

Abstract
Microglial associations with both the major Alzheimer’s disease (AD) pathognomonic entities, β‐amyloid‐positive plaques and tau‐positive neurofibrillary tangles, have been noted in previous investigations of both human tissue and mouse models. However, the precise nature of their role in the pathogenesis of AD is debated; the major working hypothesis is that pro‐inflammatory activities of activated microglia contribute to disease progression. In contrast, others have proposed that microglial dystrophy with a loss of physiological and neuroprotective activities promotes neurodegeneration. This immunohistochemical study sought to gain clarity in this area by quantifying the morphological subtypes of microglia in the mildly‐affected primary visual cortex (PVC), the moderately affected superior frontal cortex (SFC) and the severely affected inferior temporal cortex (ITC) of 8 AD cases and 15 age and gender‐matched, non‐demented controls with ranging AD‐type pathology. AD cases had increased β‐amyloid and tau levels compared to controls in all regions. Neuronal loss was observed in the SFC and ITC, and was associated with atrophy in the latter. A major feature of the ITC in AD was a decrease in ramified (healthy) microglia with image analysis confirming reductions in arborized area and skeletal complexity. Activated microglia were not associated with AD but were increased in non‐demented controls with greater AD‐type pathology. Microglial clusters were occasionally associated with β‐amyloid‐ and tau‐positive plaques but represented less than 2% of the total microglial population. Dystrophic microglia were not associated with AD, but were inversely correlated with brain pH suggesting that agonal events were responsible for this morphological subtype. Overall these novel findings suggest that there is an early microglial reaction to AD‐type pathology but a loss of healthy microglia is the prominent feature in severely affected regions of the AD brain.
Keywords: Alzheimer’s disease, brain pH, microglia, post‐mortem human brain, quantitative neuropathology
Introduction
The clinical diagnosis of Alzheimer’s disease (AD) is probabilistic and typically supported by an abbreviated form of a neuropsychological evaluation such as the Clinical Dementia Rating (CDR) 36. Confirmation of AD can only be made at autopsy where the most comprehensive diagnostic schema is provided by the National Institute of Aging‐Alzheimer’s Association work‐group 35. This diagnostic schema incorporates previously described semi‐quantitative criteria for (A) β‐amyloid (Aβ) immunostaining 60, (B) Braak staging of neurofibrillary tangles (NFTs) 6 and (C) CERAD neuritic plaque score 33 to generate an “ABC” score. An intermediate or high “ABC” score being considered sufficient to confirm AD in a demented individual. Yet, this pathological diagnosis is also probabilistic caused by the common finding of AD‐type pathology, albeit at lower levels, in similarly aged but non‐demented individuals 4. Aβ pathology appears first in the cortex before spreading more ventrally; whereas neurofibrillary pathology, including NFTs, begins in the medial temporal lobe, then spreads more dorsally and laterally throughout the limbic system and cortex with relative sparing of primary cortices 6.
Along with Aβ plaques and NFTs, the pathology of AD is characterized by neuronal loss and gliosis 59. Microglial activation has been considered a key pathomechanism in AD, with activated microglia associating with neuritic plaques and subsequently producing a neurotoxic milieu that contributes to neuronal degeneration 30, 41, 46. Single‐nucleotide polymorphisms in genes exclusively or largely expressed in microglia are associated with increased AD risk including TREM2, CD33, CR1, ABCA7 and SHIP1 31. Microglial activation in AD appears to be Aβ‐dependent; with Aβ binding to NLR Family Pyrin Domain Containing 3 16, and pattern recognition receptors such as RAGE, scavenger receptors 44 and toll‐like receptors (TLR2, TLR4 and TLR6) 8 being sufficient to cause microglial activation in mouse models of AD.
In the past, microglia were regarded as quiescent cells that only responded to brain injury. It is now known that microglia are physiologically active, surveying their territory by continually extending and retracting their processes 38. Extensive in vitro and animal studies of microglia demonstrate their ability to sample their surrounding microenvironment and are particularly involved in synaptic homeostasis 26 throughout life by pruning of synapses in the developing CNS 47 and mediating remodeling in the adult brain 40. Microglia are functionally dynamic cells as evidenced by the wide range of morphologies observed in the brain under normal and pathological states 11. There have been a number of attempts to categorize microglia into distinct subtypes but there is little consensus on what constitutes each subtype or the nomenclature used to describe them.
Briefly, in studies using human post‐mortem brain tissue, ramified (healthy) microglia (previously called “resting” or “quiescent” microglia) are characterized by equally distributed, long, thin, ramified processes attached to a small, spherical soma 5. The terms “activated,” “reactive,” “bushy” and “hypertrophic” have been used to describe microglia that are responding to injury or potentially injurious events. Activated microglia display significantly hypertrophic soma and processes with some degree of deramification (hence the use of the term “deramified” by some authors) 3, 45. Fully activated microglia, with enlarged soma and lacking processes entirely, are termed “amoeboid” and have enhanced phagocytic capabilities 52. “Dystrophic” microglia are characterized by highly tortuous, asymmetrical and truncated processes and may demonstrate discontinuous or beaded immunostaining. Such dystrophic microglia may also be termed “pseudo‐fragmented” as electron microscopy studies have revealed that apparent discontinuities in their cellular processes represent a redistribution of the selected marker, with the processes remaining contiguous when visualized using multiple markers 61. Other features of dystrophic microglia that occur variably include the formation of spheroids and condensation within the nucleus, possibly indicative of pyknosis 56. Streit and colleagues found that dystrophic microglia preceded neurofibrillary pathology in AD 54 raising the possibility that a loss of microglial function may be as, or more, important than a gain of toxic function in AD pathophysiology.
In this immunohistochemical investigation, microglia were identified by their expression of ionized calcium binding adaptor molecule 1 (IBA1), which is known to be an effective pan‐microglial marker that is independent of “activation” states compared to other commonly used markers 23, 54. In our previous work, we had demonstrated greater densities of dystrophic microglia in severely affected cortical areas of the AD brain, namely the inferior temporal cortex (ITC) and the anterior cingulate cortex 9. Here, this work has been extended by quantifying the morphological subtypes of microglia by manual and automated methods in AD cases and controls with variable amounts of AD‐type pathology in three neocortical areas – the primary visual cortex (PVC), superior frontal cortex (SFC) and ITC. AD pathology and particularly tau pathology are known to spread in a stereotypical fashion through the cortex 2, 6, 13, 60. Furthermore, the density of NFTs is inversely correlated with surviving neurons in the AD cortex 14. These three regions were chosen to reflect the reported spread of tau pathology, with the PVC being one of the last regions to be affected 6. This is also reflected by a volumetric study where the severely affected ITC undergoes ~35% atrophy and the PVC ~ 15% in AD cases compared to gender‐matched controls 17, a scenario that potentially allows regional comparisons to model disease progression within cases 58. The aim here was to gain a greater understanding of spatiotemporal relationships between the morphological subtypes of microglia and AD progression.
Methods
This immunohistochemical study of microglial subtypes using human post‐mortem brain tissue from AD patients and non‐demented controls was approved by the University of Sydney’s Human Research Ethics Committee (HREC #2015/477). All tissue samples for this study along with demographics, clinical and pathological diagnosis were supplied by the New South Wales Brain Tissue Resource Centre (NSW BTRC) and the Sydney Brain Brank (SBB), collectively referred to as NSW Brain Banks (NSWBB) following clearance from their Scientific Advisory Committee. APOE ε4 genotyping was also carried out as previously described 32. NSWBB measure brain pH on a frozen (‐80oC) segment of the lateral cerebellar hemisphere which has previously been shown to be representative of the whole brain pH 51 and may be used as a marker of the length and severity of events in the pre‐mortem period 12, 34. The demographic and clinicopathological characteristics of the cohort are listed in Table 1.
Table 1.
Cohort details.
| Case ID (n = 23) | Age | Sex | Status | Cause of death | Disease duration (years) | PMI (hours) | Brain pH | Fixation (months) | CDR | ABC score | AD likelihood b | APOE genotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M01 | 69 | Male | Control | Cardiac failure | – | 16 | 6.6 | 97 | – | A3 B1 C3 | Low | E3/E4 |
| M02 | 74 | Female | Control | Cancer | – | 20 | 6.59 | 19 | – | A3 B1 C3 | Low | E3/E4 |
| M03 | 76 | Female | AD | Cardiorespiratory failure | 11 | 3 | 5.98 | 35 | 3 | A3 B2 C3 | Intermediate | E3/E4 |
| M04 | 77 | Male | AD | Aspiration pneumonia | 9 | 26 | 6.34 | 12 | 2 | A3 B3 C2 | High | E4/E4 |
| M05 | 78 | Female | Control | Respiratory failure | – | 11 | 6.3 | 78 | – | A0 B0 C0 | Low | E3/E3 |
| M06 | 78 | Female | Control | Toxicity | – | 45 | 6.05 | 58 | – | A0 B1 C0 | Low | E3/E4 |
| M07 | 81 | Male | Control | Cardiac failure | – | 29 | 6.57 | 26 | – | A0 B2 C0 | Low | E3/E3 |
| M08 | 80 | Male | Control | Respiratory failure | – | 12 | 6.5 | 9 | – | A0 B0 C0 | Low | E3/E3 |
| M09 | 80 | Female | AD | Alzheimer's disease | 10 | 32 | 6.54 | 25 | 3 | A3 B3 C3 | High | E3/E4 |
| M10 | 82 | Female | Control | Respiratory failure | – | 7.5 | 6.45 | 102 | – | A3 B1 C2 | Low | E3/E3 |
| M11 | 83 | Male | AD | Cerebrovascular | 5 | 25 | 6.26 | 70 | 3 | A3 B3 C3 | High | E4/E4 |
| M12 | 83 | Female | AD | Uraemia | 7 | 3 | 5.88 | 61 | 3 | A3 B3 C2 | High | E3/E4 |
| M13 | 84 | Female | AD | Aspiration pneumonia | 13 | 6 | 6.32 | 64 | 3 | A3 B3 C3 | High | E3/E4 |
| M14 a | 85 | Male | Control | Cancer | – | 9 | 6.57 | 61 | – | A2 B2 C3 | Intermediate | E3/E3 |
| M15 | 85 | Female | Control | Respiratory failure | – | 10 | 6.63 | 34 | – | A2 B1 C0 | Low | E3/E3 |
| M16 | 85 | Female | Control | Pneumonia | – | 23 | 6.44 | 73 | 0 | A2 B0 C1 | Low | E3/E3 |
| M17 | 85 | Female | AD | Cardiorespiratory failure | 5 | 10 | 5.91 | 15 | 3 | A3 B3 C3 | High | E3/E3 |
| M18 a | 87 | Female | Control | Cancer | – | 5 | 6.38 | 34 | – | A2 B2 C0 | Intermediate | E3/E4 |
| M19 a | 92 | Female | Control | Pancytopenia | – | 5 | 6.08 | 34 | 0 | A2 B2 C2 | Intermediate | E3/E3 |
| M20 | 93 | Female | Control | Cardiac failure | – | 21 | 6.96 | 81 | 0.5 | A1 B0 C0 | Low | E3/E3 |
| M21 a | 87 | Female | Control | Acute peritonitis | – | 24 | – | – | 0 | A3 B2 C1 | Intermediate | – |
| M22 | 98 | Female | AD | Cerebrovascular | 6 | 11 | 6.11 | 18 | 3 | A3 B3 C2 | High | E3/E3 |
| M23 a | 102 | Female | Control | Acute renal failure | – | 5 | 5.92 | 35 | 0 | A2 B2 C2 | Intermediate | E2/E3 |
High‐pathology controls.
