

Abstract
Deposition of amyloid‐β (Aβ) is central to Alzheimer's disease (AD) pathogenesis and associated with progressive neurodegeneration in traumatic brain injury (TBI). We analyzed predisposing factors for Aβ deposition including monomeric Aβ40, Aβ42 and Aβ oligomers/protofibrils, Aβ species with pronounced neurotoxic properties, following human TBI. Highly selective ELISAs were used to analyze N‐terminally intact and truncated Aβ40 and Aβ42, as well as Aβ oligomers/protofibrils, in human brain tissue, surgically resected from severe TBI patients (n = 12; mean age 49.5 ± 19 years) due to life‐threatening brain swelling/hemorrhage within one week post‐injury. The TBI tissues were compared to post‐mortem AD brains (n = 5), to post‐mortem tissue of neurologically intact (NI) subjects (n = 4) and to cortical biopsies obtained at surgery for idiopathic normal pressure hydrocephalus patients (iNPH; n = 4). The levels of Aβ40 and Aβ42 were not elevated by TBI. The levels of Aβ oligomers/protofibrils in TBI were similar to those in the significantly older AD patients and increased compared to NI and iNPH controls (P < 0.05). Moreover, TBI patients carrying the AD risk genotype Apolipoprotein E epsilon3/4 (APOE ε3/4; n = 4) had increased levels of Aβ oligomers/protofibrils (P < 0.05) and of both N‐terminally intact and truncated Aβ42 (P < 0.05) compared to APOE ε3/4‐negative TBI patients (n = 8). Neuropathological analysis showed insoluble Aβ aggregates (commonly referred to as Aβ plaques) in three TBI patients, all of whom were APOE ε3/4 carriers. We conclude that soluble intermediary Aβ aggregates form rapidly after TBI, especially among APOE ε3/4 carriers. Further research is needed to determine whether these aggregates aggravate the clinical short‐ and long‐term outcome in TBI.
Keywords: Alzheimer's disease, amyloid‐β, amyloid β oligomers, traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI), a major cause of long‐term disability 26, is associated with persistent neuroinflammation, white matter degeneration and progressive cortical atrophy at long‐term following the initial impact 19. Additionally, epidemiological evidence has linked TBI to the development of various neurodegenerative disorders 6, 11, 20, including a fourfold increased risk for Alzheimer's disease (AD) with a younger age at AD onset 20. Moreover, brains from TBI patients often display deposition of amyloid‐β (Aβ), the peptide which accumulates as insoluble aggregates (commonly referred to as Aβ plaques) in the AD brain 21. Apart from post‐mortem assessments, TBI‐induced insoluble Aβ aggregates can be visualized by PET using recently developed amyloid binding tracers 25, including Pittsburgh compound B (PiB). When PiB‐PET was used in TBI patients, increased retention signals in the cortex and striatum, similar to what is usually seen in AD patients, were observed within one year following the initial injury 17, 42.
Available evidence suggests that TBI‐related Aβ pathology can appear very rapidly and insoluble Aβ aggregates are present within hours after the injury in approximately 30% of the cases 18, 38. In addition, accumulation of both aggregated Aβ 46 and amyloid‐β precursor protein (AβPP) 22, the protein from which Aβ is formed, could be observed in axons from resected TBI brain tissues.
Novel analytical tools have now enabled the investigation of oligomeric/protofibrillar Aβ species. Emerging evidence suggests that such intermediary, soluble prefibrillar, forms of Aβ are particularly neurotoxic and can cause synaptic dysfunction 1, 34, 40, 48. These species can be detected in affected parts of the AD brain 31 and they may also form in response to TBI as suggested by the reported increase in CSF Aβ oligomer levels in TBI patients 12.
Here, we analyzed the relationship between TBI and Aβ pathology in humans and can show that not only insoluble Aβ aggregates and monomers, but also oligomers/protofibrils, are rapidly induced by TBI. Additionally, we show that the AD risk genotype ApolipoproteinE ε3/4 (APOE ε3/4) is correlated to increased brain tissue levels of all investigated Aβ species in TBI patients.
MATERIALS AND METHODS
Ethics statement
All clinical and experimental research described herein was approved by the regional ethics committee in Uppsala, Sweden (decision numbers 2005/103, 2008/303, 2009/89 and 2010/379). Written informed consent was obtained from the TBI patients' closest relatives and from the patients themselves had they sufficiently recovered from their injury at >6 months post‐injury. Informed consent was also obtained from the idiopathic normal pressure hydrocephalus (iNPH) patients as well as the neurologically intact (NI) subjects and the AD patients (or their relatives) for post‐mortem brain donations.
Traumatic brain injury cohort
Twelve patients with severe TBI, defined as post‐resuscitation Glasgow Coma Scale (GCS) scores ≤8, were included. Demographics and clinical characteristics are shown in Table 1. Patients were >16 years of age and no patient had any other known neurological disorder or Down's syndrome. Patients were endotracheally ventilated and sedated and continuous measurements of intracranial pressure (ICP) and cerebral perfusion pressure (CPP) were performed (for details, see 7). Patients included in this study were subjected to surgical focal decompression due to life‐threatening elevations of intracranial pressure (ICP) and/or the presence of a space‐occupying brain swelling or hemorrhage.
Table 1.
TBI patients‐characteristics.
| Pat # | Age | Gender | APOE genotype | Cause of injury | Other injuries | Time post‐injury (h) | Region of surgery | Surgery | mGCS pre‐op | Aβ‐IR |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 22 | M | ε3/3 | MVA | Thi | 9 | LT | Ccx + DC | 4 | na |
| 2 | 72 | M | ε3/3 | Fall | None | 4 | LFr | Ccx + DC | 2 | na |
| 3 | 40 | M | ε3/3 | MVA | None | 4 | RFr | Ccx | 5 | na |
| 4 | 74 | M | ε3/4 | Fall | None | 4 | RT | Ccx | 5 | Aß |
| 5 | 58 | M | ε3/3 | Fall | Efx | 9 | LT | Ccx | 5 | no |
| 6 | 49 | M | ε3/4 | Fall | None | 84 | RT | Ccx* | 5 | no |
| 7 | 19 | F | ε2/4 | Fall | None | 16 | RFrT | Ccx + DC† | 3 | no |
| 8 | 65 | M | ε3/4 | Fall | Ffx, Thi | 180 | LT | Ccx | 3 | Aß |
| 9 | 25 | M | ε3/3 | SPR | None | 24 | LFrP | Ccx + DC‡ | 2 | AAS |
| 10 | 67 | M | ε3/4 | SBO | None | 4 | LT | Ccx | 5 | Aß |
| 11 | 51 | M | ε3/3 | Fall | None | 53 | RFr | Ccx + DC | 5 | no |
| 12 | 52 | M | ε3/3 | Fall | None | 42 | RT | Ccx§ | 4 | no |
*Coagulopathy.
†DC prior to Ccx, performed at resurgery.
