

Abstract
Altered levels of steroids have been reported in the brain, cerebral spinal fluid and plasma of patients with mood disorders. Neuroimaging studies have reported both functional and structural alterations in mood disorders, for instance in the anterior cingulate cortex (ACC) and dorsolateral prefrontal cortex (DLPFC). In order to determine whether the endogenous production of steroids is altered in the ACC and DLPFC of patients with major depressive disorder (MDD) or bipolar disorder (BPD), quantitative real‐time PCR was performed to detect mRNA expression level of key enzymes in the steroid biosynthetic pathways. In MDD, a significant decrease in mRNA level of cytochrome P450 17A1 (CYP17A1, synthesizing C19 ketosteroids) in the ACC and a significant increase in mRNA levels of hydroxysteroid sulfotransferase 2A1 [SULT2A1, catalyzing the sulfate conjugation of dehydroepiandrosterone (DHEA)] were observed in the DLPFC, suggesting alterations in DHEA and its sulfate metabolite DHEAS levels. Decreased intensity and distribution of CYP17A1 immunohistochemical staining was found in the ACC of MDD patients. Interestingly, there was a significant positive correlation between the mRNA levels of CYP17A1 and tyrosine‐related kinase B (TrkB) full length isoform. In a unique post‐mortem human brain slice culture paradigm, BDNF mRNA expression was found to be significantly increased following incubation with DHEA. Together, these data indicate a close relationship between DHEA and BDNF‐TrkB pathways in depression. Furthermore, in the DLPFC, higher mRNA levels of 11β‐hydroxysteroid dehydrogenase‐1 (HSD11B1, reducing cortisone to the active hormone cortisol) and steroidogenic acute regulatory protein (STAR, facilitating the shuttle of cholesterol through the intermembrane space) were found in the MDD patients and BPD patients, respectively. In conclusion, this study suggests the presence of a disturbance in the endogenous synthesis of DHEA and DHEAS in mood disorders, which has a close relationship with BDNF‐TrkB signaling.
Keywords: anterior cingulate cortex, bipolar disorder, cytochrome P450 17A1, dorsolateral prefrontal cortex, hydroxysteroid sulfotransferase 2A1, major depressive disorder
Introduction
The anterior cingulate cortex (ACC) and dorsolateral prefrontal cortex (DLPFC) are of importance for mood regulation and are reported to be involved in the pathogenesis of mood disorders. Several neuroimaging studies have shown both structural and functional changes in these brain areas in depression, that is, abnormal volume or grey matter density and altered glucose metabolism or blood flow, and reversed changes in metabolism of such brain regions after pharmacological treatment or deep brain stimulation 9. In addition, neuropathological studies have revealed both neuronal and glial alterations in the ACC/DLPFC of mood disorder patients, that is, reduced neuronal and glial densities 12, 36. Moreover, we have observed changes in the expression levels of neurotransmitter‐related and stress‐related molecules 11, 33, 57, 58 in these brain areas in mood disorders.
Steroids are not only synthesized from cholesterol in the periphery by the adrenals and gonads, but also in the brain 4. The latter products are called “neurosteroids” and are thought to regulate gene expression and to modulate neuronal excitability through binding to receptors in the brain 39. Accumulating evidence suggests that steroids are implicated in the pathophysiology and treatment of mood disorders. For example, pregnenolone (PREG) and dehydroepiandrosterone (DHEA) levels are increased in the posterior cingulate and parietal cortex of patients with bipolar disorder (BPD) 28. Reduced concentrations of allopregnanolone (ALLO), a metabolite of progesterone, have been observed in the cerebral spinal fluid (CSF) of a clinical population of depressed patients 37, 38. On the other hand, the antidepressant fluoxetine normalizes ALLO levels in the mouse brain 32. Several steroids (ie DHEA and ALLO) were shown to have clear antidepressant‐ and anxiolytic‐like effects 42. Some observations further indicate that hormone replacement therapy is beneficial in mood disorder in peri‐ and post‐menopausal women 19 and in elderly depressed men 2. The possible involvement of steroids in depression is further supported by the observation that DHEA, PREG and their sulfate esters (DHEAS and PREGS), along with ALLO can modulate the activity of the hypothalamo‐pituitary‐adrenal (HPA) axis 29 whose hyperactivity is a major characteristic of depression 3. Although their pathological roles and mechanisms of action are at present far from clear, there is a growing amount of evidence regarding steroid alterations in mood disorders. In order to determine whether the endogenous production of steroids is altered in the ACC and DLPFC of patients with depressive disorders, we determined the expression level of key enzymes in the steroid biosynthetic pathways.
Steroids exert some neuroprotective and neurotrophic effects. For example, DHEA promotes spine synapse formation in hippocampal neurons 24 which is similar to brain‐derived neurotrophic factor (BDNF). A wide range of basic and clinical studies revealed that BDNF and its receptor tyrosine‐related kinase B (TrkB) play critical roles in depression and antidepressants 7, 27. In our previous study, we also found decreased BDNF and TrkB mRNA level in the ACC/DLPFC in the depressed cohort 34. However, there are so far no systematic studies on the changes in expression patterns of the steroidogenic enzymes and related regulatory molecules in the ACC/DLPFC from patients with mood disorders. Even though they have similar neurotrophic effects, the relationship between the neurosteroids and BDNF and TrkB in mood disorders has not been well explored.
In the present study, quantitative real‐time PCR (qPCR) was carried out to detect mRNA expression levels of key steroidogenic enzymes and related regulatory molecules in the isolated grey matter from ACC or DLPFC of elderly non‐suicidal mood disorder patients and their matched controls. Both major depressive disorder (MDD) and BPD were included in the diagnosed group. Alterations in mRNA expression levels of the enzymes were observed suggesting a role for steroids in the pathogenesis of mood disorder. Immunohistochemistry (IHC) in post‐mortem human ACC provided additional evidence for changes in protein levels. In addition, the mRNA expression levels of cytochrome P450 17A1 (CYP17A1, synthesizing C19 ketosteroids, including DHEA) appeared to be positively correlated to those of TrkB in the ACC of the MDD patients, suggesting a close relationship between CYP17A1 or the products of CYP17A1 and TrkB in MDD. To address this putative relationship between CYP17A1 and TrkB we treated post‐mortem human brain slices in short‐term cultures with DHEA (a major product of CYP17A1) or DHEAS. The use of post‐mortem human brain slice cultures was pioneered by our group and it has been established that many cells in the cultured slices remain viable for long periods in vitro 49, 50, 53, 54. Here, we show that treatment with DHEA increases BDNF mRNA levels in the human brain slices, compared to vehicle‐treated slices. Thus in ACC/DLPFC alterations in local steroid production are present in mood disorders and this may influence the BDNF‐TrkB pathway. Furthermore, in the DLPFC, higher mRNA levels of 11β‐hydroxysteroid dehydrogenase‐1 (HSD11B1, which reduces cortisone to the active hormone cortisol) and steroidogenic acute regulatory protein (STAR, inducing steroidogenesis by transporting cholesterol into the mitochondria) were found in the MDD patients and BPD patients, respectively.
Material and Methods
Subjects
Post‐mortem brain specimens from elderly non‐suicidal patients with chronic mood disorders and their matched controls without a primary psychiatric or neurological disease, as described before in detail 33, 34, 43, were obtained from the Netherlands Brain Bank (NBB), following informed written consent from the patients or their next of kin for the autopsy and use of brain material and clinical files for research purposes. The patients with mood disorders were clinically diagnosed with either MDD or BPD. The diagnosis was made in Dutch psychiatric hospitals on the basis of the presence and severity of the symptoms of either MDD or BPD as well as the exclusion of other psychiatric and neurological disorders according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV), and confirmed by a board certified psychiatrist (Drs. W.J.G. Hoogendijk, E. Vermetten or G. Meynen) according to the DSM‐IV criteria using the extensive medical records that are present in the NBB. The absence of neuropathological changes was confirmed in all subjects by systematic neuropathological investigation 47. Patients with mood disorders and controls were pairwise matched for sex and group‐matched for age, post‐mortem delay, clock time and month of death, cerebrospinal fluid (CSF) pH and brain weight. The samples were obtained from ACC of 12 depressed patients (5 MDD patients/ 7 BPD patients) and 12 matched controls (5 controls for MDD group/ 7 controls for BPD group) and DLPFC of 14 patients with mood disorders (5 MDD patients/ 9 BPD patients) and 14 matched controls (5 controls for MDD group/ 9 controls for BPD group). Detailed clinico‐pathological information and P‐values of matched parameters are given in Supporting Information Table S1.
