

Abstract

Transient receptor potential canonical channel 6 (TRPC6) has been implicated in many kinds of malignant tumors, but very few potent TRPC6 antagonists are available. In this study, a benzothiazole amide derivative 1a was discovered as a TRPC6 activator in a cell-based high-throughput screening. A series of benzothiazole amide derivatives were designed and synthesized. The docking analyses indicated that the conformations of the compounds bound to TRPC6 determined the agonistic or antagonistic activity of the compounds against TRPC6, and compound 1s with the tetrahydronaphthalene group in R1 position fit well into the binding pocket of the antagonist-bound conformation of TRPC6. Compound 1s showed an inhibitory potency order of TRPC3 (IC50 3.3 ± 0.13 μM) ≈ C6 (IC50 4.2 ± 0.1 μM) > C7 with good anti-gastric cancer activity in a micromolecular range against AGS and MKN-45, respectively. In addition, 1s inhibited the invasion and migration of MKN-45 cells in vitro.
1. Introduction
Transient receptor potential canonical (TRPC) channels, which belong to the TRP family, are nonselective Ca2+ permeable cation channels expressed in various tissues.1−5 Based on the amino acid sequence, TRPCs are grouped into TRPC1, TRPC2, TRPC3/6/7, and TRPC4/5 in which the TRPC3/6/7 shares 65–78% sequence identity.4,6 TRPC6 has been reported to involve the regulation of cardiac hypertrophy, pulmonary vascular tone and permeability, blood pressure, renal fibrosis, and Alzheimer’s disease.7−13 TRPC6 also plays a key role in the carcinogenesis and is overexpressed in gastric cancer, breast cancer, and glioblastoma. However, only a low level of TRPC6 is expressed in the corresponding gastric normal tissues, indicating that TRPC6 channels might be a novel anti-cancer drug target against gastric cancer.14−18
Due to the high sequence conservation among TRPCs, it is very challenging to design selective small-molecule TRPC6 antagonists. Very few compounds have been reported as TRPC6 inhibitors.5,11,19−25 1H-Imidazole, 1-[2-(4-methoxyphenyl)-2-[3-(4-methoxyphenyl)propoxy]ethyl] (SKF-96365), a nonselective TRPC6 antagonist with an IC50 value of 4.9 μM has been reported to inhibit the Ca2+ elevation regulated by TRPC6 channels.14,26,27 The IC50 values of specific TRPC6 antagonists, 2-(phenylamino)thiazol-4-yl)(piperidin-1-yl)methanone derivatives (SAR-7334, Figure 1), [4-(6-aminopyridazin-3-yl)piperidin-1-yl]-[4-[4-(trifluoro-methy-l)phenoxy] phenyl]methanone (BI 749327), 4-(((1R,2R,3aR,7aS)-2-((R)-3-aminopiperidin-1-yl)-3a,7a-dime-thyl-5-oxooctahydro-1H-inden-1-yl)oxy)benzonitrile (DS88790512), and larixol congener SH045 were 7.9, 13, 11, and 5.8 nM, respectively.11,28−31 Although a number of TRPC6 antagonists were reported in a nanomolar range, very few of them were utilized in vivo, partially due to the low potency and poor bioavailability.11 Our group has reported a selective TRPC3 agonist HDM with the pyrazolopyrimidine skeleton, demonstrating high affinity with EC50 (TRPC3) ∼19 nM and EC50 (TRPC6) ∼1.385 μM.32 One methyl group was added into HDM to produce a high selective TRPC6 antagonist HQR with an IC50 value of ∼1 μM, which suppressed the gastric cancer cells proliferation and the growth of xenograft tumors.33
Figure 1.

Active compounds targeted TRPC6.
A variety of benzothiazole amides analogs from the Molecular Libraries Small Molecule Repository (MLSMR) were screened using the fluorescence Ca2+ assay. Compound 1a (PubChem: 24761863, Figure 1) was first founded as a TRPC6 agonist with an EC50 value of ∼70.79 μM as the primary hit. The replacement of methoxyl group with methyl group (1b) at position 4 and 1-methoxyl substitution of the naphthalene at position 1 (1c) led to the loss of agonist activity of the source lead compound, suggesting that the substitution at position 1 or 4 may play an important role in the structure–activity relationship study.
2. Results and Discussion
2.1. Rational Design
Subtle difference in the compound structure induces the activity alternation, which may attribute to the different binding modes of the compounds against the antagonist-bound or agonist-bound conformation of TRPC6. The mutation of Glu509 on S2 and Asp530 on S3 almost completely abolished the response of TRPC6 to the antagonist AM-1473, and the side chains of His446 on S1 and Arg758 on the reentrant loop moved for adapting to the antagonist-binding (PDB: 6uza) compared to no major structural changes in the side chains of agonist-bound conformation (PDB: 6uz8),31 which characterized the important roles of these residues bound to the antagonists. Molecular docking analyses were carried out to analyze the difference between the antagonists and agonists against TRPC6. In the case of SAR-7334 and BI 749327, the TRPC6 inhibitory activities could be explained by the possible interactions between the compounds and the antagonist-bound conformation of TRPC6. SAR-7334 and BI 749327 fit much deeper into the residues of S1-S4, the TRP helix and the membrane-reentrant loop, which formed the basis for the inhibitory study (Figure S1). SAR-7334 engaged in one hydrogen-bond interaction with Glu509 and one van der Waals contact with Asp530 (Figure 2A). Moreover, SAR-7334 was involved one π–π interaction with His446 as well as one π-cation with Arg758. BI 749327 made a hydrogen-bond interaction with Glu509, a π-alkyl interaction with His446 and a weak van der Waals contact with Arg758 (Figure 2B), which may explain the decreased antagonistic effect of BI 749327. However, the agonists HDM and 1a lacked intermolecular interactions with these key residues and even formed unfavorable bumps with the residues of TRPC6 (Figure 2C,D), which induced the loss of antagonistic activities of these two compounds.