‡Initial surgery for acute subdural hematoma and DC in primary hospital, Ccx + revised DC secondary surgery.
§Initial surgery for aSDH, Ccx at re‐surgery.
AAS = axonal swelling seen by IHC using antibodies directed to amyloid precursor protein; Aß‐IR = ß‐amyloid‐ immunoreactivity seen by immunohistochemistry (IHC); APOE= Apolipoprotein E; Ccx = removal of cortical contusion; DC = Decompressive craniectomy; Efx = extremity fracture; F = female; Fr = frontal; Ffx = facial fracture; L = left; M = male; MVA = motor‐vehicle accident; mGCS = motor component of the Glasgow Coma Scale; na= not available; P = parietal; pre‐op = preoperative; R = right; SBO = struck by object; SPR = sports‐related; T = temporal; Thi = thoracic injury.
Sampling and preparation of brain tissues from TBI patients
All surgically removed brain tissue (Figure 1) was after surgical decompression placed in a sterile prelabeled container and stored in −80°C freezer until analyzed. In nine of the twelve TBI patients, half of the tissue was put in a routinely used fixative, 4% buffered formalin (HistoLab Products AB, Gothenburg, Sweden, cat no 02176). The samples were fixed for 24–72 h and then paraffin‐embedded and processed by hardware Tissue tek VIP (Sakura, CA, USA).
Figure 1.
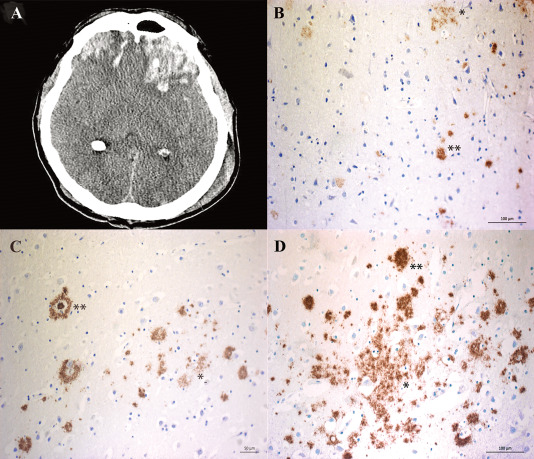
Amyloid‐β (Aβ) aggregates in surgically resected brain tissue in a subset of patients with severe TBI. A. An example of patients with a focal left frontal contusion injury following severe TBI causing midline shift, obliteration of the basal cisterns and life‐threatening mass effect to the surrounding brain tissue. Immediate surgical removal of the lesion was mandated, allowing immunohistochemical and biochemical analysis of the removed and injured brain tissue. B–D. Immunohistochemical image from the three TBI patients demonstrating accumulation of diffuse (*) and compact (**) insoluble Aβ aggregates.
From the fresh frozen contused brain tissue, samples of approximately 5 mm2 were taken for the biochemical analysis by ELISA. The samples were homogenized on ice using Dounce homogenizer (2 × 10 strokes) in 1:10 weight:volume Tris buffered saline (20 mM Tris, 137 mM NaCl) with the addition of protease inhibitors (CompleteMini, Roche). The samples were centrifuged at 16 000 × g for 1 h at +4°C and the supernatants were defined as TBS extracts, and stored at −80°C until analyzed.
Sampling and preparation of post‐mortem brain tissues from AD and neurologically intact subjects
Brain tissue from twelve post‐mortem subjects was included. The diagnosis of the five deceased dementia cases (AD) were given at the time of their death and followed the recommendations from 1991 (3, 32). Four of these cases fulfilled the neuropathological criteria for definite AD, of which one was an APP Swedish mutation (K670N/M671L) carrier. In one case, the primary alterations were Lewy body‐related (Dementia with Lewy bodies), in addition to a concomitant AD‐related pathology. Seven subjects without neurological deficits during life (NI) were neuropathologically assessed 9, and in all these cases sparse subtentorial hyperphosphorylated tau pathology was observed (Braak stages b to 2). Concomitant cortical Aβ pathology (Thal phase 1) was observed in three out of the seven cases, whom were excluded from the study. From the remaining NI subjects (n = 4) temporal cortex specimens were available. The specimens from NI subjects (n = 4) and biopsies from iNPH patients (n = 4) did not differ in their Aβ levels and their data were pooled and included as control brain tissues (n = 8, Tables 3 and 4). No control subjects had sustained a previous TBI. All samples were homogenized on ice as described above.
Table 3.
Demographics of the NI cohort.
| Pat # | Age at death (years) | Gender | APOE genotype | Time post‐mortem (h) | HPtau‐pathology Braak stage | Aβ pathology Thal phase |
|---|---|---|---|---|---|---|
| 1 | 88 | Male | ε2/3 | 39 | 1 | 0 |
| 2 | 88 | Female | ε3/3 | 22 | 2 | 0 |
| 3 | 63 | Male | ε3/3 | 30 | b | 0 |
| 4 | 92 | Male | ε3/3 | NA | 1 | 0 |
APOE = Apolipoprotein E; Braak= Braak neurofibrillary tangle stage; NA = not available; Thal = Thal beta‐amyloid stage; HPtau = hyperphosphorylated tau; Aβ = amyloid β; IR = immunoreactivity.
Table 4.
Demographics of the iNPH cohort.
| Pat # | Age (years) | Gender | APOE genotype | In Cerebrospinal fluid | In brain tissue | ||||
|---|---|---|---|---|---|---|---|---|---|
| Tau (ng/L) | HPtau (ng/L) | Aß 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42 (ng/L) | NFL (ng/L) | Aβ‐IR | HPtau‐IR | ||||
| 1 | 67* | Male | ε3/4 | <75 | 16 | 368 | 320 | ns | ns |
| 2 | 82 | Male | ε3/3 | 173 | 29 | 776 | 1310 | ns | ns |
| 3 | 76 | Female | ε3/3 | 192 | 24 | 729 | 1270 | ns | sparse |
| 4 | 76 | Male | ε3/3 | 242 | 34 | 598 | 1130 | ns | ns |
Reference interval: CSF‐Tau <400 ng/L, CSF‐HPtau <80 ng/L, CSF‐ Aß 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42 >550 ng/L, CSF‐NFL <1850 ng/L.
*= Biopsy containing only white substance.
APOE = Apolipoprotein E; Aß= Amyloid ß; HPtau= Hyperphosphorylated tau; NFL= Neurofilament‐light; IR‐immunoreactivity; ns = not seen; iNPH = idiopathic normal pressure hydrocephalus.
Sampling and preparation of cerebrospinal fluid and brain tissues from idiopathic normal pressure hydrocephalus patients
Four iNPH patients, planned for ventriculoperitoneal shunt insertion, were included. Lumbar CSF samples were obtained prior to surgery and analyzed for Aβ1–42, total tau, hyperphosphorylated tau (HPtau) and neurofilament‐light (NFL) using commercial ELISA kits (Table 3).
