

Abstract
While clusterin is reportedly involved in Alzheimer's disease (AD) pathogenesis, how clusterin interacts with amyloid‐β (Aß) to cause Aß neurotoxicity remains unclear in vivo. Using 5×FAD transgenic mice, which develop robust AD pathology and memory deficits when very young, we detected interactions between clusterin and Aß in the mouse brains. The two proteins were concurrently upregulated and bound or colocalized with each other in the same complexes or in amyloid plaques. Neuropathology and cognitive performance were assessed in the progeny of clusterin‐null mice crossed with 5×FAD mice, yielding clu −/−;5×FAD and clu +/+;5×FAD. We found far less of the various pools of Aß proteins, most strikingly soluble Aß oligomers and amyloid plaques in clu −/−;5×FAD mice at 5 months of age. At that age, those mice also had higher levels of neuronal and synaptic proteins and better motor coordination, spatial learning and memory than age‐matched clu +/+;5×FAD mice. However, at 10 months of age, these differences disappeared, with Aß and plaque deposition, neuronal and synaptic proteins and impairment of behavioral and cognitive performance similar in both groups. These findings demonstrate that clusterin is necessarily involved in early stages of AD pathogenesis by enhancing toxic Aß pools to cause Aß‐directed neurodegeneration and behavioral and cognitive impairments, but not in late stage.
Keywords: Alzheimer's disease, amyloid pathology, apolipoprotein J, Aß oligomers, chaperone protein
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease and the most common type of dementia in the elderly. Early investigation of the cause of AD supported the amyloid hypothesis of pathogenesis in which accumulation of amyloid‐β (Aβ), derived from proteolytic cleavage of amyloid precursor protein (APP), leads to neurodegeneration. Recent efforts have aimed to find risk factors other than Aβ that may contribute to late‐onset AD, which is the phenotype in most cases of the disease.
Clusterin, also known as apolipoprotein J (Apo J), has been identified as an important risk factor for late‐onset AD by large‐scale GWAS 17, 24. Clusterin is a ubiquitously expressed glycoprotein and is present in very high levels in the brain. In patients with AD, clusterin levels are even higher in the brain 14, 33, 40, serum and cerebrospinal fluid (CSF) 8, 13. Clusterin is found to colocalize with Aβ in the amyloid deposits of brains of AD patients 8, 22, 34.
Clusterin, the first reported extracellular mammalian chaperone protein 20, 42, binds to biologically active hydrophobic residues exposed on misfolded or aggregated Aβ proteins 43, 44, including toxic oligomers, to prevent further aggregation, render them susceptible to degradation, or suppress their toxicity. Accordingly, clusterin was shown to decrease Aβ‐induced neurotoxicity in rodent hippocampal neurons 4 and attenuate the toxic effects of Aβ oligomers in a novel in vivo model of Caenorhabditis elegans 2. Other studies have demonstrated that Aβ‐bound clusterin facilitates the clearance of Aβ across the blood–brain barrier (BBB) 3, 57. Aβ oligomers pre‐incubated with clusterin induced less neuronal death and learning and memory impairment in rats compared with Aβ oligomers without clusterin 7. These findings support protective mechanisms by which clusterin may inhibit AD pathogenesis.
However, increasing evidence suggests that clusterin also specifically interacts with Aβ oligomers to exacerbate their toxicity. When clusterin binds to toxic Aβ oligomers, it sequesters and stabilizes the oligomeric structures, interfering with their further aggregation into less toxic fibril aggregates 41. Clusterin binding has been shown to shield Aβ oligomers from enzymatic degradation 32, 37, 38. Clusterin augmented Aβ toxicity in PC12 cells or organotypic mouse brain slice cultures in parallel with the formation of clusterin‐induced and ‐stabilized Aβ oligomers 25, 40, 41. Knockdown of clusterin in primary neurons reduced Aβ toxicity 23, and the genetic ablation of clusterin (clu −/−) in PDAPP mice that develop AD pathology caused significantly fewer amyloid deposits and less neuritic dystrophy with no alteration of the total Aβ deposition 10. More interestingly, a clinical longitudinal study 11 found significant associations between levels of clusterin and Aβ in CSF and entorhinal cortex atrophy seen on MRI, implying that clusterin‐Aβ interaction is important in the earliest stages of AD pathogenesis.
In the present study, we evaluated the effect of genetic deletion of clusterin on amyloid pathogenesis and motor and cognitive behavioral performance in 5×FAD transgenic mice, which co‐express five familial hAPP/PS1 mutations and develop severe AD pathology at an early age 39. The results provide the first in vivo evidence that clusterin is an important endogenous factor facilitating the early stage of AD pathogenesis, but its influence is attenuated or lost in the late stage when amyloid deposition is already quite advanced.
Materials and Methods
Animal studies
This animal study was approved by the Institutional Animal Care and Use Committee of the Asan Institute for Life Sciences, Asan Medical Center (Seoul, Korea).
Clusterin‐deficient (clu −/−;5×FAD) and their littermate 5×FAD mice (clu +/+;5×FAD) were obtained by breeding clusterin‐null mice (clu −/−) 10, 35 with 5×FAD transgenic mice, which co‐express hAPP695 with three familial AD mutations (K670N/M671L, I716V and V717I) and presenilin (PS1) with two familial mutations (M146L and L286V) 39. Experiments were performed with 5‐ and 10‐month‐old male and female mice. All animals were bred and housed with free access to food and water at 22.0°C ± 1°C and with a 12‐h light–dark cycle.
Tissue preparation and histologic or immunohistochemical examination
Shortly after completing the behavioral tests described below, the animals were killed under deep anesthesia. The cerebral hemispheres were divided, snap‐frozen in liquid nitrogen and immediately stored at −80°C. Sagittal 12‐μm thick sections of the left hemisphere were serially mounted on 1% poly‐L‐lysine‐coated glass slides using a cryostat (HM550; Microm, Walldorf, Germany).
Brain Aβ deposition was evaluated by immunohistochemistry using an anti‐Aβ(17–24) antibody (4G8; BioLegend, San Diego, CA, USA). The sagittal sections were fixed with 4% paraformaldehyde and briefly incubated in 70% formic acid (FA). Endogenous peroxidase activity in the tissue was depleted by treatment with 3% hydrogen peroxide, and the sections were blocked with 3% normal serum and 0.3% Triton X‐100 in phosphate buffered saline (PBS) and reacted with the primary antibody (4G8; dilution, 1:200) and then with a biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA). Finally, the sections were treated with avidin horseradish peroxidase (Vector Laboratories) and developed with 0.015% 3,3′‐diaminobenzidine (DAB) and 0.001% hydrogen peroxide (Vector Laboratories). For immunofluorescent examination, the 4G8‐immunoreacted tissues were visualized with Alexa Fluor‐conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA).
To detect colocalization of clusterin and Aβ in amyloid deposits, the brain tissues were immunologically co‐stained with the primary antibodies, anti‐Aβ (4G8) and anti‐clusterin/Apo J (Novus Biologicals, Littleton, CO, USA; dilution, 1:100) antibodies and then with two corresponding fluorescent secondary antibodies, Alexa Fluor 488‐ and 555‐conjugated antibodies.
To examine congophilic compact amyloid plaques, brain sections were stained with Hematoxylin QS (a modification of Mayer's hematoxylin; Vector Laboratories) and Accustain® Congo red amyloid staining solution (Sigma‐Aldrich, St. Louis, MO, USA) 26. After washing with absolute ethanol, the stained tissue sections were examined for congophilic plaques using a light or polarized microscope (Eclipse 800i; Nikon, Tokyo, Japan). For further analysis, microscopic images of stained tissues were captured with a DS‐Fi1 digital camera connected to NIS‐Elements image processing software (Nikon).
Because compact amyloid plaques emit blue fluorescence in situ under UV illumination 26, we examined blue fluorescent amyloid deposits in the brain tissues under a fluorescence microscope (Eclipse 800i) with a UV‐2A filter (dichroic, 400 nm; excitation, 330–380 nm; barrier, 420 nm).
The load of amyloid deposits or plaques in the brain was expressed as the per cent area of 4G8‐immunoreactive deposits or the number of congophilic plaques per mm2 in regions of interest from the hippocampal, cortical and thalamic areas.
