

Abstract
Astroblastoma is a rare, enigmatic tumor of the central nervous system (CNS) which shares some clinicopathologic aspects with other CNS tumors, especially ependymoma. To further clarify the nature of astroblastoma, we performed clinicopathologic and molecular genetic studies on eight cases of astroblastoma. The median age of the patients was 14.5 years, ranging from 5 to 60 years, and seven of the patients were female. All tumors arose in the cerebral hemisphere and radiologically appeared to be well‐bordered, nodular tumors often associated with cystic areas and contrast‐enhancement. Six of the seven patients with prognosis data survived without recurrences during the follow‐up periods ranging from six to 76 months. One patient had multiple recurrences and died six years later. All tumors exhibited salient microscopic features, such as being well demarcated from the surrounding brain tissue, perivascular arrangement of epithelioid tumor cells (represented by “astroblastic” pseudorosettes, trabecular alignment, and pseudopapillary patterns), and hyalinized blood vessels. Immunoreactivity for GFAP, S‐100 protein, Olig2, and EMA was variably demonstrated in all tumors, and IDH1 R132H and L1CAM were negative. Array comparative genomic hybridization revealed numerous heterozygous deletions on chromosome X in the four tumors studied, and break‐apart fluorescence in situ hybridization demonstrated rearrangement of MN1 in five tumors with successful testing. The characteristic clinicopathologic and genetic findings support the idea that astroblastoma is distinct from other CNS tumors, in particular, ependymoma. In addition, MN1 rearrangement and aberrations of chromosome X may partly be involved in the pathogenesis of astroblastoma.
Keywords: astroblastoma, chromosome X, MN1
Introduction
Astroblastoma is a rare glial neoplasm included in the category of “other gliomas,” in addition to chordoid glioma of the third ventricle and angiocentric glioma, in the 2016 WHO Classification of Tumours of the Central Nervous System (CNS) 1. This enigmatic tumor preferentially arises in the cerebral hemispheres of children, adolescents, and young adults, and is characterized by astroblastic pseudorosettes (perivascular arrangement of GFAP‐positive glial cells with broad processes) and vascular hyalinization.
Bailey and Cushing first coined the term “astroblastoma” in 1926 4, and Bailey and Bucy reported the first series of 25 cases in 1930 3. However, the definition of this rare tumor has been obscure and their series may have contained other glial tumors 6, 7, 11, 24. Therefore, there has been skepticism about the existence of astroblastoma 39. Bonnin and Rubinstein in 1989 redefined the clinicopathologic characteristics of astroblastoma, which were almost compatible with those of the current criteria, and subdivide their series into low‐grade and high‐grade types 5. In 2000, Brat et al. also analyzed the clinicopathologic features of astroblastoma with a study of 20 cases and revealed chromosomal alterations, such as gains of 20q and 10 and losses of 9q, 10, and X by conventional comparative genomic hybridization (CGH) 7.
Even after the publications of Bonnin and Rubinstein and Brat et al 5, 7, there still remains some controversies about astroblastoma 18. First, the clinicopathologic features of astroblastoma overlap with those of ependymoma—both tumors arise in young patients and share some pathologic features, such as well‐circumscribed tumor formation, the presence of perivascular pseudorosettes, and GFAP‐immunoreactivity 7. Second, astroblastic pseudorosettes are not always specific to astroblastoma and are sometimes observed in other glial tumors including anaplastic astrocytoma and glioblastoma 11, 18, 26. Therefore, differential diagnosis may be challenging between astroblastoma and other glial tumors, especially ependymomas 8. The characterization of specific genetic aberrations may be necessary to reveal the distinctiveness of astroblastoma.
Recently, a comprehensive molecular genetic analysis on CNS primitive neuroectodermal tumor (PNET) revealed a unique subset of tumors with a characteristic genetic abnormality, MN1 alteration 30. Interestingly, these genetically defined tumors exhibited astroblastoma‐like clinicopathologic features. In the present study, we investigated the clinicopathologic features and molecular profiles of astroblastoma of eight cases in order to confirm the distinctiveness of astroblastoma from other glial neoplasms, especially ependymomas, and demonstrated that X chromosomal abnormalities and the rearrangement of MN1 gene may partly be involved in the genesis of astroblastoma.
Materials and Methods
Materials
Eight cases, microscopic features of which meet the diagnostic criteria of astroblastoma defined by the 2016 WHO CNS tumor classification 1, were selected from the pathology files of the institutes of the authors. Two (Cases 3 and 6) have already been reported elsewhere 9, 38. This research was approved by the Ethics Committee of Gunma University.
Methods
Clinical study
Clinical data and images were obtained from the charts and the neurosurgeons of each hospital.
Light microscopy
Several known microscopic features of astroblastoma were histologically evaluated. These characteristics included the margin between tumors and surrounding tissue, tumor cell arrangement around the vessels, perivascular hyalinization and degenerative changes 1, 5, 7. Tumors were divided into low‐grade and high‐grade based on the following criteria: tumors with cellular atypia and a high‐proliferative activity—5 or more mitoses/10 high power fields (HPF) and/or 20% or more labeling indices (LI) of Ki‐67 were classified as high‐grade, and tumors composed of bland, uniform cells with low‐proliferative activity (less than 5 mitoses/10HPF and less than 20% LI of Ki‐67) were classified as low‐grade.