Brain tissue biopsies from iNPH patients were obtained at the time of ventriculoperitoneal shunt insertion 8, according to our clinical routine. The ca 2.11 mm diameter biopsies included both cortical and subcortical tissues and the procedure was as previously described 8. A first sample was placed in a prelabeled 1.5 mL Eppendorf tube and stored in a −80°C freezer until analyzed and the second sample was placed in 4% buffered formalin. Both the frozen and the fixed tissues were then processed following the same protocol as described above.
Immunohistochemistry for Aβ aggregates
Seven μm thick sections were cut and placed on SuperFrost® plus slides (Menzel‐Gläser, Vienna, Austria) for immunohistochemical (IHC) analysis. Aβ aggregates were visualized using Aβ antibodies (6F/3D, M0872; dilution 1:100, Dako, Glostrup, Denmark) and for visualization, Dako Autostainer plus with Dako EnVision FLEX detection system was used.
In vitro generation of Aβ protofibrils
Amyloid‐β protofibrils were defined as soluble Aβ aggregates >100 kDa, which eluted in the void volume on a Size Exclusion Chromatography Superdex 75 column. Lyophilized Aβ1–42 peptide (American Peptide Company, CA, USA, cat no 62–0‐80, lot no 12077006T) was dissolved to 100 µM in 10 mM NaOH. Aβ1–42 protofibrils were prepared by diluting the Aβ1–42 peptide to 50 µM in 0.1 M phosphate buffer. The preparation was incubated for 30 minutes at 37°C and then centrifuged at 16 000 × g for 5 minutes to pellet potential large aggregates. The supernatants were further purified from monomers by size exclusion chromatography (Superdex 75 column, GE Healthcare, Sweden) in 0.05 M Phosphate buffer, 0.15 M NaCl, pH 7.4, and protofibrils were collected in the void fraction as previously described 34, 43. The generated Aβ protofibrils were used for the standard curves in the oligomer/protofibril assays.
Biochemical measurements of Aβ oligomers
As previously described, the mAb82E1 sandwich ELISA detects both smaller (<100 kDa) and larger soluble aggregates of Aβ 47 (Figure 2A). Briefly, the mAb82E1 antibody (IBL, Fujioka, Japan), detecting the N‐terminus of β‐secretase cleaved AβPP, was used for capture (0.25 µg/mL). Next, the TBS brain extracts were diluted fivefold and incubated in duplicates for 2 h at 22°C. A biotinylated mAb was used for detection (0.25 µg/mL). Sample concentrations were calculated from a standard curve based on the in vitro generated Aβ42 protofibrils using a 4‐parameter equation.
Figure 2.

TBI increases the levels of Aβ oligomers and protofibrils in surgically resected brain tissue. A. Individual oligomer Aβ levels in patient samples detected with the mAb82E1 sandwich ELISA. The absence of a bar indicates that the value was below the limit of detection. B. Dot‐plot of individual and median Aβ oligomer levels in TBI, Alzheimer's disease (AD) and in the control group (CTRL) consisting of idiopathic iNPH patients and non‐injured, NI individuals. Oligomers were significantly elevated (*) by TBI in comparison to the control group. C. Individual Aβ protofibril levels in patient samples detected with the mAb158 sandwich ELISA. D. Dot‐plot of individual and median Aβ protofibril levels in TBI, AD and in the control group. Aβ protofibril levels were significantly elevated (*) by TBI in comparison to the control group. (A,C) Each bar represents a mean value and standard deviation (SD) from two ELISA experiments performed on two different occasions. E,F. There was a significant positive correlation between Aβ1–42 (E), Aβx‐42 (F), and Aβ protofibril levels in TBI patients. Aβ = Amyloid β, # = TBI patients with Amyloid β aggregates, * = Significant difference (P < 0.05), Strep‐HRP = Streptavidin‐Horseradish Peroxidase; TMB = Tetramethylbenzidine.
The limit of detection was estimated to 1.5 pM oligomers. Data is presented as pg oligomers per mg total protein.
Biochemical measurements of Aβ protofibrils
The previously described mAb158 sandwich ELISA specifically detects Aβ protofibrils 10, 43 (Figure 2B). For this assay, the protofibril selective monoclonal mouse antibody mAb158 (IgG2a, BioArctic, Stockholm, Sweden) was used for capture (2 µg/mL). The TBS brain extracts were diluted fivefold and incubated in duplicates for 2 h, followed by detection with biotinylated mAb158 (0.5 µg/mL). Sample concentrations were calculated from a standard curve based on the in vitro generated Aβ42 protofibrils using a 4‐parameter equation. All samples were measured on two separate occasions.
The limit of detection of the mAb158 sandwich ELISA was 1.5 pM protofibrils. Data is presented as pg protofibrils per mg total protein.
Biochemical measurements of total Aβ
Two different immunoassay kits in a three‐in‐one (triplex) well format were used to measure Aβ38, Aβ40 and Aβ42 (Meso Scale Diagnostics, Rockville, MD, USA). The first kit utilized a detection antibody (mAb6E10, cat no K15200E‐2) with an N‐terminal binding epitope (aa 3–8), herein referred to as recognizing “full‐length Aβ 1–38, 1–40, 1–42,” even though it also can recognize molecules lacking the first two amino acids. The second kit detected both full‐length and N‐terminally truncated Aβ forms by the mid‐region (aa 17–24) detection antibody (mAb4G8, cat no K15199E‐2), herein referred to as recognizing Aβ “X‐38, X‐40, X‐42.”
TBS extracts were subjected to denaturation and monomerization by boiling in 1% SDS prior to a tenfold dilution followed by dispension of the samples into duplicate ELISA plate wells. Determination of total protein content was performed with the BCA protein assay kit (Pierce, cat no 23227).
The limit of detection for Aβ38, Aβ40 and Aβ42 was 5, 1.2 and 0.2 pM, respectively, similar for both assays. Data is presented as pg Aβ38, Aβ40 and Aβ42 per mg total protein.
APOE genotyping
APOE genotyping was performed essentially as described by Hixson and Vernier 15. In brief, genomic DNA was amplified using forward APOE primer (AGACGCGGGCACGGCTGTCCAAGGAGC) and reverse APOE primer (TCGCGGGCCCCGGCCTGGTACACTGC) with the addition of 5% DMSO. The PCR product was used for Restriction Fragment Length Polymorphism (RFLP) analysis by Hha I digestion, and fragments were separated on a high resolution MetaPhor™ agarose gel (Lonza, cat no 50181). The PCR amplification and RFLP were repeated three times.
HAMA analysis
Human Anti‐Mouse Antibodies (HAMA) are found in 10–20% of naive serum samples and can potentially cross‐link the antibodies in a sandwich ELISA setting, resulting in false positive signals 24. To address this issue, HAMA was analyzed in the patient with the highest levels of oligomers and protofibrils on the mAb82E1 and mAb158 ELISAs with or without HAMA buffer (Mabtech, Sweden cat no 3652‐J2). In addition, the samples were run on the same ELISAs, but where the plate instead had been coated with irrelevant mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA cat no 015‐000‐003).