Figure 2.
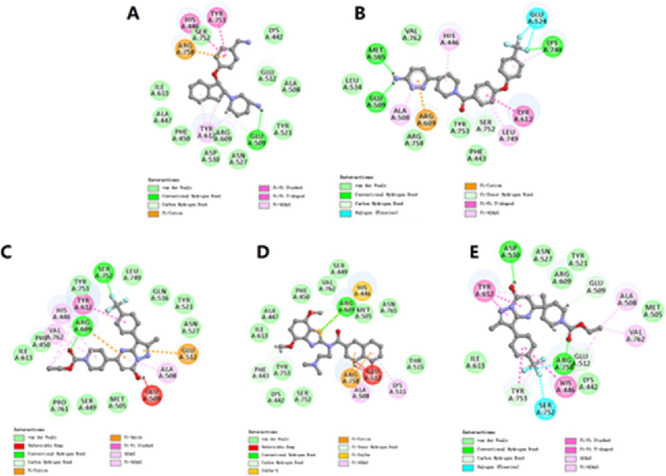
Detailed interactions of SAR-7334 (A), BI 749327 (B), HDM (C), 1a (D), and HQR (E) interacted with the antagonist-bound conformation of TRPC6 (PDB ID: 6uza). The 3D binding modes of TRPC6 and the compounds are shown in Figure S1.
HDM had the agonistic activity against TRPC6 (EC50 = 1.385 μM), and its methylated derivative (HQR) showed moderate antagonistic activity against TRPC6 (IC50 = 1.0 μM). The docking analyses indicated that the methyl group induced the conformational change of HQR by producing the steric effect and fit well into the antagonist-bound conformation of TRPC6 (Figure S1), which was similar with SAR-7334 and BI 749327. HQR formed a hydrogen bond with Asp530 and a weak van der Waals contact with Glu509 (Figure 2E). This indicated that the conformations of the compounds bound to TRPC6 determined the agonistic or antagonistic activity of the compounds against TRPC6.
2.2. Structure–Activity Relationship (SAR) Analysis
Based on the activity alteration from HDM to HQR, the possibility of converting the agonist 1a into an antagonist was explored. Benzothiazole amides 1d-1af by further diversification of 1a at position R1–R5 were designed, synthesized, and evaluated in the HEK293 cellular assay system with a stable expression of the mouse TRPC6.
A series of compounds were first designed and synthesized through rational drug design and structure–activity relationship. Compounds 1d-1v containing 4-methoxyben-zo[d]thiazol-2-yl heterocycle with various aromatic substituents and 4-dialkylamino substituents at position R1 and R2 were synthesized and evaluated, respectively. No in vitro potency was observed when the 2-chloro-5-thiophene group at position R1 replaced with 2-methoxypyridin-4-yl (1d-1g), styrene (1h-1k) or 2-naphthalene (1r, 1t-1v, Table 1). Surprisingly, incorporation of 4-(trifluoromethyl) benzene (1m-1o), benzo[d][1,3]dioxole (1p-1q) at position R1 with the dialkylamino group at position R2 (1m-1q) resulted in analogs with the TRPC6 inhibitory potency. The steady increase in potency of the described compounds (1o > 1n > 1m > 1l, 1q > 1p) was identified by the introduction of a larger size of the amino substituents at position R2 from dimethylamino, diethylamino, 1-pyrrolidine to 1-piperidine group. It is interesting that the naphthalene group in compound 1r was replaced with 1,2,3,4-tetrahydronaphthalene group (1s), resulting in the improved antagonistic activity of 1s against TRPC6 (IC50 = 4.2 ± 0.1 μM by Ca2+ assay). Therefore, the binding modes of 1r and 1s interacted with the TRPC6 were further analyzed for explaining the activity alteration.
Table 1. Inhibitory Effects of 1d-1v against TRPC6 Channels.

Effects on the activity of TRPC6 or the IC50 value against the activity of TRPC6 activated by the HDM at a concentration of 10 μM,32 which was calculated based on one experiment (N = 3); NA: No activity.
The compounds with the naphthalene substituents at position R1 (1a-1c, 1r, 1t-1v) had no antagonistic potency against TRPC6. The naphthalene group in 1a formed an unfavorable bump with Glu512 in the antagonist-bound conformation of TRPC6 by docking analyses. The demethoxy derivative (1r) also showed similar conformation against TRPC6 (Figure 3A,C). Therefore, we hypothesized that the naphthalene group may not be an optimal design, and the group should be substituted to reduce the unfavorable bump with the target. The naphthalene group was replaced with the tetrahydronaphthalene (1s), which made a distortion in this position, alternated the stacking angle, and eliminated unfavorable bumps. Moreover, the N,N-dimethylamino ethyl moiety of 1s had polar interactions with Glu509 and Glu512, respectively (Figure 3B,D), and the benzo[d]thiazole core of 1s was involved in a π-cation interaction with Arg609, confirming the stable binding pattern in vitro assay.