Quantitative real‐time polymerase chain reaction (PCR) assay
Total RNAs were extracted from the right hemispheres using the RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) and cDNAs were generated using the iScript™ cDNA synthesis kit (Bio‐Rad, Hercules, CA, USA). The quantitative RT‐PCR was conducted with the Bio‐Rad CFX connect Real‐time PCR machine using sense and antisense primers (250 nM each; Macrogen, Seoul, Korea) with 5× HOT FIREPol® EvaGreen® qPCR Supermix (Solis BioDyne, Tartu, Estonia). The sequences of sense and anti‐sense primers used were as follows: for clusterin (Apo J; NM_013492), 5′‐CAG TTC CCA GAC GTG GAT TT‐3′ and 5′‐TCC TGG CAC TTT TCA CAC TG‐3′, for Apo E (BC083351), 5′‐TGT CCT GCA ACA ACA TCC AT‐3′ and 5′‐TAT TAA GCA AGG GCC ACC AG‐3′, for Dickkopf Wnt signaling pathway inhibitor 1 (DKK‐1; NM_010051), 5′‐CCT TCG GAG ATG ATG GTT GT‐3′ and 5′‐GTT CTT GAT CGC GTT GGA AT‐3′ and for GAPDH (NM_008084), 5′‐AGG TCG GTG TGA ACG GAT TTG‐3′ and 5′‐TGT AGA CCA TGT AGT TGA GGT CA‐3′, respectively. Finally, mRNA expression levels were calculated relative to that of GAPDH using the ΔΔCt method 27.
Quantitative enzyme‐linked immunosorbent assay (ELISA)
After the right brain hemispheres were weighed, they were homogenized in PBS (pH 7.4; 1:10, w/v) containing protease inhibitors (cOmplete™ Protease Inhibitor Cocktail; 1 tablet/50 mL; Roche Diagnostics, Mannheim, Germany) and centrifuged at 100 000 × g; the supernatant was then collected (the PBS‐soluble fraction). The pellets were sonicated in 2% SDS in PBS, and the SDS‐soluble fraction was collected by centrifugation at 100 000 × g. Finally, the SDS‐insoluble pellets were solubilized in 70% FA and the FA‐soluble fraction was collected by centrifugation at 100 000 × g.
We subjected the PBS‐, SDS‐ and FA‐soluble fractions to sandwich ELISA for quantification of human Aβ40 and Aβ42 (Thermo Fisher Scientific). Analysis of FA‐soluble fractions was preceded by neutralization with 1 M Tris (adjusted to pH 11.0 using 5 M NaOH). In addition, the quantification of oligomeric Aβs (oAβ) in PBS‐ and SDS‐soluble fractions and Aβ aggregates (aAβ) in SDS extracts was performed using an Oligomeric Aβ ELISA kit (BioSensis, Thebarton, SA, Australia) and an Aβ aggregate ELISA kit (Thermo Fisher Scientific), respectively.
We also measured the amount of clusterin in the brain tissue (SDS extracts) using a SimpleStep® ELISA kit for mouse clusterin (Abcam, Cambridge, UK).
All measurements were taken at 450 nm using the Synergy H1 Hybrid microplate reader (BioTek, Winooski, VT, USA).
Immunoblotting and co‐immunoprecipitation analysis
The following primary antibodies were used for immunoblotting: anti‐Aß(1–16) (6E10; BioLegend; dilution, 1:2000), anti‐clusterin/Apo J (Novus Biologicals; 1:1000), anti‐apolipoprotein E (Apo E; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:1000), anti‐DKK‐1 (Dickkopf‐1; Boster, Pleasanton, CA, USA: 1:1000), anti‐NeuN (Merck Millipore, Darmstadt, Germany; 1:500), anti‐nonphosphorylated neurofilament protein SMI‐32 (BioLegend; 1:2000), anti‐synaptophysin (Santa Cruz Biotechnology; 1:5000), anti‐ankyrin G (Santa Cruz Biotechnology; 1:2000), anti‐PSD95 (postsynaptic density protein 95; Applied Biological Materials, Richmond, Canada; 1:2000), anti‐VAMP (vesicle‐associated membrane protein)/synaptobrevin (Santa Cruz Biotechnology; 1:1000), anti‐ProSAP2 (postsynaptic scaffold protein)/Shank 3 (Santa Cruz Biotechnology; 1:500) and anti‐ß‐actin (Sigma‐Aldrich; 1:2000) antibodies.
Brain hemispheres were homogenized in PBS (pH 7.4; 1:10, w/v) containing protease inhibitors (cOmplete™ Protease Inhibitor Cocktail), and the lysates were collected by centrifugation at 15 000 × g. The amount of protein in the lysate was measured using a bicinchoninic acid assay (Bio‐Rad).
For Western blot analysis, proteins were prepared in sample buffer [62.5 mM Tris (pH 6.8), 2% SDS, 10% glycerol, 0.01% bromophenol blue, 5% mercaptoethanol and 50 mM dithiothreitol], and then separated by SDS‐polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore) using semidry blotters (TE70 PWR; Amersham Biosciences, Uppsala, Sweden). To detect Aß proteins, protein lysates in tricine sample buffer [200 mM Tris‐HCl (pH 6.8), 40% glycerol, 2% SDS and 0.04% Coomassie Blue G‐250; Bio‐Rad] were electrophoresed on 10%–20% Mini‐PROTEAN™ Tris‐Tricine Precast Gel (Bio‐Rad) under nonreducing conditions. After blocking with 5% skim milk and 1% BSA in TBS‐T buffer (25 mM Tris, 150 mM NaCl, 0.1% Tween 20; pH 7.4), the blot was reacted with a primary antibody and subsequently with a horseradish peroxidase‐conjugated secondary antibody (Santa Cruz Biotechnology).
To determine interactive complexes of Aß peptides and clusterin in the brain, we immunoprecipitated brain lysates (containing 800 μg of total protein) with anti‐Aß(1–16) antibody (6E10) plus Protein G‐sepharose beads (GE Healthcare, Buckinghamshire, UK) or anti‐clusterin/Apo J antibody (Novus Biologicals) plus Protein G‐sepharose beads. After washing in PBS, the protein/antibody‐bound beads were eluted in tricine sample buffer [200 mM Tris‐HCl (pH 6.8), 40% glycerol, 2% SDS and 0.04% Coomassie Blue G‐250]. Shortly thereafter, the supernatant protein samples were collected by brief centrifugation and fractionated by electrophoresis on 10%–20% Tris‐Tricine Precast Gel (Bio‐Rad) under nonreducing conditions. Finally, the proteins were detected by Western blot using anti‐Aß antibody (6E10) or anti‐clusterin/Apo J antibody.
Immunoreactive proteins were visualized using ECL™ Prime Western Blotting Detection Reagent (GE Healthcare) with the Davinch‐Western™ Chemiluminescence Imaging System (CAS‐400SM; Davinch‐K, Seoul, Korea). The band intensities were measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA), and protein levels were normalized to those of β‐actin.
Cognitive and motor behavioral analysis
Five‐ or 10‐month‐old mice were tested for spatial learning and memory in a Morris water maze (MWM) consisting of a circular plastic pool (120‐cm in diameter) with a cylindrical escape platform (10‐cm in diameter) in the northwest quadrant of the pool, 0.5‐cm under the surface of opaque water (maintained at 21.0°C ± 1.0°C) and with three visual cues on the wall. For 5 consecutive days, the animals were given daily training to swim and find the hidden escape platform. Each day, they had three 60‐s trials, each starting at a different quadrant (northeast, southeast and southwest quadrant). Three hours after the final training, the mice performed a probe trial for 60‐s starting from the southeast quadrant with the escape platform removed. The time, path and distance spent swimming in the pool as well as the frequency the target zone was crossed trying to locate the platform were monitored with SMART Video Tracking System (Harvard Apparatus, Holliston, MA, USA).
For the following 3 days, the same mice were given a balance beam task to evaluate balance and motor coordination. The beam was a flat wooden bar (1.2‐cm wide, 80‐cm long) connected to two 40‐cm high support columns. A dark escape chamber was located at the opposite end from where the mice were placed on the beam. The test was conducted in dim light with brighter illumination condensed on the starting point of the beam. The mice were encouraged to move from the starting point to the escape chamber on the other end. They were trained with three 90‐s trials per day with a 15‐min interval between trials. The time it took for the mice to traverse the beam and enter the chamber was recorded. If a mouse fell off the beam, we excluded that trial and conducted another trial. If a mouse stayed on the beam but did not reach the chamber within 90‐s, the maximum time was recorded as 90‐s.
Statistics
Data are presented as the means ± standard errors of the mean. Statistical comparisons between the groups were performed with an unpaired t‐test or one‐way ANOVA with Student–Newman–Keuls post hoc test, using Prism (GraphPad Software, La Jolla, CA, USA). Differences were considered significant at P < 0.05.
Results
Interactive expression of Aß and clusterin in the brains of 5×FAD mice
We evaluated the expression levels of clusterin in the brains of 5×FAD mice and their wild‐type littermates. In qRT‐PCR or Western blot analysis (Figure 1A–C), we found that 5×FAD mice expressed higher levels of clusterin than their littermates at the same age (5 or 10 months of age) and had a significant increase in clusterin levels from 5 to 10 months of age. In the wild‐type littermates, there was no significant difference in clusterin levels between 5 and 10 months of age. The increases in clusterin levels in the 5×FAD mice were also detectable by quantitative ELISA measurement of mouse clusterin in the brain lysates (Figure 1D). In addition, there were similar patterns of elevation of apolipoprotein E (Apo E) (Figure 1A, E and F) and DKK‐1 (Supplementary Figure S1) in 5×FAD mice. These findings indicate that Aß deposition leads to the increased expression of clusterin, Apo E or DKK‐1 in the brain.
