

Abstract
Aicardi–Goutières syndrome (AGS) is an early‐onset, autoimmune and genetically heterogeneous disorder with severe neurologic injury. Molecular studies have established that autosomal recessive mutations in one of the following genes are causative: TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1 and IFIH1/MDA5. The phenotypic presentation and pathophysiology of AGS is associated with over‐production of the cytokine Interferon–alpha (IFN‐α) and its downstream signaling, characterized as type I interferonopathy. Astrocytes are one of the major source of IFN in the central nervous system (CNS) and it is proposed that they could be key players in AGS pathology. Astrocytes are the most ubiquitous glial cell in the CNS and perform a number of crucial and complex functions ranging from formation of blood‐brain barrier, maintaining ionic homeostasis, metabolic support to synapse formation and elimination in healthy CNS. Involvement of astrocytic dysfunction in neurological diseases—Alexander's disease, Epilepsy, Alzheimer's and amyotrophic lateral sclerosis (ALS)—has been well‐established. It is now known that compromised astrocytic function can contribute to CNS abnormalities and severe neurodegeneration, nevertheless, its contribution in AGS is unclear. The current review discusses known molecular and cellular pathways for AGS mutations and how it stimulates IFN‐α signaling. We shed light on how astrocytes might be key players in the phenotypic presentations of AGS and emphasize the cell‐autonomous and non‐cell‐autonomous role of astrocytes. Understanding the contribution of astrocytes will help reveal mechanisms underlying interferonopathy and develop targeted astrocyte specific therapeutic treatments in AGS.
Keywords: Aicardi–Goutières syndrome, astrocytes, interferon, type I interferonopathy
Introduction
Aicardi–Goutières syndrome (AGS) is a genetic disorder, first described by Jean Aicardi and Françoise Goutières in 1984 in the context of work on congenital infections, when they reported eight infants from five different families with a progressive neurological disorder 1. Observations of familial cases further suggested a genetic disorder and ultimately several genes (TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1 and IFIH1/MDA5) were identified as causative of disease 6, 26, 43, 48, 63. AGS is characterized by early‐onset encephalopathy, progressively resulting in loss of motor and cognitive skills with severe intellectual disability 63. Onset of AGS can occur as early as birth with hepatosplenomegaly, elevated liver enzymes, abnormal neurologic findings and thrombocytopenia. Most affected individuals, however, show normal development before the onset of AGS symptoms, which includes a subacute encephalopathy with seizures, extreme irritability, sterile pyrexias, loss of achieved skills, spasticity, dystonia and slowing of head growth. Over time, disease manifestations include multisystem involvement, most prominently chilblain skin lesions, present on the fingers, toes and ears in over 40% of affected individuals 16. Neuroimaging shows calcification of basal ganglia predominantly in putamen, globus pallidus and thalamus along with white matter changes seen primarily in the frontotemporal region of the brain 1, 36, 43, 63. Cerebral and brain stem atrophy, bilateral striatal necrosis and intracerebral vasculopathy are some of the other distinguishable features observed using imaging studies. Most affected individuals present with symptoms within the first year of life, with a correlation of decreased survival in patients less than 5 years of age and the highest mortality rate associated with TREX1 patients 13.
Affected infants show abnormal cerebrospinal fluid (CSF) with sterile lymphocytosis 1 and elevated neopterin and tetrahydrobiopterin, suggesting auto‐inflammation that mimics the sequelae of congenital viral infection. IFN‐α levels are increased in serum and CSF, with the latter being higher 37. In fact, the presence of increased CSF IFN levels, neopterin and tetrahydrobiopterin without evidence of congenital infection should raise diagnostic consideration for AGS 14. In AGS, the innate immune response stimulates peripheral upregulation of IFN and downstream end‐organ damage, however questions remain about the source of IFN in the central nervous system (CNS). This review provides an overview of the genetic and cellular pathways contributing to AGS, with a focus on involvement of CNS glia particularly astrocytes.
Genetic and Molecular Mechanisms Underlying AGS
While the clinical manifestation of AGS have overlapping features, particularly the overproduction of IFN, AGS is a genetically heterogeneous disease. Crow and colleagues have identified mutations in seven genes—TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1 and IFIH1/MDA5, that contribute to AGS and provide a molecular basis for understanding the disease. The inheritance pattern of most of these mutations is autosomal recessive leading to loss of function; however, there are rare de novo autosomal dominant mutations, in particular with gain of function such as IFIH1 mutations. We discuss below the function of each of the gene, downstream signaling pathways and manifestation of disease due to their genetic aberrations, also represented in Figure 1.
Figure 1.