Tissue collection, RNA isolation, cDNA synthesis and quantitative real‐time PCR
Grey matter containing all six layers was isolated from 50 μm thick cryostat sections cut from snap‐frozen DLPFC/ACC tissue as described before 6. RNA isolation, cDNA synthesis and quantitative real‐time PCR were performed as described before 20, 21. RNA integrity value (RIN), an indicator of isolated RNA quality, was determined and no difference was found between the mood disorder and control groups (see Supporting Information Table S2). QPCR reactions and analysis were performed as published before. The absolute amount of target genes was calculated by 1010 × E ‐Ct (E = 10–(1/slope)) 15. The geometric mean of the absolute amount of the stably expressed housekeeping genes was calculated as a reliable normalization factor to control for sampling differences. The absolute amount of the transcript obtained was divided by the normalization factor to get the normalized relative values (mRNA relative values in the figures) for the final statistical analysis. Detailed protocol and information on the primers for the target genes involved in steroid pathways are shown in Supporting Information Methods S3 and Supporting Information Table S4, respectively.
IHC for CYP17A1 and SULT2A1 in post‐mortem human ACC
We used IHC to examine the cellular localization and perform semi‐quantification of the cellular expression of the steroidogenic enzymes, SULT2A1 and CYP17A1, two genes that changed strongly in depression. The antibody CYP17A1 (Santa Cruz, sc‐46085) was a goat polyclonal IgG preparation against a peptide mapping at the N‐terminus of CYP17A1 of human origin. The specificity of CYP17A1 antibody was confirmed by IHC in fetal human dorsal root ganglia. There was no detectable signal using antisera immunodepleted with pure, bacterially expressed human CYP17A1, while there was clear signal with antisera without immunodepletion 41. The antibody SULT2A1 (Santa Cruz, sc‐18725) was a goat polyclonal IgG preparation against a peptide mapping within an internal region of SULT2A1 of human origin, and its specificity has been demonstrated in human liver and adrenal tissues using both western blot and immunohistochemical evaluations 14.
The IHC procedure on paraffin ACC sections was as follows. The sections were deparaffinized and rehydrated, followed a wash in TBS (0.05 M Tris, 0.15 M NaCl, pH 7.6). After antigen unmasking by heating the sections for 10 minute to a maximum 90°C in 0.01 M citrate buffer at pH 6.0, sections were allowed to cool down to room temperature. Subsequently sections were pre‐incubated in TBS‐milk (5% w/v in TBS) for 1 h to reduce non‐specific staining. Incubation with the first antibody (CYP17A1, 1:1000; SULT2A1, 1:500) in TBS containing 0.5% (v/v) Triton‐X 100, 0.25% (w/v) gelatin and 5% (w/v) milk (SUMI) at room temperature for 1 hour was followed by overnight incubation at 4°C. The next day, after rinsing with TBS, sections were incubated with anti‐goat IgG biotin‐labeled antibody (1:400; Vector laboratories) in SUMI at room temperature for 1 h. This was followed by rinsing in TBS and incubation with the avidin‐biotin complex (1:800; Vectastain ABC kit; Vector laboratories) for 1 h. Finally, sections were stained with 3.3′‐diaminobenzidine (DAB, Sigma) at 0.5 mg/ml in TBS, containing 0.23% (w/v) nickel‐ammoniumsulfate (Merck) and 0.01% H2O2. The reaction was stopped by washing in TBS and sections were dehydrated, cleared in xylene and coverslipped using Entellan (Merck). Photographs were collected with a Zeiss Axioskop microscope (Zeiss) with Neofluar objectives (Zeiss), a motorized XYZ stage and a camera (Sony). For semi‐quantitative analysis of each molecule of interest, the photographs for each region were coded and ranked from higher to lower intensity. A score were assigned to similar group categories as: – = no staining (absent); + = not widespread and transparent staining (weak); ++ = widespread staining with individual granules of the reaction product distinguishable (moderate); and +++ = widespread homogenous staining (strong); ++++ = widespread and intense opaque staining (very strong) 23.
Post‐mortem brain slice culture and steroid treatment
Fresh post‐mortem human brain material from prefrontal cortex of five controls subjects who died without a neurological disease was obtained from the NBB. Tissue, comprising cortical grey matter and a small amount of white matter, was cut in 300 µm thick slices using a McIlwain tissue chopper. Slices were plated in a 24‐well plate and were maintained for a short period (5 days) in culture as previously described 49, 50, 53, 54 with small modifications.
Neurobasal A medium (Invitrogen) was used and complemented with gentamycin (50 µg/ml; Invitrogen), HEPES (2 mM; Invitrogen), glutamax (2 mM; Invitrogen) and B27 medium supplement (1:50; Invitrogen). After plating, slices were treated with DHEA or DHEAS (Sigma Aldrich) dissolved in ethanol at four different concentrations: 0.1 nM, 1 nM, 10 nM and 100 nM, or ethanol only. After 2 days in culture, the medium was refreshed with 100 µl of new medium containing the appropriate concentration of steroids. After 5 days in culture, tissue was collected and quickly frozen in liquid nitrogen. Samples were stored at −80°C until use for RNA isolation and qPCR. qPCR was performed as described above and analyzed gene expression levels of BDNF and TrKB.FL which were normalized with the geometric mean of mRNA expression level of EF1a and GAPDH as housekeeping genes.
In a previous study, we found that the cells in the slices recovered from the agonal state of death during the first few days in vitro and that viability started to decline after 2–3 weeks 50. In order to confirm presence of viable cells in medium or medium plus 0.01% ethanol (in which steroids where diluted), some cultured slices were kept at the end of first experiment and viability staining with a Live/Dead kit (Life Technologies) was performed.
Statistical analysis
Statistical analysis was carried out with SPSS (version 17.0, SPSS Incorporation). To compare gene expression levels in ACC/DLPFC areas, the nonparametric Mann‐Whitney test was used because Shapiro‐Wilk test showed that the expression levels were not normally distributed. The difference between MDD or BPD group and their respective matched controls were analyzed in ACC and DLPFC separately. Correlations for gene expression levels were tested by the Spearman's correlation coefficient. For human brain slice culture qPCR analysis, data were log‐transformed and compared using a linear mixed model with patient as a random effect, and weights to correct for heteroscedasticity (following Levene's test which indicated this). Dunnett's post hoc test was used with the vehicle‐only condition as control. All tests were two‐sided.
Results
Altered mRNA expression levels in ACC and DLPFC in MDD
In ACC, the mRNA expression level of CYP17A1 was significantly decreased (–1.85‐fold difference, P = 0.009, Figure 1B, Table 1) while that of SULT2A1 was significantly increased (2.59‐fold difference, P = 0.028, Figure 1C, Table 1) in MDD patients. There were no changes in the BPD group. In DLPFC, a statistically significant increase was found in the expression level of SULT2A1 (1.34‐fold difference, P = 0.009, Figure 1D, Table 1). Furthermore, in DLPFC, higher mRNA levels of HSD11B1 (2.13‐fold difference, P = 0.028, Figure 1E, Table 1) in the MDD group and STAR (1.23‐fold difference, P = 0.038, Figure 1F, Table 1) in the BPD group were also found. The function of CYP17A1, SULT2A1, HSD11B1 and STAR is shown in Figure 1A.
Figure 1.