Statistical analysis
Statistica 12.0 (Stat Soft, Tulsa, OK) was used for descriptive and analytical statistics. Since the data were not normally distributed, evaluated using the Kolmogorov‐Smirnov test, non‐parametric statistics were used. Age was expressed as means ± standard deviation (SD), while non‐normally distributed data were expressed as medians and range. For all analyses, data were similar in iNPH and NI patients and therefore the two groups were pooled and used as controls in group‐wise comparison. Kruskal‐Wallis test was used for skewed distributions followed by Mann‐Whitney U‐test to determine significant differences between groups. The Wilcoxon signed‐rank test was used to compare related variables. Spearman's rank‐order correlations were used for analyses of the relationships. Two‐tailed P‐values were used and a P‐value < 0.05 was considered statistically significant.
RESULTS
Patient characteristics
Detailed clinical and radiological data of the twelve TBI patients are provided in Table 1. The mean age of the patients was 49.5 ± 19 (age ± SD) years. The contusions were surgically evacuated at a median of 13 h post‐injury.
The mean age of the AD patients was 79 ± 11 years, of the iNPH patients 75 ± 6 years and of the NI subjects 79 ± 10 years. The TBI cohort was significantly younger than the AD (P = 0.004) and the control (P = 0.0005) groups. The median post‐mortem time for AD patients was 20 h (Table 2) and for the NI subjects of the control group 30 h (Table 3).
Table 2.
Demographics of the dementia cohort.
| Pat # | Age at death (years) | Gender | APOE genotype | Age at onset (years) | Time post‐mortem (h) | Neuropathological diagnosis |
|---|---|---|---|---|---|---|
| 1 | 61* | Female | ε3/4 | 58 | 48 | AD |
| 2 | 84 | Female | ε3/4 | 81 | 20 | AD |
| 3 | 85 | Female | ε2/3 | 83 | 21 | AD |
| 4 | 77 | Female | ε3/4 | 71 | 7 | DLB/AD |
| 5 | 90 | Female | ε3/4 | 84 | 11 | AD |
APOE = Apolipoprotein E; DLB= Dementia with Lewy bodies; AD = Alzheimer's disease.
*APP Swedish mutation carrier.
Insoluble Aβ aggregates/plaques are present in a subgroup of TBI patients
Of the nine TBI patients for whom brain tissues were available also for immunohistochemical analysis, insoluble Aβ aggregates were visualized in three whereas in the remaining six no Aβ pathology was observed (Figure 1). All of the three patients displayed both diffuse and compact Aβ aggregates (Figure 1B–D). None of the iNPH biopsies showed any Aβ aggregates (Table 4). Among the NI subjects, sparse tau pathology was evident (Braak stage b to 2) (Table 3) although no Aβ pathology could be seen (Thal 0).
Elevated levels of Aβ oligomers in TBI brain tissue
Oligomeric Aβ, as measured by the mAb82E1 sandwich ELISA that can recognize both smaller (<100 kDa) as well as larger Aβ aggregates 47, was detected in all twelve TBI patients, in three out of five AD patients, in two out of four iNPH patients and in two out of the four NI subjects. In the TBI group the median level was 129 pg/mg total protein (range 29–342), significantly higher than the control group for which the median was 16.5 pg/mg total protein (range 0–126, P = 0.0015, Figure 2A,B). The levels in the TBI group did not differ significantly from the AD group for which the median level was 54 pg/mg total protein (range 0–399, P = 0.44, Figure 2A,B). No correlation between age and Aβ oligomer levels was seen in any of the groups (data not shown).
Elevated levels of Aβ protofibrils in TBI brain tissue
Protofibrillar Aβ, as measured by the mAb158 sandwich ELISA, could be detected in seven out of twelve TBI patients, in two out of five AD patients and in none of the iNPH or NI control subjects (Figure 2C,D). In TBI patients the median Aβ protofibril level was 15 pg/mg total protein (range 0–629), which was significantly higher than for the control group (where no protofibrils could be detected) (P = 0.03). No differences in the protofibril levels could be seen between the TBI and AD groups, for which the median level was 0 pg/mg total protein (range 0–111, P = 0.80, Figure 2C,D). No correlation between age and Aβ protofibril levels could be seen for any of the groups (data not shown).
Levels of Aβ monomers are not elevated in TBI brain tissue
When assessing Aβ1‐40, there was no difference between TBI, the AD group and controls (P = 0.056, Kruskal‐Wallis test, Figure 3B,C). As for Aβx‐40, the levels were lower in TBI than in both AD (P = 0.0003) and controls (P = 0.0007, Figure 3B,C). Neither the Aβ1‐40 nor the Aβx‐40 levels correlated with age or with Aβ oligomers/protofibrils (data not shown).
Figure 3.
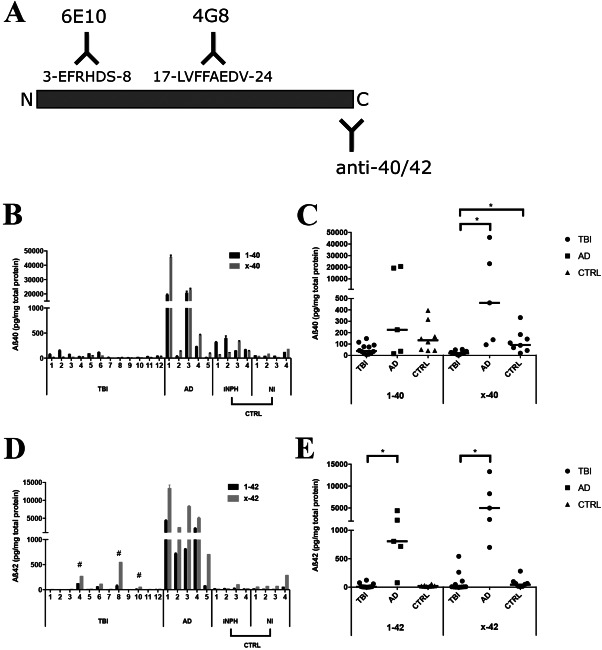
No elevations of Aβ monomers in TBI brain tissue A. Detection of Aβ monomers with mAb6E10 and mAb4G8 antibodies. The mAb6E10 antibody detects more N‐terminally intact Aβ40 and 42 as compared to mAb4G8 that detects Aβ40 and Aβ42 by mid‐region binding. B. Individual full‐length (Aβ1‐40) and N‐truncated (Aβx‐40) Aβ40 monomer levels detected by mAb6E10 and mAb4G8, respectively, in TBI, Alzheimer's disease (AD) and in the control group (CTRL) consisting of idiopathic iNPH patients and NI individuals. C. Dot‐plot of individual and median Aβ1‐40 and Aβx‐40 levels. Levels of Aβx‐40 were significantly lower (*) in TBI patients than in both AD and the control group. D. Individual full‐length (Aβ1‐42) and truncated (Aβx‐42) Aβ42 monomer levels detected by mAb6E10 and mid‐region mAb4G8, respectively. Of the four TBI patients with markedly higher levels of Aβx‐42 than Aβ1‐42, the three patients with immunohistochemical evidence of Aβ aggregates are indicated with #. E. Dot‐plot of individual and median Aβ1‐42 and Aβx‐42 levels in TBI, AD and in the control group (CTRL). Levels of both Aβ1‐42 and Aβx‐42 were significantly lower (*) in brain tissue from TBI than from AD patients. (B,D); Data shown as the mean from one ELISA experiment with error bars showing variation between duplicate ELISA wells. Aβ = Amyloid β, * = Significant difference (P < 0.05).