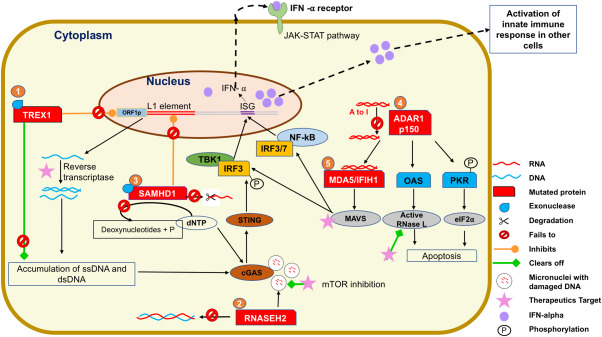
Molecular pathways and activation of innate immune signaling pathway in AGS. 1 In AGS individuals, the mutated form of TREX1 loses its ability to inhibit expression of the protein ORF1P which is essential for assembly of the retro element L1 and hence mutant TREX1 cannot block the L1 activity. Additionally, altered TREX1 loses 3′ exonuclease activity and fails to degrade products of reverse transcriptase single‐stranded (ss) and double‐stranded (ds) DNA, which starts accumulating and stimulates the innate immune system via cGAS‐STING pathway. This leads to serine‐threonine protein kinase (TBK1) mediated phosphorylation of IFN regulator 3 (IRF3) triggering Interferon stimulated genes (ISGs) to produce IFN‐α. Targeting reverse transcriptase could be a good therapeutic target for TREX1 AGS individuals. 2 RNASEH2, a complex of RNASEH2A, RNASEH2B and RNASEH2C degrades RNA : DNA hybrid. However, in AGS individuals, the exact mechanism of how it induces IFN‐α stimulation is unknown. Rnaseh2b deficient cells accumulate damaged cytosolic DNA aggregates in form of micronuclei. These micronuclei stimulate expression of ISGs via binding to cGAS. This mechanism could be operating in AGS individuals as it is believed that there is presence of endogenous damaged DNA molecules. Additionally, micronuclei associated with Rnaseh2b deficiency were cleared by induction of autophagy through pharmacological inhibition of the mechanistic target of rapamycin (mTOR) pathway and could be a feasible therapeutic option for RNASEH2B‐dependent disease. 3 In SAMHD1 AGS individuals, a likely source of endogenous nucleic acid could be due to failure to regulate L1 retro transposition. Moreover, mutated SAMHD1 cannot clear the dNTPs and ssRNA resulting in activation of IFN‐α via cGAS‐STING pathway. 4 In ADAR1 mutants, there is an accumulation of dsRNA and this dsRNA binds to MDA5/IFIH1 and activates the MAVS pathway further triggering IFN‐α production. In addition, the anti‐viral pathways PKR and OAS‐RNAse L system are also activated in the absence of ADAR1 editing. 5 MDA5/IFIH1 mutation is another gene in AGS individuals which causes increase of IFN‐α production by MAVS pathway. Targeting of the downstream signaling MDA5/MAVS pathway could also be a promising therapeutic option in AGS individuals with ADAR1 and MDA5 mutation.
TREX1
Three‐prime repair exonuclease 1 (TREX1) is the best studied AGS causative gene. TREX1 functions as an editor for DNA polymerase and plays a crucial role in DNA repair mechanism 9, 10, 47. TREX1 mutations causing AGS are generally autosomal recessive 62; however, there are reports of autosomal dominant heterozygous TREX1 mutations 62. When TREX1 was first identified, Rice et al proposed that this enzyme is involved in the DNA processing pathway to cleave and clear DNA from the cell. They also proposed that mutations in this enzyme can result in accumulation of intracellular DNA and its intermediates, which are sensed as foreign material triggering an inflammatory immune response 14. Yang et al verified this hypothesis and provided evidence for accumulation of ssDNA in Trex1 null mouse fibroblasts 76. Subsequent studies showed that TREX1 is not only a DNA exonuclease, but also functions as an exoribonuclease, indicating that mutations in TREX1 can also lead to accumulation of RNA along with DNA 77.
Mouse models were developed for AGS to examine the role of Trex1 in CNS inflammation. Trex1 knock‐out mice have a reduced lifespan with a median age of 6 months and displayed a cardiac inflammatory response along with autoimmune activation. These mice failed to show the CNS inflammation seen in AGS individuals 50. Trex1 D18N knock‐in mice similarly manifested systemic inflammation, lymphoid hyperplasia, vasculitis and kidney abnormalities with detection of immune complexes and autoantibodies in corresponding tissues but did not show a CNS phenotype 27. Further, in vitro studies have provided mechanistic insights about how TREX1 dysfunction contributes to an immune response in AGS. TREX1 regulates several retroelements including the long interspersed element‐1 (LINE1 or L1), observed to be accumulating as ssDNA retroelements in the heart of Trex1 knockout mice 69. L1 is highly expressed in the nervous system 11 and there are multiple regulatory mechanisms to inhibit L1, TREX1 being one of them. TREX1 can alter the cellular localization and reduce expression of the open reading frame (ORF)1p protein, which is crucial for assembly of L1 elements. Thus, TREX1 has the ability to deplete ORF1p and block L1 activity; however, mutations in TREX1 leads to increased ORF1p levels and fails to suppress expression of L1 elements 39. Treatment with reverse transcriptase inhibitors can block the accumulation of L1 elements and may be a therapeutic option for TREX1 patients 70.
TREX1 regulates and prevents self‐activation of the cGAS‐STING pathway, a downstream signaling pathway of the innate immune system that senses presence of cytosolic DNA. cGAS (cyclic GMP‐AMP synthase) binds to the detected cytosolic DNA and produces cyclic GMP‐AMP (cGAMP) from GTP and ATP as substrates respectively 25. cGAMP then binds to stimulator of interferon genes (STING) leading to phosphorylation of the transcription factor IFN regulator 3 (IRF3), which then enters the nucleus and stimulates transcription of type I IFN genes 9, 74. The release of IFN and binding to its cognate receptor induces expression of IFN‐stimulated genes (ISG) known as the “IFN signature,” leading to a cGAS‐STING pathway mediated antiviral response. The deletion of type I IFN receptor in the Trex1 knockout mice abolishes the IFN signature completely and rescues the peripheral immune phenotype, emphasizing the critical role of IFN signaling in TREX1 mediated AGS pathology 69.
RNASEH2A, RNASEH2B and RNASEH2C
RNASEH2 is a trimeric enzyme consisting of three subunits A, B and C, encoded by RNASEH2A, RNASEH2B and RNASEH2C genes respectively, which represent the corresponding catalytic, scaffolding and protein interaction domains 15. Biallelic mutations in RNASEH2A, RNASEH2B and RNASEH2C result in partial loss‐of‐function in AGS, which accounts for over half of all AGS cases 16, 63. RNASEH2 functions along with RNASEH1 to degrade RNA:DNA hybrids that arise during DNA replication and R‐loop formation 8. RNASEH2 complex is also involved in ribonucleotide excision repair mechanism and removes single ribonucleotides embedded in DNA 22, 60, 68.