Figure 1.
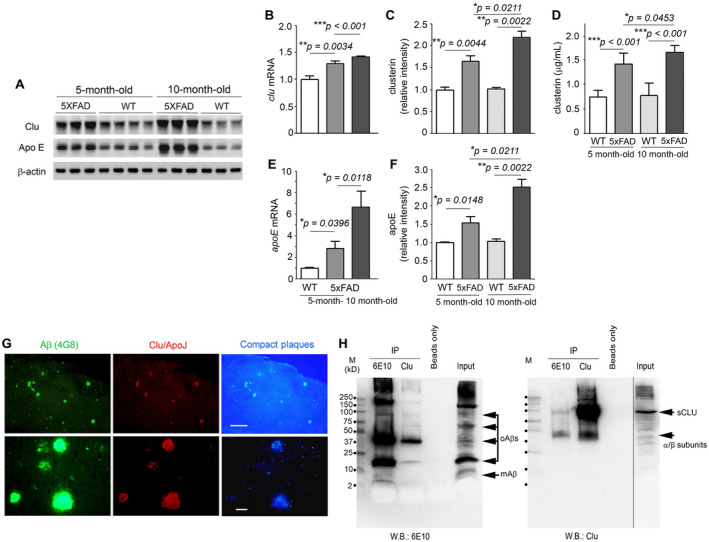
Interactions between clusterin and Aß in the brains of 5×FAD mice. A. Representative Western blots show expression or deposition of clusterin (Clu), Apo E and ß‐actin (as a loading control) in the brains of 5‐ or 10‐month‐old 5×FAD mice and their wild‐type littermates (WT). B and E. Clusterin (B) or Apo E (E) mRNA levels were measured by quantitative real time PCR. Values were calculated relative to that of GAPDH and bars are presented as fold changes relative to control values (5‐month‐old wild‐type mice) (n = 6–10 for all groups). C and F. Graphs show densitometric quantification of clusterin (C) or Apo E (F) from representative blots (A). After normalization with ß‐actin, the fold of change is presented as compared to control (5 month‐old wildtype mice) (n = 3–4 for all groups). D. ELISA quantification of mouse clusterin from SDS extracts (n = 6–9 for all groups). Data represent mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, or ***P < 0.001. G. Fluorescence microphotographs show colocalization of Aß (green) and clusterin (red) in compact amyloid plaques (blue). After double immunofluorescence staining with anti‐Aß(17–24) (4G8) and anti‐clusterin (Clu)/Apo J antibodies, the brain tissues were examined at lower (×40, top row) and higher (×400, bottom row) magnifications under the appropriate fluorescence filters. Scale bars, 500 μm (top row) and 50 μm (bottom row). H. Brain lysates (800 μg) were immunoprecipitated (IP) using either anti‐Aß(1–16) (6E10) or anti‐clusterin (Clu) antibody, and the precipitates were subjected to Western blot (W.B.) with anti‐Aß(1–16) (6E10) (left panel) or anti‐clusterin antibody (Clu; right panel). Beads only and input samples (50 μg as total protein) show negative and positive signals, respectively. The numbers along the 1st lane (M, molecular marker) of each blot denote the molecular sizes (kD). mAß, Aß monomers; oAßs, Aß oligomers; sCLU, secretory clusterin; α/ß subunits, α or ß subunits of clusterin.
5×FAD mice not only produce abundant levels of Aß but also develop Aß deposits or amyloid plaques in their brain at a relatively young age 39. Aß‐specific 4G8‐immunohistochemistry detected a number of amyloid plaques throughout the brain tissues of 5‐month‐old 5×FAD mice, primarily identified as compact core amyloid plaques by their in situ emission of blue fluorescence under ultraviolet (UV) illumination 26 (Figure 1G). In line with studies of human brains affected with AD 6, 8, 22, we found close colocalization of clusterin and Aß immunoreactivities in compact core amyloid plaques (Figure 1G).
To explore in vivo interactions between clusterin and Aß, we performed co‐IP experiments with brain lysates of 5×FAD mice (Figure 1H). When proteins co‐immunoprecipitated with anti‐Aß antibody (6E10) were fractionated under nonreducing conditions and then immunoblotted with anti‐clusterin antibody, we found two sizes of clusterin, 40‐kD α/β subunits and 80‐kD mature secretory clusterin (sCLU). Additionally, reverse co‐IP, which used anti‐clusterin antibody and anti‐Aß antibody (6E10) for IP and Western blot, respectively, detected multiple forms of Aß (monomers, dimers, oligomers and high‐molecular‐weight Aß aggregates). These results thus suggest that clusterin and Aß interact with each other in vivo, where clusterin interacts or binds with different Aß structures.
Effects of genetic deletion of clusterin on amyloid pathogenesis
To investigate whether clusterin is implicated in amyloid pathogenesis, we compared the deposition of Aß in the brains of clu +/+;5×FAD and clu −/−;5×FAD mice at 5 or 10 months of age. Western blot analysis revealed that Aß was prominently upregulated with age in both clu genotypes of 5×FAD mice as their entire blot signals became more intense at 10 than at 5 months of age (Figure 2A). In contrast, the Aß signals were generally attenuated in clu −/−;5×FAD mice compared with those in clu +/+;5×FAD mice at 5 months of age, whereas the clu genotype‐related differences in Aß intensities were undetectable at 10 months of age. Notably, significant differences among the groups were observed in band intensities of Aß monomers, dimers and oligomers (with molecular sizes ranging from approximately 4.0 kD to 50.0 kD).
Figure 2.
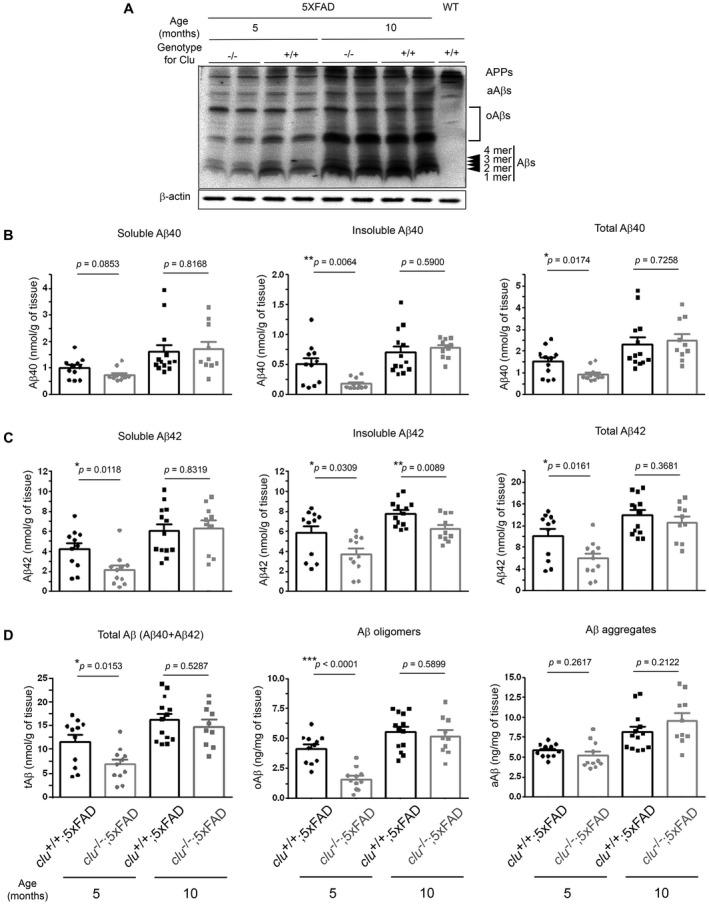
Deposition of various pools of brain Aß in clu+/+;5×FAD and clu−/−;5×FAD mice. A. Representative Western blot shows expression or deposition of multiple sizes of Aß proteins in the brains of clu +/+;5×FAD and clu −/−;5×FAD mice at 5 or 10 months of age. After immunoblotting with anti‐Aß(1–16) antibody (6E10) (top panel), the blot was deprobed and reacted again with anti‐ß‐actin antibody as a loading control (bottom panel). Blot signals from a wild‐type littermate mouse are shown on the right end of the blot (lane 9). oAßs, Aß oligomers; aAßs, Aß aggregates. B–D. ELISA quantification shows the deposition of various pools of Aß proteins. Soluble (1st column) and insoluble (2nd column) Aß40 (B) or Aß42 (C) were analyzed from PBS‐ and SDS‐soluble fractions, and FA‐soluble fractions of brain lysates, respectively. Total levels of Aß40 (B, 3rd column), Aß42 (C, 3rd column) and Aß (D, 1st column) were calculated as the sum of soluble and insoluble Aß40, Aß42 and total Aß40 and total Aß42, respectively. Soluble Aß oligomers (oAß) were quantified from PBS‐ and SDS‐soluble fractions (D, 2nd column), whereas Aß aggregates (aAß) were analyzed from SDS extracts without ultracentrifugal fractionation (D, 3rd column). All ELISA analyses were performed in duplicate. Numbers (males and females) of animals included in each group were 11 (5 and 6; for 5‐month‐old clu +/+;5×FAD), 11 (5 and 6; for 5‐month‐old clu −/−;5×FAD), 13 (6 and 7; for 10‐month‐old clu +/+;5×FAD) or 10 (5 and 5; for 10‐month‐old clu −/−;5×FAD mice). *P < 0.05, **P < 0.01, or ***P < 0.001.