Immunohistochemistry
Immunostainings were performed on formalin‐fixed and paraffin‐embedded (FFPE) tissue sections, using an automated immunohistochemical slide staining system (BenchMark XT, Ventana Medical Systems, Tucson, AZ, USA). Primary antibodies used were as follows: glial fibrillary acidic protein (GFAP) (clone 6F2, 1:100, M0671, Dako, Agilent Technologies, Santa Clara, CA, USA), S‐100 protein (polyclonal, 1:2,000, Z0311, Dako), Olig2 (polyclonal, 1:500, 18953, IBL, Fujioka, Gunma, Japan), epithelial membrane antigen (EMA) (clone E29, 1:2,000, M0613, Dako), podoplanin (clone D2‐40, 1:50, M3619, Dako), IDH1 R132H (clone H09, 1:40, DIA H09, Dianova, Hamburg, Germany), CD34 (clone QBEND‐10, 1:100, M7165, Dako), L1CAM (clone UJ127.11, 1:100, L4543, Sigma‐Aldrich, St. Louis, MO, USA), and Ki‐67 (clone MIB1, 1:50, M7240, Dako). GFAP, S‐100 protein, Olig2, EMA, and Ki‐67 were studied in all tumors, but CD34, IDH1, podoplanin, and L1CAM were studied in seven, seven, six, and six tumors, respectively (Table 2). Results of immunostaining were semi‐quantitatively scored as follows: negative (positive cells, 0%), 1+ (1–25%), 2+ (26–50%), and 3+ (51–100%). Ki‐67 labeling indices were evaluated at the hot spots, using a free application software (Gunma LI, Gunma University, Japan) 33.
Table 2.
Immunohistochemical profiles, proliferative activity, grades, and FISH status of astroblastomas.
| Case | GFAP | S‐100 | Olig2 | EMA | CD34 | IDH1 | Podoplanin | L1CAM | Ki‐67 (%) | Mitosis | Grade | MN1 FISH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2+ | 2+ | 3+ | 1+ | – | 3+ | – | 2 | 2 | Low |
Fused red/green: 1 Red: 1 Green: 1 |
|
| 2 | 1+ | 3+ | 3+ | 1+ | – | – | 3+ | – | 2 | 1 | Low |
Fused red/green: 1 Red: 2 Green: 1 |
| 3 | 1+ | 2+ | 2+ | 3+ | – | – | 2+ | – | 11 | 1–2 | Low |
Fused red/green: 1 Red: 1 Green: 1 |
| 4 | 1+ | 3+ | – | 1+ | – | – | 1+ | – | 17 | 7 | High |
Fused red/green: 1 Red: 1 Green: 0 |
| 5 | 1+ | 1+ | – | 3+ | – | – | 1+ | – | 30 | 10 | High | NA |
| 6 | 2+ | 3+ | 2+ | 1+ | ND | ND | 1+ | ND | 57 | 10 | High | ND |
| 7 | 3+ | 3+ | 3+ | 3+ | – | – | ND | ND | 16 | 6 | High | NA |
| 8 | 1+ | 1+ | 3+ | 2+ | – | – | ND | – | 24 | 8 | High |
Fused red/green: 3 Red: 1–3 Green: 1–3 |
Abbreviations: FISH = fluorescence in situ hybridization; GFAP = glial fibrillary acidic protein; EMA = epithelial membrane antigen; ND = not done; NA = data not available.
Array CGH
DNA was extracted from FFPE tissue sections, as previously described 20. Array CGH analysis was carried out using a 4 × 180K CGH oligonucleotide microarray (Agilent Technologies) as described previously 20. The sizes of gains and losses were refined by manual inspection of probe intensity plots. The log2 ratio of <–1.0 at the region of interest represented homozygous deletion, and a value of −1.0 to −0.2 represented heterozygous deletion 32.
Fluorescence in situ hybridization analysis
Dual‐probe hybridization using an intermittent microwave irradiation method was applied to 4 μm thick FFPE tissue sections, as described previously 36. MN1 break‐apart FISH probes were prepared from bacterial artificial chromosome (BAC) clones RP11‐72G21 and RP11‐432I9 labeled with ENZO Orange‐dUTP and ENZO Green‐dUTP (Abbott Molecular Inc., Des Plaines, IL, USA), respectively, with the former encompassing exon 1 of MN1, which represents most of its coding region. Metaphase FISH to verify clone mapping positions was performed using peripheral blood cell cultures of a healthy donor. Signals were scored in at least 100 non‐overlapping, intact nuclei. A cutoff of >20% nuclei with abnormal signal was considered positive for rearrangement.
Direct DNA sequencing for the BRAF mutation
Genomic DNA extracted from FFPE sections was amplified and sequenced using the primers described previously 28. PCR products were sequenced on a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with the Big Dye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems) following standard procedures.
Results
Clinical findings
The clinical features of each case are shown in Table 1. There were seven females and one male. Their ages ranged from 5 to 60 years (median, 14.5 years; mean, 20.9 years). All tumors affected cerebral hemispheres: frontal lobe (5 cases), occipital 2, and parietal 1. The representative symptoms were headache, convulsions, nausea, vomiting, and paralyses.
Table 1.