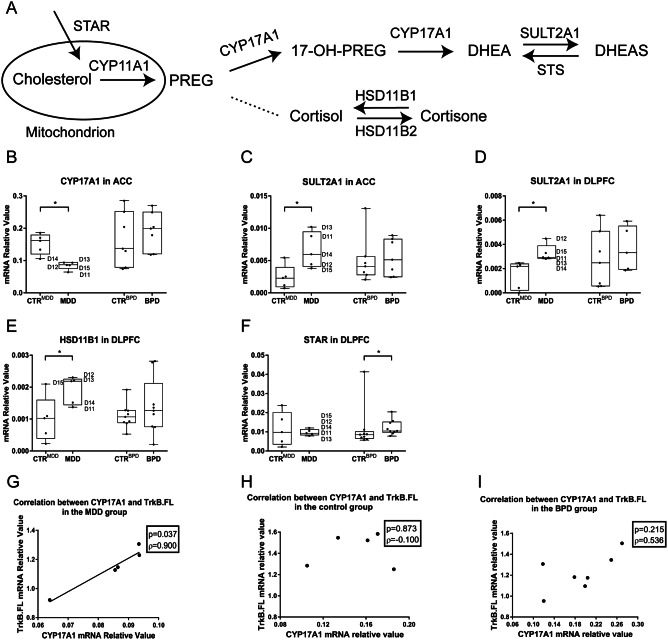
Alterations in the mRNA expression levels of target genes in anterior cingulate cortex/dorsolateral prefrontal cortex. Scheme is showing CYP17A1, SULT2A1, HSD11B1 and STAR in the steroid synthesis pathway, A. Box plots are showing the median, 25th–75th percentiles and the total range of the mRNA expression of target genes in ACC/DLPFC. In ACC, mRNA expression levels of CYP 17A1 was significantly decreased in the MDD patients, B (*P = 0.009, −1.85‐fold difference). While an increase in the mRNA expression level of SULT2A1 was found in the MDD patients, C (*P = 0.028, 2.59‐fold difference). In DLPFC, significantly higher mRNA expression level of SULT2A1 was found in the MDD patients, D (*P = 0.009, 1.31‐fold difference). Higher mRNA levels of HSD11B1, E (2.13‐fold difference, *P = 0.028) in the MDD group and STAR, F (1.23‐fold difference, *P = 0.038) in the BPD group were also found. D11–D15 represents individual MDD patients. In contrast to a positive correlation between the mRNA expression levels of CYP17A1 and TrkB.FL in ACC was found in the MDD patients—G, no correlation was observed in control subjects—H or in BPD patients—I. Abbreviations: ACC = anterior cingulate cortex; BPD = bipolar disorder group; CTRBPD = control group matched for bipolar disorder group; CTRMDD = control group matched for major depressive disorder group; CYP11A1 = cytochrome P450 family 11 subfamily A member 1; CYP17A1 = cytochrome P450c17A1; DHEA = dehydroepiandrosterone; DLPFC = dorsolateral prefrontal cortex; HSD11B1 = hydroxysteroid 11‐β dehydrogenase type 1; MDD = major depressive disorder group; PREG = pregnenolone; STS = steroid‐sulfatase; STAR = steroid acute regulator; SULT2A1 = hydroxysteroid sulfotransferase 2A1; TrkB.FL = tropomyosin‐related kinase B (full length isoform).
Table 1.
Results of expression of target genes in the ACC and DLPFC between the diagnostic groups and their matched control groups.
| A | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ACC area | Medians of relative expression levels of target genes | Fold changes | Z‐values (Mann‐Whitney test) | P‐values | ||||||
| Gene | CtrMDD | MDD | CtrBPD | BPD | MDD/CtrMDD | BPD/CtrBPD | MDD vs. CtrMDD | BPD vs. CtrBPD | MDD vs. CtrMDD | BPD vs. CtrBPD |
| AKR1C1 | 0.0194 | 0.0178 | 0.0159 | 0.0201 | 0.91 | 1.26 | −0.104 | −1.597 | 0.917 | 0.110 |
| AKR1C2 | 0.1441 | 0.0934 | 0.1212 | 0.2345 | 0.65 | 1.94 | −1.149 | −0.575 | 0.251 | 0.565 |
| AKR1C3 | 0.1783 | 0.1224 | 0.1360 | 0.1453 | 0.69 | 1.07 | −0.94 | −0.064 | 0.347 | 0.949 |
| AROM | 5.26 E–03 | 3.82 E–03 | 3.58 E–03 | 3.92 E–03 | 0.73 | 1.09 | −0.731 | −0.958 | 0.465 | 0.338 |
| CYP11B1/2 | 5.14 E–09 | 4.81 E–09 | 5.94 E–10 | 5.47 E–09 | 0.94 | 9.21 | −0.731 | −1.725 | 0.465 | 0.085 |
| CYP17A1 | 0.1614 | 0.0865 | 0.1369 | 0.1992 | 0.54 | 1.46 | −2.611 | −0.319 | 0.009 ↓ | 0.749 |
| CYP21A2 | 2.72 E–05 | 1.19 E–05 | 8.93 E–05 | 7.01 E–05 | 0.44 | 0.78 | −0.731 | −0.429 | 0.465 | 0.668 |
| DBI | 0.2893 | 0.1776 | 0.2612 | 0.2868 | 0.61 | 1.10 | −1.776 | −0.575 | 0.076 | 0.565 |
| HSD3B1/2 | 1.14 E–04 | 1.13 E–04 | 9.24 E–05 | 2.65 E–04 | 0.99 | 2.87 | −0.731 | −1.597 | 0.465 | 0.110 |
| HSD11B1 | 5.46 E–04 | 6.50 E–04 | 5.53 E–04 | 9.82 E–04 | 1.19 | 1.78 | −0.940 | −1.286 | 0.347 | 0.199 |
| HSD11B2 | 0.0067 | 0.0046 | 0.0060 | 0.0109 | 0.68 | 1.83 | −0.104 | −0.571 | 0.917 | 0.568 |
| HSD17B1 | 1.4579 | 0.6305 | 0.6553 | 2.1958 | 0.43 | 3.35 | −1.358 | −1.342 | 0.175 | 0.180 |
| HSD17B10 | 0.0118 | 0.0099 | 0.0105 | 0.0119 | 0.84 | 1.13 | −0.731 | −0.703 | 0.465 | 0.482 |
| SDR5A1 | 0.1846 | 0.1898 | 0.2083 | 0.2004 | 1.03 | 0.96 | −0.313 | −0.192 | 0.754 | 0.848 |
| STAR | 8.89 E–03 | 7.99 E–03 | 8.13 E–03 | 9.24 E–03 | 0.90 | 1.14 | −1.776 | −1.469 | 0.076 | 0.142 |
| STS | 5.40 E–03 | 4.19 E–03 | 4.78 E–03 | 4.45 E–03 | 0.77 | 0.93 | −0.940 | −1.342 | 0.347 | 0.180 |
| SULT2A1 | 2.31 E–03 | 5.98 E–03 | 4.11 E–03 | 5.14 E–03 | 2.59 | 1.25 | −2.193 | −0.447 | 0.028 ↑ | 0.655 |
| SULT2B1 | 2.46 E–05 | 3.13 E–05 | 5.81 E–05 | 1.08 E–04 | 1.27 | 1.85 | −0.940 | −0.961 | 0.347 | 0.337 |
| TSPO | 0.1039 | 0.0475 | 0.0520 | 0.0935 | 0.46 | 1.80 | −1.358 | −1.342 | 0.175 | 0.180 |
| B | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DLPFC area | Medians of relative expression levels of target genes | Fold changes | Z‐values (Mann‐Whitney test) | P‐values | ||||||
| Gene | CtrMDD | MDD | CtrBPD | BPD | MDD/CtrMDD | BPD/CtrBPD | MDD vs. CtrMDD | BPD vs. CtrBPD | MDD vs. CtrMDD | BPD vs. CtrBPD |
| AKR1C1 | 0.0143 | 0.0289 | 0.0179 | 0.0152 | 2.02 | 0.85 | −1.149 | −1.443 | 0.251 | 0.149 |
| AKR1C2 | 0.0925 | 0.1566 | 0.1393 | 0.1523 | 1.69 | 1.09 | −0.522 | −1.104 | 0.602 | 0.270 |
| AKR1C3 | 0.1972 | 0.1679 | 0.1329 | 0.2133 | 0.85 | 1.60 | −0.313 | −1.104 | 0.754 | 0.270 |
| AROM | 1.90 E–03 | 1.98 E–03 | 2.39 E–03 | 2.67 E–03 | 1.04 | 1.12 | −0.731 | −0.309 | 0.465 | 0.757 |
| CYP11B1/2 | 2.84 E–08 | 2.28 E–08 | 1.79 E–08 | 2.67 E–08 | 0.80 | 1.49 | −0.104 | −0.751 | 0.917 | 0.453 |
