181012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr., Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.
- Dennington R.; Keith T.; Millam J.. GaussView, Version 5; Semichem, Inc.: Shawnee Mission, KS, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.