Figure 3.
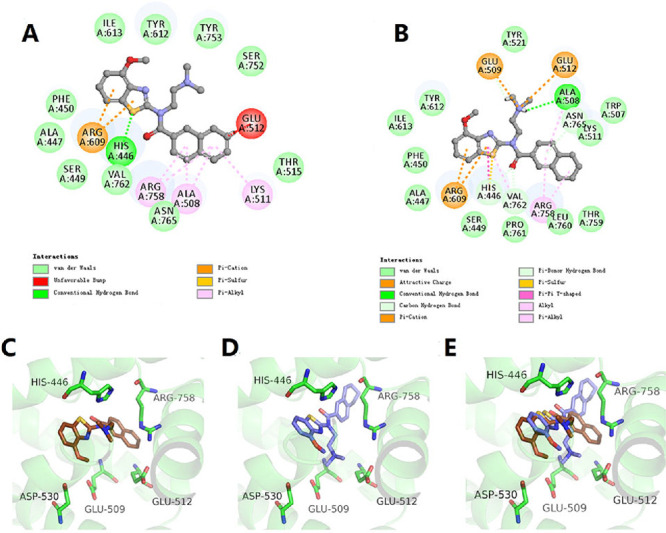
The binding modes of 1r (A, C) and 1s (B, D) interacted with the antagonist-bound conformation of TRPC6 (PDB: 6uza). 1r and 1s were shown in brown and blue sticks in 3D mode. (E) Superimposed docking structures of TRPC6 in complex with 1s and 1r.
Based on the characteristics of the metal ion in the TRPC6 channel, we tried to introduce sulfur atoms and designed a series of compounds. The importance of the substitution at the R2 position of the benzothiazole ring on the activity of TRPC6 was explored with compounds 1w-1ab. When R1 was 2-chloro-5-thiophene, benzothiazole amides 1w-1ab demonstrated varying levels of antagonist activity against TRPC6 (Table 2). Without the methoxyl substitution at position R3–R5 on the phenyl ring has led to analogs 1aa-1ab with good antagonist potency at the TRPC6 receptor with a mean IC50 value of 15.1 ± 1.4 and 8.8 ± 1.3 μM, respectively. The potency of the dimethyl amino group (1w, 1y, and 1aa) at position R2 was ∼2–4 folds lower than that of the prototypical pyrrolidine substituent (1x, 1z, and 1ab). However, the 2-bromo-5-thiophene group at position R1 had a deleterious effect on the TRPC6 antagonist potency (1 ac-1ad) as did the 2-bromo-5-furan group at position R1 (1ae-1af).
Table 2. Effect of Compounds 1w−1af on TRPC6 Channels.

Effects on the activity of TRPC6 or the IC50 value against the activity of TRPC6 activated by the HDM at a concentration of 10 μM,32 which was calculated based on one experiment (N = 3); NA: No activity.
2.3. Functional Characterization of 1s on the Activities of TRPC3-TRPC7
Compound 1s inhibited the [Ca2+]i increases evoked by HDM (10 μM) with an IC50 value of 4.2 ± 0.1 μM in TRPC6-expressing cells (n = 3) (Figure 4A,B) without self-fluorescence, and 1s was chosen to further explore. 1s immediately suppressed the TRPC6 currents evoked by the GSK1702934A (1 μM) in the whole-cell voltage clamp assay (Figure 4C). Based on the currents at +80 mV, an IC50 value of 1s for the monovalent cation currents of TRPC6 was calculated as 4.19 ± 0.04 μM (Figure 4D, n = 6–8), which was comparable to that obtained in the Ca2+ assay (Table 1).
Figure 4.
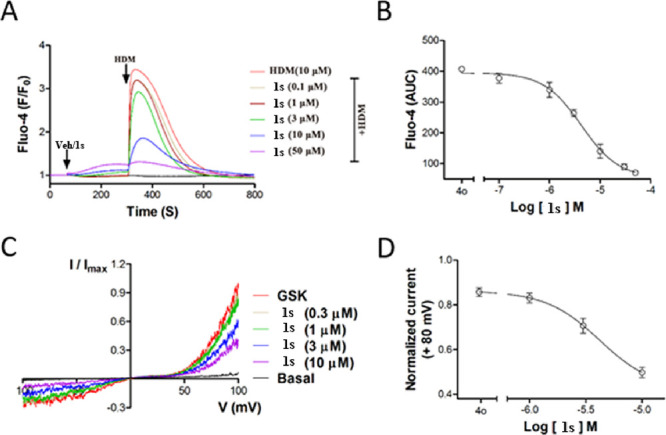
Compound 1s inhibited TRPC6 expressed in HEK293 cells. (A) 1s inhibited the Ca2+ response in TRPC6-HEK293 cells induced by HDM. Inoculated cells in the 96-well plate were loaded with Fluo-4, and fluorescence changes were read in a microplate reader while adding 1s and HDM, as shown by horizontal bars at the top of the traces. The traces represent the reading (F0) of three repeated measurements of an experiment at the beginning, normalized to the average fluorescence changes (ΔF) of fluorescence, which was repeated three times. (B) Concentration-response curves for compound 1s inhibiting TRPC6 was determined by Ca2+ assay (Fluo-4). The solid lines indicated the fitting of the Hill equation, which obtained the IC50 values. (C) By changing the concentration of compound 1s in the TRPC6 cell, the current–voltage (I–V) relationships were acquired through the voltage ramps. GSK1702934A (1 μM) activated current and 1s inhibited current. (D) Concentration-response curves for compound 1s determined by electrophysiology recording to inhibit TRPC6. The solid lines were represented by the Hill equation, which produced the IC50 value.
