

Abstract
Objectives
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is characterized by strategic white matter (WM) hyperintensities on MRI. Pathological features include WM degeneration, arteriolosclerosis, lacunar infarcts, and the deposition of granular osmiophilic material. Based on the hypothesis that the gliovascular unit is compromised, we assessed the nature of astrocyte damage in the deep WM of CADASIL subjects.
Methods
We evaluated post‐mortem brains from CADASIL, cerebral small vessel disease, similar age cognitively normal and older control subjects. Standard immunohistochemical, immunofluorescent, and unbiased stereological methods were used to evaluate the distribution of astrocytes, microvessels, and autophagy markers in five different brain regions.
Results
Compared to the controls, the deep WM of CADASIL subjects overall showed increased numbers of glial fibrillary acidic protein (GFAP)‐positive clasmatodendritic astrocytes (P=0.037) and a decrease in the percentage of normal appearing astrocytes (P=0.025). In accord with confluent WM hyperintensities, the anterior temporal pole contained abundant clasmatodendritic astrocytes with displaced aquaporin 4 immunoreactivity. Remarkably, we also found strong evidence for the immunolocalization of autophagy markers including microtubule‐associated protein 1, light chain 3 (LC3), and sequestosome 1/p62 and Caspase‐3 in GFAP‐positive clasmatodendritic cells, particularly within perivascular regions of the deep WM. LC3 was co‐localized in more than 90% of the GFAP‐positive clasmatodendrocytes.
Conclusions
Our novel findings show astrocytes undergo autophagy‐like cell death in CADASIL, with the anterior temporal pole being highly vulnerable. We propose astrocytes transform from normal appearing type A to hypertrophic type B and eventually to clasmatodendritic type C cells. These observations also suggest the gliovascular unit of the deep WM is severely impaired in CADASIL.
Keywords: Astrocytes, autophagy, CADASIL, stroke, vascular dementia, white matter
Introduction
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common widely occurring hereditary stroke disorder leading to cognitive impairment and dementia 4, 38. It is caused by over 250 distinct mutations in the NOTCH3 gene. In addition to the presence of severe arteriopathy, lacunar infarcts, and deep white matter (WM) changes, CADASIL is characterized by the presence of aggregated NOTCH3 extracellular domain fragments within in granular osmiophilic material (GOM) 38, 39. Hypomorphic NOTCH3 function, causing a partial loss of NOTCH3 protein function in vascular smooth muscle cells is also characteristic of CADASIL 2. However, WM hyperintensities identified on magnetic resonance imaging (MRI) in the anterior temporal pole and external capsule are key radiological signatures of CADASIL. We previously demonstrated that WM hyperintensities in the anterior temporal pole largely align with perivascular spaces and highly rarefied tissue 40. The degeneration and axonal disconnectivity in the WM 9 is associated with abnormalities in oligodendrocytes and accumulation of degraded myelin basic protein. Oligodendrocytes together with astrocytes and microglia also form the gliovascular unit. We recently showed that astrocytes transform to clasmatodendrocytes in the deep WM of elderly post‐mortem stroke survivors, who develop dementia 6. This implicates disruption of the gliovascular unit and loss of integrity of the blood–brain barrier (BBB) in the WM. The cellular mechanisms involved in astrocytic transformation and whether any protective mechanisms are implicated are unknown 3. We therefore reasoned that CADASIL in which there is severe WM degeneration will be pivotal to examine cellular mechanisms involved astrocyte pathology.