As per NIA‐AA diagnostic criteria 35.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on free‐floating 45‐μm fixed sections from the PVC, SFC and ITC of cognitively normal individuals with variable AD‐type pathology (CDR = 0–0.5; n = 15) and AD patients (CDR 2–3; n = 8) to quantify the morphological subtypes of microglia. Heat‐induced epitope retrieval was performed by baking sections in a 60oC oven overnight using a 0.95 mmolL‐1 sodium citrate (pH 8.5) buffer. Sections were washed with 50% ethanol followed by endogenous peroxidase block using 0.9% H2O2 (50% ethanol diluent). Standard blocking was performed for 30 minutes in 10% normal goat serum (NGS; Gibco, Life Technologies Australia Pty Ltd., Mulgrave, Australia) diluted using a 0.05 molL‐1 tris‐buffered saline (1 × TBS; pH 7.4) wash buffer solution with additional Triton‐X100 detergent (0.1%). Sections underwent primary antibody (IBA1; 1:1000, rabbit monoclonal; 019‐1974, Wako Pure Chemical Industries, Japan) incubation at 4oC overnight. All incubations were followed by three washes with gentle agitation for five minutes using 1 × TBS. Negative control sections incubated without primary antibodies were run concomitantly during each staining procedure. Secondary antibody incubation (biotinylated anti‐rabbit IgG (H+L), 1:200; BA‐1000, Vector Laboratories, Burlingame, CA, USA) was performed at room temperature (RT) for one hour followed by an hour in avidin‐biotin‐peroxidase complex solution (1:100; Vectastain Elite ABC, Universal). DAB (0.7 mg/mL; SigmaFastTM 3, 3’‐diaminobenzidine tablets, Merck & Company, Inc., Kenilworth, NJ, USA) in an aqueous solution containing urea hydrogen peroxide tablet (0.67 mg/mL; H2O2 equivalence, 0.24 mg/mL) was applied for 15 seconds. Regressive counterstaining involving sequential immersion into hematoxylin 28, followed by acid alcohol (two second immersion) and Scott’s blueing solution (45 seconds) was performed prior to cover slipping using DPX mounting media (06522‐Sigma Pharmaceuticals, Rowville, VIC, Australia).
Quantification of microglia
Microglia were quantified from images acquired by an Olympus VS‐120 slide scanner following an adapted stereological approach 27. Briefly, images of three 500‐μm wide strips of the full cortical thickness were acquired from areas where the gray matter was at its thinnest and strictly parallel to the gray matter‐white matter boundary. An eyepiece graticule at 200 × magnification (0.25 mm2) with defined inclusion and exclusion lines was used as an unbiased counting frame. IBA1 + cells were counted if a nucleus and surrounding soma could be identified. Inter‐rater reliability was established between two experienced raters on 10% of sections with >95% concordance for both microglial and neuronal counts. All counts were expressed as densities and adjusted to account for cortical atrophy in AD by multiplying raw densities by the fraction of the mean cortical width of each case by the mean cortical width of all controls for each region. Total microglia were enumerated and microglia also subdivided into one of three subtypes based on their morphology: ramified, activated or dystrophic. Ramified cells were defined on the basis of their thin, highly branched processes and spherical soma. Activated microglia were defined by somal enlargement with either hyper‐ramification or a reduced number of thickened processes. Microglia that were ameboid in shape, large and lacking processes were counted as activated cells as they occurred infrequently. Dystrophic cells were identified by spheroidal swellings, fragmentation of cellular processes and reduced ramification 57. In addition, microglial clusters were also counted; arbitrarily defined as three or more cell bodies that could fit within a 50 μm2 graticule sub‐region.
Immunofluorescence
Immunofluorescent (IF) double labeling was performed to investigate relationships between microglia and Aβ‐ and tau‐positive pathology. Antigen retrieval was as described above, followed by a 12‐minute formic acid (90%) incubation at RT to optimize Aβ immunoreactivity. All incubations were followed by three washes for five minutes with gentle agitation using 0.01molL‐1 PBS (pH 7.4). Sections were co‐incubated in antibodies for IBA1 (1:1000) and Aβ (1:500; mouse monoclonal; M0872, Dako, Santa Clara, CA, USA) at 4oC overnight. Sections were co‐incubated in secondary antibodies (1:200; AlexaFluor 488, goat anti‐mouse IgG (H+L); A11001, Invitrogen, Waltham, MA, USA); (1:200; AlexaFluor 568, goat anti‐rabbit IgG (H+L); A11011, Invitrogen) for 72 hours with gentle agitation at 4oC in the dark and wrapped in aluminum foil. Autofluorescence caused by lipofuscin and aldehydes was minimized by washing sections for 10 minutes in 0.1% Sudan Black B (B.D.H Laboratory Chemicals Group) (70% ethanol diluent). Sections were mounted with DAPI FluoroshieldTM medium (F6057; Merck & Company, Inc., Kenilworth, NJ, USA) before coverslipping. Tau immunolabeling was performed as previously described 9. Briefly, Heat‐induced epitope retrieval was performed with sodium citrate (pH 6.0), before permeabilizing, blocking with BSA and overnight incubation in primary antibodies: total tau (1:500; Dako, K9JA) and IBA1 (1:50; Millipore, MABN92). Sections were co‐incubated in secondary antibodies (1:200; AlexaFluor 555 and 647, anti‐mouse and anti‐rabbit IgG (H+L), respectively; A21424 and A21244, respectively, Invitrogen). The total tau antibody has been previously shown to immunostain NFTs, neuritic plaques and neuropil threads and is comparable to phospho‐tau (Ser262; 12E8) immunoreactivity 42. Hoechst 33342 was added to label nuclei and sections were mounted in Prolong Gold (Invitrogen).
Aβ and tau imaging
Researchers were blind to section status throughout the staining and imaging process. Aβ IF slides were imaged using a Zeiss LSM 510 Meta confocal microscope at the Advanced Microscopy Facility (AMF), Bosch Institute, The University of Sydney. Aβ was quantified in three cortical strips using a 20×/0.8 objective. These regions of interest (ROIs) were located at random while visualizing DAPI staining. Cortical strips were constructed of serial images (450 μm2) from the surface to the gray‐white boundary; moving inward sequentially. Individual images were maximum intensity projections (MIPs) of three z‐slices (z‐step = 5 μm) centered at the area with the most intense level of staining and were thresholded manually. Widefield images of total tau IF slides were captured using an Olympus VS‐120 slide scanner. Whole section overviews using DIC and fluorescence were taken with the 10 × objective to clearly identify gray matter and overall level of staining. Four ROIs of 500 μm2 (totaling 1 mm2 per section) were systematically mapped out within mid‐cortical layers (II‐V) distributed throughout the entire section. A 6‐μm deep z‐stack comprised of seven z‐slices of each ROI was obtained using a 40×/0.95 objective. ROIs chosen were representative of the overall level of tau staining throughout the gray matter, allowing for an accurate assessment of tau pathology within the cortex. All slide scanner image files (.vsi) were opened in Fiji using the BioFormats Importer. A MIP was produced and saved as a .tif file for subsequent thresholding and positive pixel analysis. Tau‐positive plaques and tangles within all ROIs of each case were manually counted.
Neuronal staining and quantification
Neurons were quantified on 10‐μm formalin‐fixed paraffin‐embedded (FFPE) sections stained with 0.1% cresyl violet acetate. Sections were dewaxed in xylene and hydrated through a series of graded ethanol solutions. Cresyl violet solution was applied for eight minutes followed by a deionized water rinse. Differentiation was achieved by briefly washing sections in absolute ethanol. The appropriate level of staining was confirmed by light microscopy before sections were cleared in xylene for nine minutes and mounted with DPX for cover slipping. Neuronal counts were performed using an Olympus BX50 microscope at 200 × magnification.