Clinical data of astroblastomas.
| Case | Age | Sex | Location | Symptoms | Images | Treatment | Prognosis |
|---|---|---|---|---|---|---|---|
| 1 | 6 | F | Lt. frontal | Generalized convulsion | Well‐demarcated, contrast‐enhancing tumor with calcified foci and peripheral edema; 3.7 × 2.7 × 2.5 cm; low on T1 and low on T2 | Gross total resection | NED (3 years and 3 months) |
| 2 | 6 | F | Occipital | Headache | Well‐demarcated, solid tumor showing contrast enhancement and edema | Gross total resection | NED (6 years and 4 months) |
| 3 * | 18 | F | Rt. frontal | Headache, nausea | Well‐demarcated, solid and cystic tumor with enhancement and little edema; low to iso on T1 and low to iso on T2 | Gross total resection | NED (6 months) |
| 4 | 24 | F | Lt. frontal | Muscle weakness of rt. side, partial seizures of rt. upper arm | Well‐demarcated tumor with contrast enhancement; low on T1 and low and high on T2 | Gross total resection, radiation (50Gy) | NED (5 years) |
| 5 | 5 | F | Lt. parietal | Headache, nausea, rt. hemiparesis | Well‐demarcated, lobular tumor with cystic component | Excision, chemoradiotherapy for recurrent tumors | Recurrences (9 times), DOD (6 years later) |
| 6 | 11 | M | Lt. frontal | No data | Multicystic tumor | Gross total resection | No data |
| 7 † | 60 | F | Lt. frontal | Muscle weakness of lt. extremities | Well‐demarcated, lobulated tumor containing small cysts | Gross total resection | NED (2 years) |
| 8 | 37 | F | Lt. occipital | Headache, vomiting | Ill‐defined, contrast‐enhancing tumor with mild edema; low on T1 and low on T2 | Gross total resection, chemoradiotherapy | NED (2 years) |
Imaging features
All eight tumors studied had similar radiographic findings. The tumors were well‐demarcated, lobular or nodular, and were superficially located in the cerebral hemisphere (Figure 1). Four of eight possessed cystic components and one had calcified foci (Table 1). The tumors had low‐signal intensity on T1‐weighed images and low‐to‐high intensity on T2 by MRI. Most of them were enhanced with gadolinium administration. Some tumors were associated with mild edema in the surrounding areas.
Figure 1.
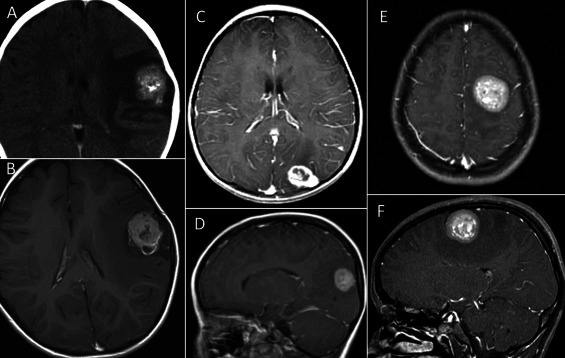
Radiologic images of astroblastomas. Astroblastomas are generally located in the superficial portion of the cerebral hemispheres (A–F). They are well‐demarcated, contrast‐enhancing tumors often associated with mild edema, cystic components (B), and calcification (A). (A, B: Case 1; C, D: Case 2; E, F: Case 4; A, computed tomography; B–F, magnetic resonance imaging).
Treatment and prognosis
Gross total resection was performed in all patients but one. In addition, adjuvant radiation and chemotherapy were performed in three and two patients, respectively. Follow‐up data could be obtained from seven cases. During the follow‐up periods, ranging from 6 to 76 months (median, 39 months; mean, 43 months), all patients but one were alive without recurrences or metastases (Table 1). One patient died of the disease six years after the onset, and had nine recurrences in spite of repeated surgeries (seven times), radiation, and chemotherapy.
Light microscopy
Most surgical samples were removed en bloc instead of in small pieces. All eight tumors were well‐circumscribed from surrounding brain parenchyma without infiltrative borders (Figure 2A). All tumors had characteristic perivascular arrangement of tumor cells, which appeared to be perivascular (astroblastic) pseudorosettes, trabecular/cord‐like alignment and pseudopapillary patterns depending on the direction of tissue section and cleft artifacts (Figure 2B–D). The vasculatures were well‐developed and their walls were variously associated with fibrosis. In some tumors and areas, the perivascular fibrosis was so extensive that the tumor cells were degenerated and replaced by densely hyalinized stroma (Figure 2E). There also were some degenerative changes, such as calcification (2 cases) and the formation of cholesterol crystals (2 cases) (Figure 2F). Perivascular pseudorosettes and hyalinization were salient microscopic features found in all tumors, but some tumors contained areas composed of a sheet‐like growth of monomorphic tumor cells devoid of hyalinized vessels. In addition, small cystic areas were observed in two tumors.
Figure 2.
