

Sir, we review two autopsy cases of dystrophy type 1 (DM1) with severe tau pathologies. Central nervous system involvement commonly occurs in DM1 9, with some cases reporting overexpression of 3‐repeat tau (especially fetal tau isoform) in DM1 brain 5, 10. Unlike these previous observations, here we report a DM1 patient with predominant 4‐repeat tauopathy. We briefly discuss the distinct clinicopathological features.
Case 1 showed distal weakness of the upper limbs and grip myotonia at age 17. Other family members were also diagnosed with muscular dystrophy. He was admitted to our hospital at age 30. Amplification of the CTG triplet yielded approximately 1100–1200 repeats. His overall IQ score, as measured by the Wechsler Adult Intelligence Scale‐revised (WAIS‐R), was 111 at age 50. His score on the Hasegawa dementia scale‐revised [HDS‐R; a mental examination method used in Japan with a scale of 0–30 (<21 representing dementia)] was 30/30 at age 54. He died at 57 years old because of pneumonia.
Case 2 had shown muscle weakness and mental retardation since childhood. Family members were also diagnosed with muscular dystrophy, but no other neurological disorders such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) or frontotemporal lobar degeneration‐17 (FTLD‐17). He came to our hospital at age 45. Amplification of the CTG triplet yielded approximately 4700–5700 repeats. His HDS‐R score was 18/30 at age 50. He was unable to answer questions involving serial subtractions of seven or recite digits backward. He also exhibited hypersomnia and hypoactivity. However, there were no signs of parkinsonism or aphasia. He developed pneumonia, had a stroke and died during an episode of recurrent pneumonia when he was 64 years old.
Macroscopically, the brain of case 1 weighed 1450 g, with no obvious brain atrophy. For case 2, the brain weighed 1079 g. The cerebral cortex showed left‐dominant atrophy due to stroke. Microscopically, we observed numerous phosphorylated tau‐positive structures in the CA1 region, subiculum, entorhinal cortex, ambient gyrus, amygdala, subthalamic nucleus, oculomotor nucleus, substantia nigra, limbic system, cerebral neocortex and brainstem in both cases. As both patients were around 60, this is a significantly different pathology from primary age‐related tauopathy. The tau isoforms detected were different between the two cases, especially in the brainstem (Figure 1). RD3 antibody‐positive structures, such as globose type neurofibrillary tangles (NFTs) and glial tauopathies were noted in case 1 (Figure 1A), but not case 2 (Figure 1B). Conversely, in case 1, cytoplasmic staining with RD4 antibody was not observed (Figure 1C), although nuclear and neuropil staining of RD4 antibody was confirmed in case 2 (Figure 1D). Consistent with these RD3‐ and RD4‐immunoreactivity results, AT‐8‐immunopositive neurons with severe neuropil threads and glial structures were observed in both cases (Figure 1E,F). Gallyas stained‐positive structures were also detected in both cases (Figure 1G,H). We did not detect any amyloid plaques.
Figure 1.
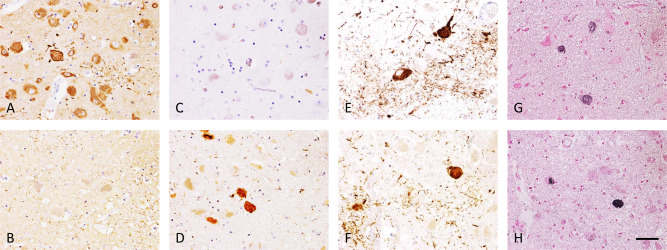
Histological findings in the midbrain of DM1 cases. Immunohistochemistry with RD3 antibody reveals many 3‐repeat tau positive neurons with occasional globose type neurofibrillary tangles (NFTs) and glial tauopathies in case 1 (A), but no positive neurons in case 2 (B). Immunohistochemistry with RD4 antibody shows no positive neurons in case 1 (C), yet several 4‐repeat tau positive neurons with globose type NFTs in case 2 (D). Immunohistochemistry with anti‐phosphorylated tau antibody (AT‐8) shows positive neurons with NFTs, neuropil threads and glial inclusions in both cases (E, case 1; F, case 2). In both cases, globose type NFTs are argyrophilic by Gallyas silver impregnation (G, case 1; H, case 2). Scale bar = 50 µM.