Image analysis
Image analysis was performed using Fiji (NIH, Bethesda, MD, USA). Aβ and tau areal fraction, a more accurate indicator of pathology load than quantifying individual entities because of size variation, was determined by expressing a positive pixel count as a percentage of total pixels (Supplementary Figure S1). However, to enumerate the percentage of Aβ‐immunopositive plaques (Aβ plaques) that were associated with a microglial cluster, the density of Aβ plaques was also calculated for each region. Given the subjective nature of microglial subtype classification, automated morphometric image analyses were also performed to complement traditional, qualitative cell counts. Cortical strip images of IBA1‐stained IHC sections were used and underwent color deconvolution and manual thresholding where two primary parameters were measured: (i) “arborised area” which was determined by summing the area of all convex hull polygons overlaying each cell mask and (ii) “structural complexity” which was assessed using the Skeletonize 2D/3D ImageJ plugin 1 which provided total branch length, total number of branches and total number of junctions (Supplementary Figure S2).
Statistical analyses
The normality of all data was tested using the Shapiro‐Wilk test. Group differences were determined using either a Welch’s T test or a Mann–Whitney U test where data were not normally distributed, or chi‐squared test. Regional differences were compared using either one‐way analysis of variance (ANOVA) with Games‐Howell test for pairwise comparisons or Kruskal‐Wallis test with Dunn’s test for pairwise comparisons using SPSS statistics 24 (SPSS Inc., Chicago, IL, USA). P‐values < 0.05 were considered statistically significant. Potential confounders including brain pH, APOE genotype, post‐mortem interval (PMI) and fixation period along with age and sex were investigated by univariate analysis. Factors that were significantly associated with microglial subtypes, arborized area or skeletal analysis parameters were included in multivariate models along with case‐control status using JMP 10 (SAS Institute Inc., Cary, NC, USA). Microglial parameters that survived multivariate testing were further investigated by calculating Pearson’s r or Spearman’s ρ for relationships with tau or Aβ. All graphs presented here show significant findings following multivariate testing and were produced using GraphPad Prism 7.00 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Clinicodemographic characteristics
Pathologically confirmed AD cases and non‐demented controls were matched for age and sex. The AD cases were more likely to carry the APOE ε4 allele (P = 0.04), had greater AD‐type pathology according to the ABC scoring system 35 and Braak staging 7 and had lower brain pH (P = 0.03) and brain weight (P = 0.04) (Table 2).
Table 2.
Demographic, clinical and pathological characteristics of cohort. a
| Control (n = 15) | AD (n = 8) | P‐value | |
|---|---|---|---|
| Sex (male/female) | 4, 11 | 2, 6 | 0.9 |
| Age (years) | 83.9 ± 8.1 | 83.3 ± 6.8 | 0.9 |
| Disease duration (years) | – | 8.3 ± 3.0 | – |
| ABC score (Low, intermediate, high) | 10, 5, 0 | 0, 1, 7 | <0.0001 |
| Braak stages (0/I/II, III/IV, V/VI) | 11, 4, 0 | 0, 2, 6 | 0.0002 |
| Brain pH | 6.4 ± 0.27 | 6.2 ± 0.24 | 0.03 |
| PMI (hours) | 16.2 ± 11.1 | 14.5 ± 11.5 | 0.8 |
| Fixation (months) | 52.9 ± 29.7 | 37.5 ± 23.9 | 0.2 |
| Brain weight (g) | 1241 ± 150.7 | 1086 ± 163 | 0.04 |
| APOE ε4 (no/yes) | 10, 4 b | 2, 6 | 0.04 |
Mean ± standard deviation throughout.
APOE genotype of one control case missing (M21).
Neuropathology
Cortical thickness differed between AD cases (2.1 ± 0.3 mm) and controls (2.6 ± 0.5 mm, P = 0.01) in the ITC, whereas cortical thinning was not observed in the PVC (2.2 ± 0.2) or SFC (2.8 ± 0.4) of AD cases compared to controls (PVC = 2.2 ± 0.2, P = 0.6; SFC = 2.7 ± 0.4, P = 0.8). Neuronal loss was observed in the SFC (145.8 ± 63.5 cells) and ITC (132.9 ± 29.6) of AD cases compared to controls (SFC = 207.2 ± 35.1; ITC = 220.7 ± 69.2) (Figure 1A). Aβ areal fraction in the PVC (1.2 ± 0.5%), SFC (1.6 ± 1.5%) and ITC (1.8 ± 0.6%) of AD cases was greater compared to controls (PVC = 0.3 ± 0.5%, P = 0.0004; SFC = 0.5 ± 0.6%, P = 0.02; ITC = 0.6 ± 0.8%, P = 0.002) (Figure 1B). Similarly, the density of Aβ plaques in the PVC (22.2 ± 17.8 plaques/mm2), SFC (37.1 ± 28.6) and ITC (40.6 ± 19) of AD cases was higher compared to controls (PVC = 6.6 ± 12.8, P = 0.003; SFC = 11.5 ± 15.8, P = 0.01; ITC = 14.4 ± 20.9, P = 0.008) (Figure 1C). Tau areal staining in the mid‐cortical layers was significantly greater in AD cases (PVC = 1.4 ± 0.6%; SFC = 1.9 ± 0.8%; ITC = 1.8 ± 0.6%) compared to controls (PVC = 0.01 ± 0.03%, P < 0.0001; SFC = 0.07 ± 0.2%, P < 0.0001; ITC = 0.1 ± 0.2%, P < 0.0001) across all three regions (Figure 1D). The density of cortical tau‐positive neuritic plaques was significantly higher in the PVC (18.8 ± 12.5 plaques/mm2), SFC (8.4 ± 6.0) and ITC (7.4 ± 9.4) of AD cases compared to controls (PVC = 0.2 ± 0.8, P < 0.0001; SFC = 0.4 ± 1.4, P = 0.0001; ITC = 0.4 ± 0.8, P = 0.002) (Figure 1E). Cortical NFT density was significantly greater in AD cases (PVC = 9.4 ± 8.7 NFTs/mm2; SFC = 25.4 ± 13.9; ITC = 21.9 ± 20.4) compared to controls (PVC = 0.2 ± 0.6, P < 0.0001; SFC = 0.8 ± 3.1, P < 0.0001; ITC = 0.7 ± 1.9, P < 0.0001) in all regions (Figure 1F). Pathological load did not differ significantly between any of the three regions in AD or control cases.
Figure 1.
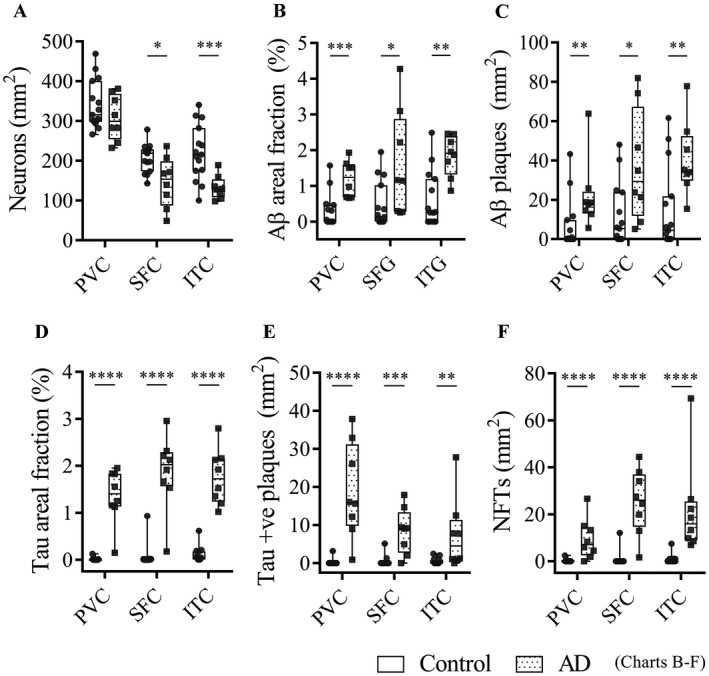
Neuropathological characterization. Box and whisker plots show mean values for: cortical neuronal density (A), cortical Aβ areal fraction (percentage of positive pixels stained) (B), Aβ plaque density (C), cortical tau areal fraction (D), tau‐positive neuritic plaque density (E) and NFT density (F). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Error bars indicate SD throughout.
Loss of healthy microglia in the ITC of AD brains
Total IBA1‐immunopositive microglia and commonly defined morphological subtypes: ramified (Figure 2A), activated and dystrophic were quantified in the three cortical regions of AD cases and controls. A range of phenotypes within the activated (Figure 2B,C) and dystrophic (Figure 2D,E) subtypes were seen within all individuals as well as clusters made up of activated microglia in those individuals with AD pathology (Figure 2F). Total microglia were lower in the ITC of AD cases compared to controls. This was exclusively caused by a reduction in the number of ramified microglia in the ITC of AD cases (Table 3). In contrast, neither activated nor dystrophic microglia differed in total number, or as a percentage of total microglia, between AD cases and controls in any of the three regions. Microglial clusters were relatively rare in all regions but were seen more frequently in the PVC of AD cases compared to controls. There was no difference in the density of clusters in the SFC or ITC between AD cases and controls.
Figure 2.