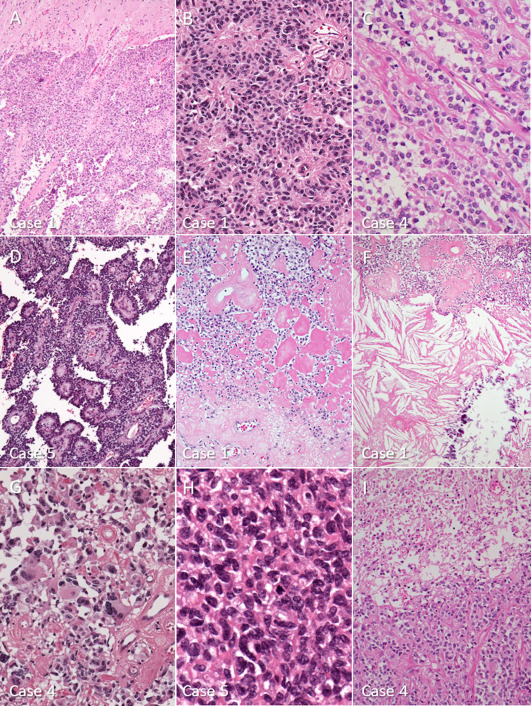
Microscopic features of astroblastomas. A. a well‐circumscribed border between tumor tissue (lower part) and brain parenchyma (upper part). Case 1. B. perivascular arrangement of columnar tumor cells exhibiting characteristic astroblastic pseudorosettes. Case 1. C. trabecular or cord‐like alignment of cuboidal tumor cells beside parallel arrays of fibrous connective tissue. Case 4. D. conspicuous pseudopapillary patterns, created by the cleavage between perivascular tumor arrangements. Case 5. E. thick fibrosis around the vessels and densely hyalinized areas replacing tumor cells. Case 1. F. degenerative changes, such as cholesterol crystals and calcification, are sometimes seen. Case 1. G. pleomorphic tumor cells, focally seen in some tumors, do not necessarily indicate malignancy. Case 4. H. high‐grade astroblastoma showing anaplasia and mitotic features. Case 5. I. necrosis (upper part) seen in a high‐grade astroblastoma. Case 4.
Tumor cells were relatively uniform in size, but varied in shape, ranging from cuboidal, columnar, and epithelioid to short‐spindle. They possessed ovoid, irregular nuclei with occasional indentations and pseudoinclusions, and pale to eosinophilic cytoplasm. Tumor cells extended their monopolar, broad processes to vessels, forming typical astroblastic pseudorosette (Figure 2B), but lacked fibrillary processes. Some pleomorphic cells including multinucleated cells, were found in two tumors (Figure. 2G).
In the present study, the eight astroblastomas were subdivided into three low‐grade and five high‐grade tumors. In the high‐grade group, tumor cells had hyperchromatic, atypical nuclei, and scant cytoplasm. Mitotic counts from the high‐grade tumors were 6–10/10HPF (Figure 2H) and Ki‐67 LI were 16–57% (Table 2). Necrosis and microvascular proliferation (MVP) were noted in three tumors and one tumor, respectively (Figure 2I). Pleomorphism was observed in one tumor. On the other hand, mitotic counts from low‐grade tumors were 1‐2/10 HPF, and K‐67 labeling indices were 2%, 2%, and 11%. Nuclear atypia was mild and one tumor exhibited nuclear pleomorphism. Necrotic areas were also noted in one tumor of the low‐grade group.
Immunohistochemistry
Astroblastomas showed varied immunoreactivity for GFAP, S‐100 protein, Olig2, EMA, and podoplanin, and were negative for IDH1 R132H, CD34, and L1CAM (Table 2).
GFAP and S‐100 protein were confirmed to be positive in all tumors. GFAP‐positive cells, however, were less conspicuous and scored (1+) in five tumors, (2+) in two and (3+) only in one (Figure 3A,B). On the other hand, S‐100 protein‐positive cells were more numerous, scored as (3+) in four tumors (Figure 3C). Olig2‐immunoreactivity was detected in six tumors and the number of positive cells was more than those of GFAP (Figure 3D).
Figure 3.
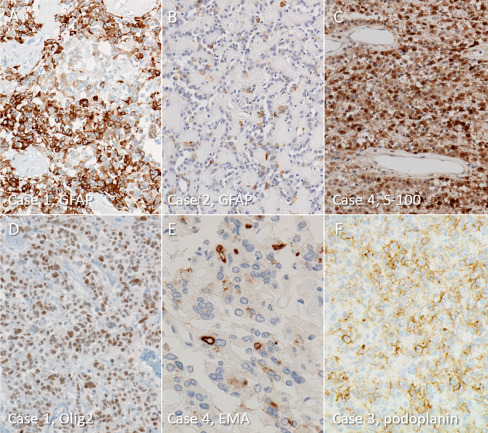
Immunohistochemical findings of astroblastomas. A. a large number of GFAP‐positive tumor cells are present in only one tumor. Case 1. B. five tumors contain only scattered GFAP‐positive cells, which makes diagnosis challenging. Case 2. C. most astroblastomas have diffuse immunoreactivity for S‐100 protein. Case 4. D. many tumor cells express intranuclear Olig2‐immunoreactivity. Case 1. E. dot‐like and small ring‐like positivity for EMA is occasionally noted. Case 4. F. membranous and small dot‐like immunoreactivity for podoplanin is also seen in astroblastomas. Case 3.
EMA and podoplanin were also positive in all tumors studied: EMA in 8/8 and podoplanin in 6/6 cases. Positive cells were primarily immunoreactive on membranes with an occasional intracytoplasmic dot‐like pattern (Figure 3E,F).