| CYP17A1 | 0.0702 | 0.0909 | 0.0772 | 0.0943 | 1.29 | 1.22 | −0.731 | −1.104 | 0.465 | 0.270 |
| CYP21A2 | 2.49 E–05 | 2.96 E–05 | 3.80 E–06 | 7.47 E–06 | 1.19 | 1.97 | −0.577 | −0.265 | 0.564 | 0.791 |
| DBI | 0.1845 | 0.2489 | 0.1737 | 0.2813 | 1.35 | 1.62 | −0.940 | −1.192 | 0.347 | 0.233 |
| HSD3B1/2 | 1.29 E–04 | 1.58 E–04 | 1.22 E–04 | 1.41 E–04 | 1.22 | 1.16 | −0.104 | −0.289 | 0.917 | 0.773 |
| HSD11B1 | 1.03 E–03 | 2.19 E–03 | 1.07 E–03 | 1.26 E–03 | 2.13 | 1.18 | −2.193 | −0.662 | 0.028 ↑ | 0.508 |
| HSD11B2 | 0.0059 | 0.0052 | 0.0041 | 0.0083 | 0.89 | 2.02 | −0.522 | −1.104 | 0.602 | 0.270 |
| HSD17B1 | 1.4383 | 0.9572 | 0.8459 | 1.4868 | 0.67 | 1.76 | −0.940 | −1.192 | 0.347 | 0.233 |
| HSD17B10 | 0.0093 | 0.0106 | 0.0090 | 0.0089 | 1.14 | 0.98 | −0.522 | −0.309 | 0.602 | 0.757 |
| SDR5A1 | 0.2594 | 0.1708 | 0.1633 | 0.2150 | 0.66 | 1.32 | −0.522 | −1.280 | 0.602 | 0.200 |
| STAR | 9.86 E–03 | 9.31 E–03 | 8.54 E–03 | 1.05 E–02 | 0.94 | 1.23 | −0.104 | −2.075 | 0.917 | 0.038 ↑ |
| STS | 7.19 E–03 | 6.60 E–03 | 4.76 E–03 | 7.92 E–03 | 0.92 | 1.66 | −0.313 | −1.015 | 0.754 | 0.310 |
| SULT2A1 | 2.18 E–03 | 2.94 E–03 | 2.49 E–03 | 3.32 E–03 | 1.34 | 1.33 | −2.611 | −0.731 | 0.009 ↑ | 0.465 |
| SULT2B1 | 1.07 E–04 | 3.08 E–05 | 2.75 E–05 | 1.05 E–04 | 0.29 | 3.81 | −0.522 | −1.280 | 0.602 | 0.200 |
| TSPO | 0.0648 | 0.0598 | 0.0519 | 0.0926 | 0.92 | 1.78 | −0.940 | −0.839 | 0.347 | 0.402 |
Abbreviations: AKR1C1, 2 and 3 = aldoketoreductase C1, 2 and 3; metabolize progesterone into 20‐alpha‐hydroxy‐progesterone and allopregnanolone; AROM = aromatase; transforms from testosterone and androstenedione to estradiol and estrone, respectively; CYP11B1/2 = cytochrome P450 family 11 subfamily B member 1 and 2; involved in the synthesis of glucocorticoids and mineralcorticoids; CYP17A = cytochrome P450 17A1; synthesizing C19 ketosteroids; CYP21A2 = cytochrome P450c21A2; involved in the synthesis of glucocorticoids and mineralocorticoids; DBI = diazepam binding inhibitor; inducing steroidogenesis by transporting cholesterol into the mitochondria; HSD3B1/2 = hydroxysteroid 3‐β dehydrogenase 1 and 2; catalyze the oxidative conversion of delta‐5‐3‐beta‐hydroxysteroid precursors to delta‐4‐ketosteroids; HSD11B1 = hydroxysteroid 11‐β dehydrogenase type 1; synthesizes cortisol; HSD11B2 = hydroxysteroid 11‐β dehydrogenase type 2; synthesizes cortisone; HSD17B1 = hydroxysteroid 17‐β dehydrogenase type 1; converts estrone into estradiol; HSD17B10 = hydroxysteroid 17‐β dehydrogenase type 10; transforms allopregnanolone into dehydroprogesterone or DHP; STAR = steroid acute regulator; inducing steroidogenesis by transporting cholesterol into the mitochondria; SRD5A1 = steroid 5 α‐reductase type 1; involved in testosterone and progesterone metabolism; STS = steroid sulfatase; hydrolyzes 3‐beta‐hydroxysteroid sulfates, i.e. from pregnenolone‐sulfate, DHEA‐sulfates, estradiol‐sulfate to pregnenolone, DHEA and estradiol; SULT2A1 = hydroxysteroid sulfotransferase 2A1; catalyzing the sulfate conjugation of dehydroepiandrosterone; SULT2B1 = hydroxysteroid sulfotransferase 2B1; transforms pregnenolone and DHEA into their sulfate esters; TSPO = 18 kDa translocator protein; inducing steroidogenesis by transporting cholesterol into the mitochondria.
Other genes analyzed in our study did not show any significant changes in ACC or DLPFC (see in Table 1): AKR1C1, 2 and 3; AROM; CYP11B1/2; CYP21A2; HSD3B1/2; HSD11B2; HSD17B1; HSD17B10; SRD5A1; STS; SULT2B1; DBI and TSPO.
Localization and semi‐quantitative evaluation of CYP17A1 and SULT2A1 protein in ACC in MDD
In order to determine whether protein levels were altered in MDD which would be consistent with the changes of mRNA expression levels of CYP17A1 and SULT2A1 (two genes that changed strongly in depression), IHC was performed and showed that in the post‐mortem human ACC CYP17A1 protein was present throughout all six layers of the grey matter as well as the white matter (Figure 2A), in both neurons and glia‐cells. Higher magnification images revealed the presence of CYP17A1 in the cytoplasm of neurons in the grey matter (Figure 2C–E) and glia‐cells in layer 1 (Figure 2B) and white matter (Figure 2F) where it was observed in a punctate pattern. A similar immunohistochemical staining pattern was found for SULT2A1 (Figure 3) while the positive staining was less strong than that of CYP17A1, indicating lower protein expression level of SULT2A1 than that of CYP17A1 in ACC. Our data also suggested lower SULT2A1 mRNA level than CYP17A1 (data not shown).
Figure 2.
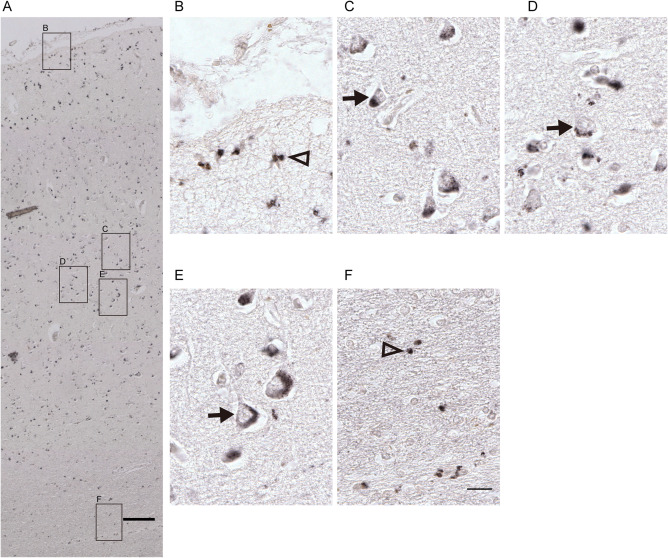
Representative illustrations of CYP17A1 IHC in the human anterior cingulate cortex. Immunohistochemical staining in the human cingulate cortex (ACC) showed that CYP17A1 protein was localized from the pia to the white matter in ACC, A, and it was present in both, nucleolus containing larger neurons (arrows in C–E in the grey matter) and in smaller glia‐cells (triangle in B in the non‐neuron containing layer 1 and F in the white matter). Scale bars: A = 200 µm; B–F = 25 µm.