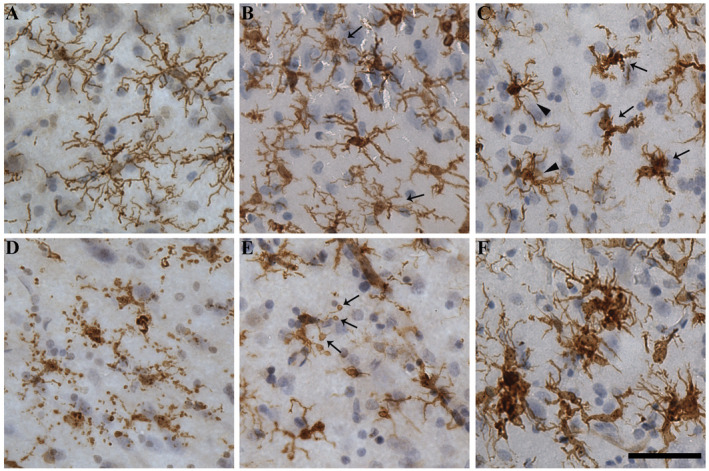
Microglial morphologies. Representative photomicrographs from the ITC show exemplars of microglial subtypes quantified here. A. Ramified microglia seen in a control case (M20) have small, spherical soma and symmetrically distributed, thin, highly branched processes. B. Activated microglia in an AD case (M09) that are hypertrophied and have reduced branching compared to ramified microglia. Arrows indicate ramified microglia for comparison. C. Activated microglia that are ameboid in morphology (arrows) are seen among other activated cells (arrow heads) in another AD case (M11). D. Dystrophic microglia that are pseudo‐fragmented with discontinuous IBA1 staining of processes in the same AD case (M11). E. A dystrophic microglia that is deramified and has spheroidal swellings at the end of its processes (arrows) from an AD case (M13). F. Clusters of activated microglia seen in an AD case (M09). Nuclei counterstained with hematoxylin. Scale bar = 50 μm.
Table 3.
Quantification of the morphological subtypes of microglia.
| Control (cells/mm2) | % | AD | % | P‐value | |
|---|---|---|---|---|---|
| PVC | |||||
| Total Microglia | 140.7 ± 45.2 | 139.6 ± 43.1 | 0.95 | ||
| Ramified microglia | 55.3 ± 34 | 39.3 | 39.5 ± 25 | 28.3 | 0.26 |
| Activated microglia | 55 ± 29.3 | 39.1 | 64 ± 27.4 | 45.8 | 0.48 |
| Dystrophic microglia | 30.4 ± 17.3 | 21.6 | 36 ± 19.9 | 25.8 | 0.48 |
| Microglial clusters | 0.7 ± 1.9 | 2.8 ± 2.3 | 0.03 | ||
| SFC | |||||
| Total Microglia | 188.4 ± 59.6 | 187.1 ± 62 | 0.96 | ||
| Ramified microglia | 70.8 ± 36.9 | 37.6 | 49.1 ± 23.8 | 26.2 | 0.15 |
| Activated microglia | 81.6 ± 47 | 43.3 | 90.4 ± 42.9 | 48.3 | 0.67 |
| Dystrophic microglia | 36 ± 23.4 | 19.1 | 47.6 ± 24.3 | 25.4 | 0.28 |
| Microglial clusters | 0.8 ± 1.1 | 3.2 ± 3 | 0.05 | ||
| ITC | |||||
| Total Microglia | 205 ± 66 | 128 ± 52.2 | 0.01 | ||
| Ramified microglia | 77.3 ± 47.2 | 37.7 | 13.8 ± 8.9 | 10.8 | 0.001 |
| Activated microglia | 86.6 ± 42.4 | 42.2 | 70.6 ± 60.4 | 55.2 | 0.46 |
| Dystrophic microglia | 41.1 ± 23.3 | 20.0 | 43.7 ± 30.5 | 34.1 | 0.82 |
| Microglial clusters | 1.1 ± 1.3 | 1.9 ± 2.1 | 0.28 |
Non‐demented, higher pathology‐controls have increased activated microglia
The control cases were sub‐divided into five “high‐pathology control cases” (HPCs), whose levels of AD‐type pathology were similar to the AD cases and 10 “low‐pathology control cases” (LPCs) with low or no AD pathology (Table 2). ITC gray matter of HPCs was thicker compared to LPCs (P = 0.01; Figure 3A). Aβ positive immunostaining (P = 0.02; Figure 3B) and cortical tau immunostaining (P = 0.01; Figure 3C) was also increased compared to LPCs. This also coincided with more total (P = 0.02) and activated microglia (P = 0.01) in HPCs compared to LPCs (Figure 3D). Group differences presented here survived multivariate testing (see Supplementary tables ST1 and ST2 for univariate and multivariate tests that included data for control cases only).
Figure 3.
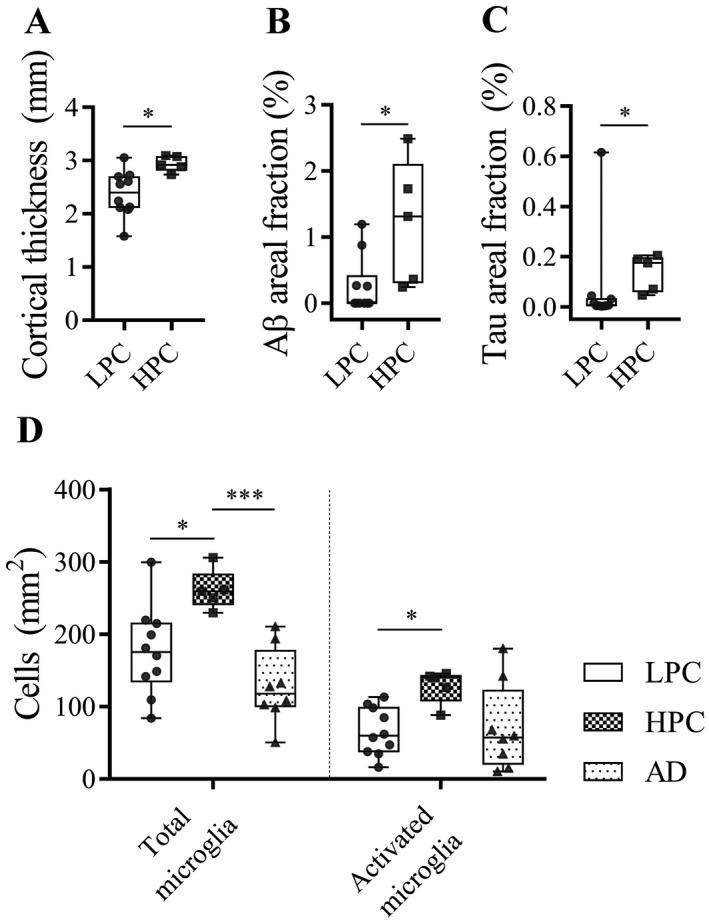
Comparisons between low pathology controls (LPCs) and high‐pathology controls (HPCs). A. HPCs had thicker cortices, elevated Aβ (B) and tau areal staining (C) compared to LPCs in the ITC. D. Total and activated microglia were also higher in HPCs compared to LPCs.
Reduced microglial arborized area and structural complexity in AD
A reduction in the arborized area was observed in the SFC and ITC of AD cases compared to controls, but not in the PVC (Figure 4A; Table 4). Similarly, a standard positive pixel analysis showed these same effects (Figure 4B). A skeletal analysis showed that the total branch length, the total number of branches and the total number of junctions were significantly reduced in AD cases compared to controls in the ITC (Table 4). Total junctions were also significantly reduced in the SFC of AD cases compared to controls, but there were no changes in any of these parameters in the PVC.
Figure 4.
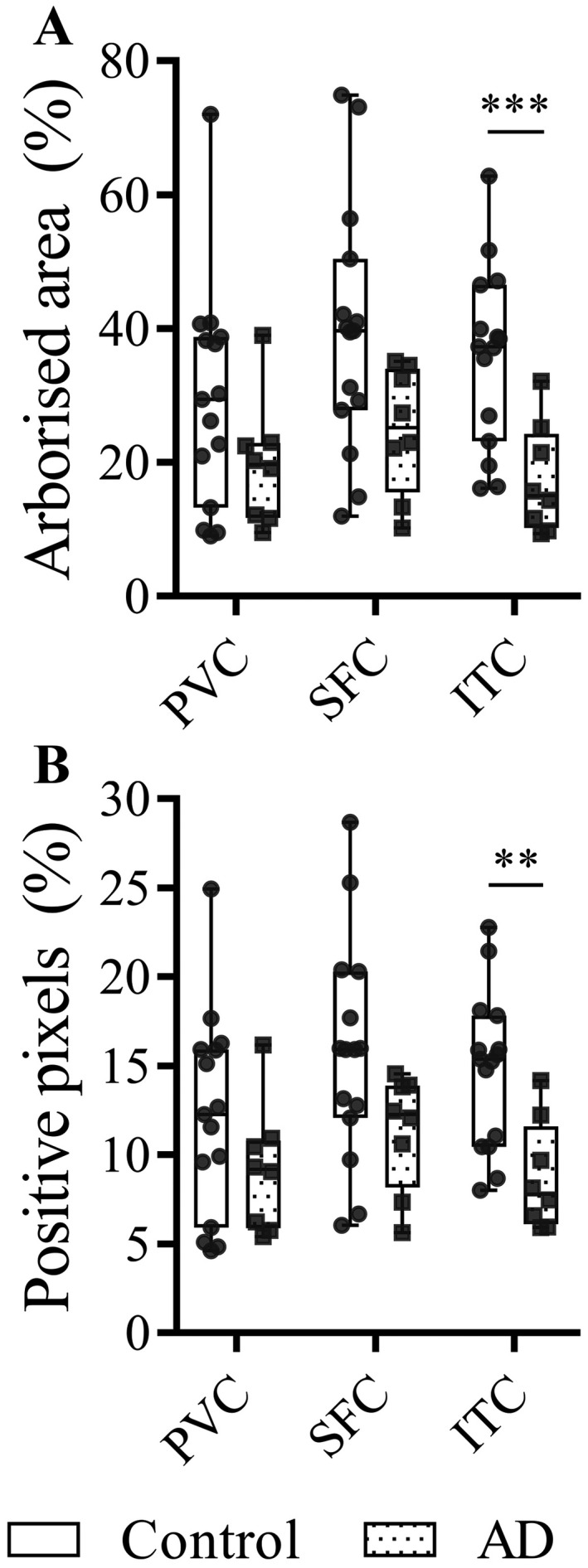
Microglial arborized area. Box and whisker plots demonstrating the area of tissue covered by microglial processes as determined using the convex hull analysis (A) and IBA1 positive pixel analysis (B) in the PVC, SFC and ITC of AD cases and controls.