Array CGH
Cases with sufficient quality and quantity of DNA (Cases 1, 3, 4, and 8) were analyzed by array CGH. The results are shown in Figure 4, Supporting Information Tables S1 and S2. The array CGH demonstrated numerous, small and large, heterozygous deletions in the X chromosomes of all four tumors studied (Figure 4A, Supporting Information Table S2). The deletions were distributed throughout both the short and long arms of the X chromosome, and oscillating copy‐number states were compatible with chromothripsis (Figure 4A). In addition, all four cases had multiple deletions (Cases 1, 3, and 4) or gains (Case 8) near the MN1 locus on 22q12.1, in which focal chromothripsis might also be involved (Figure 4B, Supporting Information Table S1). There were several other recurrent abnormalities in autosomal chromosomes including heterozygous deletions of 1p36.22 (Cases 4, 8) and 14q (Cases 4, 8). On the other hand, no evidence of chromothripsis involving chromosome 11q13.1 was observed, a characteristic feature of supratentorial ependymomas with C11orf95‐RELA fusions 21.
Figure 4.

Representative images of comparative genomic hybridization. A. numerous heterozygous deletions are observed throughout both the short and long arms of the X chromosome. The oscillating copy‐number patterns are compatible with chromothripsis. The arrows indicate the position of BEND2. B. Multiple deletions (Cases 1 and 3) or gains (Case 8) are seen near the MN1 locus on 22q12.1, in which focal chromothripsis may also be involved. The arrows indicate the position of MN1.
FISH analysis for MN1 rearrangement
The results are summarized in Table 2. All cases but Case 6 were tested. Although the FISH probe did not hybridize in Cases 5 and 7, the assay was successful in the remaining five cases. All harbored MN1 gene rearrangement using break‐apart FISH (Table 2, Figure 5A–D). Four different patterns of abnormal signals were observed. Cases 1 and 3 showed a classic split pattern—a combination of one fused red/green (yellow) signal, one red signal, and one green signal (Figure 5A). One fused red/green signal, two red signals, and one green signal were observed in tumor cells of Case 2 (Figure 5B). In Case 4, one fused red/green signal and one isolated red signal were seen, with isolated green signals lost (Figure 5C). Most tumor cells in Case 8 showed three fused red/green signals, one red signal, and one green signal, and some cells showed one or two additional red and/or green signals (Figure 5D,E).
Figure 5.
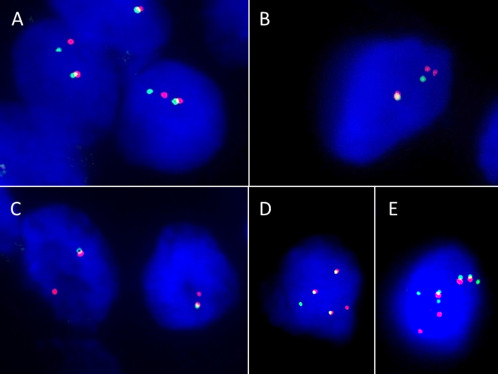
FISH results for MN1 rearrangement of astroblastomas. A. a classic split pattern, a combination of one fused red/green signal, one red signal, and one green signal is observed in Case 3. B. Case 2 shows one fused red/green (yellow) signal, two red signals, and one green signal. C. in Case 4, one fused red/green signal and one isolated red signal are seen, and isolated green signals are lost. D and E. most tumor cells in Case 8 show three fused red/green signals, one red signal, and one green signal (D), and some cells show one or two additional red and/or green signals (E).
Direct DNA sequencing for the BRAF mutation
Cases 1–4, and 8 were tested for mutation in exon 15 of BRAF, and no mutation was observed in any of the cases.
Discussion
The clinical features of the eight cases collected based on the current criteria of astroblastoma were similar to those of the cases reported by Bonnin and Rubinstein 5, Brat et al 7, and others 2, 17, 19, 34. All patients, but one, were children or young adults. Female preponderance (female:male = 7:1) was also confirmed by the present data. Most astroblastomas reported, including in this report, affected the cerebral hemispheres and the most common site was the frontal lobe followed by the occipital lobe 7. Astroblastomas appeared to be well‐demarcated tumors associated with cyst and enhancement in the superficial portion of the cerebral hemisphere 7. A bubbly appearance in the solid component on MRI was reported to be characteristic of this rare tumor 23.
Salient microscopic features of astroblastoma are astroblastic pseudorosettes and perivascular hyalinization, and the tumor is well‐circumscribed from the surrounding brain tissue 1, 5, 7, 8. Astroblastic pseudorosettes are emphasized as a diagnostic criterion, but they may vary between areas and cases, and are sometimes indiscernible. Trabecular arrangement of cuboidal or columnar tumor cells or pseudopapillary patterns may represent other aspects of pseudorosettes. Perivascular fibrosis and hyalinization are prominent in this tumor, but may also vary. Tumor cells are cuboidal or columnar in shape and lack fibrillary processes, as suggested by Brat et al 7.
Immunohistochemical profiles of astroblastomas corresponded to those of glial neoplasms ‐ immunoreactive for GFAP, S‐100 protein, and Olig2. All tumors studied were positive for GFAP, but as stated in previous studies, reactivity varied greatly 5. On the other hand, S‐100 protein immunostainings may show more positive tumor cells than that of GFAP 24. The present study demonstrated Olig2‐positive tumor cells in most cases, as reported by Lehman et al 17. Furthermore, in the present study no astroblastomas expressed immunoreactivity for L1CAM, which has been reported to be positive in most supratentorial ependymomas with C11orf95‐RELA fusion 22.