The effect of 1s on the TRPC3 and TRPC7 channels was examined by the fluorescence Ca2+ assay. Similar as TRPC6, 1s caused a detectable change in intracellular Ca2+ concentration ([Ca2+]i) in TRPC3-HEK293 cells in a concentration-dependent manner (Figure 5A,E). Treatment with 1s inhibited the response to subsequent application of HDM (10 μM), obtaining the IC50 values of 3.3 ± 0.13 μM (Figure 5B). Different from TRPC3/6, 1s almost did not cause a discernible Ca2+ response in TRPC7-HEK293 cells (Figure 5F). These data indicate that 1s is an effective antagonist of TRPC3/6 channels, and its potency order is TRPC3 ≈ C6 > C7.
Figure 5.
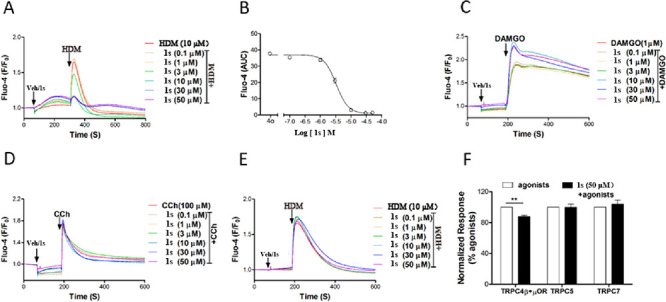
Compound 1s inhibited agonist-evoked TRPC3/4/5/7 activity. 1s inhibited the increase of [Ca2+]i in HEK293 cells that expressed human TRPC3 (A, induced by 10 μM HDM), mouse TRPC4 (C, induced by 1 μM DAMGO), mouse TRPC5 (D, induced by 100 μM CCh), and mouse TRPC7 (E, induced by 10 μM HDM). (B) Concentration-response curves for compound 1s inhibiting TRPC3 was determined by Ca2+ assay (Fluo-4). (F) Compound 1s had little or no effect on Ca2+ response in TRPC7-HEK293 cells.
The above data indicate that the benzothiazole amide 1s has antagonistic activity on the TRPC3/6 subgroup of TRPC channels. To verify the selectivity, the Ca2+ assay was used to detect the effects of 1s on stable HEK293 cell line that co-expressed TRPC4/5 and μ-opioid receptor and on HEK293 cell lines expressing TRPA1, TRPM8, TRPV1, and TRPV3. For TRPC4, 1s only partially inhibited the membrane depolarization induced by DAMGO (1 μM), indicating a very weak inhibitory activity (Figure 5C). For TRPC5-, TRPA1-, TRPM8-, TRPV1-, and TRPV3-expressing cells planted in 96-well plates and wells loaded with Fluo-8, 1s (50 μM) did not significantly affect the Ca2+ response induced by the agonist of the corresponding channels (Figure 5D). Therefore, compound 1s significantly inhibited TRPC3/6, had a weak effect on TRPC4, and no effect on TRPC5 and other TRP channels.
2.4. Anti-Proliferation Activity of 1s against Gastric Cancer Cells in vitro
The anti-gastric cancer activity of compound 1s was tested with SKF-96365 as a positive control. 1s exhibited remarkable cytotoxicity against the two human gastric cancer cell lines. It inhibited cell proliferation of both MKN-45 and AGS in a dose-dependent manner, with IC50 values of 8.00 ± 0.51 and 12.03 ± 1.01 μM, respectively, which was better than SKF-96365 with the IC50 values of 15.34 and 37.03 μM in the same condition (Figure 6).
Figure 6.

Cytotoxicity of 1s against MKN-45and AGS cells. MKN-45 or AGS cells were treated with the indicated concentrations of compound 1s or SKF-96365 for 72 h, and then the cell viability was detected by the MTT assay. Data were normalized to the same trial without drug treatment and to the means ± SEM of the three trials.
2.5. Anti-Cell Migration and Invasion of 1s against MKN-45 Cells
The wound-healing migration assay and Boyden chamber assay were applied to evaluate the effect of 1s on the migration and invasion of MKN-45 cells. Treatment with 1s at a concentration of 5 μM inhibited the MKN-45 cell migration (Figure 7A,C). At the same time, 1s impaired the activity of MKN-45 cells to move through the pores toward the chemoattractant below (Figure 7B,D).
Figure 7.