Figure 3.
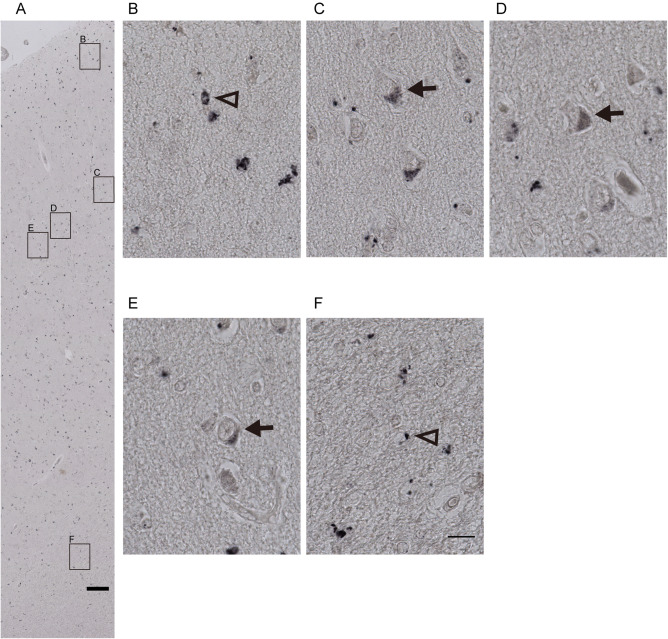
Representative microphotographs of SULT2A1 IHC in the human anterior cingulate cortex. Immunohistochemical staining in the human anterior cingulate cortex (ACC) showed that SULT2A1 protein was localized from the pial layer to the white matter in ACC, A, and was present in both, nucleolus containing larger neurons (arrows in C–E in the grey matter) and in smaller glia‐cells (triangle in B in the non‐neuron containing layer 1 and F in the white matter). Scale bars: A = 200 µm; B–F = 25 µm.
Semi‐quantitative analysis of immunohistochemical results showed that compared to the control group, the intensity and distribution of CYP17A1 staining were significantly decreased in the ACC of MDD patients (Supporting Information Table S5). However, there was no significant difference in the intensity and distribution of SULT2A1 staining in ACC of MDD patients to that in the controls.
Correlations between mRNA expression levels of CYP17A1 and TrkB.FL in ACC of MDD patients
In order to determine whether there was a close relationship between steroid and BDNF‐TrkB pathway as they share similar neuroprotective and neurotrophic effects, Spearman's correlation between the mRNA levels of steroid synthesizing enzymes or related regulatory molecules and BDNF/TrkB was performed. In ACC of MDD patients, a positive correlation was found between the mRNA levels of CYP17A1 and TrkB full length isoform (TrkB.FL) (Figure 1G; ρ = 0.900, P = 0.037), both of which showed a significant decrease in depression [for TrkB.FL, see our previous paper 34]. No such correlation was found in the control group (Figure 1H) or in the BPD group (Figure 1I).
Increased BDNF expression level in post‐mortem human cortex culture slice after DHEA treatment
After 5 days in culture, we still detected many viable cells either in slices treated with medium only or medium plus ethanol (Figure 4) as we have found in previous studies 50, 54.
Figure 4.
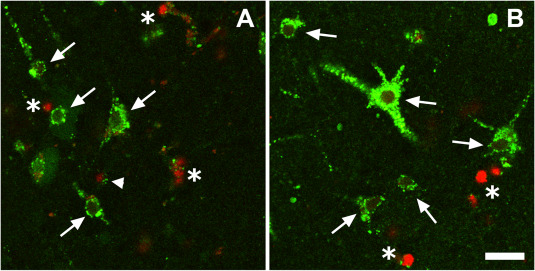
Illustration of cell viability of cortical slides cultured for 5 days. Images are from a control slice maintained in medium only, A, and a slice maintained in medium plus 0.01% ethanol, B. Cells with a green cytoplasm and a dark nucleus had an intact cell membrane and active esterases indicating that they were viable (arrows). Green cells with a red nucleus had active esterases, but also a leaky plasma membrane. This indicated that they were in some state intermediate between viable and dead (arrow heads). Dead cells only had a red nucleus showing a leaky plasma membrane and a lack of active esterases (asterisk). There is no difference for cell viability between the control medium and the treated medium. Scale bar = 25 μm.
Since we found a significant correlation (P = 0.037) between DHEA synthetic enzyme CYP17A1 and TrkB.FL (the BDNF receptor) in MDD patients, we determined the effects of DHEA and DHEAS treatment on BDNF and TrkB.FL in a unique post‐mortem human brain slice culture. Although the functioning of the brain as an organ stops at death, many cells in brain slices from deceased people can survive in vitro conditions for several weeks 49, 50 and serve as a model to reflect in vivo phenomena after certain treatments. Compared to the vehicle treated group, there was a clear increase in the mRNA expression level of BDNF after DHEA treatments for 5 days (Figure 5A). The P‐values were 0.4623 for 0.1 nM DHEA, 0.0205 for 1 nM DHEA, 0.9628 for 10 nM DHEA and 0.0432 for 100 nM DHEA, respectively. DHEA did not significantly change the expression level of TrkB.FL (Figure 5B). No changes were found in the expression levels of BDNF (Figure 5C) or TrkB.FL (Figure 5D) after DHEAS treatment.
Figure 5.
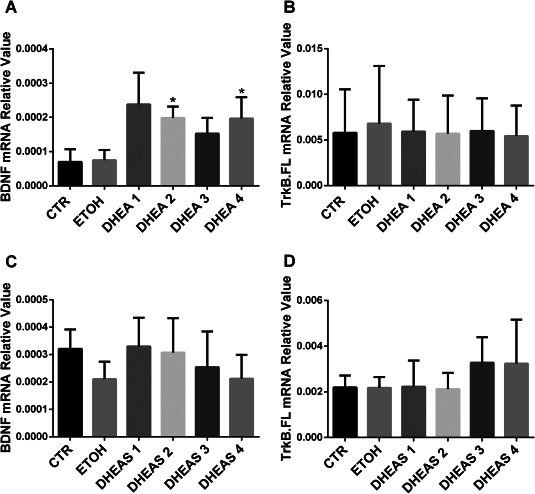
BDNF and TrkB expression after DHEA and DHEAS treatment. The mRNA expression level of BDNF was clearly increased after DHEA treatment, but only 1 nM and 100 nM might be considered significant, A. There was no difference in the mRNA expression level of TrkB.FL after DHEA treatment, B. No changes were found in the expression levels of BDNF—C or TrkB.FL—D after DHEAS treatment. *P < 0.05. DHEA 1= 0.1 nM; 2 = 1 nM; 3 = 10 nM; 4 = 100 nM. DHEAS 1 = 0.1 nM; 2 = 1 nM; 3 = 10 nM; 4 = 100 nM.
Discussion
In the present study, we have shown for the first time that in the post‐mortem human ACC, CYP17A1 mRNA levels were decreased while SULT2A1 mRNA levels were increased in elderly non‐suicide MDD patients. In addition, a reduction in CYP17A1 immunocytochemical staining in neurons and glial cells was observed in those patients. A significant increase of SULT2A1 mRNA level in DLPFC was also observed in MDD. We found a positive correlation between the mRNA levels of CYP17A1 and TrkB.FL, in ACC of MDD patients but not in control subjects. The in vitro human brain slice study showed that treatment with DHEA (a major product of CYP17A1) indeed induced an upregulation of BDNF, the ligand for TrkB.FL, which suggested the presence of a functional connection between the CYP17A1 biosynthetic pathway and BDNF‐TrkB pathway that may underlie the pathogenesis of depression. Furthermore, we found enhanced mRNA levels of HSD11B1 in the MDD group and STAR in the BPD group.