The most interesting finding of the present molecular analyses is the numerous heterozygous deletions in the X chromosome, which were detected by array CGH in the four tumors studied. The wide distribution of deletions found in almost the entire X chromosome may suggest that chromothripsis is involved in the pathogenesis of the aberrations 29. Brat et al. reported loss of X chromosome in two of seven tumors with conventional CGH analysis 7. In addition, multiple deletions (Cases 1, 3, and 4) or gains (Case 8) near the MN1 locus on 22q12.1 were observed in the present study, and the pattern of these copy‐number changes is reminiscent of focal chromothripsis. The present array CGH results were different from the aberrations reported by Brat et al 7 and Jay et al 14. The chromosomal alterations Brat et al. reported were 20q gain (4 of 7), 19 gain 3, 9q loss 2, and 10 loss 2, 7. Those of Jay et al. were losses of chromosomes 10, 21, and 22 14. Compared with array CGH, conventional CGH and classical cytogenetic analyses are less sensitive and less specific. Thus, the methodological differences used may be related to the discordance between the data reported and ours.
Another important genetic aberration was MN1 rearrangement demonstrated by FISH in five astroblastomas with successful hybridization in the present study. Four of five also had aberrations of the X chromosome. Recently, MN1 rearrangement was found in a small population of CNS embryonal tumors 30. Based on comprehensive genetic analyses, Sturm et al. found four new, unrecognized tumor types in so‐called CNS‐PNETs: CNS neuroblastoma with FOXR2 activation, CNS Ewing sarcoma family tumor with CIC alteration, CNS high‐grade neuroepithelial tumor with MN1 alteration (CNS HGNET‐MN1), and CNS high‐grade neuroepithelial tumor with BCOR alteration 30. Further characterization of the HGNET‐MN1 tumors revealed that about 40% exhibited features compatible with astroblastomas as follows: female predominance, occurrence in older children or young adults, and microscopic characteristics including pseudopapillary patterns and perivascular hyalinization. The rest (60%) of the HGNET‐MN1 tumors were histologically classified as CNS‐PNET, ependymoma, embryonal tumor with multilayered rosettes (ETMR), and so on. RNA sequencing demonstrated two specific fusion genes in the HGNET‐MN1: MN1‐BEND2 in three tumors and MN1‐CXXC5 in one. Interestingly, BEND2 is located on Xp22.13. Considering the data presented here, chromothripsis of the X chromosome may be involved in the formation of the MN1‐BEND2 fusion gene. Although the number of tumors (four) with MN1 rearrangement and X chromosomal aberrations was too small to reach indisputable conclusions, MN1 rearrangement with fusion partner genes, such as BEND2 and, less frequently, CXXC5, and other unidentified genes, may drive genetic alteration of astroblastomas 30. More recently, Wood et al. also reported MN1 alteration in four of eight CNS tumors that exhibited astroblastoma‐like morphology 35.
The present FISH analysis demonstrated four different patterns of split signals of MN1. In the original report by Sturm et al., FISH showing MN1 rearrangement exhibited one fused red/green signal, two or three red signals, and two green signals 30, which was not observed in the present study. Different FISH patterns may exist in MN1 rearrangement as in other rearranged genes such as ALK and ROS1 10, 37.
Recently, Lehman et al. reported that the BRAF V600E mutation was found in 8 of 21 astroblastomas (38%) in a study in which MN1 rearrangement was not investigated 17. The present study showed that all five cases tested did not harbor the mutation. There seems to be no reasonable explanation for the differences between the data of Lehman et al. and ours. Astroblastic pseudorosettes, however, are rather non‐specific structures, and they may occasionally be observed in other high‐grade gliomas including epithelioid glioblastoma, which often has the BRAF mutation. More cases need to be assessed to clarify whether the BRAF V600E mutation is involved in the pathogenesis of astroblastomas.
Because of microscopic similarities, such as perivascular pseudorosettes, papillary patterns and fibrosis, astroblastomas should be distinguished from several mimickers including ependymoma and other infiltrating gliomas 8. In particular, ependymomas are the most important for differential diagnosis, in that, as stated previously, both tumors share several clinicopathological features. Ependymomas, however, have fibrillary cell processes surrounding the vasculature, instead of stub‐like cell processes and generally lack perivascular fibrosis/sclerosis 8. GFAP is diffusely positive in perivascular “acellular” zones and Olig2 is usually negative in ependymomas 12. In addition, two‐thirds of supratentorial ependymomas exhibit L1CAM immunoreactivity 21. Because whole genome sequencing or RNA sequencing failed to show MN1 rearrangement, genetic aberrations of ependymomas seem to be different from the data shown in the present study 21, 22. Therefore, astroblastoma is considered to be distinct from ependymoma in both clinicopathologic and genetic aspects, but there may be some relationship in cellular differentiation and histogenesis 2, 8, 9, 16, 25.
Astroblastomas are generally divided into low‐grade (well‐differentiated) and high‐grade (malignant) subtypes 2, 5, 7, 17, 19, 34, although WHO grades are not provided 1. The present cases also could be divided into three low‐grade and five high‐grade tumors based on atypical features and proliferative activities. Similarly, Bonnin and Rubinstein designated the high‐grade type as astroblastomas associated with cellular atypia, compact cellularity, high mitotic rate and vascular endothelial hyperplasia 5. In the series by Brat et al., malignant astroblastomas had more than 5 mitoses/10HPF and palisading necrosis 7. Astroblastomas with high‐grade (malignant) histology tend to be associated with a high rate of recurrence and aggressive biological behaviors, but some patients may have long survival 5, 34. However, recurrences were also reported in low‐grade astrocytomas 2, 13, 15, 27. In the present study, six of seven patients survived without recurrence or metastasis during the follow‐up periods with a mean of 43 months. One patient had multiple recurrences and died six years later. Considering that the tumors were well‐demarcated from the surrounding tissue, gross total resection may be feasible for astroblastomas and, therefore, a favorable prognosis can be expected irrespective of microscopic grade 31. It may be interesting to know the biological behavior of HGNET‐MN1 reported by Sturm et al. This tumor group is histologically heterogeneous, and includes astroblastoma, CNS‐PNET, ependymoma, ETMR, etc. Unfortunately, follow‐up data were not provided in the study 30. Further clinical studies based on the molecular profiles are mandatory to clarify the biological behavior of astroblastomas.