Next, to further elaborate the differences in Aß deposition, we measured the amounts of various Aß peptides in the brains of clu +/+;5×FAD and clu −/−;5×FAD mice. We used ELISA to quantify the levels of soluble Aß40 or Aß42 and Aß oligomers in PBS‐ and SDS‐soluble fractions and insoluble Aß40 or Aß42 in FA‐soluble fractions in serially fractionated brain lysates. Total Aβ40 or Aβ42 amounts were calculated as the sum of the soluble and insoluble fractions of Aß40 or Aβ42. Aß aggregates were also analyzed in SDS extracts collected by brief centrifugation at 10 000 × g but without ultracentrifugal fractionation. The levels of soluble, insoluble and total Aß40 or Aß42 were lower in 5‐month‐old clu −/−;5×FAD mice than in clu +/+;5×FAD mice (by 24.4%–63.8%; P = 0.0064–0.0853) (Figure 2B and C), demonstrating that total Aß (sum of Aß40 and Aß42) level was significantly lower in clu −/−;5×FAD mice (by 41.0%; P = 0.0153) (Figure 2D). Differences in Aß42 levels were even more striking, with soluble Aß42 levels in clu −/−;5×FAD mice 49.5% lower than those in clu +/+;5×FAD mice (P = 0.0118) and insoluble Aß42 35.7% lower (P = 0.0309) (Figure 2C). Soluble Aß oligomers were also markedly lower in clu −/−;5×FAD mice (by 60.9%; P < 0.0001), but Aß aggregates were only slightly lower (by 11.1%; P = 0.2617) (Figure 2D). From 5 months to 10 months of age, the levels of all pools of Aß proteins (total, soluble and insoluble Aß40 or Aß42, Aß oligomers and Aß aggregates) were significantly or slightly increased in both clu genotypes of 5×FAD mice (Figure 2B–D), resulting in no differences in the levels of all pools of Aß proteins between clu −/−;5×FAD and clu +/+;5×FAD mice at 10 months of age (Figure 2B–D), except that insoluble Aß42 was 20.0% lower in clu −/−;5×FAD mice (P = 0.0089) (Figure 2C).
From these results for Aß, we calculated the proportion of each pool or form of Aß within the total amount of Aß (sum of Aß40 and Aß42). First, we found neither clu genotype‐related nor age‐related differences in the ratio of total soluble or insoluble Aß to total Aβ (Supplementary Figure S2A and S2B). However, compared with clu +/+;5×FAD mice, clu −/−;5×FAD mice at 5 months of age had a significantly lower ratio of soluble Aß oligomers to total Aβ or to Aß aggregates (by 34.6% or 52.9%; P = 0.0145 or 0.0002), whereas those ratios in both clu genotypes were not significantly different at 10 months of age (Supplementary Figure S2C and S2E). The proportion of Aß oligomers in the soluble fraction of total Aβ (sum of Aß40 and Aß42) was also marginally lower in clu −/−;5×FAD mice at 5 months of age (by 27.3%; P = 0.0994) (Supplementary Figure S2D). However, the ratio of Aß aggregates to total Aβ tended to be higher in clu −/−;5×FAD mice than that in clu +/+;5×FAD mice at each age (Supplementary Figure S2F). Further, when we compared the levels of Aß40 and Aß42, the ratio of total Aß40 to total Aß42 was somewhat higher in clu −/−;5×FAD mice than that in clu +/+;5×FAD mice at each age (by 32.7% or 23.7%; P = 0.1603 or 0.0695) (Supplementary Figure S2G), whereas the ratio of soluble Aß40 to soluble Aß42 was about twice higher in clu −/−;5×FAD mice at 5 months of age (by 91.0%, P = 0.0229), with the lack of clu genotype‐related difference at 10 months (by 5.1%, P = 0.7502) (Supplementary Figure S2H).
We also measured the loads of 4G8‐immunoreactive or Congo red‐positive amyloid deposits or plaques (Figure 3A). At 5 months of age, compared with clu +/+;5×FAD mice, clu −/−;5×FAD mice had a lower load of 4G8‐immunoreactive amyloid deposits and fewer Congo red‐positive amyloid plaques (by 40.4% and 43.1%; P = 0.0154 and 0.0004, respectively), which reflect the deposition of all forms of APP and Aß, and the fraction of insoluble Aß fibrils, respectively (Figure 3B). Thereafter, the loads substantially increased in the clu −/−;5×FAD mice so that by 10 months of age, there were no clu genotype‐related differences (Figure 3B). Finally, the ratios of Congo red‐positive amyloid plaques to 4G8‐immunoreactive amyloid deposits did not significantly differ between the two clu genotypes at 5 or 10 months of age. Instead, the ratios were significantly decreased with age in both clu genotypes (P = 0.0015 for clu −/−;5×FAD mice or P = 0.0004 for clu +/+;5×FAD mice) (Supplementary Figure S2I).
Figure 3.
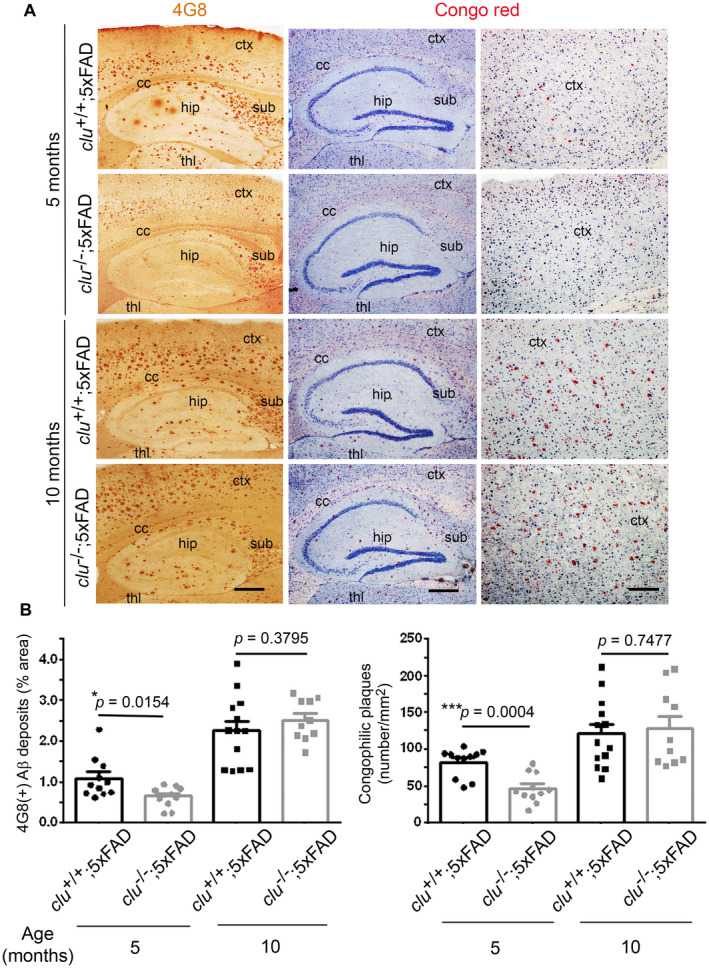
Quantification of amyloid deposits or plaques by immunohistochemical or Congo red staining. A. Sagittal brain sections of 5‐ or 10‐month‐old clu +/+;5×FAD and clu −/−;5×FAD mice were stained with anti‐Aβ(17–24) antibody (4G8) or Congo red dye to detect pan‐amyloid precursor protein (APP)/amyloid‐β (Aß)‐immunoreactive deposits (1st column, brown dots), or compact amyloid plaques (2nd and 3rd column, red dots). hip, hippocampus; ctx, cortex; thl, thalamus; cc, corpus callosum; sub, subiculum. Scale bars, 500 μm (1st and 2nd column) or 200 μm (3rd column). B. The per cent area of 4G8‐immunoreactive amyloid deposits (left graph) or the total number of congophilic amyloid plaques (right graph) in the same brain region of interest was measured in five brain sections per animal, taken every 200 μm lateral from the midline. Numbers (males and females) of animals in each group were 11 (5 and 6; for 5‐month‐old clu +/+;5×FAD), 11 (5 and 6; for 5‐month‐old clu −/−;5×FAD), 13 (6 and 7; for 10‐month‐old clu +/+;5×FAD), or 10 (5 and 5; for 10‐month‐old clu −/−;5×FAD mice). *P < 0.05, **P < 0.01, or ***P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Considered together, these quantitative results indicate that clusterin expression is correlated with amyloid deposition at the early age in 5×FAD mice, but the effects decline or disappear with age.