The median levels of Aβ1‐42 in TBI were similar to the median levels of the controls (P = 0.08) but lower than in AD (P = 0.0013). For Aβx‐42, the median levels in TBI did not differ to the median Aβx‐42 levels of controls (P = 0.08) but were lower than in AD (P = 0.0003, Figure 3D,E).
For TBI, there was a positive correlation between levels of Aβ1‐42 and age (Spearman's r = 0.77, P = 0.003; data not shown), although not between Aβx‐42 and age (P = 0.12). In contrast, for the AD group Aβ1‐42 levels were negatively correlated with age (Spearman's r = −0.90, P = 0.037; data not shown). For the control group, neither Aβ1‐42 nor Aβx‐42 levels correlated with age (data not shown).
Aβ1‐42 and Aβx‐42 levels in TBI (Spearman's r = 0.60, P = 0.038 and r = 0.70, P = 0.01; Figure 2E,F) and Aβ1‐42 levels in AD (Spearman's r = 0.89, P = 0.04) were positively correlated with the levels of Aβ protofibrils (data not shown).
The levels of Aβ1‐38 were lower in TBI compared to AD cases (P = 0.019), whereas they were similar to the controls (P = 0.24). Aβx‐38 could be detected in AD and control tissues but only in one TBI patient. Neither the Aβ1‐38 nor the Aβx‐38 levels correlated with age or with Aβ oligomer/protofibril levels (data not shown).
The APOE ε3/4 genotype leads to elevated levels of Aβ42 and Aβ oligomers/protofibrils in TBI patients
Among the TBI patients, four were found to have the APOE ε3/4 genotype (APOE ε3/4+), whereas seven were APOE ε3/3 and one APOE ε2/4 (collectively referred to as APOE ε3/4‐; Table 1). In the AD group, APOE ε3/4 was found in four out of five patients (Table 2), whereas there was one APOE ε3/4 carrier in the control group of iNPH patients and NI subjects (Tables 3 and 4).
The age of the APOE ε3/4+ TBI patients (mean age 64 ± 11 years) did not differ significantly from the APOE ε3/4‐ patients (mean age 42 ± 19 SD, P = 0.11) nor from the age of the AD patients (mean age 79 ± 11 SD; P = 0.11). The TBI patients with APOE ε3/4 were younger than the control group (P = 0.048). Insoluble Aβ aggregates were found in three out of the nine TBI tissues for which immunohistochemistry was performed (Figure 1), all of whom were APOE ε3/4+.
The Aβ oligomer, protofibril and Aβ42 levels are increased in APOE ε3/4+ TBI patients
The Aβ oligomer levels in APOE ε3/4+ TBI patients were significantly higher than in the APOE ε3/4‐ patients (P = 0.048) and higher than in controls (P = 0.008, Figure 4E), while no difference was seen when comparing APOE ε3/4+ TBI patients to AD (P = 0.41 data not shown). The APOE ε3/4‐ TBI patients also had significantly higher Aβ oligomer levels than controls (P = 0.01, Figure 4E).
Figure 4.
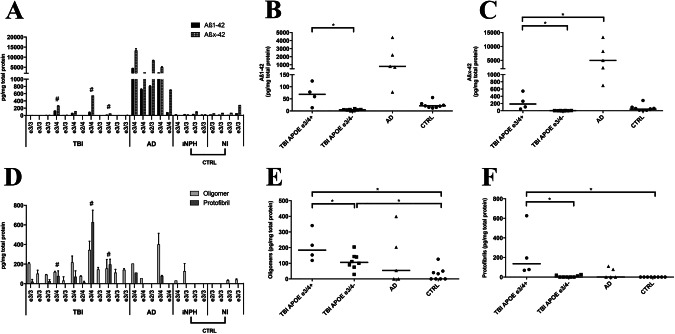
The APOE ε3/4 genotype is associated with increased levels of Aβ42, Aβ oligomers and Aβ protofibrils following TBI. A. Individual full‐length (Aβ1‐42) and truncated (Aβx‐42) Aβ42 monomer levels detected by N‐terminal mAb6E10 and mid‐region mAb4G8, respectively, in TBI, Alzheimer's disease (AD) and in the control group (CTRL) consisting of idiopathic iNPH patients and NI individuals. The individual APOE genotype for each individual is shown. # indicates immunohistochemical evidence of Aβ aggregates. B. Dot‐plot of individual and median Aβ1‐42 in TBI patients with (APOE ε3/4+) and without (APOE ε3/4‐) the APOE ε3/4 genotype, in AD and in the control group. In TBI patients, the Aβ1‐42 levels were significantly elevated (*) in APOE ε3/4+ patients compared with APOE ε3/4‐ patients. C. Dot‐plot of individual and median Aβx‐42 levels in APOE ε3/4+ and APOE ε3/4‐ TBI patients, in AD and in the control group. In APOE ε3/4+ TBI patients, levels of Aβx‐42 were significantly lower than in Alzheimer's disease patients although elevated (*) in comparison to APOE ε3/4‐. D. Amyloid‐β (Aβ) oligomer and Aβ protofibril levels in TBI patients, in AD and in a control group consisting of iNPH and NI patients. The individual APOE genotype for each individual is shown. # indicates immunohistochemical evidence of Aβ aggregates. E. Dot‐plot of individual and median Aβ oligomer levels in APOE ε3/4+ and APOE ε3/4‐ TBI patients, in AD and in the control group. Compared to controls, Aβ oligomers were significantly elevated (*) in both APOE ε3/4+ and APOE ε3/4‐ TBI patients, while in TBI Aβ oligomers were significantly elevated (*) in APOE ε3/4+ when compared to APOE ε3/4‐ patients. F. Dot‐plot of individual and median Aβ protofibril levels in APOE ε3/4+ and APOE ε3/4‐ TBI patients, in AD and in the control group. Aβ protofibrils were significantly elevated (*) in APOE ε3/4+ TBI patients when compared both to APOE ε3/4‐ and controls. APOE = Apolipoprotein E, Aβ = Amyloid β, * = Significant difference (P < 0.05).