Table 4.
Microglial structural complexity.
| Control | AD | P‐value | |
|---|---|---|---|
| Arborized area (%) | |||
| PVC | 29.3 ± 16.7 | 19.7 ± 9.4 | 0.09 |
| SFC | 39.6 ± 18.5 | 24.8 ± 9.4 | 0.02 |
| ITC | 35.8 ± 13.4 | 17.5 ± 8.1 | 0.0006 |
| Positive pixels (%) | |||
| PVC | 12.2 ± 5.7 | 9.2 ± 3.6 | 0.2 |
| SFC | 15.8 ± 6.2 | 11.3 ± 3.3 | 0.03 |
| ITC | 14.8 ± 4.4 | 8.8 ± 3.1 | 0.001 |
| Summary of skeletal analyses (mm2) | PVC | ||
| Total branch length (mm) | 47.2 ± 33.8 | 32.9 ± 29.3 | 0.06 |
| Total #branches | 3873 ± 1406 | 3042 ± 805 | 0.14 |
| Total #junctions | 1114 ± 524.2 | 756.1 ± 312.1 | 0.09 |
| SFC | |||
| Total branch length (mm) | 78.6 ± 68.6 | 44.6 ± 36 | 0.1 |
| Total #branches | 5385 ± 1873 | 3949 ± 1139 | 0.06 |
| Total #junctions | 1762 ± 854.5 | 1031 ± 426.2 | 0.03 |
| ITC | |||
| Total branch length (mm) | 65.7 ± 50.9 | 21.9 ± 8.8 | 0.0002 |
| Total #branches | 5480 ± 2091 | 2665 ± 1110 | 0.002 |
| Total #junctions | 1685 ± 757.6 | 625.2 ± 386.6 | 0.001 |
Microglial clustering was not a major feature of the AD cortex
Clusters of microglia (three or more cell bodies within a 50‐μm2 graticule field) were occasionally found near cored and diffuse Aβ plaques and tau‐positive plaques in both the control (Figure 5A–F) and AD groups (Figure 5G–O). These microglia were predominantly activated or, less commonly, dystrophic in morphology (Figure 5G–I). There was no difference between the percentage of Aβ plaques associated with a microglial cluster between the AD and control groups in any of the three regions: PVC (AD = 16.8 ± 14.4%, control = 12.2 ± 23.4; P = 0.15), SFC (AD = 12.4 ± 12.7, control = 5.8 ± 5.5; P = 0.28) and ITC (AD = 5.4 ± 6.5, control = 17.2 ± 19.9; P = 0.25) (Figure 5J–L) or tau‐positive plaques (Figure 5M–O) in either the AD cases or controls.
Figure 5.
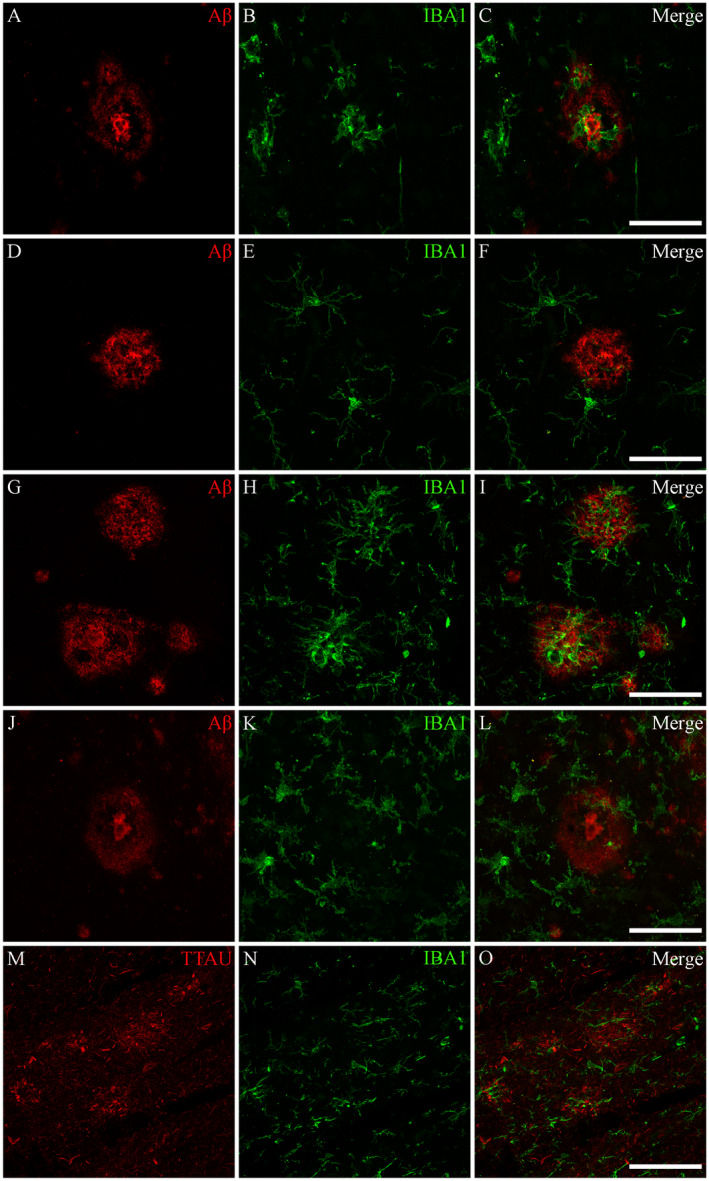
Associations between microglia and AD plaques. Microglia were found to only occasionally cluster around tau‐positive plaques and Aβ‐immunopositive diffuse and cored plaques in both AD and control cases. Photomicrographs of immunofluorescent labeling for Aβ (A), IBA1 (B) and the merged image (C) from a control case with elevated AD‐type pathology (M23) demonstrates a cluster of ameboid microglia associated with a cored Aβ plaque. D–F. Ramified microglia and no microglial activation associated with a cored plaque of another control case (M19). Similarly, for AD cases, microglia were found to only occasionally cluster around Aβ plaques; as seen in the photomicrographs from an AD case (M09) demonstrating both significant clustering around diffuse and cored Aβ plaques (G–I), and an absence of microglial clustering (J–L). M–O. Tau‐positive plaques from the same AD case demonstrating an absence of microglial clustering. All photomicrographs were acquired using a Zeiss LSM 800 confocal microscope (40×/1.3 Oil). Scale bars = 50 μm.
Morphological changes of microglia correlate with tau pathology
Potential confounders of microglial parameters including brain pH, PMI and fixation time were investigated by univariate analyses (Supplementary table ST3) and those that were or approached significance were included in multivariate analyses with case status for the three regions. Neuronal loss in the SFC and ITC, and reduced arborized area, IBA1 positive pixels, total branch length, total number of branches and number of ramified microglia in the ITC, as well as increased density of microglial clusters in the PVC remained significantly different following multivariate testing (Table 5; refer to Supplementary table ST4 for all multivariate statistics).
Table 5.
Summary of multivariate analyses (case‐control differences).
| PVC | SFC | ITC | ||||
|---|---|---|---|---|---|---|
| R 2 | P‐value | R 2 | P‐value | R 2 | P‐value | |
| Neurons | 0.26 | 0.01 | 0.36 | 0.003 | ||
| Arborized area | 0.36 | 0.003 | ||||
| IBA1 positive pixels | 0.35 | 0.004 | ||||
| Total branch length | 0.22 | 0.03 | ||||
| Total #branches | 0.12 | 0.04 | ||||
| Ramified microglia | 0.39 | 0.002 | ||||
| Microglial clusters | 0.2 | 0.04 | ||||
Those microglial parameters associated with AD status were further investigated for associations with either tau or Aβ pathology (Table 6). In the ITC, neurons, arborized area, IBA1 positive pixels and ramified microglia were all inversely correlated with the level of tau staining. There was also an inverse correlation between total branch length and Aβ in the ITC. Microglial clusters were positively correlated with both Aβ and tau staining in the SFC, and only tau staining in the PVC.
Table 6.
Neuropathological correlations. a
| PVC | SFC | ITC | |||||
|---|---|---|---|---|---|---|---|
| Pathology | R 2 | P‐value | R 2 | P‐value | R 2 | P‐value | |
| Neurons | Tau b | 0.27 | 0.01 | ||||
| Arborized area | Tau | 0.2 | 0.03 | ||||
| IBA1 positive pixels | Tau | 0.18 | 0.04 | ||||
| Total branch length | Aβ b | 0.2 | 0.03 | ||||
| Ramified microglia | Tau | 0.27 | 0.01 | ||||
| Microglial clusters | Aβ b | 0.4 | 0.001 | ||||
| Tau b | 0.27 | 0.01 | 0.12 | 0.04 | |||
All correlations presented in this table include variables that are significantly different between groups and remained so following multivariate analysis (excluding microglial clusters in the SFC, where P = 0.05).
Indicates most significant correlation determined by multivariate analysis that included both Aβ and tau areal fractions (following univariate analyses demonstrating correlations with both pathologies).
Brain pH is an important effector of microglial dystrophy
Among the potential confounders, brain pH was inversely correlated with a number of different microglial parameters; most notably the presence of dystrophic microglia (Table 7). The exception was ramified microglia in the SFC where a positive correlation was observed. A combined analysis of cases and controls showed that dystrophic microglia were inversely correlated with brain pH in all three regions (Figure 6).