Major mechanisms of cell death after ischemia are apoptosis and necrosis, and both have been implicated in delayed neuronal cell death after hypoxic–ischemic injury 14. Macroautophagy, a degradation pathway for organelles and long‐lived proteins too large to be degraded by the ubiquitin–proteasome system, is also triggered in cells after hypoxic and excitotoxic injury, and excessive or imbalanced induction can contribute to cell death 7, 20. Although there have been numerous reports on the role of autophagy in neurodegenerative diseases, there is lack of autophagy studies in relation to cerebrovascular disorders including in CADASIL or post‐stroke dementia. Growing evidence suggests autophagy is enhanced following cerebral ischemia, and is stimulated in response to in vivo or in vitro instigated energy deficits, hypoxia, endoplasmic reticulum stress, and oxidative stress 20, 28, 37. The two most commonly studied proteins involved in autophagy are LC3 (microtubule‐associated protein 1, light chain 3) and Beclin‐1. During the initiation of autophagy, LC3‐I becomes anchored to the autophagic vacuole membrane to form LC3‐II, a specific marker for autophagosomes. Beclin‐1 is involved in the recruitment of the membranes which form the autophagosomes, and also interacts with anti‐apoptotic protein B‐cell lymphoma 2 as an upstream gatekeeper of apoptosis 27. Another protein which has been suggested to have a pathogenic role in autophagy dysfunction is sequestosome 1 (SQSTM1), or more commonly known as p62, a regulatory protein involved in protein homeostasis and DNA repair 17. p62 binds directly to LC3 and can target protein aggregates and organelles for autophagic degradation, and has a role in regulating the degradation of ubiquitinated tau 26. In an effort to evaluate the integrity of the gliovascular unit and mechanisms of astrocytic cell death, we aimed to study the distribution and quantify the expression of GFAP immunoreactive cells and protein markers of autophagy in different regions of the WM in CADASIL against similar age controls.
Methods
Subjects and tissues
Demographic details and diagnoses of the subjects are shown in Table 1. The mean age of the CADASIL and young control subjects were not different. Available case notes and radiological reports indicated CADASIL subjects showed extensive WM changes consistent with small vessel disease and met the minimum criteria for cognitive impairment 1. CADASIL diagnosis was confirmed by the presence of NOTCH3 gene mutations or granular osmiophilic material (GOM) in arteries within skin biopsies 40. None of the controls had neurological or pathological evidence for cerebrovascular disease or neurodegenerative disorder. Tissue blocks from brains of CADASIL subjects and similar age controls were collected from four sources. In addition to the Newcastle Brain Tissue Resource (NBTR), Newcastle University, Campus for Ageing and Vitality, we obtained cases from the MRC London Brain Bank for Neurodegenerative Diseases, the MRC Sudden Death Brain and Tissue Bank, University of Edinburgh and Neurology Department, Ludwig Maximilians University, Germany. Tissue from older controls was obtained from the NBTR. Use of brain tissue was approved by the local research ethics committee of the Newcastle upon Tyne Hospitals NHS Foundation Trust, the NBTR committee, and the ethics committees overseeing the Brain Banks at the other respective sites.
Table 1.
Demographic details of CADASIL cases and controls. Abbreviations: GI = gastrointestinal; SVD = small vessel disease; WM = white matter
| Group (n) | Age (years) | Gender | Mutation site | Duration (years) | Key clinical features and risk factors |
|---|---|---|---|---|---|
| CAD1 | 44 | F | Arg153Cys | 8 | Cardiac arrhythmias |
| CAD2 | 53 | F | Arg133Cys | 6 | No vascular risk |
| CAD3 | 55 | M | Arg558Cys | 11 | Brief history of gout |
| CAD4 | 58 | M | Arg985Cys | 13 | No vascular risk |
| CAD5 | 59 | M | Arg169Cys | 12 | No vascular risk |
| CAD6 | 61 | M | Arg169Cys | 10 | Obesity (55 years‐) |
| CAD7 | 66 | F | D239_D253del | 23 | No vascular risk, obesity |
| CAD8 | 68 | F | Arg133Cys | 18 | Smoking history |
| CAD9 | 68 | M | Arg153Cys | 28 | Smoking, prostate tumor |
| CAD10 | 52 | M | Arg141Cys | 15 | No vascular risk |
| CAD11 | 74 | M | Arg141Cys | 12 | No vascular risk |
| CADASIL 11, a, b | 58.8 ± 7.4 | 7M/4F | — | 10.8 | Mean age at onset 46 years |
| Young Controls 10 | 65.7 ± 8.1 | 3M/7F | — | — | No significant cerebrovascular or neurodegenerative disorder. No pathological diagnosis |
| SVD 10 | 82.4 ± 8.1 | 4M/6F | — | 7.9 | Vascular dementia, heart failure, cancer, GI bleed, sudden death |
| Older controls 12 | 84.0 ± 8.0 | 3M/9F | — | — | No significant cerebrovascular or neurodegenerative disorder. Heart failure, cancer, ischemic bowel infection. |
Length of fixation ranged 1–19 months for CADASIL, young controls and SVD samples and 2–68 months for old controls.