We classified neuronal density into three categories based on RD3‐ and RD4‐immunopositive neurons at a magnification of ×200: 1 or 2 immunopositive neurons were characterized as low degree (+), 3 or 5 immunopositive neurons as moderate degree (++) and >6 immunopositive neurons as high degree (+++) (Figure 2). We used these criteria to examine the hippocampus, cerebral neocortex, basal ganglia, thalamus, substantia nigra and dorsal vagal nucleus. The results are summarized in Table 1. Specifically, in case 1, there was a tendency for 3‐repeat tau dominant deposition not only in the brainstem but also in the limbic system and cerebral neocortex. In contrast, in case 2, there was a tendency for 4‐repeat tau deposition. We performed western blotting to detect sarkosyl‐insoluble tau isoforms. Four‐repeat tau was detected in both cases (Figure S1).
Figure 2.
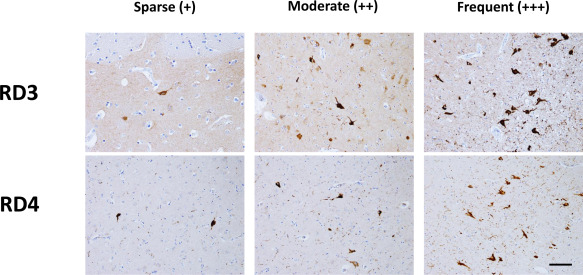
Scoring for RD3 and RD4 immunopositive neuron densities at ×200 magnification. (−), absent; (+), 1 or 2 immunopositive neurons; (++), 3–5 immunopositive neurons; (+++), >6 immunopositive neurons. Scale bar = 100 µM.
Table 1.
Evaluation of DM1 neuropathology based on density and regional distribution of tau‐immunopositive neurons. Neuron density was scored as: none (−), low degree (+), moderate degree (++) and high degree (+++) (see Figure 2).
| Three‐repeat tau | Four‐repeat tau | |||
|---|---|---|---|---|
| Case 1 | Case 2 | Case 1 | Case 2 | |
| CA1 | (+) | (+) | (+) | (++) |
| Entorhinal cortex | (+++) | (+) | (++) | (+++) |
| Amygdala | (+++) | (+) | (+) | (++) |
| Frontal lobe cortex | (+) | (−) | (−) | (+) |
| Temporal lobe cortex | (+) | (−) | (−) | (+) |
| Parietal lobe cortex | (−) | (−) | (−) | (+) |
| Basal ganglia | (+) | (−) | (−) | (+) |
| Subthalamic nucleus | (++) | (−) | (−) | (+) |
| Oculomotor nucleus | (+++) | (−) | (−) | (++) |
| Substantia nigra | (++) | (−) | (−) | (+) |
| Dorsal vagal nucleus | (+) | (−) | (−) | (+) |
DM1 can be considered to show aspects of tauopathy 1, 10. NFTs are mainly observed in the hippocampus, entorhinal cortex and most temporal areas in DM1 brain 1. In the human adult brain, six tau isoforms are expressed by alternative splicing of exons 2, 3 and 10 1. Exon 10 generates 4‐repeat tau. Some cases report fetal tau isoform overexpression in DM1 5, 10, but the mechanism is still poorly understood. Mis‐splicing of exons 2, 3 and 10 is suggested to promote fetal expression of the 3‐repeat tau isoform 5. RNA binding proteins such as muscleblind‐like protein (MBNL)‐1, MBNL2 and CUGBP Elav‐like family member 2 (CELF2) are also thought to be involved 2, 4, 6, 7. Case 1 showed typical distribution and 3‐repeat phosphorylated tau. However, in case 2, 4‐repeat tau was mainly observed in the hippocampus and brainstem. This is a very characteristic pathological change, which might be induced by the microtubule‐associated protein tau (MAPT) gene haplotype, or alternatively gene mutations involving exon 10. However, we were unable to obtain consent for genetic testing from the patients or their families. Selective accumulation of 4‐repeat tau in the brain of case 2 suggests multiple factors contribute to DM1 tauopathy.