Table 7.
Brain pH effects on microglial morphology.
| PVC | SFC | ITC | ||||
|---|---|---|---|---|---|---|
| R 2 | P‐value | R 2 | P‐value | R 2 | P‐value | |
| Arborised area | 0.24 | 0.02 | 0.37 | 0.003 | ||
| IBA1 positive pixels | 0.22 | 0.03 | 0.4 | 0.002 | ||
| Total #branches | 0.44 | 0.0008 | ||||
| Total #junctions | 0.4 | 0.002 | 0.45 | 0.0006 | ||
| Ramified microglia | 0.64 | <0.0001 | ||||
| Dystrophic microglia | −0.56 a | <0.0001 | −0.55 a | 0.008 | −0.56 a | 0.006 |
Spearman’s ρ.
Figure 6.
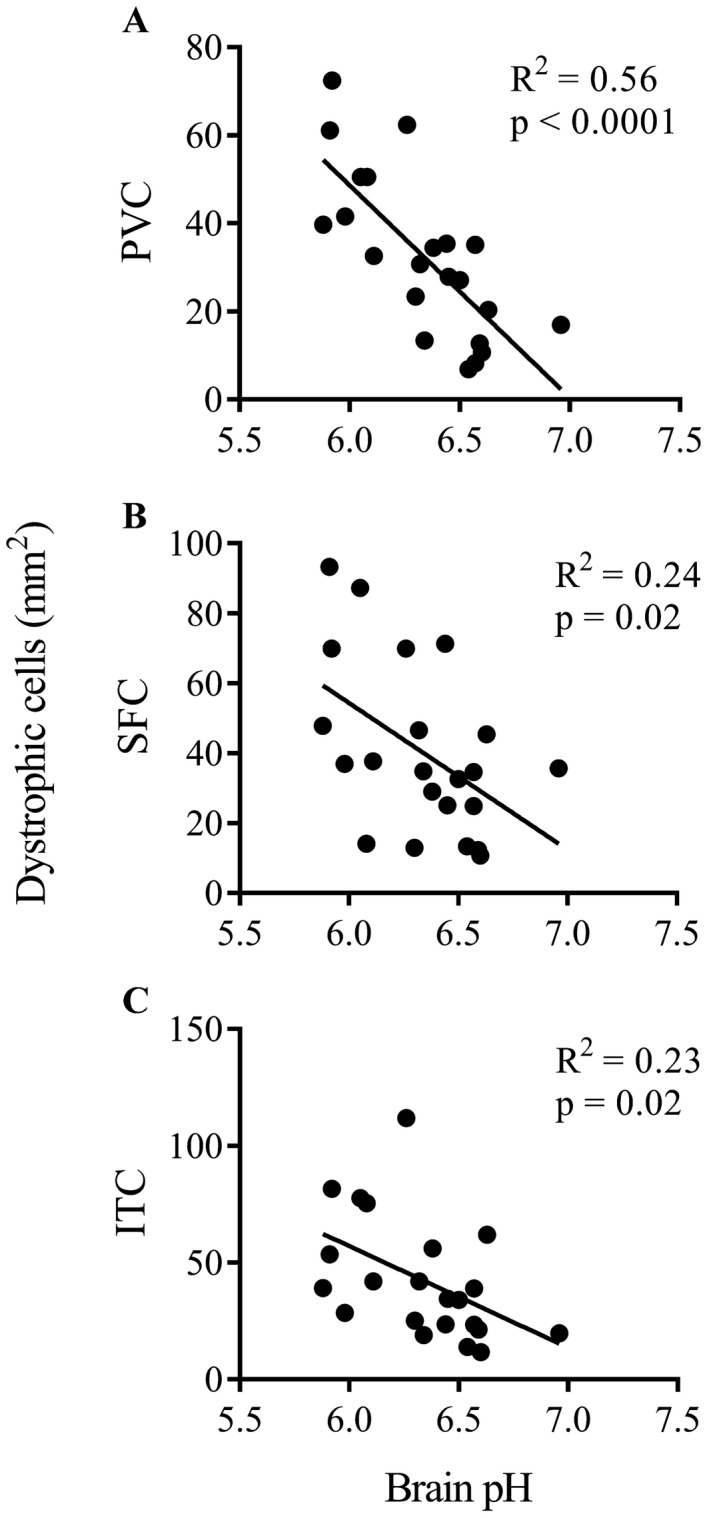
Dystrophic microglia and brain pH. Scatter plots and regression lines show the inverse relationships between dystrophic microglia and brain pH in the PVC (A), SFC (B) and ITC (C).
Discussion
This study undertook to provide clarity on how microglia relate temporally to the progression of Alzheimer’s disease by comparing morphological subtypes in differentially affected areas of post‐mortem brain tissue. Unlike previous studies, our findings showed that activated microglia were only increased in high‐pathology controls while clustering of microglia with Aβ plaques was uncommon and highest in the mildly affected PVC (~17%), suggesting that there is an early microglial reaction to Aβ but this is prior to the onset of dementia. Whereas only 12% and 5% of Aβ plaques were associated with a microglial cluster in the SFC and ITC, respectively. Second, the severely affected ITC, was characterized by a reduced number of ramified (healthy) microglia. Third, dystrophic microglia were not associated with AD status or pathology, but rather brain pH.
As expected, AD cases had greater quantities of Aβ and tau‐positive plaques and neurofibrillary pathology compared to non‐demented controls. The ITC showed a reduction in the number of neurons and AD‐related cortical thinning, consistent with a more advanced stage of AD than the SFC, which had reduced neurons but no cortical atrophy and the PVC in which neither of these measures was affected. Therefore, changes seen in the PVC can be viewed as representative of earlier disease mechanisms; with previous pathological and molecular studies also suggesting that the amount of tau pathology in the PVC has minimal effects on the neurons in this region 22, 28. Intra‐regional differences in cortical pathology was not as extensive as predicted here from staging schema and volumetric studies. The commonly used ABC staging criteria for AD is semi‐quantitative in nature, stressing the spread of pathology rather than the quantity of pathology, specifically as it pertains to each defined phase or stage of the criteria 21. Indeed, it is quite rare to find studies that have systematically quantified AD pathology across cortical regions, with those that have, describing significant heterogeneity in the accumulation of plaques and NFTs within regions and limited regional differences 37, 64.
The working hypothesis for how microglia contributes to AD pathogenesis has been that activated cells associate with plaques and produce a neurotoxic milieu that culminates in neuronal degeneration 30, 41, 46. Others have suggested that astrocytes may be equally or more prominent in this area 48, 49, 63, while Liddelow and colleagues recently showed that the interplay between microglia and astrocytes is critical by demonstrating that microglia‐derived cytokines induce a neurotoxic phenotype in astrocytes 29. In contrast, work begun by Streit, Graeber and Kreutzberg 55 and continued by Stevens, Barres and colleagues opined that a loss of microglial function and particularly a deficit in their role in synaptic maintenance may be more important in AD pathogenesis 18. Streit and colleagues suggested that dystrophic microglia were an early and prominent feature of AD 54, an idea which may also be relevant to other neurodegenerative diseases such as Dementia with Lewy bodies 3.
Yet, rather than confirming the predominance of either activated or dystrophic microglia in the AD cortex, the major finding here was a reduction in ramified microglia in the ITC only, and that this loss occurred in the absence of a commensurate increase in either the activated or dystrophic subtypes. In contrast, activated microglia were significantly greater in controls with sufficient AD pathology 35 to be pathologically diagnosed with AD. If these “high pathology” controls are representative of the proposed preclinical AD phenotype 53 then microglial activation may be an early driver of the disease. However, the fact remains that microglial reactivity seen in this group was not associated with dementia and so could alternatively be considered a protective mechanism against the neurotoxic effects of relatively high AD‐type pathology, an idea consistent with transition events of protective disease‐associated microglia (DAM) occurring at early stages of the disease process 25. Furthermore, as DAM are known to localize closely around plaques, the possibility that this activation is associated with neuroprotective mechanisms is further strengthened by the observation of increased microglial clustering in PVC, an area that may be considered representative of earlier stages in the disease process caused by the milder effects of AD observed here.
Given the turnover of microglia in the adult human brain 43, the loss of microglia could represent a decrease in proliferation or increase in cell death. Alternatively, it may represent a loss of IBA1 immunoreactivity caused by the downregulation of this calcium binding protein. The latter has important implications for microglial functionality as reduced IBA1 would result in impaired mobility and phagocytosis 24. Additional pan‐microglial markers are required to differentiate between these two scenarios. Indeed, previous investigations have observed significant increases in other markers, such as MHC II, with at least one identifying an IBA1‐/MHC II+ subpopulation 48. However, this phenotypic subpopulation represented a minority of cells in that study. Alternatively, this question could be approached with proliferative and apoptotic markers but our experience with the proliferative marker Ki‐67 10 and the apoptotic marker, cleaved caspase 3 (unpublished data) are that these are rare events in the adult brain and a single “point in time” assay may not be informative for a chronic disease such as AD.
As Aβ is widely regarded as a precursor to tau pathology, further temporal information on the role of microglial subtypes in AD pathogenesis was sought by determining the relative associations with Aβ or tau load. Microglial clusters were correlated with both Aβ and tau pathology in the PVC and SFC (univariate testing), representing early microglial reactivity that may be associated with neuroprotective processes and potentially corresponding with DAM that have been investigated elsewhere 25. Microglial arborized area, IBA1 positive pixels and the number of ramified microglia were inversely correlated with the level of tau in the ITC, which is indicative of the neurodegenerative stage of the disease and is consistent previous reports of microglial degeneration following phagocytosis of AT8 and/or AT100‐postive phospho‐tau species 45.