WM Score; white matter pathology score assessed using scale from 10. The MMSE scores for the patients ranged from 12 to 21. Controls, mean age was not significantly different to mean age of CADASIL group (P > 0.05).
Mode of WM scores were 3 in young controls, 1 in young controls and 2 in older controls 9.
Immunohistochemistry and immunofluorescence (IF) labeling
Formalin‐fixed paraffin‐embedded post‐mortem WM brain tissue from five different regions including the pre‐frontal lobe (PF), temporal pole (TP), temporal lobe (TL), parietal lateral lobe (PL), and occipital inferior lobe (OL) were examined. These extended along the rostro‐caudal axis of the cerebrum, per atlas of Perry and Oakley 24. The blocks were cut serially at 10‐μm thickness for routine tinctorial staining and immunohistochemistry as described previously 40. Macroscopic and microscopic pathology was assessed using standardized protocols as described 10, 19. Haemotoxylin and eosin (H&E), luxol fast blue (LFB) and cresyl fast violet (CFV) were used as standard stains for describing neuropathological changes and for detection of infarcts, presence of WM rarefaction, and extent of arteriopathy. Cerebrovascular lesions including SVD pathology were assessed using a grading system as described previously 10. Vascular sclerotic index (SI) was assessed as described previously 40.
Ten‐micrometer‐thick paraffin wax embedded coronal sections were immunostained with various primary mouse monoclonal and rabbit or goat polyclonal antibodies including, aldehyde dehydrogenase 1 family, member L1 (ALDH1L1; 1:100 in 0.1% Trion X‐100‐PBS for IF, clone 7G8, mouse, 14‐9595, eBioscience), aquaporin 4 (AQP4; 1:50, rabbit, 16473‐1‐AP, Proteintech), Beclin‐1 (1:100, mouse, AM1818a, Abgent), Caspase‐3 (Cleaved Caspase‐3, 1:100, Asp175, rabbit; product #9661, Cell Signaling Technology), Collagen IV (COL4, 1:1000, mouse, C1926, Sigma); Delta Like‐1 (DLL1; 1:1000, rabbit, Ab# 76655 Abcam), fibrinogen (1:2000, rabbit, A0080, Dako), GFAP (1:1000, rabbit, Z0334, Dako), GFAP mouse antibody (1:50, clone 6F2, monoclonal M0761, Dako), glucose transporter‐1 (GLUT‐1, 1:200, rabbit, #21041, Thermo Scientific), Intercellular adhesion molecule‐1 (ICAM‐1, CD54, 1:300, mouse, F1743, Dako), LC3 (autophagic vacuoles; 1:100, rabbit, AP1801a, Abgent), p62 (1:300, mouse, #610832, BD Biosciences). Tissue sections first underwent antigen retrieval by heating in the microwave with citrate buffer for 12 min before being quenched with TBS and 3% hydrogen peroxide. Sections were then blocked with serum derived from the species in which the secondary antibody was generated, before being immunostained with the primary antibody overnight at 4°C. Specificities of the GFAP, LC3, p62, and Caspase‐3 antibodies were either verified per manufacturer's datasheet or immunostaining of cortical tissue or sections without primary antibody as described previously 8, 9.