Effects of clusterin deficiency on Aß‐induced neuronal or synaptic loss
The neuropathology of AD includes loss of neurons and synapses as well as Aß deposition. Thus, to determine the involvement of clusterin in Aß‐induced neuronal or synaptic degeneration, we compared the expression of neuronal or synaptic marker proteins using Western blot analysis (Figure 4). The proteins with striking changes in expression included neuronal specific NeuN, nonphosphorylated neurofilament protein SMI‐32 52, presynaptic synaptophysin, axon initial segment‐specific and voltage‐gated sodium channels‐associated ankyrin G 47, postsynaptic PSD‐95, vesicle‐associated membrane protein (VAMP; synaptobrevin) and postsynaptic scaffold protein ProSAP2/Shank 3 15. The levels of the neuron‐ and synapse‐specific proteins were significantly lower in 5‐month‐old clu +/+;5×FAD mice than those in nontransgenic wild‐type mice, but were significantly increased in the age‐matched clu −/−;5×FAD mice. However, the protein expressions in clu −/−;5×FAD mice were decreased by 10 months of age to the levels as those in clu +/+;5×FAD mice.
Figure 4.
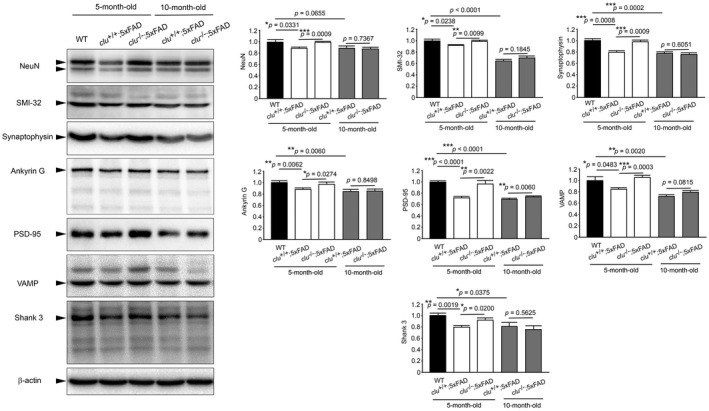
Effect of clusterin and age on expression of neuronal or synaptic proteins in the brain of 5×FAD mouse. A. Representative Western blots show expression of neuronal or synaptic proteins in 5‐ and 10‐month‐old clu +/+;5×FAD and clu −/−;5×FAD mice and 5‐month‐old nontransgenic wild‐type (WT) mice, representing only results with significant differences in expression; neuronal specific NeuN (molecular size, 46‐48 kD), nonphosphorylated neurofilament SMI‐32 (200 kD), presynaptic synaptophysin (38 kD), axon initial segment‐specific and voltage‐gated sodium channels‐associated ankyrin G (190 kD), postsynaptic PSD‐95 (95 kD), vesicle‐associated membrane protein (VAMP, synaptobrevin; 16 kD), postsynaptic scaffold protein ProSAP2/Shank3 (180 kD) and ß‐actin (42 kD). B–H. Densitometric quantification of indicated proteins after normalization to ß‐actin. Graphs show significant differences in the levels of the indicated proteins (arrows in blots) between nontransgenic wild‐type and 5×FAD mice and between 5‐month‐old clu +/+;5×FAD and clu −/−;5×FAD mice but not between 10‐month‐old clu +/+;5×FAD and clu −/−;5×FAD mice. Protein levels are shown as values relative to a control (black bars, wild‐type mice). Bars denote mean ± SEM of three independent experiments. Each group includes 6 mice (3 males and 3 females). *P < 0.05, **P < 0.01, or ***P < 0.001.
Cognitive and motor performance in clusterin‐null 5×FAD mice
Finally, to determine whether clusterin is involved in cognitive and behavioral impairment during the development of AD, we evaluated the effect of genetic deletion of clusterin on cognitive and motor performance in 5×FAD mice.
To evaluate the effects of clusterin on spatial learning and memory performance, the mice were trained in a Morris water maze (MWM) with 3 trials every day for 5 days (Figure 5A). Unexpectedly, both 5‐month‐old clu −/−;5×FAD mice and nontransgenic wild‐type mice found the platform significantly more quickly than the clu +/+;5×FAD mice of same age on the first day of training (P = 0.0021 and 0.0183, respectively). Thereafter, the 5‐day repeated trainings led to significant improvement in locating the hidden platform for every group of 5‐month‐old mice, although the nontransgenic wild‐type mice and clu −/−;5×FAD mice were consistently and significantly faster than clu +/+;5×FAD mice. On the last trial day, clu −/−;5×FAD mice were able to locate the platform more rapidly than clu +/+;5×FAD mice (P = 0.0004), indicating that clusterin deficiency promoted better spatial learning performance in 5‐month‐old 5×FAD mice. Three hours later, the animals underwent a free swim probe test to evaluate recall for the previous location of the target platform, a measure of spatial reference memory retention in trained animals (Figure 5B–F). Comparing the speed or the mean distance in reaching the target zone where the escape platform had been located, the number of times the target zone was crossed, and the time spent in the target quadrant, we found that 5‐month‐old clu +/+;5×FAD mice had impaired performance relative to their nontransgenic wild‐type littermates. Age‐matched clu −/−;5×FAD mice had significantly better spatial memory performance than the clu +/+;5×FAD mice. However, this good level of performance did not persist till 10 months of age in the mice with genetic deletion of clusterin. There were no differences between 10‐month‐old clu −/−;5×FAD and clu +/+;5×FAD mice in performance of MWM and probe trials (Figure 5A–F).
Figure 5.
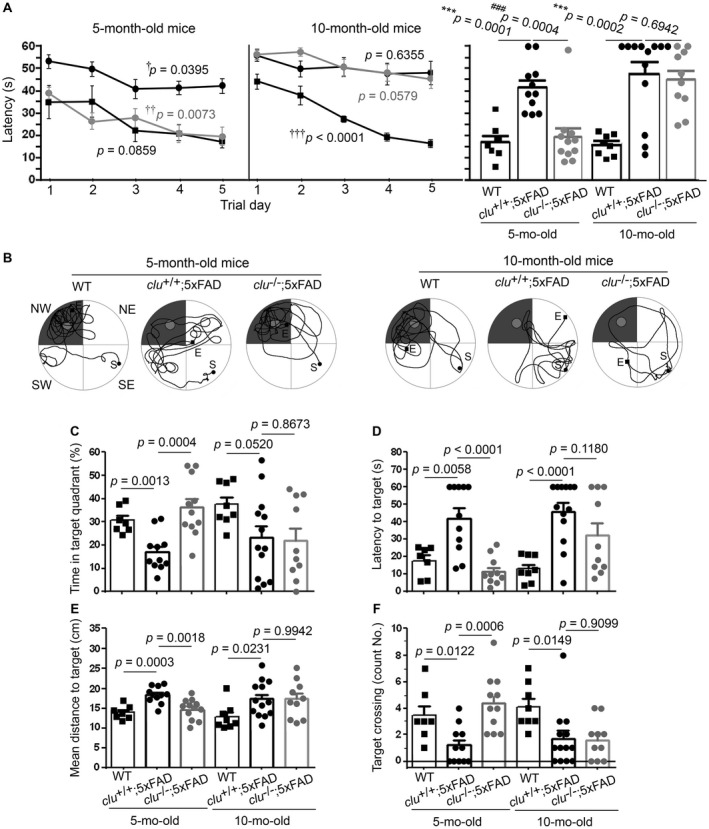
Effect of clusterin on performance of spatial learning and memory in 5×FAD mice. A. Animals were trained daily to find a hidden platform in a Morris water maze (MWM). The time taken to find the platform was significantly longer over 5 days in clu +/+;5×FAD mice (black circles) compared with their nontransgenic wild‐type littermates (rectangles). The clu −/−;5×FAD mice (gray circles) were significantly faster than clu +/+;5×FAD mice at 5 months of age (left graph), but at 10 months of age, they performed no better than clu +/+;5×FAD mice (right graph). Crosses (†) denote the effect of 5 days‐trainings on the time to reach the target platform in each group (by one‐way ANOVA with Student–Newman–Keuls post hoc test). † P < 0.05, †† P < 0.01, or ††† P < 0.001. Vertical bars on the right panel denote the values of latency time taken by each animal group on the last trial day. B–F. After the training, 60 s‐probe trials were performed to evaluate the entire path taken (B; from points “S” to “E”) and the percentage of time spent in the target zone (northwest shadow quadrant in B) searching for the platform that had been removed (small circular area in target quadrant in B) (C) as well as time (D) and mean distance (E) taken to get the first touch to the target location, and the frequency at which the target location was crossed (F). The clu +/+;5×FAD mice performed poorly on all evaluations compared with nontransgenic wild‐type mice. The clu −/−;5×FAD mice at 5 months of age performed as well as the wild‐type littermates at 5 months of age, but at 10 months of age, their performance was as poor as the clu +/+;5×FAD mice. Numbers (males and females) of animals in each group were 11 (5 and 6; for 5‐month‐old clu +/+;5×FAD), 11 (5 and 6; for 5‐month‐old clu −/−;5×FAD), 13 (6 and 7; for 10‐month‐old clu +/+;5×FAD), or 10 (5 and 5; for 10‐month‐old clu −/−;5×FAD mice). Asterisk (*) and pound (#) denote the comparisons between clu +/+;5×FAD and nontransgenic wild‐type mice and between clu +/+;5×FAD and clu −/−;5×FAD mice, respectively (by an unpaired t‐test). *,# P < 0.05, **,## P < 0.01, or ***,### P < 0.001.