The other major finding here was an inverse correlation between brain pH and dystrophic microglia in all three regions. A low brain pH is often indicative of a severe and prolonged agonal period 12, 34, common in patients with neurodegenerative diseases. The antemortem factors and events that comprise a patient’s agonal period impact a patient’s brain pH. These factors include: duration of hospitalization, coma, respiratory illness and need for ventilation, systemic inflammation and duration of terminal phase. Unlike the work of Streit and colleagues, there were no differences in the number of dystrophic microglia, including in a sub‐cohort excluding samples with pH < 6.0. It is unclear whether previous studies proposing a link between microglial dystrophy and progression of AD considered differences in brain pH 54; however, the loss of healthy microglia in the ITC here still supports a loss of function hypothesis for the role of microglia in AD. More widely the prominence of immune signaling pathways in omic studies using post‐mortem brain tissue could similarly represent the influence of brain pH rather than a general inflammatory mechanism in brain disease 15, 50, 62, 65.
This investigation had three important limitations. Namely, the subjective categorization of the morphological subtypes of microglia, the use of a single microglial marker, IBA1 and the small cohort size. Microglial activation does not follow a monophasic (“all‐or‐nothing”) process, rather these cells respond to changes in their surroundings in a calibrated manner, thereby allowing for a wide range of morphological presentations that can be difficult to categorize. Microglia are rapidly responsive cells operating in time scales of hours and days unlike AD, the archetypal chronic brain disease. This response time may well account for the relationship between dystrophic microglia and brain pH, a proxy for agonal period. Although IBA1 is recognized as an effective pan‐microglial marker 23, more subtle changes in activation including phagocytic status may have been missed. For example, other human post‐mortem studies have demonstrated upregulation of major histocompatibility complex class II (MHC II) and cluster of differentiation 68 (CD68), markers of activation and phagocytosis, respectively; see review 19.
Study strengths include the use of well‐characterized tissue and a previously verified stereological approach that ensured a quantitative exploration across the entire cortical strip for pathology, residual neurons and microglia 27. This stereological approach ensured that the sampling area is sufficient to generate data that is representative of the tissue section under investigation and by selecting cortical strips at areas where the cortical thickness is at its thinnest, oblique cuts made during tissue sectioning do not inflate thickness measurements. Furthermore, the largely subjective morphological characterization of microglia was corroborated by automated “skeletal” analysis which validated our previous observations of reductions in structural complexity and arborized area in the more severely affected ITC 9. A validated approach for normalization of cortical atrophy 39 was also employed, although this may still have underestimated the true loss of cells in the ITC of AD patients as atrophy in the rostrocaudal dimension is not taken into consideration. Reassuringly, a previous study has indicated that shrinkage in this dimension is minimal 20.
Overall, the novel findings here suggest that ramified microglia are lost in more advanced stages of AD. The loss of the surveillance and support of microglia may cause neurons to be more susceptible to tau‐related degeneration. While activation, as seen in higher pathology controls and as an increase in the clustering of microglia in the PVC of AD cases, may be seen as key processes in preventing conversion to dementia. Future studies on morphological changes in microglia in human AD brain tissue must account for the patient’s brain pH as a potential confounder for dystrophic microglia. Further immunohistochemical studies with functional markers will be required to determine the more nuanced aspects of microglia dysfunction in AD, but the current findings suggest that strategies aiming to augment microglial function are likely to be more effective as a treatment than those attenuating their pro‐inflammatory activities, particularly in earlier phases of the disease.
Data Accessibility Statement
All data pertinent to this study is contained within the manuscript or supplementary tables and figures.
Supporting information
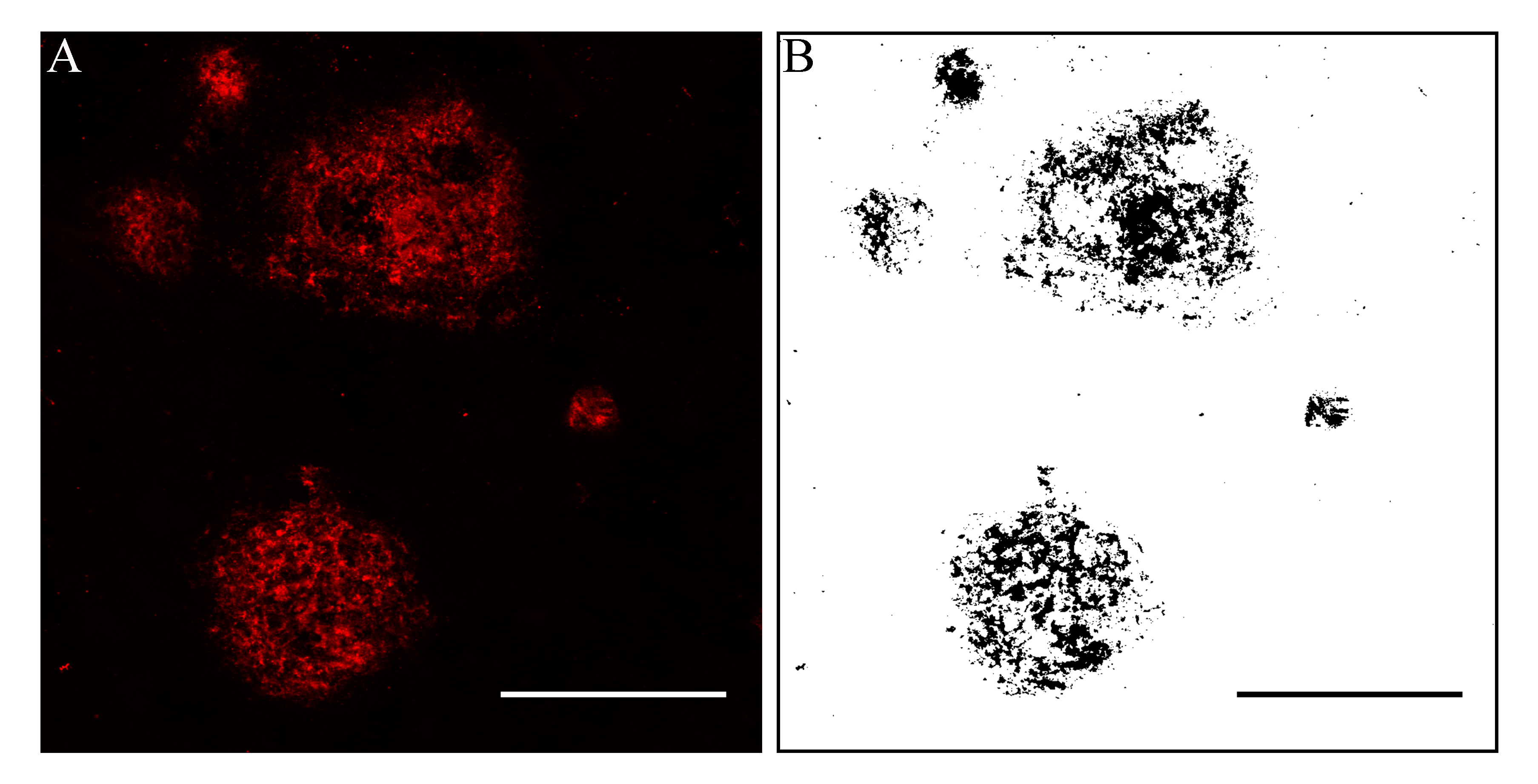
Figure S1. Quantification of Aβ and tau areal fraction. A. Confocal photomicrograph showing Aβ positive immunofluorescent plaques in the ITG of an AD case (M09). B. The corresponding digitally generated mark up used to calculate areal fraction with an ImageJ positive pixel algorithm. Scale bars = 50 μm.
{kind=link}
Figure S2. Quantification and analysis of microglial territoriality and skeletal structure. A. Photomicrograph demonstrating a single field of view with IBA1 + microglia and hematoxylin counterstained nuclei from an AD case (M09). ImageJ color deconvolution plugin was used to separate DAB (B) and hematoxylin (C) stains; with the third component (D) representing the remainder of the subtractive mixing algorithm. The image of the isolated DAB staining was manually thresholded, with the digitally generated mark up (E) allowing for the convex hull area to be calculated by summing all polygons bounded in green (F) and the production of tagged skeletal frames of each cell mask (G) that could be used to calculate total branch length, total number of branches and total number of junctions. Scale bar = 100 μm.
{kind=link}
Acknowledgments
The authors would like to thank the donors and their families for their kind gift. This work was funded by a Sydney Medical School Foundation Fellowship (GS). Brain tissue was received from the NSW Brain Tissue Resource Centre and Sydney Brain Bank. These brain banks are supported by the NHMRC of Australia, The University of New South Wales, Neuroscience Research Australia and the National Institute of Alcohol Abuse and Alcoholism [NIH (NIAAA) R28 AA012725].