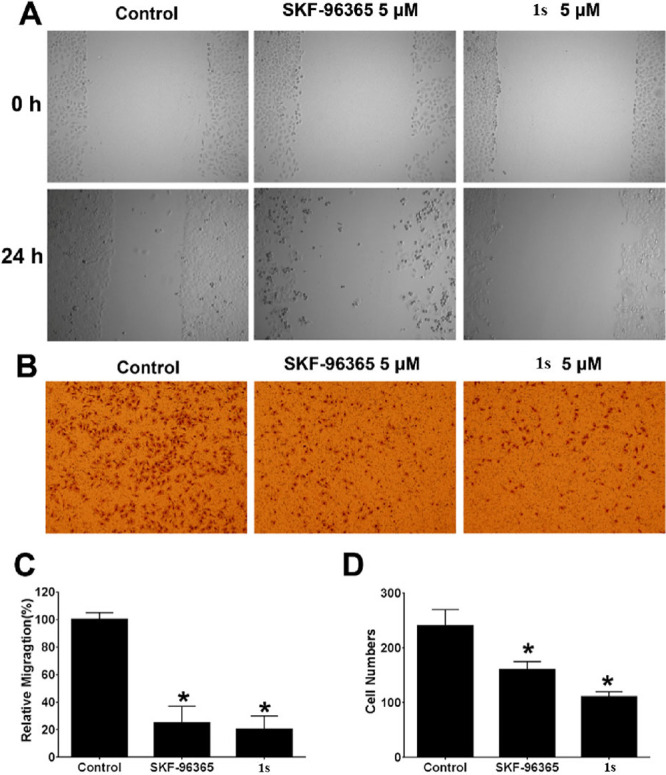
Compound 1s inhibited the migration and invasion of MKN-45 cells. (A, C) MKN-45 cells were treated with SKF-96365 and 1s at the indicated concentrations. Scratches were made and images were taken at 0 and 24 h, and the migration distances were summarized. (B, D) MKN-45 cells in serum-free medium were plated at the top uncoated membranes with SKF-96365 and 1s at specified concentrations, and the lower chamber was filled with a complete medium and cells were then allowed to migrate for 24 h, and the cells on the bottom part of the membrane were stained with crystal violet and summarized. *p < 0.05, compared with the control group.
3. Conclusions
In summary, the pharmacological properties of newly synthesized benzothiazole amides compounds as novel TRPC3/6 antagonists were studied by [Ca2+]i-imaging and electrophysiology recording. A lead compound 1a was identified as a TRPC6 activator with an EC50 of ∼70.79 μM as a starting point. Chemical modifications on the benzothiazole amide scaffold focused on the introduction of aromatic groups at R1 and amino groups at R2. Twenty-nine benzothiazole amides were designed and synthesized. Compound 1s has an IC50 value of 4.2 ± 0.1 μM in TRPC6-expressing cells. Compared with other TRP families, 1s has a reasonable selective high potency against TRPC3/6 and has no effect on the TRPC5, TRPC7, and other TRP channels. Furthermore, compound 1s has a strong inhibition effect on the activation of TRPC6 mediated by HDM and has an inhibitory effect on the proliferation of gastric cancer cells, MKN-45, and AGS cells with the IC50 values of 8.00 ± 0.51 and 12.03 ± 1.01 μM, respectively. Further experiments are currently underway to evaluate its pharmacokinetic profile, in vivo anti-tumor activity as well as its mechanism of anti-gastric cancer activity and potential therapeutic utilities, and the results will be reported in the near future.
4. Experimental Section
4.1. General Methods
All commercially available starting materials and solvents were reagent grade and used without further purification. Unless otherwise specified, all reactions were performed under an argon atmosphere. The reaction progress was monitored by thin-layer chromatography (TLC). The purification of all compounds was purified by silica gel column chromatography. 1H and 13C NMR spectra were recorded on a Bruker AV400 spectrometer (1H-400 MHz, 13C-101 MHz). High-resolution mass spectrometry (HRMS) was performed with a Thermo LTQ XL Orbitrap instrument. Low-resolution mass spectrometry (ESI-MS) was performed on AB Sciex 4000 Qtrap.
4.2. Synthesis
The synthesis route (Scheme S1) and NMR spectra of benzothiazole amides derivatives 3d-7af are provided in the Supporting Information.
4.3. Channel Inhibition
Antagonism of synthesized TRPC6 ligands were evaluated in vitro, as measured by inhibition of receptor activation by the TRPC6 agonist HDM as previously reported.32 The human embryonic kidney HEK293 cell line that co-expressed mouse TRPC6 and the Gq/11-PLC-coupled M5 muscarinic receptor was detected by fluorometric assay. These cells showed a strong persistent membrane depolarization respond to the muscarinic agonist, carbachol (CCh), which could be detected by the FLIPR membrane potential (FMP) dye as an increase in fluorescence. In transfected HEK293 cells, the tested compounds did not significantly activate the TRPC6-mediated Ca2+ increase. On the other hand, the TRPC6-HEK293 cells were preincubated with different doses of the tested compounds (5 min) and then incubated with the agonist HDM (10 μM), and Ca2+ elevation in most cells was inhibited due to the response of TRPC6 to the agonist HDM.