After completion of the MWM test, the animals underwent 3 days of training on a balance beam task (Figure 6). On the first trial day, nontransgenic wild‐type littermates more rapidly reached the other end, followed by clu −/−;5×FAD mice, with the clu +/+;5×FAD mice being the slowest; these differences, however, were not statistically significant. However, by the third day, the 5‐month‐old wild‐type mice and clu −/−;5×FAD mice moved into the escape chamber increasingly quickly (P = 0.0363 and 0.0036, respectively), spending a significantly shorter time on the beam compared with the age‐matched clu +/+;5×FAD mice (P = 0.0011 and 0.0008, respectively). In contrast, at 10 months of age, clu −/−;5×FAD and clu +/+;5×FAD mice both failed to acquire the capability to run along the beam with the balance and motor coordination achieved by nontransgenic wild‐type mice (P = 0.6391, 0.2042 and 0.0036, respectively). Thus, there were no differences between the two clu genotypes of 5×FAD mice on the third day of balance beam task (P = 0.9351) (Figure 6).
Figure 6.
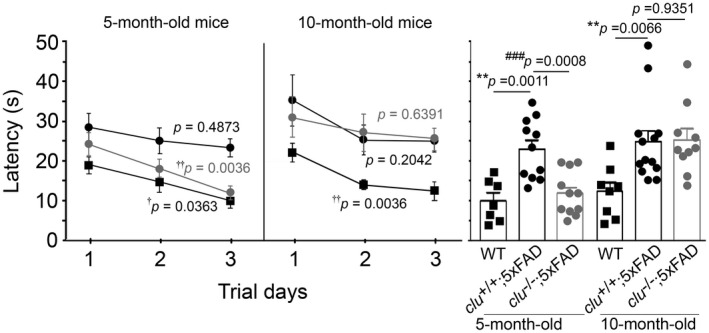
Effect of clusterin on balance and motor coordination in 5×FAD mice. At 5 months of age, clu −/−;5×FAD mice (gray circles) and wild‐type mice (rectangles) reached the escape chamber at the opposite end of the beam significantly faster over 3 days of trials than clu +/+;5×FAD mice (black circles) (left graph). However, by 10 months of age, the clu −/−;5×FAD mice took significantly longer than before and were now as slow as clu +/+;5×FAD mice (right graph), both genotypes of 5×FAD mice being significantly slower than 10‐month‐old wild‐type mice. Vertical bars on the right panel denote the values of latency time taken by each animal group on the last trial day. Numbers (males and females) of animals in each group were 11 (5 and 6; for 5‐month‐old clu +/+;5×FAD), 11 (5 and 6; for 5‐month‐old clu −/−;5×FAD), 13 (6 and 7; for 10‐month‐old clu +/+;5×FAD), or 10 (5 and 5; for 10‐month‐old clu −/−;5×FAD mice). Asterisk (*) and pound (#) signs denote comparisons between clu +/+;5×FAD and wild‐type mice and between clu +/+;5×FAD and clu −/−;5×FAD mice, respectively (by an unpaired t‐test). *,# P < 0.05, **,## P < 0.01, or ***,### P < 0.001. Crosses (†) denote the effect of 3 days of training on the time to reach the escape chamber in each group (by one‐way ANOVA with Student–Newman–Keuls post hoc test). † P < 0.05 or †† P < 0.01.
Discussion
Since clusterin can maintain Aß solubility by preventing Aß aggregation 32 and promote its clearance from brain by mediating BBB transport 3, 57, early interest in clusterin was centered on defining its protective role against AD. Experimental support for this possibility came from findings that clusterin alleviated Aß toxicity in neuronal cells 4, 29, 56. However, growing evidence suggests that clusterin interacts with Aß to enhance its neurotoxicity and cause neurodegeneration 23, 25, 40, 41. A previous study with hAPP transgenic PDAPP mice 10 provided the first evidence of an in vivo functional role for clusterin, with its absence leading to fewer amyloid plaques and lesser neuritic dystrophy despite a consistent level in total Aß. Similarly with our present study of 5×FAD mice, a recent study 55 also found a marked decrease in total amyloid deposition containing soluble and insoluble Aβ as well as parenchymal amyloid plaques in clusterin‐null APP/PS1 mice. Although the different findings concerning the deposition of total Aß likely came from the different background features of hAPP transgenic mice used 55, it is important that clusterin deficiency prevented amyloid deposition and Aβ‐induced neurodegeneration in the hAPP transgenic mouse models of AD.
In the present study, we observed interactions between clusterin and Aß proteins in the brains of 5×FAD mice. Both mRNA and protein levels of clusterin were higher in 5×FAD mice than in age‐matched wild‐type littermates, a finding similar to that in the brains of human patients with AD compared with controls without dementia 14, 33, 40. While clusterin levels did not significantly change with age in wild‐type mice, they substantially increased with age in 5×FAD mice. In addition, the expression of DKK‐1, a Wnt antagonist mediating clusterin‐driven Aß toxicity 23, 45, was also significantly higher and increased with age in 5×FAD mice. These findings indicate that Aß deposition causes increased expressions of clusterin and DKK‐1 in the hAPP‐transgenic mouse models 46. Next, we detected colocalization of Aß and clusterin in nearly all compact amyloid plaques in 5×FAD mice, which is similar to that found in the brains of AD patients 8, 22, 34. Furthermore, we found that Aß and clusterin were in the same complex as they were co‐immunoprecipitated with each other in brain lysates of 5×FAD mice. Because clusterin was detected together with Aß proteins of various sizes in our co‐IP experiment, it is thought that clusterin has the propensity to bind or interact with different forms of Aß, such as monomers, oligomers, or fibril aggregates in vivo. Using purified or synthetic proteins, many studies have shown that clusterin binds to Aβ proteins with high affinity 2, 13, 31, 37.
This study provides the first in vivo evidence that clusterin‐directed Aß deposition and oligomerization augment Aß neurotoxicity and underlie AD pathogenesis in hAPP transgenic mice. Although knockdown of clusterin was previously shown to reduce Aβ toxicity in primary neurons 23, we further elucidated how genetic loss of clusterin influences AD pathology in 5×FAD mice. Clusterin deficiency led to less Aß (total, soluble and insoluble Aß40 and Aß42) and fewer amyloid plaques in the brains of 5×FAD mice at 5 months of age. Clusterin binding could render Aß resistant to proteolytic degradation and maintain its stability 32, 37; therefore, clu +/+;5×FAD mice would exhibit higher amyloid deposition than clu −/−;5×FAD mice. Alternatively, the reduced level of Aß likely is due to the decrease in Aß generation in clu −/−;5×FAD mice.
However, we specifically paid attention to Aß oligomers and Aß aggregates because Aβ neurotoxicity is primarily attributed to its oligomer forms rather than its monomer or fibril forms 25, 40, 41. Aß42 is considered to be more neurotoxic than Aß40 because of its propensity to more readily aggregate into oligomers 16, 30, 48, 53. Clusterin, then, could sequester or stabilize toxic Aβ oligomers so that they are less degradable, or inhibit further aggregation of Aβ oligomers into nontoxic fibrils 32, 37. When compared the results of ELISA quantification in clu −/−;5×FAD mice with those in clu +/+;5×FAD mice, level of soluble Aß42, in which include Aß42 oligomers, was more reduced than that of soluble Aß40 or insoluble Aß42, with the most dramatic reduction in soluble Aß oligomers. The difference in Aß aggregates composed of Aß oligomers, protofibrils and fibrils, was not statistically significant. Next, we found no clu genotype‐related difference in the ratio of soluble Aß/total Aß or insoluble Aß/total Aß, whereas the ratio of soluble Aß oligomers/total Aß, soluble Aß oligomers/total soluble Aß, or soluble Aß oligomers/Aß aggregates was markedly lower in clu −/−;5×FAD mice. The ratio of Aß aggregates/total Aß became rather somewhat higher. We further found that lack of clusterin caused lower loads of 4G8‐immunoreactive plaques and fewer Congo red‐positive fibrillar amyloid plaques, which are representative of total Aß deposition and insoluble Aß aggregates, respectively. The ratios of Congo red‐positive plaques/4G8‐immunoreactive plaques were similar in both clu genotypes of 5×FAD mice. Finally, 5‐month‐old clu −/−;5×FAD mice had less neuronal and synaptic loss as evaluated by the expression of neuronal or synaptic proteins. These mice also had significantly better motor and cognitive performances. Collectively, these results suggest that clusterin is involved in amyloid pathogenesis responsible for neurodegeneration and behavioral impairment seen in the 5×FAD mouse model of early AD, perhaps by facilitating Aß production and deposition or by shifting Aß composition toward more toxic soluble Aß oligomers.