In AGS a common mechanism of disease is thought to result from an accumulation of nucleic acids, triggering an immune response 31, 60. A likely source of nucleic acid accumulation in RNASEH2A, B and C mutations include retroelements, DNA replication/repair and DNA fragments generated during cell death 59. In AGS, RNASEH2A, B and C‐compromised cells are postulated to accumulate RNA:DNA hybrids via retroelements 16; however, Bartsch et al recently reported that instead of suppressing retrotransposition, RNASEH2 in fact mobilizes retroelements 3. This makes it unlikely that the accumulating nucleic acids are derived from retroelements in RNASEH2A, B and C mutations. Both mice with complete loss of RNASEH2B activity (Rnaseh2b−/−) and Rnaseh2b “knock out first” mice (Rnaseh2bKOF), which have reduced RNASEH2B levels (6% of normal values) result in embryonic and perinatal death, respectively 4, 31, 60. The embryonic tissues showed extensive presence of ribonucleotides in genomic DNA validating the role of RNASEH2 in DNA repair and maintaining genomic stability 8, 31, 68. Although a DNA damage phenotype was observed in Rnaseh2bKOF mice, an immune response was not detected in this model 4.
In human AGS individuals, there is reduced activity of the RNASEH2 complex 63 but it remains challenging to know if reduced activity of RNASEH2 complex as seen in Rnaseh2bKOF mouse model leads to DNA damage and accumulation, triggering an immune response. A knock‐in mouse model of Rnaseh2b, Rnaseh2bA174T/A174T was developed to study the frequently occurring human mutation A177T that provides some insight into development of an immune response 44. The mice had reduced RNASEH2B activity (30% of wild type), but again failed to produce clinical features of human AGS. However, studies on mouse embryonic fibroblasts from Rnaseh2bA174T/A174T mice illustrated that RNASEH2B, like TREX1, triggers an immune response via the cGAS‐STING pathway, serving as a common signaling pathway in AGS 44. Consistent with this observation, Rnaseh2b deficient cells accumulated aggregates of cytosolic DNA fragments resembling micronuclei, which are associated with genomic instability 3. These cytoplasmic membrane‐bound DNA co‐localized with the innate immune sensor cGAS, providing direct evidence for DNA damage mediated activation of cGAS‐STING pathway in RNASEH2 mutations 3. Interestingly, these micronuclei were cleared by induction of autophagy through pharmacological inhibition of the mechanistic target of rapamycin (mTOR) pathway and could be a novel therapeutic approach in AGS patients with RNASEH2B mutations. There are potentially multiple mechanisms regulating activation of innate immunity since the RNASEH2 complex can have several different mutations in different subunits, localized to different domains of the enzyme 23.
ADAR1
Adenosine deaminase acting on RNA (ADAR) catalyzes the hydrolytic deamination of adenosine (A) to inosine (I) in double stranded (ds) RNA. This A‐to‐I editing is believed to be most abundant in the brain compared with other tissues 33. Site‐specific RNA editing generates new isoforms of encoded proteins, making ADAR1 essential for regulating coding regions, microRNAs and RNA transcripts. There are two isoforms of ADAR1: the constitutively expressed ADAR1 p110, which is localized to the nucleus and edits dsRNA before nuclear export; and the inducible ADAR1 p150, a full‐length isoform which is IFN inducible and localized to the cytoplasm. Heterozygous and homozygous mutations in ADAR1 can lead to AGS and stimulate IFN production as observed in other AGS mutations. ADAR1 mutations cause dystonia due to bilateral striatal neurodegeneration 42 and can also result in dyschromatosis symmetrica hereditaria (DSH1) in East Asian population 29.
The Adar1 mouse studies to model AGS have been challenging as deletion of Adar1 in mice is embryonically lethal at E 12.5 due to defective hematopoiesis and overproduction of type I IFN 71. However, when Adar1 null mice are crossed with knock‐out mice of IFN receptor subunit (Ifnr−/−), they live an additional 3–4 days albeit still being embryonically lethal.
ADAR1 modifies the host RNA to help distinguish between the foreign and host nucleic acid, thus preventing an abnormal activation of the innate immune system 46. The downstream molecular pathways of ADAR1 involve signaling through the cytoplasmic viral sensor or pattern recognition receptor melanoma differentiation‐associated protein 5 (MDA5), which binds to viral dsRNA in the cytoplasm during a viral infection. In the absence of ADAR, MDA5 binds to the accumulating host dsRNA and activates the innate immune mitochondrial antiviral signaling protein (MAVS) pathway in the Adar1 knockouts. This turns on the transcription factors NF‐kB, IRF3/7 and AP1 and further induces production of type I IFN and ISGs. While ADAR p150 regulates IFN stimulation, increased IFN levels can also enhance ADARp150 expression through a feedback loop. Pestal et al have further demonstrated that in Adar1 knock‐out mice, it is the MAVS pathway and not the cGAS‐STING pathway that contributes to embryonic death 56. Recent evidence demonstrates activation of other innate immune sensors including Protein Kinase R (PKR) and the Oligoadenylate‐synthetase (OAS)‐Ribonculease L (RNase L) system in the absence of ADAR editing 11, 38.
MDA5/IFIH1
MDA5, the pattern recognition receptor (described above) is transcribed by the IFIH1 (interferon induced with helicase C domain 1) gene in humans and heterozygous mutations in IFIH1 are also reported to cause AGS 51, 65. A mouse model of Ifih1 was generated using spontaneous mutagenesis using N‐ethyl‐N‐nitrosourea (ENU) leading to the missense mutation G821S. These mice developed lupus‐like nephritis and systemic autoimmune symptoms with elevated IFN levels 24; however, no CNS phenotype was reported. ADAR1 negatively regulates MDA5 (Ifih1) mediated antiviral response as double knockouts of Adar−/− Ifih1−/− survived postnatally with decreased ISG levels; although the mice died 2 days postnatally. A knock‐in mouse model of catalytically inactive Adar1E861A/E861A was developed and also found to be embryonically lethal. However, Adar1E861A/E861A Ifih1−/− mice were viable and looked fairly normal, indicating that the loss of enzymatic activity of ADAR1 can be overcome by removing the MDA5 sensor 40. Thus, ADAR and MDA5 signaling are intimately connected and targeting these downstream signaling pathways can be a promising mechanism for blocking mutant ADAR1 mediated inflammation.