The levels of Aβ protofibrils in APOE ε3/4+ TBI patients were similar to those of AD patients (P = 0.19), but higher than in APOE ε3/4‐ TBI patients (P = 0.004) and in controls (P = 0.004, Figure 4F). Moreover, the APOE ε3/4‐ TBI samples did not differ in levels of Aβ protofibrils when compared to controls (P = 0.23, Figure 4F).
The levels of Aβ1‐38 and Aβx‐38 did not differ between APOE ε3/4+ and APOE ε3/4‐ TBI cases, but Aβx‐38 were lower in APOE ε3/4+ TBI patients in comparison to AD (P = 0.016, data not shown). Also Aβ1‐40 and Aβx‐40 levels did not differ between the two APOE subgroups, but were lower in APOE ε3/4+ TBI patients than in the control group (P = 0.028 for both comparisons, data not shown), while Aβx‐40 levels were lower in comparison to AD (P = 0.016, data not shown).
The Aβ1‐42 levels in APOE ε3/4+ TBI patients were significantly higher than in APOE ε3/4‐ TBI patients (P = 0.004) and were similar to the levels in the AD (P = 0.06) and control groups (P = 0.15, Figure 4A–C). Similarly, the Aβx‐42 levels in APOE ε3/4+ TBI patients were significantly higher than in APOE ε3/4‐ TBI patients (P = 0.004), but lower than in AD cases (P = 0.016) and similar to controls (P = 0.15, Figure 4C). Also, APOE ε3/4 carriers across all groups (n = 9) had significantly higher levels of Aβx‐42 than Aβ1–42 (P = 0.011).
HAMA analysis
When controlling for HAMA activity in the mAb158 and mAb82E1 ELISAs, with either HAMA buffer and/or irrelevant mouse IgG coating, no signs of any such activity could be seen (data not shown).
DISCUSSION
In this report, the first evidence of rapid accumulation of potentially toxic Aβ oligomers and protofibrils in human brain tissue after TBI is provided. The buildup of such Aβ molecules, known to be centrally involved in AD pathogenesis, seems to occur rapidly as we could detect markedly elevated levels in surgically resected tissues already by four hours post‐injury. Thus, cells in the injured brain areas seem to respond to the damaging insult either by an increased generation or by a decreased degradation of these prefibrillar Aβ aggregates.
Based on these observations, we hypothesize that the TBI‐induced increase of these potentially toxic Aβ species may aggravate the clinical outcome both in the shorter and longer perspective. A local increase in Aβ oligomers/protofibrils may impede cellular functions and also result in impaired tissue recovery, whereas a more sustained presence of these species could lead to spreading of pathology into anatomically connected brain areas. Such effects have been demonstrated on various AD animal models and are likely to occur also in the human brain. Thus, propagation of pathology between interconnected brain regions may well explain the documented increase in the incidence of AD in patients who have previously suffered from TBI 20.
Our findings further illustrate that TBI patients carrying the APOE ε3/4 genotype (APOE ε3/4+), in addition to an increased deposition of insoluble Aβ aggregates, display even higher levels of Aβ oligomers and protofibrils as compared to APOE ε3/4‐ negative patients. A similar response could be seen also for truncated and full length forms of Aβ42, the Aβ species with the highest aggregation propensity. The aggravated pathological response in APOE ε3/4+ TBI patients is in line with previous observations suggesting that patients with this genetic background are more prone to respond with amyloid pathology following TBI 30. Since the only APOE ε3/4+ control patient displayed low levels of oligomers and no protofibrils, it suggests that the increased levels in APOE ε3/4+ TBI patients were a consequence of the injury.
The underlying mechanisms for the increase of oligomers and protofibrils in TBI are not known. Although the mechanisms resulting in Aβ deposits are plausibly multifactorial, axonal injury may be a key contributor due to the progressive accumulations of the Aβ substrate AβPP as well as its cleaving enzymes 22. In severe TBI, hypoxia and oxidative stress may also potentiate Aβ aggregation. In AD brains, cognitive symptoms are preceded by reductions of cerebral blood flow 14, 39 and compelling evidence suggest that hypoxia activate β‐ and γ‐secretases, causing augmented AβPP cleavage and subsequent Aβ production 37. Reduced cerebral blood flow is common following TBI, and in addition, all TBI patients in this study suffered from raised ICP mandating surgical treatment. We cannot exclude that cerebral ischemia, suggested by immunohistochemistry in some of the evaluated samples, also resulted in a degree of tissue acidosis contributing to the accumulations of various Aβ species.
Previous studies have suggested that the brain interstitial fluid (ISF) levels of monomeric Aβ are associated with the clinical outcome in TBI patients 4 and may be more pronounced in diffuse axonal injuries than in focal TBI 29. These previous microdialysis studies 4, 27, 29 cannot firmly establish a TBI‐induced alteration in Aβ levels since the ISF levels of healthy individuals are unknown. However, analyses of Aβ in CSF demonstrated decreased levels of Aβ42 in TBI compared to control subjects 33, a feature that is similar to what is typically seen in AD patients and believed to reflect the increased brain deposition of these molecular species. Collectively, these studies support the notion that TBI can result in raised Aβ levels in injured brain regions. Presumably, the increased presence of Aβ monomers can then lead to the formation of oligomeric/protofibrillar aggregates as well as to the deposition of ready‐formed fibrils in the injured brain tissues.
Moreover, this study is the first to evaluate the presence of truncated Aβ in TBI. Mainly, we found an increase of N‐truncated Aβ42 species in APOE ε3/4+ TBI patients. Previous studies have suggested that N‐truncated Aβ species, particularly Aβ4‐42 and pyroglutamate‐modified (pE) AβpE3‐42, are abundant in Aβ aggregates, induce neuronal toxicity 2 and are prone to rapidly oligomerize 41. Interestingly, we could observe a correlation between the levels of N‐truncated Aβ42 and Aβ protofibrils suggesting that this Aβ form could be particularly responsible for the increased formation of oligomers and protofibrils.
An important limitation of this study is the small patient number. In Western societies the incidence of severe TBI is decreasing. In addition, in the management of severe TBI decompressive craniectomy is increasingly performed and the removal of contusions, the prerequisite for this study, is becoming rare 23. In fact, the samples included in our present report are obtained from several years' surgery for severe TBI. Despite the small patient number, the findings of increases in various forms of soluble Aβ are statistically robust. In addition, to our knowledge no direct comparisons have been made between biopsies from surgery and subsequent post‐mortem samples in terms of Aβ measurements. Consequently, the AD post‐mortem samples used here are not ideal controls to the TBI samples, but should more be seen as relevant positive control material.