However, it should be noted that any influence of clusterin deficiency on AD pathology declined or disappeared with age. By 10 months of age, all Aß pools in clu −/−;5×FAD mice had significantly increased to levels similar to those in clu +/+;5×FAD mice. There were no detectable differences in amyloid plaque deposition, neuronal and synaptic loss, or motor and cognitive impairment between the two clu genotypes of 5×FAD mice. These findings reminded us of the previous human brain studies that demonstrated an association of clusterin with AD pathology in early stage. In longitudinal cohort analyses combining volumetric MRI scans with quantification of clusterin and Aβ proteins, plasma clusterin levels were associated with atrophy in brain regions affected in AD 21, 50. More importantly, an interaction between Aβ42 and clusterin has been exclusively related with atrophy of the entorhinal cortex, a region that is selectively affected early in the pathogenesis of AD 5, 51. In cognitively normal older individuals and those with mild cognitive impairment (MCI), decreased levels of CSF Aβ42 (related to increased brain Aβ42 deposition) and elevated CSF clusterin levels were associated with higher entorhinal cortex volume loss but not with hippocampal atrophy 11. The Aβ‐clusterin association with entorhinal cortical atrophy was stronger in those individuals than in patients with advanced AD. Together with these human cohort studies, our findings that clusterin deficiency was associated with a significantly lower degree of amyloid pathology, neuronal and synaptic loss and behavioral and cognitive impairment in 5‐ month‐old mice but not in 10‐ month‐old mice suggest that clusterin may facilitate amyloid pathogenesis and induce Aβ‐directed neurodegeneration in the early stages of AD 11, 28, 50.
Currently, it is unclear how clusterin can have disparate effects on the early and late stages of AD pathogenesis. One possible explanation is that the effect of clusterin on Aß neurotoxicity may depend upon a substoichiometric ratio of clusterin and Aß 54. Earlier in vitro studies showed that clusterin modulates amyloid pathogenesis in a biphasic manner to promote or inhibit its neurotoxicity. A higher proportion of clusterin in an Aß‐clusterin complex could cause relatively less Aß aggregation and toxicity, whereas a lower proportion could facilitate Aß aggregation and increase toxicity 4, 19, 41, 56. Our present study supports that hypothesis because we observed that, with age, levels of Aß increased to a greater extent than those of clusterin. In the brains of 10‐month‐old 5×FAD mice, total Aß (sum of Aß40 and Aß42) and Aß42 were elevated by 39.6% and 37.4%, respectively, relative to values in 5‐month‐old 5×FAD mice. While clusterin levels also increased from 5 to 10 months of age, the 17.3% increase was relatively smaller than that of Aß proteins. The resulting decline in the ratio of clusterin to Aß may affect amyloid pathogenesis, thereby aggravating neurodegeneration and behavioral or cognitive impairment 36. A further possibility is that as AD progresses into the later stages with a rapid increase in Aß deposition, the clusterin content might become insufficient to mediate amyloid pathogenesis and other more amyloidogenic factors such as phosphorylated tau (p‐tau) or Apo E may become more prominent. Corroborating this hypothesis, we noticed that, by 10 months of age, clu −/−;5×FAD mice had levels of amyloid pathology, neuronal and synaptic loss and motor and cognitive impairment comparable to those of clu +/+;5×FAD mice. A prior study in Apo E and clusterin double knockout PDAPP mice 9 showed that the concurrent absence of Apo E and clusterin caused earlier onset and larger amyloid deposition, suggesting that clusterin can modify the structure, accumulation, deposition and toxicity of Aß in cooperation with Apo E. In addition, a human brain study 11 found that interaction only between clusterin and Aβ was associated with volume loss in the entorhinal cortex in cognitively normal elderly people, whereas in those with MCI, atrophy was also related to the interaction of p‐tau and Aβ. Moreover, they showed that interaction between Aβ and p‐tau was significantly associated with later atrophy in the hippocampus or other temporal lobe regions, which was not the case with the interaction of clusterin and Aβ. Therefore, these findings support the contention that other factors such as Apo E or p‐tau have a stronger influence than clusterin on AD pathology in the later stages.
A recent study is of note because it investigated the effect of clusterin deficiency on amyloid pathology in APP/PS1 transgenic mice 55, describing the role of clusterin in Aβ transport across the BBB via low‐density lipoprotein receptor‐related protein‐2 (LRP2) 3. They found a marked decrease in amyloid deposition in the brain parenchyma but a parallel increase in cerebral amyloid angiopathy (CAA) in clu −/−;APP/PS1 mice. As CAA is likely due to amyloid deposition on the cerebral vessel walls along the perivascular drainage pathways 1, it could be indicative of an abnormal Aβ clearance in AD brain. Mounting evidence suggests that Aβ40 has a stronger propensity to promote CAA relative to Aβ42, and thus the higher Aβ40/Aβ42 ratio favors toward CAA development 12, 18. When we briefly compared the levels of Aβ40 and Aβ42 in our 5×FAD mice, the Aβ40/Aβ42 ratio was significantly increased in the soluble fractions of 5 months‐old clu −/−;5×FAD mice, supporting the previous findings 49, 55. However, there was a statistically nonsignificant increase in the ratio of total Aβ40/Aβ42, and the ratio differences disappeared at 10 months. While these differences in Aβ40/Aβ42 ratio might represent the effect of clusterin deficiency on CAA in 5×FAD mice, it would be warranted to explore the expression of CAA in clusterin‐wildtype or ‐null 5×FAD mice in order to define the roles of clusterin in Aβ clearance or transport in vivo.
In conclusion, clusterin preferentially augments the toxic Aß pools to facilitate the early stage of AD pathogenesis, and thereby promotes Aß‐directed neurodegeneration and behavioral and cognitive impairments. However, as the pathology progresses, its influences decline, suggesting that substances other than clusterin may be more critical in the later stages. Therefore, a strategy to reduce the level of clusterin or to inhibit clusterin‐Aβ interactions may provide potential therapeutic opportunities for AD in early stage, but it should be considered whether such a treatment can consistently retard the disease pathogenesis in later advanced stages.
Declarations
The authors declare no conflict of interest.
Supporting information
Figure S1. Expressions of mRNA and protein of DKK‐1 in the brains of 5‐month‐old wild‐type (WT), and 5‐ or 10‐month‐old 5×FAD mice.
Figure S2. Relative proportions of various pools of Aß proteins in the brains of clu +/+;5×FAD and clu −/−;5×FAD mice.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF‐2015R1A2A1A15052049 and NRF‐2017R1D1A1B03030567) and by the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea (2016‐396).