SAMHD1
SAMHD1 (SAM and HD domain containing deoxynucleoside triphosphate tryphosphohydrolase 1) is a deoxunucleotide triphosphohydrolase enzyme that prevents cDNA synthesis by hydrolyzing and depleting the pool of dNTPs in the cytoplasm. SAMHD1 also displays a 3′ exonuclease activity against RNA and DNA. There is evidence that deficiency of SAMHD1 in fibroblasts results in increased genomic DNA damage 35 and similar to TREX1, SAMHD1 is a negative regulator of LINE1 retrotransposition 78. As seen with ADAR1, SAMHD1 also plays an important role in regulating the immune system and functions as a viral restriction factor. SAMHD1 can bind to viral DNA and restrict human HIV‐1 infections by reducing dNTP concentrations below the levels needed for retroviral reverse transcription 16.
Murine Samhd1 displays restricted retroviral infection and while the Samhd1 null mice produced spontaneous type I IFN, they appeared normal and did not present with a CNS phenotype 5. However, a zebrafish model with samhd1 deficiency resulted in cerebrovascular phenotype accompanied by increased transcription of IFN and ISGs as seen in human individuals 33. While a good rodent model with CNS phenotype is not available for Samhd1, the fish model can potentially be employed for testing and understanding mechanisms in vivo. Cytosolic DNA sensing in the absence of SAMHD1 triggers cGAS/STING pathway inducing production of IFN and ISGs 45 and activation of the innate immune response 64. SAMHD1 also diminishes myeloid and T cell responses to HIV infections, modulating the adaptive immune response 45. While SAMHD1 acts as a checkpoint to prevent activation of IFN mediated innate response, the downstream transcription factor IRF3 in turn induces Samhd1 transcription 75. Thus, SAMHD1 keeps in check both activation of the innate and the adaptive immune system through modulation of signaling pathways and co‐stimulatory molecules but loss‐of‐function due to SAMHD1 mutations leads to a cascade of inflammatory response.
As described above, the molecular mechanisms driving IFN responses in the different genes causing AGS has been fairly well understood. While the role of peripheral immune cells and how they mount IFN response through these pathways is explored in AGS, the cellular contributions of CNS cells, particularly astrocytes is not completely clear. In the next sections, we will be reviewing the potential role of astrocytes and discuss some studies that have addressed this question.
Astrocytes Maintain CNS Homeostasis
Astrocytes are the most abundant glial cells in the CNS and conduct important homeostatic functions in the CNS, including energy supply to neurons, maintenance of the blood–brain barrier (BBB), regulation of the excitatory neurotransmitter glutamate and recent evidence involving synapse formation, elimination and synaptic plasticity 49. Under disease, trauma and/or inflammatory conditions, astrocytes become hypertrophic with upregulation of the intermediate filament glial fibrillary acidic protein (GFAP) and key inflammatory genes, a process known as reactive astrogliosis. Reactive astrocytes play an essential role in regulating CNS inflammation and can exert both harmful and beneficial effects depending on the CNS insult. As observed in spinal cord injury, reactive astrocytes play a beneficial role in confining the spread of inflammation to area adjoining the lesion and forming a barrier to inflammatory cells in CNS 66. But in neurodegenerative and autoimmune conditions, reactive astrocytes can wield devastating effects through release of cytokines, chemokines, neurotoxic molecules leading to BBB disruption with ensuing encephalopathy and eventual neuronal damage and death 66, 67.
Astrocytes as Mediators of Neuroinflammation
IFN‐α is a common biomarker for all AGS mutations, that serves as an inflammatory mediator driving an immune disease not just in the peripheral system but also in the CNS. Astrocytes and microglia are thought to be the primary cells that produce IFN‐α in CNS in response to a viral infection or when exposed to synthetic ds RNA polyinosinic–polycytidylic acid (poly I:C) treatment; although there are conflicting reports about neuronal production of IFN‐α 41, 52, 53. Screening of the cytokines CXCL10 (chemokine [C‐X‐C motif] ligand 10) and IFN‐α using multiplexed‐luminex assay exhibited increased cytokine levels in CSF and serum of AGS individuals, serving as a diagnostic biomarker 72 and supporting the presence of intrathecal accumulation of IFN‐α and related cytokines. Additional evidence of astrocytes as an IFN‐α source came from studies in post‐mortem brain tissue of AGS affected individuals, which revealed co‐localization of the astrocyte marker GFAP with the cytokines IFN‐α and CXCL10 72.
To directly address the effect of IFN‐α produced from astrocytes, Ian Campbell's group developed a transgenic mouse model expressing IFN‐α specifically in astrocytes using a GFAP promoter 2, 7. These mice developed a clinical phenotype that closely matched the AGS individuals including encephalopathy, neurodegeneration, seizures and calcium deposits in the basal ganglia. While this model is not a true genetic AGS model, it represents a phenotypic model of AGS. In an in vitro model, astrocytes differentiated from immortalized human neural stem cells (ihNSCs) were exposed to poly I:C to induce an innate immune response, which led to elevated levels of GFAP, IFN, CXCL‐10, TNF‐α (Tumor Necrosis Factor‐alpha) and other cytokines 18. When these astrocytes were treated chronically with IFN‐α for three weeks, they displayed reduced proliferation, increased reactivity and dysregulation of astrocyte‐specific genes and proteins crucial for the maintenance of white matter. Interestingly, withdrawal of IFN‐α for 7 days after chronic exposure did not rescue the effects exerted, indicating involvement of a downstream IFN‐α mediated signaling cascade in astrocytes 18. Together, the in vivo and in vitro studies serve as a proof of principle that astrocyte mediated IFN‐α release can initiate a cascade of neuroinflammatory events exerting cell autonomous and non‐cell autonomous effects.