Since the analyses in this study were performed on surgically resected brain tissue, it was not possible to ascertain if the brains were free of Aβ before the trauma or to assess temporal changes in Aβ levels following the insult. However, previous studies utilizing PET imaging have clearly demonstrated an increased amyloid deposition following TBI. In one PiB‐based study it was found that the retention signals were increased in TBI patients with longer time from initial injury to PET scan 42, whereas another study using the same PET ligand showed that the signals peaked within the first week post‐injury 17. As PiB and other similar ligands only bind insoluble fibrillar Aβ deposits they cannot be used to assess the presence of soluble Aβ oligomers/protofibrils. The development of antibody based PET imaging ligands could enable future temporal studies of such Aβ species in TBI 44.
Furthermore, it could be argued that the observed pathological changes were due to an age‐related effect. Amyloid‐β may accumulate with increasing age and insoluble Aβ aggregates can be seen in 39–82% of post‐mortem brains of elderly non‐demented subjects 9. However, in this study we did not find any correlation to age as we were able to detect comparable levels of Aβ oligomers and protofibrils in TBI patients across all age groups. In fact, oligomers could be detected in all twelve and protofibrils in seven of the TBI patients, whereas none of the eight control subjects displayed any protofibrils although they were significantly older than the TBI cases. Thus, these data strongly imply that the formation of these prefibrillar aggregates in the TBI tissues was not a result of increasing age.
Yet another possible confounding factor relates to the fact that some blood‐derived Aβ may have been measured by the ELISAs. As the samples consisted of surgical tissues, some presence of blood cannot be avoided. The homogenates from each TBI tissue sample contained blood, with no apparent excess of blood in samples with the highest levels of soluble Aβ oligomers and protofibrils, and no HAMA activity was observed. In addition, blood platelets, the main source of blood Aβ mainly produce Aβ40 5, an Aβ species that was found at low and unaltered levels in the TBI samples. Thus, it is unlikely that the increased Aβ levels in the TBI patients were derived from blood.
The properties of Aβ oligomers and protofibrils have been extensively investigated. Initially, such species were demonstrated to be formed as a consequence of the Arctic mutation (AβPP E693G), causing early‐onset familial AD 34, 40. Subsequently, Aβ oligomers/protofibrils have been demonstrated to have a toxic influence both in vivo and ex vivo 1. In particular, Aβ protofibrils have been shown to cause neurotoxicity and electrophysiological changes in rat cortical neurons 13, inhibit long‐term potentiation in mouse hippocampus 35 and induce microglial activation in vitro 36. In addition, although the low molecular weight oligomers are also claimed to confer neurotoxicity in vitro 45, 48, the neurotoxic mechanisms of soluble Aβ aggregates in vivo have not been established.
The two sandwich ELISAs applied for this study have both been well characterized. The mAb82E1 assay measures smaller oligomeric Aβ species as well as larger protofibrils and has been frequently used both for samples from transgenic mice over‐expressing human AβPP and AD brains 16, 47. Noteworthy, the mAb82E1 assay has also been shown to detect elevated oligomer levels in CSF from patients with mild cognitive impairment, a prodromal stage of AD 16.
The mAb158 assay detects larger Aβ protofibrillar species than the mAb82E1 assay and is 2000‐fold more sensitive in detecting soluble protofibrils than monomeric Aβ 10. Although the assay can detect amyloid fibrils, the sensitivity for such species is approximately 15 times lower than for protofibrils 28. However, by including a centrifugation step (at 16K rpm) prior to analysis, fibrils were removed from the samples. Moreover, the risk of measuring other amyloid forming proteins is small, as it has been demonstrated that mAb158 does not cross‐react with other amyloid proteins such as IAPP, α‐synuclein or medin 10. Importantly, the two sandwich ELISAs used (mAb158 and mAb82E1) should not be seen as assays measuring mutually exclusive Aβ species, but rather populations of partially overlapping soluble Aβ aggregates.
Taken together, based on several different ELISAs used here, our study indicates that severe TBI in humans results in an increase of various forms of both soluble and insoluble Aβ. Most notably, data from two separate ELISAs indicate that oligomers and protofibrils, soluble prefibrillar aggregates of Aβ, were increased in resected brain tissues as early as four hours following the TBI. The changes were more pronounced among carriers of the APOE ε3/4 genotype and indicate a shared vulnerability between TBI and AD patients for this genetic risk factor. Future prospective studies are needed to establish if these early alterations of potentially toxic Aβ molecules are contributing to the long‐term risk increase for AD following TBI.
AUTHOR CONTRIBUTIONS
SAH contributed with acquisition of data, analysis and interpretation of data and drafting of the manuscript. ERW contributed with analysis and interpretation of data and drafting of the manuscript. IA contributed with analysis and interpretation of data. NM contributed with study concept and design, study supervision and drafting of the manuscript. All authors critically revised the manuscript for intellectual content.
CONFLICT OF INTERESTS
ERW and LS are employees of BioArctic AB. CM and HB are employees of BioArctic AB and own shares in the company. LL is a co‐founder and chairman of the board of BioArctic AB and owns shares in the company. The remaining authors declare no competing financial interests.
ACKNOWLEDGMENTS
This work was financed by the Swedish Research Council under the framework of EU‐ERA‐NET NEURON CnsAFlame (to NM), Swedish Brain Foundation, Swedish Research Council (to MI), Swedish Institute and the Selander Foundation (NM + LH). We acknowledge Dr Kristina Guiliana Cesarini for help with cortical biopsies of idiopathic normal pressure hydrocephalus patients and Charlotte Sahlin for assisting in establishing the APOE genotyping assay.