References
- 1. Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cerebral amyloid angiopathy: Accumulation of amyloid‐beta 40 in affected vessels. J Neuropathol Exp Neurol 57:353–359. [DOI] [PubMed] [Google Scholar]
- 2. Beeg M, Stravalaci M, Romeo M, Carra AD, Cagnotto A, Rossi A et al (2016) Clusterin binds to Abeta1‐42 oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J Biol Chem 291:6958–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2007) Transport pathways for clearance of human Alzheimer's amyloid beta‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boggs LN, Fuson KS, Baez M, Churgay L, McClure D, Becker G, May PC (1996) Clusterin (Apo J) protects against in vitro amyloid‐beta (1‐40) neurotoxicity. J Neurochem 67:1324–1327. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 6. Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J (2000) Apolipoprotein J (clusterin) and Alzheimer's disease. Microsc Res Tech 50:305–315. [DOI] [PubMed] [Google Scholar]
- 7. Cascella R, Conti S, Tatini F, Evangelisti E, Scartabelli T, Casamenti F et al (2013) Extracellular chaperones prevent Abeta42‐induced toxicity in rat brains. Biochim Biophys Acta 1832:1217–1226. [DOI] [PubMed] [Google Scholar]
- 8. Choi‐Miura NH, Ihara Y, Fukuchi K, Takeda M, Nakano Y, Tobe T, Tomita M (1992) SP‐40,40 is a constituent of Alzheimer's amyloid. Acta Neuropathol 83:260–264. [DOI] [PubMed] [Google Scholar]
- 9. DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW et al (2004) ApoE and clusterin cooperatively suppress A beta levels and deposition: Evidence that ApoE regulates extracellular A beta metabolism in vivo . Neuron 41:193–202. [DOI] [PubMed] [Google Scholar]
- 10. DeMattos RB, O'Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR et al (2002) Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 99:10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desikan RS, Thompson WK, Holland D, Hess CP, Brewer JB, Zetterberg H et al (2014) The role of clusterin in amyloid‐beta‐associated neurodegeneration. JAMA Neurol 71:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM (2005) Human apolipoprotein E4 alters the amyloid‐beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25:2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghiso J, Matsubara E, Koudinov A, Choi‐Miura NH, Tomita M, Wisniewski T, Frangione B (1993) The cerebrospinal‐fluid soluble form of Alzheimer's amyloid beta is complexed to SP‐40,40 (apolipoprotein J), an inhibitor of the complement membrane‐attack complex. Biochem J 293(Pt 1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giannakopoulos P, Kovari E, French LE, Viard I, Hof PR, Bouras C (1998) Possible neuroprotective role of clusterin in Alzheimer's disease: A quantitative immunocytochemical study. Acta Neuropathol 95:387–394. [DOI] [PubMed] [Google Scholar]
- 15. Grabrucker AM, Schmeisser MJ, Udvardi PT, Arons M, Schoen M, Woodling NS et al (2011) Amyloid beta protein‐induced zinc sequestration leads to synaptic loss via dysregulation of the ProSAP2/Shank3 scaffold. Mol Neurodegener 6:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hardy JA, Higgins GA (1992) Alzheimer's disease: The amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- 17. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al (2009) Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD et al (2004) A beta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7:954–960. [DOI] [PubMed] [Google Scholar]
- 19. Hughes SR, Khorkova O, Goyal S, Knaeblein J, Heroux J, Riedel NG, Sahasrabudhe S (1998) Alpha2‐macroglobulin associates with beta‐amyloid peptide and prevents fibril formation. Proc Natl Acad Sci U S A 95:3275–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphreys DT, Carver JA, Easterbrook‐Smith SB, Wilson MR (1999) Clusterin has chaperone‐like activity similar to that of small heat shock proteins. J Biol Chem 274:6875–6881. [DOI] [PubMed] [Google Scholar]
- 21. Hye A, Riddoch‐Contreras J, Baird AL, Ashton NJ, Bazenet C, Leung R et al (2014) Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement 10:799–807 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kida E, Choi‐Miura NH, Wisniewski KE (1995) Deposition of apolipoproteins E and J in senile plaques is topographically determined in both Alzheimer's disease and Down's syndrome brain. Brain Res 685:211–216. [DOI] [PubMed] [Google Scholar]
- 23. Killick R, Ribe EM, Al‐Shawi R, Malik B, Hooper C, Fernandes C et al (2014) Clusterin regulates beta‐amyloid toxicity via Dickkopf‐1‐driven induction of the wnt‐PCP‐JNK pathway. Mol Psychiatry 19:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 25. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M et al (1998) Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee JY, Cho E, Seo JW, Hwang JJ, Koh JY (2012) Alteration of the cerebral zinc pool in a mouse model of Alzheimer disease. J Neuropathol Exp Neurol 71:211–222. [DOI] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 28. Malkki H (2014) Alzheimer disease: Chaperone protein clusterin is involved in amyloid‐beta‐associated entorhinal atrophy in early AD. Nat Rev Neurol 10:60. [DOI] [PubMed] [Google Scholar]
- 29. Mannini B, Cascella R, Zampagni M, Van Waarde‐Verhagen M, Meehan S, Roodveldt C et al (2012) Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc Natl Acad Sci U S A 109:12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci U S A 82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsubara E, Frangione B, Ghiso J (1995) Characterization of apolipoprotein J‐Alzheimer's a beta interaction. J Biol Chem 270:7563–7567. [DOI] [PubMed] [Google Scholar]
- 32. Matsubara E, Soto C, Governale S, Frangione B, Ghiso J (1996) Apolipoprotein J and Alzheimer's amyloid beta solubility. Biochem J 316(Pt 2):671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. May PC, Lampert‐Etchells M, Johnson SA, Poirier J, Masters JN, Finch CE (1990) Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer's disease and in response to experimental lesions in rat. Neuron 5:831–839. [DOI] [PubMed] [Google Scholar]
- 34. McGeer PL, Kawamata T, Walker DG (1992) Distribution of clusterin in Alzheimer brain tissue. Brain Res 579:337–341. [DOI] [PubMed] [Google Scholar]
- 35. McLaughlin L, Zhu G, Mistry M, Ley‐Ebert C, Stuart WD, Florio CJ et al (2000) Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J Clin Invest 106:1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miners JS, Clarke P, Love S (2017) Clusterin levels are increased in Alzheimer's disease and influence the regional distribution of Abeta. Brain Pathol 27:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Narayan P, Meehan S, Carver JA, Wilson MR, Dobson CM, Klenerman D (2012) Amyloid‐beta oligomers are sequestered by both intracellular and extracellular chaperones. Biochemistry 51:9270–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA et al (2011) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid‐beta(1‐40) peptide. Nat Struct Mol Biol 19:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al (2006) Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26:10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE (1994) Purification and characterization of brain clusterin. Biochem Biophys Res Commun 204:1131–1136. [DOI] [PubMed] [Google Scholar]
- 41. Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE et al (1995) Clusterin (apoJ) alters the aggregation of amyloid beta‐peptide (A beta 1‐42) and forms slowly sedimenting a beta complexes that cause oxidative stress. Exp Neurol 136:22–31. [DOI] [PubMed] [Google Scholar]
- 42. Poon S, Easterbrook‐Smith SB, Rybchyn MS, Carver JA, Wilson MR (2000) Clusterin is an ATP‐independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding‐competent state. Biochemistry 39:15953–15960. [DOI] [PubMed] [Google Scholar]
- 43. Poon S, Rybchyn MS, Easterbrook‐Smith SB, Carver JA, Pankhurst GJ, Wilson MR (2002) Mildly acidic pH activates the extracellular molecular chaperone clusterin. J Biol Chem 277:39532–39540. [DOI] [PubMed] [Google Scholar]
- 44. Poon S, Treweek TM, Wilson MR, Easterbrook‐Smith SB, Carver JA (2002) Clusterin is an extracellular chaperone that specifically interacts with slowly aggregating proteins on their off‐folding pathway. FEBS Lett 513:259–266. [DOI] [PubMed] [Google Scholar]
- 45. Purro SA, Dickins EM, Salinas PC (2012) The secreted Wnt antagonist Dickkopf‐1 is required for amyloid beta‐mediated synaptic loss. J Neurosci 32:3492–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rosi MC, Luccarini I, Grossi C, Fiorentini A, Spillantini MG, Prisco A et al (2010) Increased Dickkopf‐1 expression in transgenic mouse models of neurodegenerative disease. J Neurochem 112:1539–1551. [DOI] [PubMed] [Google Scholar]
- 47. Santuccione AC, Merlini M, Shetty A, Tackenberg C, Bali J, Ferretti MT et al (2013) Active vaccination with ankyrin G reduces beta‐amyloid pathology in APP transgenic mice. Mol Psychiatry 18:358–368. [DOI] [PubMed] [Google Scholar]
- 48. Snyder SW, Ladror US, Wade WS, Wang GT, Barrett LW, Matayoshi ED et al (1994) Amyloid‐beta aggregation: Selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys J 67:1216–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suzuki N, Iwatsubo T, Odaka A, Ishibashi Y, Kitada C, Ihara Y (1994) High tissue content of soluble beta‐1‐40 is linked to cerebral amyloid angiopathy. Am J Pathol 145:452–460. [PMC free article] [PubMed] [Google Scholar]
- 50. Thambisetty M, An Y, Kinsey A, Koka D, Saleem M, Guntert A et al (2012) Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. Neuroimage 59:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y et al (2010) Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 67:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thangavel R, Sahu SK, Van Hoesen GW, Zaheer A (2009) Loss of nonphosphorylated neurofilament immunoreactivity in temporal cortical areas in Alzheimer's disease. Neuroscience 160:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo . Nature 416:535–539. [DOI] [PubMed] [Google Scholar]
- 54. Wilson MR, Yerbury JJ, Poon S (2008) Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol Biosyst 4:42–52. [DOI] [PubMed] [Google Scholar]
- 55. Wojtas AM, Kang SS, Olley BM, Gatherer M, Shinohara M, Lozano PA et al (2017) Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc Natl Acad Sci U S A 114:E6962–E6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yerbury JJ, Poon S, Meehan S, Thompson B, Kumita JR, Dobson CM, Wilson MR (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J 21:2312–2322. [DOI] [PubMed] [Google Scholar]
- 57. Zlokovic BV, Martel CL, Mackic JB, Matsubara E, Wisniewski T, McComb JG et al (1994) Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloid beta. Biochem Biophys Res Commun 205:1431–1437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expressions of mRNA and protein of DKK‐1 in the brains of 5‐month‐old wild‐type (WT), and 5‐ or 10‐month‐old 5×FAD mice.
Figure S2. Relative proportions of various pools of Aß proteins in the brains of clu +/+;5×FAD and clu −/−;5×FAD mice.