The decreased CYP17A1 could lead to a decrease in DHEA levels while increased SULT2A1 mRNA levels indicate an increase of DHEAS synthesis in ACC/DLPFC. This suggests a dysregulation of endogenous production of DHEA and DHEAS in the brain in MDD. Indeed, lower DHEA in women during peri‐menopause 40 and lower DHEAS concentration in both elderly men and women 44 in plasma have been found to be related to higher depression symptoms and a higher prevalence of depression, while higher serum DHEAS significantly reduced the risk of developing depressive symptoms 48. It should be noted though that conflicting findings also exist 26, possibly due to differences in study design. In addition, lower DHEA levels were found in the ventral tegmental area in prepubertal Wistar Kyoto rats (a putative animal model of childhood depression) and DHEA was shown to have antidepressant effects in this model 25. Importantly, some double‐blind placebo‐controlled trials of DHEA treatment in depression reported significant antidepressant effects 5, 52. Although these data are encouraging, they are based on relatively small samples. More large‐scale studies are needed to establish the role of DHEA in patients with depression. The ability of DHEA or DHEAS to mediate many neurobiological functions, including neuroprotection, neurogenesis, anti‐glucocorticoid, anti‐inflammatory and anti‐oxidant properties could underlie the relationships between DHEA or DHEAS and depression, as all these biological actions are thought to be involved in the development and treatment depression 26. For example, DHEA was shown to protect hippocampal neurons from glucocorticoid‐induced neurotoxicity 18. CYP17A1 synthesizes not only DHEA but also other steroids, that is, 17OH‐pregnenolone and 17OH‐progesterone, precursors of corticosteroids and 4‐dione, a precursor of testosterone and other androgens. So, decreased CYP17A1 mRNA level in ACC might also lead to local changes in these steroids, whose roles would be interesting to look at in future studies. However, we found that other androgen‐related targets, such as androgen receptors, which is involved in the function of androgens, did not show any changes in MDD in our previous study 34.
This is, as far as we know, the first study on the distribution of CYP17A1 and SULT2A1 proteins in human brain. Previously, only mRNA expression of both genes was detected in human prefrontal cortex, caudate nucleus, putamen, hippocampus and other brain regions 22. The presence of CYP17A1 protein by IHC was shown in the rat hippocampal formation 13, hypothalamic periventricular neurons, Purkinje cells of the cerebellar cortex 55, spinal cord 17 and human dorsal root ganglia (DRG) and spinal cord 41. Western blotting and IHC showed the expression of SULT2A1 protein in rat brain, specifically in frontal cortex, hippocampus and thalamus 1. The present study showed the presence of the enzymes CYP17A1 and SULT2A1 in human post‐mortem ACC, in both grey‐ and white‐matter, in neurons and glia‐cells. In addition, the semi‐quantitative analysis of CYP17A1 and SULT2A1 IHC in ACC of MDD patients supported the findings regarding the mRNA expression of these genes. It should also be noted that alterations in the transcript/protein levels might not positively correlate with the functional changes, so a decrease in the transcript/protein level may reflect no change or even compensation to an elevation in enzyme activity leading to higher DHEA levels in depression. Future studies on the enzymatic activity or the products (ie DHEA/DHEAS) of CYP17A1 and SULT2A1 have to be performed to give insight into this.
Neuroprotective and neurotrophic activity of DHEA/DHEAS is similar to that of BDNF, which exerts biological functions by binding to TrkB and facilitates neuronal growth, survival, plasticity and neurogenesis 26. For example, DHEA induced neurogenesis and neural progenitor proliferation in rat dentate gyrus of the hippocampus 16 or human neural stem cell cultures 45 have been reported. There is evidence suggesting that the effects of DHEA/DHEAS are attributable in part to interaction with BDNF and its receptor TrkB. DHEA increased BDNF production and enhanced neuronal cell proliferation, survival and neurite outgrowth via BDNF production in rat cortical neurons 35. We wondered whether this effect could also be detected in DHEA treated human brain tissue. In principle, cultures of post‐mortem tissue 50 or resected tissue 51 might be used for this purpose. We decided to use post‐mortem brain tissue because cells in this tissue remain alive for long periods in culture, whereas neurons quickly degenerate in resected tissue 51. Consistent with the rat data, DHEA upregulated the levels of BDNF in our post‐mortem human brain slice cultures. These data support the hypothesis that there is a close interaction between DHEA and BDNF. In the co‐culture model of epidermal keratinocytes and rat DRG neurons, the stimulatory effect of keratinocytes on axonal outgrowth of DRG neurons was reported to originate from both local synthesis and action of DHEA and functional TrkB signaling 46. Furthermore, we observed a correlation between the levels of CYP17A1 and TrkB.FL in ACC of MDD patients but not in control subjects, which might suggest such a relationship contributing to the development of depression. The mechanism for the correlation in depression is still unknown. However, decreased levels of BDNF and TrkB.FL in our previous study 34 and CYP17A1 in the present study were found in ACC of MDD patients, which is in accordance with the neurotrophin hypothesis of depression 10. It is of interest to note that this correlation might be a basis for new therapeutic strategies. Direct BDNF administration seems to be a highly promising pharmacotherapy for psychiatric disorders 31. However, the use of BDNF is problematic, particularly given the difficulty in delivering BDNF to the CNS and consequently in obtaining effective BDNF local concentrations 30. Molecules which are able to stimulate endogenous BDNF function or activate its cognate receptor TrkB in disease‐specific affected brain areas, may overcome the delivery challenge of administering BDNF directly. As observed in our study, DHEA could increase BDNF expression and its synthetic enzyme CYP17A1 is positively correlated to TrkB.FL. DHEA may thus be a potential candidate for depression treatment. Intraperitoneal injection of DHEA/DHEAS of rats was reported to induce antidepressant‐like behaviors in a forced swim test, which was suggested to be mediated by altering the BDNF concentration in the brain 29.
Higher levels of HSD11B and STAR also indicated abnormalities in steroid production in depression. For example, HSD11B1 predominately amplifies intracellular glucocorticoid action by converting inert cortisone to active cortisol. The higher HSD11B1 mRNA levels in the DLPFC in BPD suggest higher local active cortisol generation in BPD. Additionally, a genetic study presented that one single‐nucleotide polymorphism in HSD11B1 is associated with increased HPA‐axis activity and a higher risk for depression 8. Exogenous corticosteroids are capable of inducing symptoms of depression. Whether enhanced endogenous corticosteroid levels produced in the brain are also contributing to the depression symptoms warrants further studies. Furthermore, it has been proposed that a decreased DHEA (DHEAS)/cortisol ratio would be a risk factor for depression 26, 56. Consistent with this, our data of increased HSD11B1 expression level and decreased CYP17A1 expression level in depression might suggest a higher cortisol level and lower DHEA level, which further resulted in decreased DHEA/cortisol ratio.
A number of putative confounding effects were prevented by matching. Since no significant difference was found between the groups in terms of age, gender, pH of the cerebrospinal fluid, brain weight, PMD, the clock time and month of death, these variables will not have affected our conclusions.
It is necessary to point out that there are some limitations in our study. First, our human sample size was relatively small. Thus, the expression pattern of such genes deserves further investigation with a larger sample size. However, it should be noted that clinically and neuropathologically well‐documented post‐mortem material from elderly depressed patients who did not die from suicide with high quality mRNA is rare. Although the sample size was small in the present study, the search for molecular differences in the cortex of MDD or BPD patients was used primarily as a screening method to identify a number of candidate compounds that were subsequently tested for their effect in a human slice culture system. Second, all patients in our study received antidepressant treatment. Possible effects of medication on the target gene expression are always a limitation of the use of post‐mortem human brains. Nevertheless, we found no indication that medication in the last three months before death had confounded our results. For instance, for the genes CYP17A1 in ACC of MDD patients (see Figure 2A) the mRNA levels of patients taking benzodiazepines (subjects D14 and D15) were generally intermingled with the other data points. Three patients who were taking antipsychotics (subjects D12, D13 and D14) showed the higher CYP17A1 level compared to that of the other MDD subjects. Consequently, in case such medication would have influenced our data, it would rather have increased and not decrease CYP17A1 levels as we observed in our present study. Note that one patient who was taking selective serotonin reuptake inhibitors (subject D11) displayed the lowest CYP17A1 expression level, which might have confounded our results. However, the final conclusion is still the same after deleting the value from this patient. Additionally, the conclusions regarding transcript levels in BPD remained the same after omitting one control subject who took testosterone.