REFERENCES
- 1. Benilova I, Karran E, De Strooper B (2012) The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci 15:349–357. [DOI] [PubMed] [Google Scholar]
- 2. Bouter Y, Dietrich K, Wittnam JL, Rezaei‐Ghaleh N, Pillot T, Papot‐Couturier S et al (2013) N‐truncated amyloid beta (Abeta) 4–42 forms stable aggregates and induces acute and long‐lasting behavioral deficits. Acta Neuropathol 126:189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 4. Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N et al (2008) Amyloid‐beta dynamics correlate with neurological status in the injured human brain. Science (New York, NY) 321:1221–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Casoli T, Di Stefano G, Giorgetti B, Grossi Y, Balietti M, Fattoretti P, Bertoni‐Freddari C (2007) Release of beta‐amyloid from high‐density platelets: implications for Alzheimer's disease pathology. Ann New York Acad Sci 1096:170–178. [DOI] [PubMed] [Google Scholar]
- 6. Crane PK, Gibbons LE, Dams‐O'Connor K, Trittschuh E, Leverenz JB, Keene CD et al (2016) Association of traumatic brain injury with late‐life neurodegenerative conditions and neuropathologic findings. JAMA Neurol 73:1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elf K, Nilsson P, Enblad P (2002) Outcome after traumatic brain injury improved by an organized secondary insult program and standardized neurointensive care. Crit Care Med 30:2129–2134. [DOI] [PubMed] [Google Scholar]
- 8. Elobeid A, Laurell K, Cesarini KG, Alafuzoff I (2015) Correlations between mini‐mental state examination score, cerebrospinal fluid biomarkers, and pathology observed in brain biopsies of patients with normal‐pressure hydrocephalus. J Neuropathol Exp Neurol 74:470–479. [DOI] [PubMed] [Google Scholar]
- 9. Elobeid A, Libard S, Leino M, Popova SN, Alafuzoff I (2016) Altered proteins in the aging brain. J Neuropathol Exp Neurol 75:316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S et al (2007) Sensitive ELISA detection of amyloid‐beta protofibrils in biological samples. J Neurochem 103:334–345. [DOI] [PubMed] [Google Scholar]
- 11. Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K (2015) Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol 77:987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gatson JW, Warren V, Abdelfattah K, Wolf S, Hynan LS, Moore C et al (2013) Detection of beta‐amyloid oligomers as a predictor of neurological outcome after brain injury. J Neurosurg 118:1336–1342. [DOI] [PubMed] [Google Scholar]
- 13. Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM et al (1999) Protofibrillar intermediates of amyloid beta‐protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19:8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hays CC, Zlatar ZZ, Wierenga CE (2016) The utility of cerebral blood flow as a biomarker of preclinical Alzheimer's disease. Cell Mol Neurobiol 36:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hixson JE, Vernier DT (1990) Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 31:545–548. [PubMed] [Google Scholar]
- 16. Holtta M, Hansson O, Andreasson U, Hertze J, Minthon L, Nagga K, et al (2013) Evaluating amyloid‐beta oligomers in cerebrospinal fluid as a biomarker for Alzheimer's disease. PLoS One 8:e66381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hong YT, Veenith T, Dewar D, Outtrim JG, Mani V, Williams C et al (2014) Amyloid imaging with carbon 11‐labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol 71:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM et al (2004) Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203. [DOI] [PubMed] [Google Scholar]
- 19. Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136:28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid‐beta pathology: a link to Alzheimer's disease?. Nat Rev Neurosci 11:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson VE, Stewart W, Smith DH (2012) Widespread tau and amyloid‐beta pathology many years after a single traumatic brain injury in humans. Brain Pathol (Zurich, Switzerland) 22:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kolias AG, Adams H, Timofeev I, Czosnyka M, Corteen EA, Pickard JD et al (2016) Decompressive craniectomy following traumatic brain injury: developing the evidence base. Br J Neurosurg 30:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koshida S, Asanuma K, Kuribayashi K, Goto M, Tsuji N, Kobayashi D et al (2010) Prevalence of human anti‐mouse antibodies (HAMAs) in routine examinations. Clin Chim Acta 411:391–394. [DOI] [PubMed] [Google Scholar]
- 25. Leuzy A, Chiotis K, Hasselbalch SG, Rinne JO, de Mendonca A, Otto M et al (2016) Pittsburgh compound B imaging and cerebrospinal fluid amyloid‐beta in a multicentre European memory clinic study. Brain 139:2540–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7:728–741. [DOI] [PubMed] [Google Scholar]
- 27. Magnoni S, Mac Donald CL, Esparza TJ, Conte V, Sorrell J, Macri M et al (2015) Quantitative assessments of traumatic axonal injury in human brain: concordance of microdialysis and advanced MRI. Brain 138:2263–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Magnusson K, Sehlin D, Syvänen S, Svedberg MM, Philipson O, Söderberg L et al (2013) Specific uptake of an amyloid‐beta protofibril‐binding antibody‐tracer in AbetaPP transgenic mouse brain. J Alzheimer's Dis 37:29–40. [DOI] [PubMed] [Google Scholar]
- 29. Marklund N, Blennow K, Zetterberg H, Ronne EE, Enblad P, Hillered L (2009) Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg 110:1227–1237. [DOI] [PubMed] [Google Scholar]
- 30. Mauri M, Sinforiani E, Bono G, Cittadella R, Quattrone A, Boller F, Nappi G (2006) Interaction between Apolipoprotein epsilon 4 and traumatic brain injury in patients with Alzheimer's disease and Mild Cognitive Impairment. Funct Neurol 21:223–228. [PubMed] [Google Scholar]
- 31. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K et al (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46:860–866. [DOI] [PubMed] [Google Scholar]
- 32. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 33. Mondello S, Buki A, Barzo P, Randall J, Provuncher G, Hanlon D et al (2014) CSF and plasma amyloid‐beta temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Sci Rep 4:6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nilsberth C, Westlind‐Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C et al (2001) The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci 4:887–893. [DOI] [PubMed] [Google Scholar]
- 35. O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE et al (2010) Amyloid beta‐protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci 30:14411–14419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paranjape GS, Gouwens LK, Osborn DC, Nichols MR (2012) Isolated amyloid‐beta(1–42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem Neurosci 3:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pluta R, Furmaga‐Jablonska W, Maciejewski R, Ulamek‐Koziol M, Jablonski M (2013) Brain ischemia activates beta‐ and gamma‐secretase cleavage of amyloid precursor protein: significance in sporadic Alzheimer's disease. Mol Neurobiol 47:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI (1994) Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol, Neurosurg, Psychiatry 57:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol 57:789–794. [DOI] [PubMed] [Google Scholar]
- 40. Sahlin C, Lord A, Magnusson K, Englund H, Almeida CG, Greengard P et al (2007) The Arctic Alzheimer mutation favors intracellular amyloid‐beta production by making amyloid precursor protein less available to alpha‐secretase. J Neurochem 101:854–862. [DOI] [PubMed] [Google Scholar]
- 41. Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Bohm G, Demuth HU (2006) On the seeding and oligomerization of pGlu‐amyloid peptides (in vitro). Biochemistry 45:12393–12399. [DOI] [PubMed] [Google Scholar]
- 42. Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M et al (2016) Amyloid pathology and axonal injury after brain trauma. Neurology 86:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, et al (2012) Large aggregates are the major soluble Abeta species in AD brain fractionated with density gradient ultracentrifugation. PLoS One 7:e32014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sehlin D, Fang XT, Cato L, Antoni G, Lannfelt L, Syvanen S (2016) Antibody‐based PET imaging of amyloid beta in mouse models of Alzheimer's disease. Nat Commun 7:10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shankar GM, Li S, Mehta TH, Garcia‐Munoz A, Shepardson NE, Smith I et al (2008) Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith DH, Chen XH, Iwata A, Graham DI (2003) Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg 98:1072–1077. [DOI] [PubMed] [Google Scholar]
- 47. Tucker S, Moller C, Tegerstedt K, Lord A, Laudon H, Sjodahl J et al (2015) The murine version of BAN2401 (mAb158) selectively reduces amyloid‐beta protofibrils in brain and cerebrospinal fluid of tg‐ArcSwe mice. J Alzheimer's Dis 43:575–588. [DOI] [PubMed] [Google Scholar]
- 48. Yang T, Li S, Xu H, Walsh DM, Selkoe DJ (2017) Large soluble oligomers of amyloid beta‐protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]