Quantification was performed following the general pattern we established previously for either parenchymal or cellular protein immunoreactivity 12, 18. Briefly for GFAP‐positive cells, at least 20 images were acquired randomly from areas of the deep WM in coronal sections (Figure 2), using a Leitz DIALUX 20 brightfield microscope coupled to a lumenera infinity digital camera at 10× magnification. Using ImageJ, the total GFAP+ cells and cells showing features of clasmatodendrosis (swollen cell body and beaded processes) were counted from frontal, temporal, parietal, and occipital deep WM in each case, with the percentage of GFAP+ clasmatodendrotic and normal astroglia per total GFAP+ cells were calculated. To verify that only astrocytic cells were targeted, we determined that GFAP cell counts per 0.5 mm2 correlated positively with GFAP staining per unit area (ρ= 0.754, P < 0.001). We also showed there was a positive correlation between the frontal and temporal total GFAP immunoreactivities (ρ = 0.500, P = 0.003).
Figure 2.
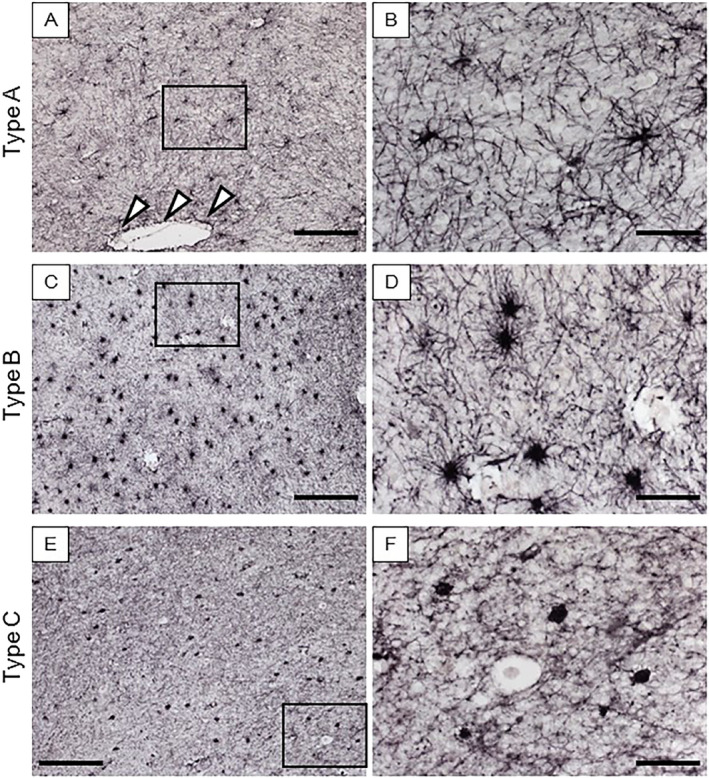
Morphology of different GFAP immunopositive cell types in the deep WM of CADASIL and control subjects. (A–B) Star‐like normal appearing type A cells. End‐feet can be seen juxtaposed to a perivascular space (arrow heads). (C–D) Abnormal type B cells with an enlarged cell body and short thick processes. (E–F) Type C cells, abnormal astrocytes with severely retracted processes and rounded cell bodies. Magnification bar = 200 μm (A, C and E) and 50 μm (B, D and F).
For immunofluorescence labeling, the primary antibodies were removed and sections were washed with PBS prior to incubation at room temperature for 1 hour with goat anti‐mouse secondary antibody, Dylight 650 conjugated (1:200, 84545, Thermo Scientific) and goat anti‐rabbit secondary antibody, Texas Red conjugated (1:200, T2767, Life Technologies). Sections were counterstained and mounted with DAPI incorporated mounting medium (Dako). A Leica TCS SP2 UV AOBS MP (upright confocal microscope) and a Life Technologies EVOS FL (LED) fluorescence microscope were used for image capture.
Statistical analysis
Data were analyzed using SPSS (V19.0, IBM) and tested for normality using the Shapiro–Wilk test. Differences between means of groups were first tested using ANOVA and where appropriate using Tukey's post hoc test. In prior analysis, data found to be not normally distributed were analyzed using non‐parametric methods. Group means such as CADASIL and controls were compared using analysis of variance (ANOVA) with post hoc Tukey tests for normal data or Kruskall‐Wallis, Newman–Keuls and the Mann–Whitney U tests for non‐normally distributed values, for example, differences between pathological variables and total GFAP positive cells or the percentage of clasmatodendrocytes in different groups. Spearman's rank ρ (rho) correlation was used to assess correlations between clinical and neuropsychometric variables or specific protein immunoreactivity measures and microvascular changes. Significant difference between means was considered when the P value was <0.05.