In summary, our current study revealed that altered endogenous steroid synthesis might constitute part of the pathogenesis of depression. The close relationship with BDNF and TrkB further supports the idea that steroids such as DHEA may provide promising novel targets for the therapeutic strategies in MDD.
Funding and Disclosure
This work was supported by China Exchange Programme of the Royal Netherlands Academy of Arts and Sciences (KNAW) (10CDP0037), National Natural Science Foundation of China (81501172) and Shanghai Municipal Commission of Health and Family Planning (20154Y0016). The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Clinico‐pathological information of patients with mood disorders and control subjects.
Table S2. P‐values for matching of subjects with mood disorders and controls.
Supporting Methods S3. RNA isolation, cDNA synthesis and quantitative real‐time PCR for postmortem human material.
Table S4. Sequence of primers, gene bank accession numbers and the length of amplified product for the target genes and reference genes.
Table S5. Semi‐quantitative sores of CYP17A1 and SULT2A1 immunohistochemical staining intensity in ACC of MDD patients.
Acknowledgments
The authors are grateful to the Netherlands Brain Bank (Director Dr. I. Huitinga) at the Netherlands Institute for Neuroscience for providing us with brain material and patient information. They also express their appreciation to Dr. M. A. Hofman for his statistical assistance, Wilma Verweij for helping with the English, R. Balesar, U. Unmehopa, B. Fisser, Drs. K. Bossers, W. Kamphuis and L. Shan for technical advice.
References
- 1. Aldred S, Waring RH (1999) Localisation of dehydroepiandrosterone sulphotransferase in adult rat brain. Brain Res Bull 48:291–296. [DOI] [PubMed] [Google Scholar]
- 2. Amanatkar HR, Chibnall JT, Seo BW, Manepalli JN, Grossberg GT (2014) Impact of exogenous testosterone on mood: a systematic review and meta‐analysis of randomized placebo‐controlled trials. Ann Clin Psychiatry 26:19–32. [PubMed] [Google Scholar]
- 3. Bao AM, Swaab DF (2010) Corticotropin‐releasing hormone and arginine vasopressin in depression focus on the human postmortem hypothalamus. Vitam Horm 82:339–365. [DOI] [PubMed] [Google Scholar]
- 4. Baulieu EE (1991) Neurosteroids: a new function in the brain. Biol Cell 71:3–10. [DOI] [PubMed] [Google Scholar]
- 5. Bloch M, Schmidt PJ, Danaceau MA, Adams LF, Rubinow DR (1999) Dehydroepiandrosterone treatment of midlife dysthymia. Biol Psychiatry 45:1533–1541. [DOI] [PubMed] [Google Scholar]
- 6. Bossers K, Wirz KT, Meerhoff GF, Essing AH, van Dongen JW, Houba P et al (2010) Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer's disease. Brain 133:3699–3723. [DOI] [PubMed] [Google Scholar]
- 7. Castren E, Rantamaki T (2010) The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol 70:289–297. [DOI] [PubMed] [Google Scholar]
- 8. Dekker MJ, Tiemeier H, Luijendijk HJ, Kuningas M, Hofman A, de Jong FH et al (2012) The effect of common genetic variation in 11beta‐hydroxysteroid dehydrogenase type 1 on hypothalamic‐pituitary‐adrenal axis activity and incident depression. J Clin Endocr Metab 97:E233–E237. [DOI] [PubMed] [Google Scholar]
- 9. Drevets WC, Price JL, Furey ML (2008) Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct 213:93–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duman RS, Monteggia LM (2006) A neurotrophic model for stress‐related mood disorders. Biol Psychiatry 59:1116–1127. [DOI] [PubMed] [Google Scholar]
- 11. Gao SF, Qi XR, Zhao J, Balesar R, Bao AM, Swaab DF (2013) Decreased NOS1 expression in the anterior cingulate cortex in depression. Cereb Cortex 23:2956–2964. [DOI] [PubMed] [Google Scholar]
- 12. Harrison PJ (2002) The neuropathology of primary mood disorder. Brain 125:1428–1449. [DOI] [PubMed] [Google Scholar]
- 13. Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT et al (2004) Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci USA 101:865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hui XG, Akahira J, Suzuki T, Nio M, Nakamura Y, Suzuki H et al (2009) Development of the human adrenal zona reticularis: morphometric and immunohistochemical studies from birth to adolescence. J Endocrinol 203:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamphuis W, Schneemann A, van Beek LM, Smit AB, Hoyng PF, Koya E (2001) Prostanoid receptor gene expression profile in human trabecular meshwork: a quantitative real‐time PCR approach. Invest Ophthalmol Vis Sci 42:3209–3215. [PubMed] [Google Scholar]
- 16. Karishma KK, Herbert J (2002) Dehydroepiandrosterone (DHEA) stimulates neurogenesis in the hippocampus of the rat, promotes survival of newly formed neurons and prevents corticosterone‐induced suppression. Eur J Neurosci 16:445–453. [DOI] [PubMed] [Google Scholar]
- 17. Kibaly C, Patte‐Mensah C, Mensah‐Nyagan AG (2005) Molecular and neurochemical evidence for the biosynthesis of dehydroepiandrosterone in the adult rat spinal cord. J Neurochem 93:1220–1230. [DOI] [PubMed] [Google Scholar]
- 18. Kimonides VG, Khatibi NH, Svendsen CN, Sofroniew MV, Herbert J (1998) Dehydroepiandrosterone (DHEA) and DHEA‐sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid‐induced neurotoxicity. Proc Natl Acad Sci USA 95:1852–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kornstein SG, Young EA, Harvey AT, Wisniewski SR, Barkin JL, Thase ME et al (2010) The influence of menopause status and postmenopausal use of hormone therapy on presentation of major depression in women. Menopause 17:828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luchetti S, Bossers K, Frajese GV, Swaab DF (2010) Neurosteroid biosynthetic pathway changes in substantia nigra and caudate nucleus in Parkinson's disease. Brain Pathol 20:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luchetti S, Bossers K, Van de Bilt S, Agrapart V, Morales RR, Frajese GV, Swaab DF (2011) Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer's disease. Neurobiol Aging 32:1964–1976. [DOI] [PubMed] [Google Scholar]
- 22. Luchetti S, Huitinga I, Swaab DF (2011) Neurosteroid and GABA‐A receptor alterations in Alzheimer's disease, Parkinson's disease and multiple sclerosis. Neuroscience 191:6–21. [DOI] [PubMed] [Google Scholar]
- 23. Luchetti S, van Eden CG, Schuurman K, van Strien ME, Swaab DF, Huitinga I (2014) Gender differences in multiple sclerosis: induction of estrogen signaling in male and progesterone signaling in female lesions. J Neuropathol Exp Neurol 73:123–135. [DOI] [PubMed] [Google Scholar]
- 24. MacLusky NJ, Hajszan T, Leranth C (2004) Effects of dehydroepiandrosterone and flutamide on hippocampal CA1 spine synapse density in male and female rats: implications for the role of androgens in maintenance of hippocampal structure. Endocrinology 145:4154–4161. [DOI] [PubMed] [Google Scholar]
- 25. Malkesman O, Asaf T, Shbiro L, Goldstein A, Maayan R, Weizman A et al (2009) Monoamines, BDNF, dehydroepiandrosterone, DHEA‐sulfate, and childhood depression‐An animal model study. Adv Pharmacol Sci 2009:405107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maninger N, Wolkowitz OM, Reus VI, Epel ES, Mellon SH (2009) Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS). Front Neuroendocrin 30:65–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martinowich K, Manji H, Lu B (2007) New insights into BDNF function in depression and anxiety. Nat Neurosci 10:1089–1093. [DOI] [PubMed] [Google Scholar]
- 28. Marx CE, Stevens RD, Shampine LJ, Uzunova V, Trost WT, Butterfield MI et al (2006) Neuroactive steroids are altered in schizophrenia and bipolar disorder: relevance to pathophysiology and therapeutics. Neuropsychopharmacol 31:1249–1263. [DOI] [PubMed] [Google Scholar]