Central nervous system involvement is an important clinical feature of DM1 9. Further, clinical variety and severity is different between congenital and juvenile‐adult forms 9. Congenital DM1 is associated with mental retardation, speech development delay and behavioral abnormalities. While overall IQ scores are frequently in the normal range in the juvenile‐adult form 9. The DM1 phenotype may have contributed to the severe mental retardation in case 2. Additionally, CTG repeat expansion size may also be related to the symptoms of case 2. In DM1, CTG repeat expansion produces mutant RNA and induces alternative splicing defects in multiple RNAs and other genes. Abnormal regulation of several alternative splicing events has been documented in DM1 brain 8.
The relationship between cerebral involvement and tauopathy in DM1 is still unclear. The 4‐repeat tauopathy is observed in some diseases, such as PSP, CBD, FTDP‐17 and argyrophilic grain disease 3, but these symptoms are different from case 2. For example, the mental faculties are relatively spared until the end‐stage in PSP and CBD.
In summary, here we have reviewed two DM1 patients, with case 2 showing severe cognitive dysfunction and atypical pathology associated with predominant 4‐repeat tauopathy. The exact mechanism of 4‐repeat tau dominancy is not yet known.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Western blotting of sarkosyl‐insoluble tau. Frozen brain tissues (about 0.2 g) were homogenized in extraction buffer containing sarkosyl and incubated for 30 min at 37 °C. After centrifugation at 20,000×g for 10 min, the supernatants were taken and ultracentrifuged at 100,000×g for 20 min. Western blots of the pellets were probed with RD3, RD4 and Tau2 antibodies. Similar band patterns including 4‐repeat tau are observed between frontal cortices of both cases.
References
- 1. Caillet‐Boudin ML, Fernandez‐Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N (2014) Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci 6:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carpentier C, Ghanem D, Fernandez‐Gomez FJ, Jumeau F, Philippe JV, Freyermuth F et al (2014) Tau exon 2 responsive elements deregulated in myotonic dystrophy type I are proximal to exon 2 and synergistically regulated by MBNL1 and MBNL2. Biochim Biophys Acta 1842:654–664. [DOI] [PubMed] [Google Scholar]
- 3. Delacourte A (2005) Tauopathies: recent insights into old diseases. Folia Neuropathol 43:244–257. [PubMed] [Google Scholar]
- 4. Dhaenens CM, Tran H, Frandemiche ML, Carpentier C, Schraen‐Maschke S, Sistiaga A et al (2011) Mis‐splicing of Tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim Biophys Acta 1812:732–742. [DOI] [PubMed] [Google Scholar]
- 5. Dhaenens CM, Schraen‐Maschke S, Tran H, Vingtdeux V, Ghanem D, Leroy O et al (2008) Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: two individual consequences of CUG trinucleotide repeats. Exp Neurol 210:467–478. [DOI] [PubMed] [Google Scholar]
- 6. Ghanem D, Tran H, Dhaenens CM, Schraen‐Maschke S, Sablonnière B, Buée L et al (2009) Altered splicing of tau in DM1 is different from the foetal splicing process. FEBS Lett 583:675–679. [DOI] [PubMed] [Google Scholar]
- 7. Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Fernández Gómez FJ et al (2015) MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep 12:1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 13:3079–3088. [DOI] [PubMed] [Google Scholar]
- 9. Meola G, Sansone V (2007) Cerebral involvement in myotonic dystrophies. Muscle Nerve 36:294–306. [DOI] [PubMed] [Google Scholar]
- 10. Vermersch P, Sergeant N, Ruchoux MM, Hofmann‐Radvanyi H, Wattez A, Petit H et al (1996) Specific tau variants in the brains of patients with myotonic dystrophy. Neurology 47:711–717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Western blotting of sarkosyl‐insoluble tau. Frozen brain tissues (about 0.2 g) were homogenized in extraction buffer containing sarkosyl and incubated for 30 min at 37 °C. After centrifugation at 20,000×g for 10 min, the supernatants were taken and ultracentrifuged at 100,000×g for 20 min. Western blots of the pellets were probed with RD3, RD4 and Tau2 antibodies. Similar band patterns including 4‐repeat tau are observed between frontal cortices of both cases.