Results
Astrocyte morphology revealed by GFAP immunohistochemistry
GFAP immunohistochemical staining of the WM of the anterior temporal pole from CADASIL subjects with WM hypersignals on MRI in life revealed abnormal astrocytes in the deeper WM (Figure 1). Screening of immunostained sections from further afield in the WM of the PF, TL, PL and OL in CADASIL subjects indicated that GFAP antibodies essentially identified three types of morphologically distinct cells (Figure 2). There were the normal appearing star‐like astrocytes with small bodies and long fibrous processes, which we designated as type A cells (Figures 1I, 2A and 2B). The other two had enlarged cell bodies with shortened or retracted fibers (type B) (Figures 1F, 2C and 2D) and rounded profiles without processes (type C) (Figures 1C, 2E and 2F). Many of the latter were found to be with smaller almost punctate cell bodies. The type B cells with enlarged cell bodies invariably exhibited thickened and shortened processes. The GFAP‐positive cells types A to C collectively likely represented a continuous spectrum from normal to abnormal. The GFAP‐positive cells were also immunostained by antibodies to ALDH1‐L1 and DLL‐1. The cytoplasmic immunoreactivities of these markers were similar to what we had observed in SVD cases from elders post‐stroke survivors 6. Similarly, several clasmatodendrocytes were immuostained by fibrinogen (data not shown).
Figure 1.
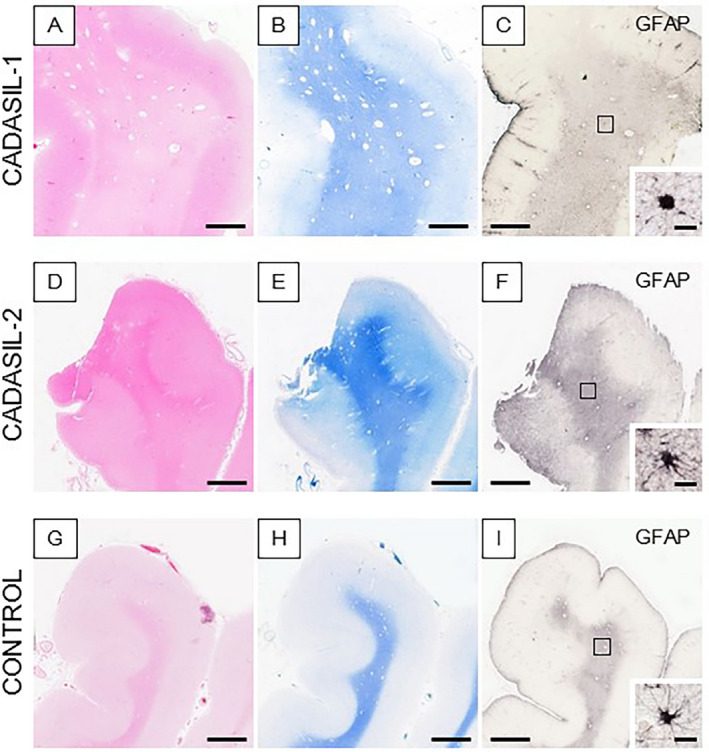
Histological features in the anterior temporal pole of CADASIL and control subjects. (A–I) Images show H&E (A, D and G), LFB (B, E and H), and GFAP (C, F and I) stained sections in two CADASIL patients with NOTCH3 mutations (R133C and R558C) (A–F) and a similar age control (G–I). Insets in C, F, and I show profiles of single GFAP immunostained astrocytes; abnormal astrocytes (C and F); and normal appearing astrocyte (I). Consistent with MRI, the WM in both CADASIL patients also revealed numerous perivascular spaces. Magnification bar = 3mm (A–I) and in insets C, F, and I = 20μm.