In summary, the present study confirmed that astroblastoma is a distinctive entity from ependymoma and other infiltrating gliomas. They have unique clinical, radiological, and pathologic features. Furthermore, aberrations of the X chromosome and MN1 rearrangement seem to be characteristic of, at least, some astroblastomas, which may be useful for diagnosis of this enigmatic tumor. To further clarify the biological behavior of astroblastomas, more studies of tumors diagnosed based on newly identified molecular markers are needed.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Table S1. Aberrations detected in autosomes by array comparative genomic hybridization (CGH).
Table S2. Aberrations detected in X chromosome by array comparative genomic hybridization (CGH).
Acknowledgments
This work was supported in part by the Collaborative Research Project (2015–2620) of the Brain Research Institute, Niigata University, Niigata, Japan. The authors thank Dr. Takashi Komori (Department of Clinical Laboratory, Tokyo Metropolitan Neurological Hospital), Dr. Junko Hirato (Department of Pathology, Gunma University Hospital), Dr. Makoto Shibuya (Department of Clinical Laboratory, Tokyo Medical University Hachioji Medical Center), Dr. Hiroyoshi Suzuki (Department of Diagnostic Pathology, Sendai Medical Center), and Dr. Hitoshi Takahashi (Department of Pathology, Brain Research Institute, Niigata University) for their valuable comments on this study. The authors have no conflicts of interest to declare.
References
- 1. Aldape KD, Rosenblum MK, Brat DJ (2016) Astroblastoma. In: WHO Classification of Tumours of the Central Nervous System, Revised 4th Edition. World Health Organization Classification of Tumours. Louis DN, Ohgaki H, Wiestler OD (eds), pp. 121–122. International Agency for Research on Cancer: Lyon. [Google Scholar]
- 2. Asha U, Mahadevan A, Sathiyabama D, Ravindra T, Sagar BKC, Bhat DI (2015) Lack of IDH1 mutation in astroblastomas suggests putative origin from ependymoglial cells? Neuropathology 35:303–311. [DOI] [PubMed] [Google Scholar]
- 3. Bailev P, Bucy P (1930) Astroblastomas of the brain. Acta Psychiat Neurol 5:439–461. [Google Scholar]
- 4. Bailey P, Cushing H (1926) A Classification of the Tumors of the Glioma Group on a Histogenetic Basis with a Correlated Study of Prognosis. Lippincott: Philadelphia, PA. [Google Scholar]
- 5. Bonnin JM, Rubinstein LJ (1989) Astroblastomas: a pathological study of 23 tumors, with a postoperative follow‐up in 13 patients. Neurosurgery 25:6–13. [PubMed] [Google Scholar]
- 6. Brat DJ (2010. ) Other glial neoplasms. In: Practical Surgical Neuropathology. A Diagnostic Approach. Perry A, Brad DJ (eds), pp. 361–369. Churchill Livingstone: Philadelphia, PA. [Google Scholar]
- 7. Brat DJ, Hirose Y, Cohen KJ, Feuerstein BG, Burger PC (2006) Astroblastoma: clinicopathologic features and chromosomal abnormalities defined by comparative genomic hybridization. Brain Pathol 10:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burger PC, Scheithauer BW (2007) Astroblastoma. In: Tumors of the Central Nervous System. AFIP Atlas of Tumor Pathology, 4th Series, Fascicle 7, pp. 170–173. Washington, DC: American Registry of Pathology. [Google Scholar]
- 9. Fu YJ, Taniguchi Y, Takeuchi S (2012) Cerebral astroblastoma in an adult: an immunohistochemical, ultrastructural and genetic study. Neuropathology 33:312–319. [DOI] [PubMed] [Google Scholar]
- 10. Gao X, Sholl LM, Nishino M, Heng JC, Jänne PA, Oxnard GR et al (2015) Clinical implications of variant ALK FISH rearrangement patterns. J Thorac Oncol 10:1648–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Husain A, Leestma JE (1986) Cerebral astroblastoma: immunohistochemical and ultrastructural features. J Neurosurg 64:657–661. [DOI] [PubMed] [Google Scholar]
- 12. Ishizawa K, Komori T, Shimada S, Hirose T (2008) Olig2 and CD99 are useful negative markers for the diagnosis of brain tumors. Clin Neuropathol 27:118–128. [DOI] [PubMed] [Google Scholar]
- 13. Janz C, Buhl R (2014) Astroblastoma: report of two cases with unexpected clinical behavior and review of the literature. Clin Neurol Neurosurg 125:114–124. [DOI] [PubMed] [Google Scholar]
- 14. Jay V, Edwards V, Squire J, Rutka J (1993) Astroblastoma: report of a case with ultrastructural, cell kinetic, and cytogenetic analysis. Pediatr Pathol 13:323–332. [DOI] [PubMed] [Google Scholar]
- 15. Kaji M, Takeshima H, Nakazato Y, Kuratsu J‐i (2006) Low‐grade astroblastoma recurring with extensive invasion. Neurol Med Chir 46:450–454. [DOI] [PubMed] [Google Scholar]