References
- 1. Arganda‐Carreras I, Fernandez‐Gonzalez R, Munoz‐Barrutia A, Ortiz‐De‐Solorzano C (2010) 3D reconstruction of histological sections: application to mammary gland tissue. Microsc Res Tech 73(11):1019–1029. [DOI] [PubMed] [Google Scholar]
- 2. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42(3 Pt 1):631–639. [DOI] [PubMed] [Google Scholar]
- 3. Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ, et al (2015) Disease‐related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA (2012) Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 72(4):599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boche D, Perry VH, Nicoll JA (2013) Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39:3–18. [DOI] [PubMed] [Google Scholar]
- 6. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82(4):239–259. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E (1995) Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging 16(3):271–278; discussion 278–284. [DOI] [PubMed] [Google Scholar]
- 8. Cameron B, Tse W, Lamb R, Li X, Lamb BT, Landreth GE (2012) Loss of interleukin receptor‐associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer's disease. J Neurosci 32(43):15112–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies DS, Ma J, Jegathees T, Goldsbury C (2017) Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer's disease. Brain Pathol 27(6):795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dennis CV, Sheahan PJ, Graeber MB, Sheedy DL, Kril JJ, Sutherland GT (2014) Microglial proliferation in the brain of chronic alcoholics with hepatic encephalopathy. Metab Brain Dis 29(4):1027–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dubbelaar ML, Kracht L, Eggen BJL, Boddeke E (2018) The kaleidoscope of microglial phenotypes. Front Immunol 9:1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Durrenberger PF, Fernando S, Kashefi SN, Ferrer I, Hauw JJ, Seilhean D, et al (2010) Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J Neuropathol Exp Neurol 69(1):70–81. [DOI] [PubMed] [Google Scholar]
- 13. Giannakopoulos P, Hof PR, Michel JP, Guimon J, Bouras C (1997) Cerebral cortex pathology in aging and Alzheimer’s disease: a quantitative survey of large hospital‐based geriatric and psychiatric cohorts. Brain Res Brain Res Rev 25(2):217–245. [DOI] [PubMed] [Google Scholar]
- 14. Gomez‐Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41(1):17–24. [DOI] [PubMed] [Google Scholar]
- 15. Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, et al (2014) Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity‐dependent genes in autism. Nat Commun 5:5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol 9(8):857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Halliday GM, Double KL, Macdonald V, Kril JJ (2003) Identifying severely atrophic cortical subregions in Alzheimer’s disease. Neurobiol Aging 24(6):797–806. [DOI] [PubMed] [Google Scholar]
- 18. Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352(6286):712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hopperton KE, Mohammad D, Trepanier MO, Giuliano V, Bazinet RP (2018) Markers of microglia in post‐mortem brain samples from patients with Alzheimer's disease: a systematic review. Mol Psychiatry 23(2):177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hyman BT, Gomez‐Isla T, Irizarry MC (1998) Stereology: a practical primer for neuropathology. J Neuropathol Exp Neurol. 57(4):305–310. [DOI] [PubMed] [Google Scholar]
- 21. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al (2012) National Institute on Aging‐Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iacono D, O'Brien R, Resnick SM, Zonderman AB, Pletnikova O, Rudow G, et al (2008) Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol 67(6):578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia‐specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57(1):1–9. [DOI] [PubMed] [Google Scholar]
- 24. Kanazawa H, Ohsawa K, Sasaki Y, Kohsaka S, Imai Y (2002) Macrophage/microglia‐specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C‐gamma ‐dependent pathway. J Biol Chem 277(22):20026–20032. [DOI] [PubMed] [Google Scholar]
- 25. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169(7):1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 26. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553. [DOI] [PubMed] [Google Scholar]
- 27. Kril JJ, Halliday GM, Svoboda MD, Cartwright H (1997) The cerebral cortex is damaged in chronic alcoholics. Neuroscience 79(4):983–998. [DOI] [PubMed] [Google Scholar]
- 28. Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, et al (2008) Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics 33(2):240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lue LF, Brachova L, Civin WH, Rogers J (1996) Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J Neuropathol Exp Neurol 55(10):1083–1088. [PubMed] [Google Scholar]
- 31. Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, et al (2015) Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol Neurodegener 10:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mills JD, Sheahan PJ, Lai D, Kril JJ, Janitz M, Sutherland GT (2014) The alternative splicing of the apolipoprotein E gene is unperturbed in the brains of Alzheimer’s disease patients. Mol Biol Rep 41(10):6365–6376. [DOI] [PubMed] [Google Scholar]
- 33. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41(4):479–486. [DOI] [PubMed] [Google Scholar]
- 34. Monoranu CM, Apfelbacher M, Grunblatt E, Puppe B, Alafuzoff I, Ferrer I, et al (2009) pH measurement as quality control on human post mortem brain tissue: a study of the BrainNet Europe consortium. Neuropathol Appl Neurobiol 35(3):329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morris JC (1997) Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr 9(Suppl. 1):173–176; discussion 177–178. [DOI] [PubMed] [Google Scholar]
- 37. Nelson PT, Abner EL, Scheff SW, Schmitt FA, Kryscio RJ, Jicha GA, et al (2009) Alzheimer's‐type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett 450(3):336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. [DOI] [PubMed] [Google Scholar]
- 39. Oorschot DE (1994) Are you using neuronal densities, synaptic densities or neurochemical densities as your definitive data? There is a better way to go. Prog Neurogibol 44(3):233–247. [DOI] [PubMed] [Google Scholar]
- 40. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al (2013) Microglia promote learning‐dependent synapse formation through brain‐derived neurotrophic factor. Cell 155(7):1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perlmutter LS, Barron E, Chui HC (1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci Lett 119(1):32–36. [DOI] [PubMed] [Google Scholar]
- 42. Rahman T, Davies DS, Tannenberg RK, Fok S, Shepherd C, Dodd PR, et al (2014) Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer's disease. J Alzheimers Dis 42(4):1443–1460. [DOI] [PubMed] [Google Scholar]
- 43. Réu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, et al (2017) The lifespan and turnover of microglia in the human brain. Cell Rep 20(4):779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009) Inflammation in Alzheimer’s disease: amyloid‐beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurogibol 87(3):181–194. [DOI] [PubMed] [Google Scholar]
- 45. Sanchez‐Mejias E, Navarro V, Jimenez S, Sanchez‐Mico M, Sanchez‐Varo R, Nunez‐Diaz C, et al (2016) Soluble phospho‐tau from Alzheimer's disease hippocampus drives microglial degeneration. Acta Neuropathol 132(6):897–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y (1997) Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 94(4):316–322. [DOI] [PubMed] [Google Scholar]
- 47. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al (2012) Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74(4):691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Serrano‐Pozo A, Gomez‐Isla T, Growdon JH, Frosch MP, Hyman BT (2013) A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol 182(6):2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Serrano‐Pozo A, Mielke ML, Gomez‐Isla T, Betensky RA, Growdon JH, Frosch MP, et al (2011) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol 179(3):1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, et al (2017) A multi‐network approach identifies protein‐specific co‐expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst 4(1):60–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sheedy D, Say M, Stevens J, Harper CG, Kril JJ (2012) Influence of liver pathology on markers of postmortem brain tissue quality. Alcohol Clin Exp Res 36(1):55–60. [DOI] [PubMed] [Google Scholar]
- 52. Sheng JG, Mrak RE, Griffin WS (1997) Neuritic plaque evolution in Alzheimer's disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol 94(1):1–5. [DOI] [PubMed] [Google Scholar]
- 53. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al (2011) Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7(3):280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118(4):475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Streit WJ, Graeber MB, Kreutzberg GW (1988) Functional plasticity of microglia: a review. Glia 1(5):301–307. [DOI] [PubMed] [Google Scholar]
- 56. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45(2):208–212. [DOI] [PubMed] [Google Scholar]
- 57. Streit WJ, Xue QS, Tischer J (2014) Bechmann I. Microglial pathology. Acta Neuropathol Commun 2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sutherland GT, Janitz M, Kril JJ (2011) Understanding the pathogenesis of Alzheimer’s disease: will RNA‐Seq realize the promise of transcriptomics? J Neurochem 116(6):937–946. [DOI] [PubMed] [Google Scholar]
- 59. Sutherland GT, Kril JJ (2012) Alzheimer's Disease: Approaches to Pathogenesis in the Genomic Age, Neuroscience ‐ Dealing With Frontiers. InTech. [Google Scholar]
- 60. Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58(12):1791–1800. [DOI] [PubMed] [Google Scholar]
- 61. Tischer J, Krueger M, Mueller W, Staszewski O, Prinz M, Streit WJ, et al (2016) Inhomogeneous distribution of Iba‐1 characterizes microglial pathology in Alzheimer’s disease. Glia 64(9):1562–1572. [DOI] [PubMed] [Google Scholar]
- 62. van Kesteren CF, Gremmels H, de Witte LD, Hol EM, Van Gool AR, Falkai PG, et al (2017) Immune involvement in the pathogenesis of schizophrenia: a meta‐analysis on postmortem brain studies. Transl Psychiat 7(3):e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vehmas AK, Kawas CH, Stewart WF, Troncoso JC (2003) Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging. 24(2):321–331. [DOI] [PubMed] [Google Scholar]
- 64. Vermersch P, Frigard B, Delacourte A (1992) Mapping of neurofibrillary degeneration in Alzheimer's disease: evaluation of heterogeneity using the quantification of abnormal tau proteins. Acta Neuropathol 85(1):48–54. [DOI] [PubMed] [Google Scholar]
- 65. Warden A, Erickson E, Robinson G, Harris RA, Mayfield RD (2016) The neuroimmune transcriptome and alcohol dependence: potential for targeted therapies. Pharmacogenomics 17(18):2081–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Quantification of Aβ and tau areal fraction. A. Confocal photomicrograph showing Aβ positive immunofluorescent plaques in the ITG of an AD case (M09). B. The corresponding digitally generated mark up used to calculate areal fraction with an ImageJ positive pixel algorithm. Scale bars = 50 μm.
Figure S2. Quantification and analysis of microglial territoriality and skeletal structure. A. Photomicrograph demonstrating a single field of view with IBA1 + microglia and hematoxylin counterstained nuclei from an AD case (M09). ImageJ color deconvolution plugin was used to separate DAB (B) and hematoxylin (C) stains; with the third component (D) representing the remainder of the subtractive mixing algorithm. The image of the isolated DAB staining was manually thresholded, with the digitally generated mark up (E) allowing for the convex hull area to be calculated by summing all polygons bounded in green (F) and the production of tagged skeletal frames of each cell mask (G) that could be used to calculate total branch length, total number of branches and total number of junctions. Scale bar = 100 μm.
Data Availability Statement
All data pertinent to this study is contained within the manuscript or supplementary tables and figures.