4.4. Cell Viability Assay
The MTT detection method is based on our previous protocol.33 Briefly, the cells (2000/well) were seeded into 96-well plates and incubated overnight and then treated with the tested compounds. After 72 h, the MTT solution was added and then incubated for 4 h. The MTT-formazan formed was dissolved in DMSO in which the absorbance was then measured at 570 nm with a microplate analyzer. The cell relative survival rate and the IC50 were calculated.
4.5. Wound Healing Assay
Following our previous protocol, the wound-healing assay was used to evaluate cell migration.33 Briefly, MKN-45 cells were seeded in a 24-well plate and cultured overnight. The wound was yielded by scraping with a sterile 200 μL pipette tip and treated it with the specified concentrations of tested compounds. After 0 and 24 h, images of the wound distance were quantified via measuring the width of the cell-free zone at six distinct positions with a digitally calibrated micrometer by microphotographs with an OLYMPUS IX 73 inverted microscope equipped with a CCD camera, and the relative migration distance was calculated using Image J software.
4.6. Transwell Cell Migration Assays
Transwell migration assays were performed using 6-well Transwell chambers (Corning Life Sciences, Lowell, MA, USA) containing 8 μm permeable pores according to the manufacturer’s instructions. The cells cultured in the serum-free medium were seeded at the top uncoated membranes with the tested compounds, and the lower chamber was filled with a complete medium containing 10% FBS. The cells were allowed to migrate for 24 h, and the cells in the upper surface of the membrane were carefully washed using PBS and removed with a cotton swab. After that, the cells on the bottom part of the membrane were fixed and stained with crystal violet in which the cells were visualized and counted from six randomly selected fields using fluorescent inverted microscope. Directional migration was quantified by cell counting using Image J.
4.7. Molecular Docking
The crystal structures of hTRPC6 in complex with AM-1473 (PDB: 6UZA and 6UZ8) was used as the receptors for docking-based studies. The geometry of proteins was first optimized with Dreiding-like force field and subsequently run through the “clean protein” from macromolecular module in Discovery Studio 2018 toolbox to standardize a detailed check. After preparing the protein complexes by monitoring the bad valence, removing all water molecules and adding hydrogens, CHARMm force field was applied to the receptors and the benzothiazole amides. The active site spheres centered on its cognate ligand was created with an automatic generated diameter of 10 Å. The remained parameters were referred to keep default settings.
4.8. Statistical Analysis
Data are expressed as the mean ± SEM. The analysis of the data obtained from in vitro experiments was performed using Origin 7.5 (Origin Lab) and GraphPad Prism (V 5.01). Statistically significant differences between two sets of data were evaluated with the Student’s t-test (p < 0.05). Statistical comparisons between multiple (>3) experimental groups were performed with analysis of variance (ANOVA) and Student’s t-test. Values with a p < 0.05 were considered statistically significant.
Acknowledgments
This work was partially supported by grants from Key Research Project of Yantai (2019XDHZ102), National Natural Science Foundation of China (82073888, 81773674), the Science and Technology Support Program for Youth Innovation in Universities of Shandong (2019KJM009), Top Talents Program for One Case Discussion of Shandong Province, the National Key R&D Program of China (2020YFA0908800), Shenzhen Science and Technology Research Grant (JCYJ20190808152019182), Hubei Province Scientific and Technical Innovation Key Project (2020BAB058), the Applied Basic Research Program of Wuhan Municipal Bureau of Science and Technology (2019020701011429), the Local Development Funds of Science and Technology Department of Tibet (XZ202001YD0028C), and the Fundamental Research Funds for the Central Universities (ZNJC201931).
Glossary
Abbreviations
- [Ca2+]i
intracellular Ca2+ concentration
- DAG
diacylglycerols
- IPAc
isopropyl acetate
- TFA
trifluoroacetic acid
- DMAP
4-dimethylaminopyridine
- DMF
N,N-Dimethylformamide
- EC50
half maximal effective concentration
- FMP
FLIPR Membrane potential
- TRPC
transient receptor potential canonical
- TRPV
transient receptor potential vanilloid
- TRPM
transient receptor potential melastatin
- SAR
structure–activity relationship
- TRPA
transient receptor potential ankyrin
- MLSMR
Molecular Libraries Small Molecule Repository
- TLC
thin-layer chromatography
- HRMS
high-resolution mass spectrometry
- ESI-MS
low-resolution mass spectrometry
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00514.