Quantitative analyses (Figure 3) in all the 5 regions showed that the total population of GFAP‐positive type C cells comprising counts from all regions was increased in CADASIL subjects compared to controls (P=0.037). Type C cells appeared as clasmatodendrocytes, exhibiting an enlarged cell body and absence of processes. At higher magnification intracellular vacuoles could be discerned in these cells. There were no significant differences in type B cells (P=0.392) which appeared abnormal but these were not necessarily changed in CADASIL. However, boxplots also revealed a trend in less number of normal astrocytes in CADASIL subjects compared to similar age controls (P=0.051). We also noted higher percentage of type C cells (P=0.046) and a significantly lower percentage of type A cells (P=0.025) in CADASIL. When we compared percentage of the sum of all the abnormal cell (types B and C) there were significant differences (P=0.046) between CADASIL and controls. This suggested that the proportions of abnormal astrocytes were substantially increased in CADASIL. We found similar profiles of astrocytes with high variability in SVD cases but these were not quantified 6. However, it was unclear whether any of the astrocytes described here coincide with the most recently described cytotoxic astrocyte type A1 21 or whether the WM astrocytes are a specialized type 5.
Figure 3.
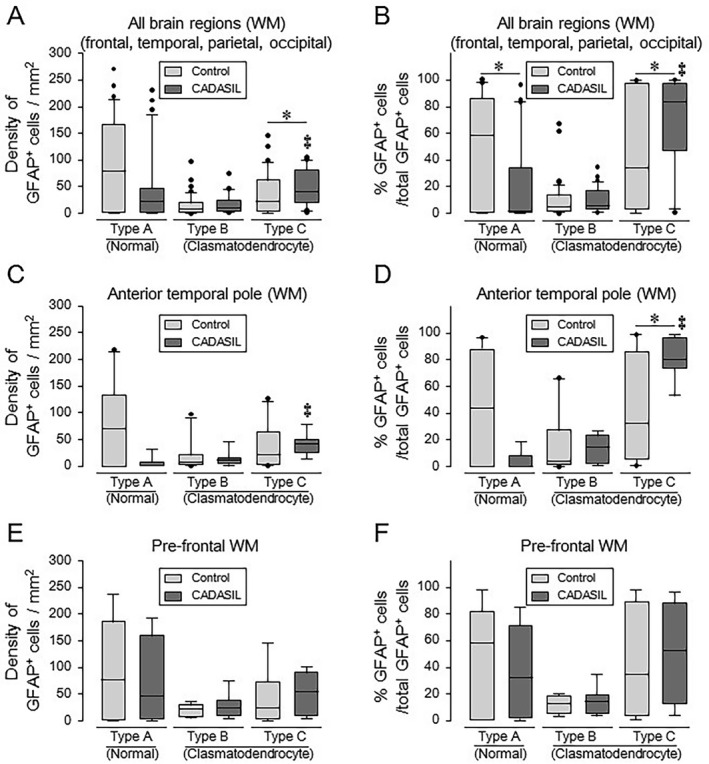
Quantitative analyses of the total population of GFAP‐positive cells in CADASIL and controls. (A–F) Box plots showing density of GFAP‐positive each type of cells averaged from all brain regions (A), anterior temporal pole (C), and pre‐frontal WM (E) as well as percentage of GFAP‐positive each type of cell per total GFAP‐positive cells averaged from all brain regions (B), anterior temporal pole (D), and pre‐frontal WM (F). Compare to controls, CADASIL subjects showed increased density of abnormal type C cells in all brain regions (A) (*P = 0.037) and increased percentage of abnormal type C cells in all brain regions (B) (*P = 0.046) and the anterior temporal pole (D) (*P = 0.011). When comparing each type of cell among CADASIL subjects, both density and percentage of abnormal type C cells were increased in all brain regions (A and B) and the anterior temporal pole (C and D) (‡ P < 0.01 throughout). However, differences between CADASIL and controls in the pre‐frontal WM did not reach statistical significance (E and F). There was relatively low abundance of type B cells in the same screened regions. Other regions including the temporal, parietal and occipital WM similarly showed differences in percentage of abnormal to normal cells compared to controls (not shown).