Impact of Peripheral Inflammation on Astrocytes
In the context of AGS, peripheral cells could also be involved in influencing astrocytic inflammatory responses and we have proposed and depicted this peripheral‐CNS cross‐talk and different cellular contributions occurring in AGS in Figure 2. Loss of function in AGS causative genes and the ensuing deficits in nucleic acid metabolism initiates activation of the innate immune cells‐ dendritic cells (DC) and macrophages 54, 55. Along with peripheral circulation of DCs, a prominent increase in DCs, particularly plasmacytoid DC (PIDC) was reported in the CSF of AGS affected individuals 28 and the PIDCs are known to be potent producers of IFN‐α 61. Thus, it is feasible that PIDCs can directly cause reactive astrogliosis and impact other CNS cells. Macrophages can similarly be activated peripherally along with DCs triggering adaptive B and T cell responses. Histological studies have shown infiltrating T cells (CD3+ cells) in the brains of AGS individuals 34, which accompanied by CSF lymphocytosis in AGS 71 could activate astrocytes. One study has directly addressed this lymphocyte–astrocyte cross talk and reported that lymphocytes can cause astrogliosis through activation of the pro‐apoptotic enzyme cathepsin D, which upon blocking is protective and decreases astrocyte activation 58.
Figure 2.
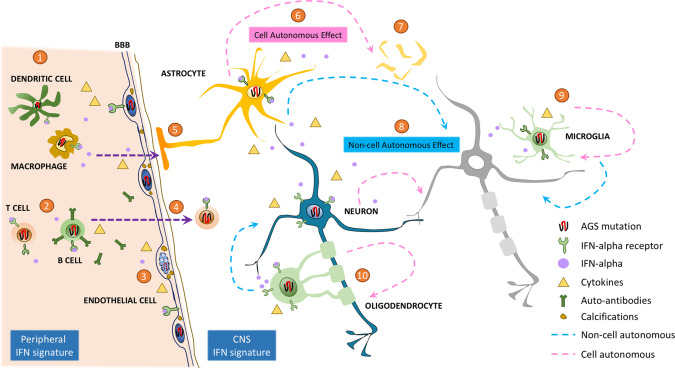
Peripheral and CNS cellular contributions toward IFN signature and its downstream effects in AGS. 1 The presence of AGS mutation causes abnormal accumulation of nucleic acids, which leads to activation of the cGAS‐STING and MDA‐MAVS pathway in the innate immune cells–dendritic cells and macrophages. They mount an inflammatory response through release of interferon alpha (IFN‐α), activation of IFN stimulated genes (ISG) and induction of other cytokines. 2 These innate immune cells further activate the adaptive immune monocytes, triggering the T and B cells to release inflammatory molecules and auto‐antibodies, respectively. 3 The peripheral IFN‐α signature causes microangiopathy and AGS mutations affect the health of endothelial cell through release of pro‐inflammatory cytokines and deposition of calcifications. 4 A clinical diagnostic marker for AGS is increased CSF IFN‐α and lymphocytosis, with supporting evidence of infiltrating T cells observed in post‐mortem brains from AGS individuals. 5 This peripheral IFN signature impinges on the underlying BBB and the adjoining astrocyte end feet, potentially contributing to astrogliosis. 6 The astrocytes harboring the AGS mutation have a cell autonomous effect leading to astrogliosis along with release of IFN‐α and stimulating production of ISGs. 7 Astrocytes with AGS mutations, particularly TREX1 mutations, have altered survival eventually leading to cell death. 8 Along with the cell‐autonomous effect of neurons on themselves, astrocyte mediated release of IFN signature can in turn exert a non‐cell autonomous effect resulting in neuronal toxicity and death. 9 While it is not tested yet, harboring AGS mutations could potentially activate microglia directly in a cell‐autonomous fashion or through release of IFN signature from astrocytes causing reactive microgliosis, which can further propagate non‐cell autonomous effects. 10 Similarly, the contribution of oligodendrocytes in AGS is not explored and could potentially worsen the ongoing pathology in the CNS due to loss of oligodendrocytes and myelinated axons.
In CSF of AGS individuals, circulating systemic and brain‐reactive auto‐antibodies were found and interestingly when the antibodies were tested, the IgGs were targeted toward astrocytes and endothelial cells in brain sections of post‐mortem AGS individuals 20. This deposition of IgG could likely lead to reactive astrogliosis as well as contribute to vascular damage and microangiopathy. In fact, knock‐down of TREX1 in endothelial cells makes them inflammatory and stimulates release of cytokines 19 and exposure to IFN‐α further increases calcification of human vascular smooth muscle cells 34.
This accumulating evidence of circulating cytokines and monocytes, DCs, auto‐antibodies and ongoing degeneration of the endothelial cells points toward a potential breach of the BBB. It is possible that the astrocyte end feet juxtaposed next to the endothelial cells and leaky BBB elicits astrogliosis. We propose that cross talk between the two arms‐ “Peripheral IFN signature” and “CNS IFN signature” together contribute to the ongoing pathogenesis observed in AGS individuals.
Cell‐Autonomous and Non‐Cell‐Autonomous Role of Astrocytes in AGS
A few studies have directly addressed the role of astrocytes as a source of IFN‐α and as mediators of inflammatory cascade in AGS. Cuadrado et al demonstrated the significance of loss‐of‐function in genes involved in AGS, using ihNSCs derived and post‐mortem tissue derived astrocytes 19. When the AGS genes ‐TREX1, SAMHD1, RNASEH2A and ADAR1 were silenced using lentivirus in these astrocytes, a significant induction of IFN‐α expression, ISG signaling and release of pro‐inflammatory cytokines and chemokines was observed in astrocytes with TREX1 knockdown. In addition, specific knockdown of TREX1 resulted in extensive astrocytic apoptosis and a heightened immune response leading to a cell‐autonomous effect in astrocytes compared to knockdown of other mutations. This TREX1 phenotype was in line with the early prognosis and severity reported in TREX1 patients 13.