- 16. Kubota T, Sato K, Arishima H, Takeuchi H, Kitai R, Nakagawa T et al (2006) Astroblastoma: immunohistochemical and ultrastructural study of distinctive epithelial and probable tanycytic differentiation. Neuropathology 26:72–81. [DOI] [PubMed] [Google Scholar]
- 17. Lehman NL, Hattab EM, Mobley BC, Usubalieva A, Schniederjan MJ, McLendon RE et al (2017) Morphological and molecular features of astroblastoma, including BRAFV600E mutations, suggest an ontological relationship to other cortical‐based gliomas of children and young adults. Neuro‐oncol 19:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mellai M, Piazzi A, Casalone C et al (2015) Astroblastoma: beside being a tumor entity, an occasional phenotype of astrocytic gliomas? Onco Targets Ther 8:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Navarro R, Reitman AJ, de León GA, Goldman S, Marymont M, Tomita T et al (2005) Astroblastoma in childhood: pathological and clinical analysis. Childs Nerv Syst 21:211–220. [DOI] [PubMed] [Google Scholar]
- 20. Nobusawa S, Lachuer J, Wierinckx A, Kim YH, Huang J, Legras C et al (2010) Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol 20:936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y et al (2014) C11orf95‐RELA fusions drive oncogenic NF‐κB signaling in ependymoma. Nature 506:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Port JD, Brat DJ, Burger PC, Pomper MG (2002) Astroblastoma: radiologic‐pathologic correlation and distinction from ependymoma. Am J Neuroradiol 23:243–247. [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenblum MK ( 2006) Astroblastoma. In: Russell and Rubinstein's Pathology of Tumors of the Nervous System, 7th edn. Hodder Arnold: London, pp. 157–165. [Google Scholar]
- 25. Rubinstein LJ, Herman MM (1989) The astroblastoma and its possible cytogenic relationship to the tanycyte. An electron microscopic, immunohistochemical, tissue‐ and organ‐culture study. Acta Neuropathol 78:472–483. [DOI] [PubMed] [Google Scholar]
- 26. Russell DS, Rubinstein LJ ( 1989) Pathology of Tumours of the Nervous System, 5th edn. Edward Arnold: London. [Google Scholar]
- 27. Salvati M, D'Elia A, Brogna C, Frati A, Antonelli M, Giangaspero F et al (2009) Cerebral astroblastoma: analysis of six cases and critical review of treatment options. J Neurooncol 93:369–378. [DOI] [PubMed] [Google Scholar]
- 28. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 29. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ et al (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 164:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sughrue ME, Choi Jay, Rutkowski MJ, Aranda D, Kane AJ, Barani IJ et al (2011) Clinical features and post‐surgical outcome of patients with astroblastoma. J Clin Neurosci 18:750–754. [DOI] [PubMed] [Google Scholar]
- 32. Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A et al (2005) Genome‐wide array‐based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 24:1348–1358. [DOI] [PubMed] [Google Scholar]
- 33. Tanaka G, Nakazato Y ( 2004) Automatic quantification of the MIB‐1 immunoreactivity in brain tumors. In: Developments in Neuroscience. Proceedings of the 3rd International Mt. Bandai Symposium for Neuroscience and the 4th Pan‐Pacific Neurosurgery Congress. International Congress Series, Watanabe K et al (eds), Vol. 1259, pp. 15–19. Elsevier B.V.: Amsterdam. doi: 10.1016/S0531-5131(03)01668-6. [DOI] [Google Scholar]
- 34. Thiessen B, Finlay J, Kulkarni R, Rosenblum MK (1998) Astroblastoma: does histology predict biologic behavior? J Neurooncol 40:59–65. [DOI] [PubMed] [Google Scholar]
- 35. Wood MD, Tihan T, Perry AJ et al (2017) Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol [Epub ahead of print; doi: 10.1111/bpa.12561]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yokoo H, Kinjo S, Hirato J et al (2006) Fluorescence in situ hybridization targeted for chromosome 1p of oligodendrogliomas (in Japanese). Rinsho Kensa 50:761–766. [Google Scholar]
- 37. Yoshida A, Kohno T, Tsuta K, Wakai S, Arai Y, Shimada Y et al (2013) ROS1‐rearranged lung cancer. A clinicopathologic and molecular study of 15 surgical cases. Am J Surg Pathol 37:554–562. [DOI] [PubMed] [Google Scholar]
- 38. Yuzawa S, Nishihara H, Tanino M, Kimura T, Moriya J, Kamoshima Y et al (2016) A case of cerebral astroblastoma with rhabdoid features: a cytological, histological, and immunohistochemical study. Brain Tumor Pathol 33:63–70. [DOI] [PubMed] [Google Scholar]
- 39. Zülch KJ (1965) Brain Tumors. Their Biology and Pathology, 2nd edn. Springer‐Verlag: New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Table S1. Aberrations detected in autosomes by array comparative genomic hybridization (CGH).
Table S2. Aberrations detected in X chromosome by array comparative genomic hybridization (CGH).