- 29. Naert G, Maurice T, Tapia‐Arancibia L, Givalois L (2007) Neuroactive steroids modulate HPA axis activity and cerebral brain‐derived neurotrophic factor (BDNF) protein levels in adult male rats. Psychoneuroendocrinology 32:1062–1078. [DOI] [PubMed] [Google Scholar]
- 30. Nagahara AH, Tuszynski MH (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 10:209–219. [DOI] [PubMed] [Google Scholar]
- 31. Peters J, Dieppa‐Perea LM, Melendez LM, Quirk GJ (2010) Induction of fear extinction with hippocampal‐infralimbic BDNF. Science 328:1288–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pinna G, Dong E, Matsumoto K, Costa E, Guidotti A (2003) In socially isolated mice, the reversal of brain allopregnanolone down‐regulation mediates the anti‐aggressive action of fluoxetine. Proc Natl Acad Sci USA 100:2035–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qi XR, Kamphuis W, Wang S, Wang Q, Lucassen PJ, Zhou JN, Swaab DF (2013) Aberrant stress hormone receptor balance in the human prefrontal cortex and hypothalamic paraventricular nucleus of depressed patients. Psychoneuroendocrinology 38:863–870. [DOI] [PubMed] [Google Scholar]
- 34. Qi XR, Zhao J, Liu J, Fang H, Swaab DF, Zhou JN (2015) Abnormal retinoid and TrkB signaling in the prefrontal cortex in mood disorders. Cereb Cortex 25:75–83. [DOI] [PubMed] [Google Scholar]
- 35. Rahmani A, Shoae‐Hassani A, Keyhanvar P, Kheradmand D, Darbandi‐Azar A (2013) Dehydroepiandrosterone stimulates nerve growth factor and brain derived neurotrophic factor in cortical neurons. Adv Pharmacol Sci 2013:506191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rajkowska G, Miguel‐Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY et al (1999) Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry 45:1085–1098. [DOI] [PubMed] [Google Scholar]
- 37. Rasmusson AM, Pinna G, Paliwal P, Weisman D, Gottschalk C, Charney D et al (2006) Decreased cerebrospinal fluid allopregnanolone levels in women with posttraumatic stress disorder. Biol Psychiatry 60:704–713. [DOI] [PubMed] [Google Scholar]
- 38. Romeo E, Strohle A, Spalletta G, di Michele F, Hermann B, Holsboer F et al (1998) Effects of antidepressant treatment on neuroactive steroids in major depression. Am J Psychiatry 155:910–913. [DOI] [PubMed] [Google Scholar]
- 39. Rupprecht R, Holsboer F (1999) Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci 22:410–416. [DOI] [PubMed] [Google Scholar]
- 40. Schmidt PJ, Murphy JH, Haq N, Danaceau MA, St Clair L (2002) Basal plasma hormone levels in depressed perimenopausal women. Psychoneuroendocrino 27:907–920. [DOI] [PubMed] [Google Scholar]
- 41. Schonemann MD, Muench MO, Tee MK, Miller WL, Mellon SH (2012) Expression of P450c17 in the human fetal nervous system. Endocrinology 153:2494–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schule C, Eser D, Baghai TC, Nothdurfter C, Kessler JS, Rupprecht R (2011) Neuroactive steroids in affective disorders: target for novel antidepressant or anxiolytic drugs? Neuroscience 191:55–77. [DOI] [PubMed] [Google Scholar]
- 43. Shan L, Qi XR, Balesar R, Swaab DF, Bao AM (2013) Unaltered histaminergic system in depression: a postmortem study. J Affect Disord 146:220–223. [DOI] [PubMed] [Google Scholar]
- 44. Souza‐Teodoro LH, de Oliveira C, Walters K, Carvalho LA (2016) Higher serum dehydroepiandrosterone sulfate protects against the onset of depression in the elderly: findings from the English Longitudinal Study of Aging (ELSA). Psychoneuroendocrinology 64:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki M, Wright LS, Marwah P, Lardy HA, Svendsen CN (2004) Mitotic and neurogenic effects of dehydroepiandrosterone (DHEA) on human neural stem cell cultures derived from the fetal cortex. Proc Natl Acad Sci USA 101:3202–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ulmann L, Rodeau JL, Danoux L, Contet‐Audonneau JL, Pauly G, Schlichter R (2009) Dehydroepiandrosterone and neurotrophins favor axonal growth in a sensory neuron‐keratinocyte coculture model. Neuroscience 159:514–525. [DOI] [PubMed] [Google Scholar]
- 47. van de Nes JA, Kamphorst W, Ravid R, Swaab DF (1998) Comparison of beta‐protein/A4 deposits and Alz‐50‐stained cytoskeletal changes in the hypothalamus and adjoining areas of Alzheimer's disease patients: amorphic plaques and cytoskeletal changes occur independently. Acta Neuropathol 96:129–138. [DOI] [PubMed] [Google Scholar]
- 48. Veronese N, De Rui M, Bolzetta F, Zambon S, Corti MC, Baggio G et al (2015) Serum dehydroepiandrosterone sulfate and incident depression in the elderly: the Pro.V.A. Study . Am J Geriatr Psychiatry 23:863–871. [DOI] [PubMed] [Google Scholar]
- 49. Verwer RWH, Baker RE, Boiten EF, Dubelaar EJ, van Ginkel CJ, Sluiter AA, Swaab DF (2003) Post‐mortem brain tissue cultures from elderly control subjects and patients with a neurodegenerative disease. Exp Gerontol 38:167–172. [DOI] [PubMed] [Google Scholar]
- 50. Verwer RWH, Hermens WT, Dijkhuizen P, ter Brake O, Baker RE, Salehi A et al (2002) Cells in human postmortem brain tissue slices remain alive for several weeks in culture. FASEB J 16:54–60. [DOI] [PubMed] [Google Scholar]
- 51. Verwer RWH, Sluiter AA, Balesar RA, Baayen JC, de Witt Hamer PC, Speijer D et al (2015) Injury response of resected human brain tissue in vitro . Brain Pathol 25:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wolkowitz OM, Reus VI, Keebler A, Nelson N, Friedland M, Brizendine L, Roberts E (1999) Double‐blind treatment of major depression with dehydroepiandrosterone. Am J Psychiatry 156:646–649. [DOI] [PubMed] [Google Scholar]
- 53. Wu L, Sluiter AA, Guo HF, Balesar RA, Swaab DF, Zhou JN, Verwer RW (2008) Neural stem cells improve neuronal survival in cultured postmortem brain tissue from aged and Alzheimer patients. J Cell Mol Med 12:1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu X, Balesar R, Lu J, Farajnia S, Zhu Q, Huang M, Bao AM, Swaab DF (2017) Increased glutamic acid decarboxylase expression in the hypothalamic suprachiasmatic nucleus in depression. Brain Struct Funct. DOI 10.1007/s00429-017-1442-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamada H, Kominami S, Takemori S, Kitawaki J, Kataoka Y (1997) Immunohistochemical localization of cytochrome P450 enzymes in the rat brain, considering the steroid‐synthesis in the neurons. Acta Histochem Et Cytochem 30:609–616. [Google Scholar]
- 56. Young AH, Gallagher P, Porter RJ (2002) Elevation of the cortisol‐dehydroepiandrosterone ratio in drug‐free depressed patients. Am J Psychiatry 159:1237–1239. [DOI] [PubMed] [Google Scholar]
- 57. Zhao J, Qi XR, Gao SF, Lu J, van Wamelen DJ, Kamphuis W et al (2015) Different stress‐related gene expression in depression and suicide. J Psychiatr Res 68:176–185. [DOI] [PubMed] [Google Scholar]
- 58. Zhao J, Verwer RW, van Wamelen DJ, Qi XR, Gao SF, Lucassen PJ, Swaab DF (2016) Prefrontal changes in the glutamate‐glutamine cycle and neuronal/glial glutamate transporters in depression with and without suicide. J Psychiatr Res 82:8–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Clinico‐pathological information of patients with mood disorders and control subjects.
Table S2. P‐values for matching of subjects with mood disorders and controls.
Supporting Methods S3. RNA isolation, cDNA synthesis and quantitative real‐time PCR for postmortem human material.
Table S4. Sequence of primers, gene bank accession numbers and the length of amplified product for the target genes and reference genes.
Table S5. Semi‐quantitative sores of CYP17A1 and SULT2A1 immunohistochemical staining intensity in ACC of MDD patients.