Previous studies have shown that TREX1 suppresses activity of the retroelement L1 using an over‐expression system and a recent study addressed how endogenous TREX1 regulates L1 in human induced pluripotent stem cell (iPSC) derived neurons and astrocytes 68. To model AGS, the iPSC lines were generated with frameshift mutations in TREX1 with an early stop codon resulting in a non‐functional TREX1 deficient cell. TREX1 deficient neural precursor cells (NPCs), neurons and astrocytes displayed accumulation of ssDNA in their cytoplasm and deep sequencing data confirmed high expression of L1 elements in NPCs. Interestingly, accumulation of L1 elements in neurons resulted in a cell‐autonomous toxicity. While increased L1 expression did not exert toxicity on TREX1 deficient astrocytes, they displayed increased levels of IFN and ISGs, which was reversed upon treatment with reverse transcription inhibitors 70. Furthermore, conditioned media from TREX1 deficient astrocytes caused neuronal toxicity over time that was mitigated with IFN receptor blockers. Interestingly, human cortical organoids were generated using control and TREX1 deficient stem cells, which resulted in microencephaly‐like size reductions due to neuronal death, similar to what is observed in AGS individuals 70. Thus, TREX1 mutation in AGS leads to both a cell autonomous and non‐cell autonomous effect on neurons due to loss of L1 regulation, downstream activation of the cGAS‐STING pathway and elevated secretion of IFN signature. This study elegantly modeled TREX1 mutation and examined the mechanisms through which retroelements activate immune responses in astrocytes and impair neuronal survival.
Considerable work remains to be done to address the contribution of astrocytes in other AGS mutations. The embryonic lethality in Adar1 mutant forms a barrier in understanding CNS pathology and contribution of astrocytes in ADAR associated AGS phenotype. However, recent work from Heraud‐Farlow et al provides evidence for significant upregulation of IFN signature in E12.5 fetal brains of the editing deficient Adar1E861A/E861A mice 30. This study implies there is a CNS source for increased IFN production, potentially contributed by astrocytes and microglia. While mechanisms of IFIH1/MDA5 mediated peripheral immune responses have been studied extensively, astrocyte specific IFIH1/MDA5 mechanisms in AGS have not been directly explored and remain to be tested. There is evidence that astrocytes respond to complexed poly I:C, a ds‐RNA mimic, by binding to IFIH1/MDA5 and activating the TLR‐3 pathway through NFκB and IRF‐3 activation leading to production of an IFN signature 21. Astrocytes have higher expression of SAMHD1 than microglia and since SAMHD1 lowers dNTP pool and infectivity of viruses, astrocytes are more resistant to virus infectivity 55. In AGS, SAMHD1 mutation results in loss of function, potentially making the astrocytes vulnerable, activation of innate immune pathways and increased production of cytokines.
In summary, so far, the role of astrocytes in AGS is demonstrated only for TREX1 mutations, which displays higher disease severity compared to other genetic mutations. The cell autonomous and non‐cell autonomous role of astrocytes in other mutations in AGS remains an open question. Similarly, the contribution of microglia as a source of the IFN mediated inflammatory cascade in AGS is also unexplored.
Perspective
AGS is a genetically heterogeneous disease caused by mutations in enzymes that cause either accumulation of immune stimulatory nucleic acids or directly instigates the innate immune sensors, which then converge on the downstream cGAS‐STING and MDA‐MAVS signaling pathways. The resulting cellular pathogenesis through induction of IFN‐α and cascade of ISGs and cytokines leads to autoinflammatory disease and tissue destruction 16. Murine models of AGS have failed to recapitulate the CNS component of the disease observed in humans. The advent of individual derived human stem cells may help circumvent the disparity observed between human and mouse models. The human TREX1 astrocyte studies indeed proved that astrocytes become dysfunctional and participate in the ongoing pathogenesis occurring in AGS through activation of the IFN pathway. Currently, there is no cure for AGS although therapies are being developed based on immuno‐modulatory drugs. Prior unsuccessful treatments for prevention of neurologic injury have included azathioprine, methylprednisolone with immunoglobulin, methylprednisolone or immunoglobulin alone 17. Other neurological conditions such as Alzheimer's disease, ALS and epilepsy have targeted astrocyte cell replacement strategies and/or astrocytic pathways based on disruption of important astrocyte homeostatic functions (metabolic support, glutamate regulation, inflammatory and signaling pathways). Similarly, understanding molecular mechanisms leading to astrocyte death, release of cytokines and ensuing inflammation in AGS can be harnessed for drug development strategies. A better understanding of mutation specific disease onset and diagnosis will also allow for timely intervention, which will be crucial in mitigating the disease and reducing tissue damage due of auto‐inflammation. While tremendous progress has been made in the last decade about AGS, future directions will require efforts in modeling the different AGS mutations to understand CNS pathology and dissect the underlying mechanisms for translational and clinical studies.