Chemistry of 1d-1af, the synthesis route of 3d-7af, docking modes of the compounds interacted with the antagonist-bound conformation of TRPC6, 1H and 13C NMR of 1d-1af, and HRMS of 1d-1af (PDF)
Author Contributions
∇ Y.W., M.Z., and Z.L. contributed equally to this work. Y.W. and F.L. performed the pharmacological experiments. M.Z. and Z.L. performed compound synthesis and optimization. T.T., M.D., X.Z., F.X., P.W., and Y.L. also contributed to compound synthesis. G.Y. and Z.C. performed the test on channel activity. J.L. performed molecular modeling. X.H. and H.W. designed the experimental idea and gave guidance. The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ong H. L.; de Souza L. B.; Ambudkar I. S. Role of TRPC Channels in Store-Operated Calcium Entry. Adv. Exp. Med. Biol. 2016, 898, 87–109. 10.1007/978-3-319-26974-0_5. [DOI] [PubMed] [Google Scholar]
- Hofmann T.; Obukhov A. G.; Schaefer M.; Harteneck C.; Gudermann T.; Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Liu H.; Luo X.; Wang Y.; Liu Y.; Jin H.; Liu Z.; Yang W.; Yu P.; Zhang L.; Zhang L. Design, synthesis and biological activities of 2,3-dihydroquinazolin-4(1H)-one derivatives as TRPM2 inhibitors. Eur. J. Med. Chem. 2018, 152, 235–252. 10.1016/j.ejmech.2018.04.045. [DOI] [PubMed] [Google Scholar]
- Guo W.; Chen L. Recent progress in structural studies on canonical TRP ion channels. Cell Calcium 2019, 83, 102075. 10.1016/j.ceca.2019.102075. [DOI] [PubMed] [Google Scholar]
- Wang H.; Cheng X.; Tian J.; Xiao Y.; Tian T.; Xu F.; Hong X.; Zhu M. X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. 10.1016/j.pharmthera.2020.107497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.; Wang C. Y.; Gou S. M.; Wu H. S.; Liu T.; Xiong J. X. Isolation and biological analysis of tumor stem cells from pancreatic adenocarcinoma. World J. Gastroenterol. 2008, 14, 3903–3907. 10.3748/wjg.14.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Eder P.; Chang B.; Molkentin J. D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7000–7005. 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef M.; Knezevic N.; Chava K. R.; Smith M.; Sukriti S.; Gianaris N.; Obukhov A. G.; Vogel S. M.; Schraufnagel D. E.; Dietrich A.; Birnbaumer L.; Malik A. B.; Mehta D. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A.; Mederos Y. S. M.; Gollasch M.; Gross V.; Storch U.; Dubrovska G.; Obst M.; Yildirim E.; Salanova B.; Kalwa H.; Essin K.; Pinkenburg O.; Luft F. C.; Gudermann T.; Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6–/– mice. Mol. Cell. Biol. 2005, 25, 6980–6989. 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Chen Q.; Ju Y.; Li W.; Li W. Palmitate induces human glomerular mesangial cells fibrosis through CD36-mediated transient receptor potential canonical channel 6/nuclear factor of activated T cell 2 activation. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2020, 1865, 158793. 10.1016/j.bbalip.2020.158793. [DOI] [PubMed] [Google Scholar]
- Lin B. L.; Matera D.; Doerner J. F.; Zheng N.; Del Camino D.; Mishra S.; Bian H.; Zeveleva S.; Zhen X.; Blair N. T.; Chong J. A.; Hessler D. P.; Bedja D.; Zhu G.; Muller G. K.; Ranek M. J.; Pantages L.; McFarland M.; Netherton M. R.; Berry A.; Wong D.; Rast G.; Qian H. S.; Weldon S. M.; Kuo J. J.; Sauer A.; Sarko C.; Moran M. M.; Kass D. A.; Pullen S. S. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 10156–10161. 10.1073/pnas.1815354116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Jia Y. C.; Cui K.; Li N.; Zheng Z. Y.; Wang Y. Z.; Yuan X. B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature 2005, 434, 894–898. 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Paez Espinosa E. V.; Murad J. P.; Ting H. J.; Khasawneh F. T. Mouse transient receptor potential channel 6: role in hemostasis and thrombogenesis. Biochem. Biophys. Res. Commun. 2012, 417, 853–856. 10.1016/j.bbrc.2011.12.058. [DOI] [PubMed] [Google Scholar]
- Cai R.; Ding X.; Zhou K.; Shi Y.; Ge R.; Ren G.; Jin Y.; Wang Y. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int. J. Cancer 2009, 125, 2281–2287. 10.1002/ijc.24551. [DOI] [PubMed] [Google Scholar]
- Aydar E.; Yeo S.; Djamgoz M.; Palmer C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: a potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23. 10.1186/1475-2867-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X.; He Z.; Zhou K.; Cheng J.; Yao H.; Lu D.; Cai R.; Jin Y.; Dong B.; Xu Y.; Wang Y. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010, 102, 1052–1068. 10.1093/jnci/djq217. [DOI] [PubMed] [Google Scholar]
- El Boustany C.; Bidaux G.; Enfissi A.; Delcourt P.; Prevarskaya N.; Capiod T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. 10.1002/hep.22263. [DOI] [PubMed] [Google Scholar]