References
- 1. Aicardi J, Goutieres F (1984) A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15:49–54. [DOI] [PubMed] [Google Scholar]
- 2. Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H et al (1998) Transgenic expression of IFN‐alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 161:5016–5026. [PubMed] [Google Scholar]
- 3. Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K et al (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26:3960–3972. [DOI] [PubMed] [Google Scholar]
- 4. Behrendt R, Roers A (2014) Mouse models for Aicardi‐Goutieres syndrome provide clues to the molecular pathogenesis of systemic autoimmunity. Clin Exp Immunol 175:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D et al (2013) Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell‐intrinsic antiviral response. Cell Rep 4:689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonnemann CG, Meinecke P (1992) Encephalopathy of infancy with intracerebral calcification and chronic spinal fluid lymphocytosis–another case of the Aicardi‐Goutieres syndrome. Neuropediatrics 23:157–161. [DOI] [PubMed] [Google Scholar]
- 7. Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T et al (1999) Structural and functional neuropathology in transgenic mice with CNS expression of IFN‐alpha. Brain Res 835:46–61. [DOI] [PubMed] [Google Scholar]
- 8. Cerritelli SM, Crouch RJ (2009) Ribonuclease H: the enzymes in eukaryotes. febs J 276:1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS‐STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149. [DOI] [PubMed] [Google Scholar]
- 10. Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B et al (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23‐H1 to degrade DNA during granzyme A‐mediated cell death. Mol Cell 23:133–142. [DOI] [PubMed] [Google Scholar]
- 11. Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL et al (2018) Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coufal NG, Garcia‐Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT et al (2009) L1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL et al (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR and IFIH1. Am J Med Genet A 167A:296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crow YJ, Jackson AP, Roberts E, van Beusekom E, Barth P, Corry P et al (2000) Aicardi‐Goutieres syndrome displays genetic heterogeneity with one locus (AGS1) on chromosome 3p21. Am J Hum Genet 67:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E et al (2006) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi‐Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38:910–916. [DOI] [PubMed] [Google Scholar]
- 16. Crow YJ, Manel N (2015) Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15:429–440. [DOI] [PubMed] [Google Scholar]
- 17. Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI (2014) Therapies in Aicardi‐Goutieres syndrome. Clin Exp Immunol 175:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuadrado E, Jansen MH, Anink J, De Filippis L, Vescovi AL, Watts C et al (2013) Chronic exposure of astrocytes to interferon‐alpha reveals molecular changes related to Aicardi‐Goutieres syndrome. Brain 136:245–258. [DOI] [PubMed] [Google Scholar]
- 19. Cuadrado E, Michailidou I, van Bodegraven EJ, Jansen MH, Sluijs JA, Geerts D et al (2015) Phenotypic variation in Aicardi‐Goutieres syndrome explained by cell‐specific IFN‐stimulated gene response and cytokine release. J Immunol 194:3623–3633. [DOI] [PubMed] [Google Scholar]
- 20. Cuadrado E, Vanderver A, Brown KJ, Sandza A, Takanohashi A, Jansen MH et al (2015) Aicardi‐Goutieres syndrome harbours abundant systemic and brain‐reactive autoantibodies. Ann Rheum Dis 74:1931–1939. [DOI] [PubMed] [Google Scholar]
- 21. De Miranda J, Yaddanapudi K, Hornig M, Lipkin WI (2009) Astrocytes recognize intracellular polyinosinic‐polycytidylic acid via MDA‐5. faseb J 23:1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eder PS, Walder RY, Walder JA (1993) Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie 75:123–126. [DOI] [PubMed] [Google Scholar]
- 23. Feng S, Cao Z (2016) Is the role of human RNase H2 restricted to its enzyme activity?. Prog Biophys Mol Biol 121:66–73. [DOI] [PubMed] [Google Scholar]
- 24. Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M et al (2014) Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40:199–212. [DOI] [PubMed] [Google Scholar]
- 25. Gao D, Li T, Li XD, Chen X, Li QZ, Wight‐Carter M, Chen ZJ (2015) Activation of cyclic GMP‐AMP synthase by self‐DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112:E5699–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giroud M, Gouyon JB, Chaumet F, Cinquin AM, Chevalier‐Nivelon A, Alison M, Dumas R (1986) A case of progressive familial encephalopathy in infancy with calcification of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Childs Nerv Syst 2:47–48. [DOI] [PubMed] [Google Scholar]
- 27. Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW (2015) Exonuclease TREX1 degrades double‐stranded DNA to prevent spontaneous lupus‐like inflammatory disease. Proc Natl Acad Sci U S A 112:5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han S, Lin YC, Wu T, Salgado AD, Mexhitaj I, Wuest SC et al (2014) Comprehensive immunophenotyping of cerebrospinal fluid cells in individuals with neuroimmunological diseases. J Immunol 192:2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayashi M, Suzuki T (2013) Dyschromatosis symmetrica hereditaria. J Dermatol 40:336–343. [DOI] [PubMed] [Google Scholar]
- 30. Heraud‐Farlow JE, Chalk AM, Linder SE, Li Q, Taylor S, White JM et al (2017) Protein recoding by ADAR1‐mediated RNA editing is not essential for normal development and homeostasis. Genome Biol 18:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A (2012) Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209:1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hwang T, Park CK, Leung AK, Gao Y, Hyde TM, Kleinman JE et al (2016) Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19:1093–1099. [DOI] [PubMed] [Google Scholar]
- 33. Kasher PR, Jenkinson EM, Briolat V, Gent D, Morrissey C, Zeef LA et al (2015) Characterization of samhd1 morphant zebrafish recapitulates features of the human type I interferonopathy Aicardi‐Goutieres syndrome. J Immunol 194:2819–2825. [DOI] [PubMed] [Google Scholar]
- 34. Klok MD, Bakels HS, Postma NL, van Spaendonk RM, van der Knaap MS, Bugiani M (2015) Interferon‐alpha and the calcifying microangiopathy in Aicardi‐Goutieres syndrome. Ann Clin Transl Neurol 2:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kretschmer S, Wolf C, Konig N, Staroske W, Guck J, Hausler M et al (2015) SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis 74:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lanzi G, Fazzi E, D'Arrigo S (2002) Aicardi‐Goutieres syndrome: a description of 21 new cases and a comparison with the literature. Eur J Paediatr Neurol 6:A9–A22. [DOI] [PubMed] [Google Scholar]
- 37. Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury‐Cukier F, Aicardi J (1988) Intrathecal synthesis of interferon‐alpha in infants with progressive familial encephalopathy. J Neurol Sci 84:201–208. [DOI] [PubMed] [Google Scholar]