- Ding X.; He Z.; Shi Y.; Wang Q.; Wang Y. Targeting TRPC6 channels in oesophageal carcinoma growth. Expert Opin. Ther. Targets 2010, 14, 513–527. 10.1517/14728221003733602. [DOI] [PubMed] [Google Scholar]
- Urban N.; Hill K.; Wang L.; Kuebler W. M.; Schaefer M. Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell Calcium 2012, 51, 194–206. 10.1016/j.ceca.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Li W.; Chen X.; Riley A. M.; Hiett S. C.; Temm C. J.; Beli E.; Long X.; Chakraborty S.; Alloosh M.; White F. A.; Grant M. B.; Sturek M.; Obukhov A. G. Long-term spironolactone treatment reduces coronary TRPC expression, vasoconstriction, and atherosclerosis in metabolic syndrome pigs. Basic Res. Cardiol. 2017, 112, 54. 10.1007/s00395-017-0643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernichtein S.; Pigat N.; Barry Delongchamps N.; Boutillon F.; Verkarre V.; Camparo P.; Reyes-Gomez E.; Mejean A.; Oudard S. M.; Lepicard E. M.; Viltard M.; Souberbielle J.-C.; Friedlander G.; Capiod T.; Goffin V. Vitamin D3 Prevents Calcium-Induced Progression of Early-Stage Prostate Tumors by Counteracting TRPC6 and Calcium Sensing Receptor Upregulation. Cancer Res. 2017, 77, 355–365. 10.1158/0008-5472.CAN-16-0687. [DOI] [PubMed] [Google Scholar]
- Urban N.; Wang L.; Kwiek S.; Rademann J.; Kuebler W. M.; Schaefer M. Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Mol. Pharmacol. 2016, 89, 197–213. 10.1124/mol.115.100792. [DOI] [PubMed] [Google Scholar]
- Diez-Bello R.; Jardin I.; Lopez J. J.; El Haouari M.; Ortega-Vidal J.; Altarejos J.; Salido G. M.; Salido S.; Rosado J. A. (−)Oleocanthal inhibits proliferation and migration by modulating Ca(2+) entry through TRPC6 in breast cancer cells. Biochim. Biophys. Acta, Mol. Cell Res. 2019, 1866, 474–485. 10.1016/j.bbamcr.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Wang Y.; Zhang C.; Yang G.; Zhang F.; Yu B.; Chai C.; Cao Z. Ribemansides A and B, TRPC6 Inhibitors from Ribes manshuricum That Suppress TGF-β1-Induced Fibrogenesis in HK-2 Cells. J. Nat. Prod. 2018, 81, 913–917. 10.1021/acs.jnatprod.7b01037. [DOI] [PubMed] [Google Scholar]
- Chen R. C.; Sun G. B.; Ye J. X.; Wang J.; Zhang M. D.; Sun X. B. Salvianolic acid B attenuates doxorubicin-induced ER stress by inhibiting TRPC3 and TRPC6 mediated Ca(2+) overload in rat cardiomyocytes. Toxicol. Lett. 2017, 276, 21–30. 10.1016/j.toxlet.2017.04.010. [DOI] [PubMed] [Google Scholar]
- Merritt J. E.; Armstrong W. P.; Benham C. D.; Hallam T. J.; Jacob R.; Jaxa-Chamiec A.; Leigh B. K.; McCarthy S. A.; Moores K. E.; Rink T. J. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 1990, 271, 515–522. 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R.; Okada T.; Onoue H.; Hara Y.; Shimizu S.; Naitoh S.; Ito Y.; Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation channel. Circ. Res. 2001, 88, 325–332. 10.1161/01.RES.88.3.325. [DOI] [PubMed] [Google Scholar]
- Maier T.; Follmann M.; Hessler G.; Kleemann H.-W.; Hachtel S.; Fuchs B.; Weissmann N.; Linz W.; Schmidt T.; Lohn M.; Schroeter K.; Wang L.; Rütten H.; Stübing C. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br. J. Pharmacol. 2015, 172, 3650–3660. 10.1111/bph.13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama K.; Nagata T.; Kobayashi J.; Nakamura A.; Miyoshi N.; Kazui M.; Sakurai K.; Sakakura T. Discovery of a bicyclo[4.3.0]nonane derivative DS88790512 as a potent, selective, and orally bioavailable blocker of transient receptor potential canonical 6 (TRPC6). Bioorg. Med. Chem. Lett. 2018, 28, 2222–2227. 10.1016/j.bmcl.2018.03.056. [DOI] [PubMed] [Google Scholar]
- Häfner S.; Burg F.; Kannler M.; Urban N.; Mayer P.; Dietrich A.; Trauner D.; Broichhagen J.; Schaefer M. A (+)-Larixol Congener with High Affinity and Subtype Selectivity toward TRPC6. ChemMedChem 2018, 13, 1028–1035. 10.1002/cmdc.201800021. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Yu X.; Chen H.; Horne D.; White R.; Wu X.; Lee P.; Gu Y.; Ghimire-Rijal S.; Lin D. C.-H.; Huang X. Structural basis for pharmacological modulation of the TRPC6 channel. eLife 2020, 9, e53311 10.7554/eLife.53311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C.; Ding M.; Zhu Y.; Lu Y.; Du J.; Miller M.; Tian J.; Zhu J.; Xu J.; Wen M.; Er-Bu A.; Wang J.; Xiao Y.; Wu M.; McManus O. B.; Li M.; Wu J.; Luo H. R.; Cao Z.; Shen B.; Wang H.; Zhu M. X.; Hong X. Pyrazolopyrimidines as Potent Stimulators for Transient Receptor Potential Canonical 3/6/7 Channels. J. Med. Chem. 2017, 60, 4680–4692. 10.1021/acs.jmedchem.7b00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M.; Wang H.; Qu C.; Xu F.; Zhu Y.; Lv G.; Lu Y.; Zhou Q.; Zhou H.; Zeng X.; Zhang J.; Yan C.; Lin J.; Luo H. R.; Deng Z.; Xiao Y.; Tian J.; Zhu M. X.; Hong X. Pyrazolo[1,5-a]pyrimidine TRPC6 antagonists for the treatment of gastric cancer. Cancer Lett. 2018, 432, 47–55. 10.1016/j.canlet.2018.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.