- 38. Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S et al (2017) Ribonuclease L mediates the cell‐lethal phenotype of double‐stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6:e25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li P, Du J, Goodier JL, Hou J, Kang J, Kazazian HH, Jr et al (2017) Aicardi‐Goutieres syndrome protein TREX1 suppresses L1 and maintains genome integrity through exonuclease‐independent ORF1p depletion. Nucleic Acids Res 45:4619–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC et al (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349:1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lieberman AP, Pitha PM, Shin HS, Shin ML (1989) Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci U S A 86:6348–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A et al (2014) A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet 51:76–82. [DOI] [PubMed] [Google Scholar]
- 43. Livingston JH, Stivaros S, van der Knaap MS, Crow YJ (2013) Recognizable phenotypes associated with intracranial calcification. Dev Med Child Neurol 55:46–57. [DOI] [PubMed] [Google Scholar]
- 44. Mackenzie KJ, Carroll P, Lettice L, Tarnauskaitė Ž, Reddy K, Dix F et al (2016) Ribonuclease H2 mutations induce a cGAS/STING‐dependent innate immune response. embo J 35:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maelfait J, Bridgeman A, Benlahrech A, Cursi C, Rehwinkel J (2016) Restriction by SAMHD1 limits cGAS/STING‐dependent innate and adaptive immune responses to HIV‐1. Cell Rep 16:1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D et al (2014) The RNA‐editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9:1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mazur DJ, Perrino FW (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′–>5′ exonucleases. J Biol Chem 274:19655–19660. [DOI] [PubMed] [Google Scholar]
- 48. Mehta L, Trounce JQ, Moore JR, Young ID (1986) Familial calcification of the basal ganglia with cerebrospinal fluid pleocytosis. J Med Genet 23:157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Molofsky AV, Deneen B (2015) Astrocyte development: a guide for the perplexed. Glia 63:1320–1329. [DOI] [PubMed] [Google Scholar]
- 50. Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G et al (2004) Gene‐targeted mice lacking the Trex1 (DNase III) 3′–>5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24:6719–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A et al (2014) Aicardi‐Goutieres syndrome is caused by IFIH1 mutations. Am J Hum Genet 95:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olson JK, Miller SD (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173:3916–3924. [DOI] [PubMed] [Google Scholar]
- 53. Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T (2007) Type I interferon response in the central nervous system. Biochimie 89:770–778. [DOI] [PubMed] [Google Scholar]
- 54. Pereira‐Lopes S, Celhar T, Sans‐Fons G, Serra M, Fairhurst AM, Lloberas J, Celada A (2013) The exonuclease Trex1 restrains macrophage proinflammatory activation. J Immunol 191:6128–6135. [DOI] [PubMed] [Google Scholar]
- 55. Peschke K, Achleitner M, Frenzel K, Gerbaulet A, Ada SR, Zeller N et al (2016) Loss of trex1 in dendritic cells is sufficient to trigger systemic autoimmunity. J Immunol 197:2157–2166. [DOI] [PubMed] [Google Scholar]
- 56. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB (2015) Isoforms of RNA‐editing enzyme ADAR1 independently control nucleic acid sensor MDA5‐driven autoimmunity and multi‐organ development. Immunity 43:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pilakka‐Kanthikeel S, Raymond A, Atluri VS, Sagar V, Saxena SK, Diaz P et al (2015) Sterile alpha motif and histidine/aspartic acid domain‐containing protein 1 (SAMHD1)‐facilitated HIV restriction in astrocytes is regulated by miRNA‐181a. J Neuroinflammation 12:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pulliero A, Marengo B, Longobardi M, Fazzi E, Orcesi S, Olivieri I et al (2013) Inhibition of the de‐myelinating properties of Aicardi‐Goutieres syndrome lymphocytes by cathepsin D silencing. Biochem Biophys Res Commun 430:957–962. [DOI] [PubMed] [Google Scholar]
- 59. Reijns MA, Jackson AP (2014) Ribonuclease H2 in health and disease. Biochem Soc Trans 42:717–725. [DOI] [PubMed] [Google Scholar]
- 60. Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA et al (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V (2011) Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol 29:163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K et al (2007) Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi‐Goutieres syndrome. Am J Hum Genet 80:811–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J et al (2007) Clinical and molecular phenotype of Aicardi‐Goutieres syndrome. Am J Hum Genet 81:713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM et al (2009) Mutations involved in Aicardi‐Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41:829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G et al (2014) Gain‐of‐function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 16:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM (2012) RNase H2‐initiated ribonucleotide excision repair. Mol Cell 47:980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell‐intrinsic initiation of autoimmunity. Cell 134:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P et al (2017) Modeling of TREX1‐dependent autoimmune disease using human stem cells highlights l1 accumulation as a source of neuroinflammation. Cell Stem Cell 21:319–331 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tolmie JL, Shillito P, Hughes‐Benzie R, Stephenson JB (1995) The Aicardi‐Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J Med Genet 32:881–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW (2008) Astrocytes produce interferon‐alpha and CXCL10, but not IL‐6 or CXCL8, in Aicardi‐Goutieres syndrome. Glia 56:568–578. [DOI] [PubMed] [Google Scholar]
- 73. Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K (2004) Stress‐induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279:4952–4961. [DOI] [PubMed] [Google Scholar]
- 74. Xiao TS, Fitzgerald KA (2013) The cGAS‐STING pathway for DNA sensing. Mol Cell 51:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yang S, Zhan Y, Zhou Y, Jiang Y, Zheng X, Yu L et al (2016) Interferon regulatory factor 3 is a key regulation factor for inducing the expression of SAMHD1 in antiviral innate immunity. Sci Rep 6:29665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang YG, Lindahl T, Barnes DE (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131:873–886. [DOI] [PubMed] [Google Scholar]
- 77. Yuan F, Dutta T, Wang L, Song L, Gu L, Qian L et al (2015) Human DNA exonuclease TREX1 is also an exoribonuclease that acts on single‐stranded RNA. J Biol Chem 290:13344–13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X et al (2013) Modulation of LINE‐1 and Alu/SVA retrotransposition by Aicardi‐Goutieres syndrome‐related SAMHD1. Cell Rep 4:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]