The regional distribution of each category of astrocytes across the five brain WM regions PF, anterior TP, TL, PL, and OL were qualitatively similar. However, there were considerable regional variations in clasmatodendritic numbers with the most robust change was in the temporal pole, consistent with WMHs upon MRI. There were consistently higher percentage of degenerated cells (type C) and lower percentage of type A (normal) cells in CADASIL compared to controls, as indicated by the median. The low percentage of type B cells with hyperplasic cell bodies and shortened processes in both disease and control groups across all brain areas were notable. Greater differences in the percentage of degenerating type C cells were apparent in the anterior temporal pole and temporal lobe with almost equivalent degree of changes in the pre‐frontal lobe (Figure 3). Type A counts tended to be lower in the temporal pole (P=0.055) with a significant increase in the degenerating type C cells in the temporal lobe. We noted there was no clear relationship between specific genotype mutations and astrocyte pathology. This suggested NOTCH 3 mutations had no specific effects on degree of astrocyte change within the WM of brain regions examined.
Localization of AQP4 in astrocytes and microvascular markers
Double‐immunofluorescence staining showed co‐localization of AQP4 and GFAP in the end‐feet of normal astrocytes (Figure 4). Clasmatodendritic astrocytes showed localization of AQP4 and GFAP in the cell bodies. There was a clear indication that numerous clasmatodendritic astrocytes had retracted end‐feet containing the water channel protein AQP4 in CADASIL and SVD cases 6. As expected this was most prominent in the anterior temporal pole of CADASIL subjects.
Figure 4.
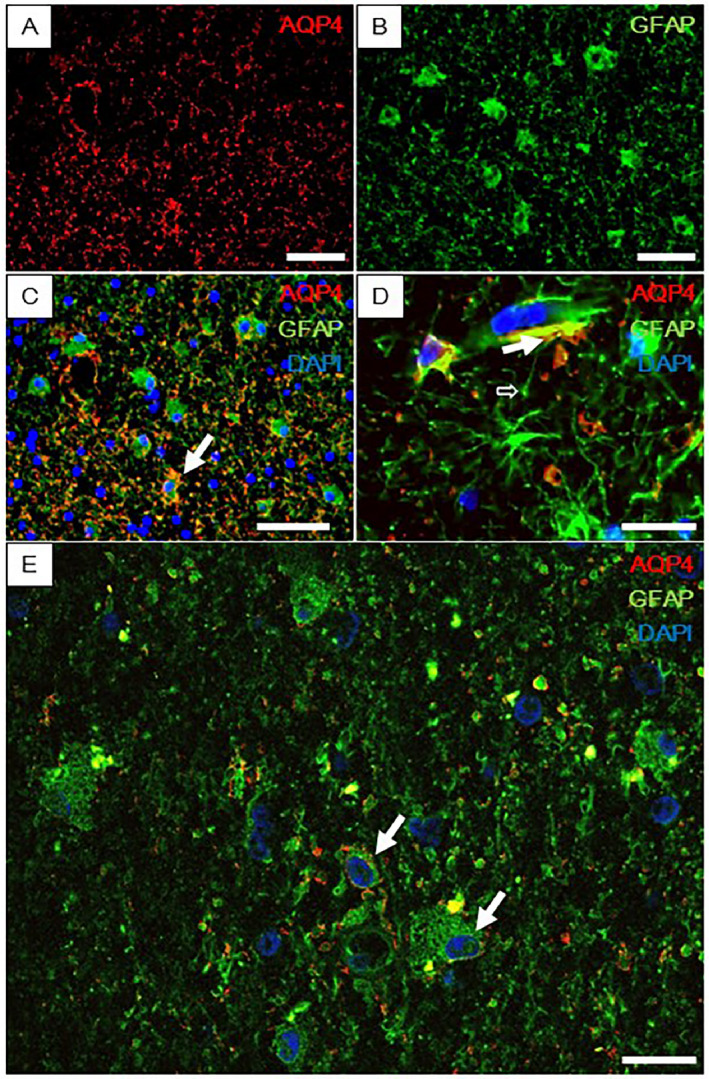
Immunofluorescence staining of type C clasmatodendrocytic astrocytes with antibodies to aquaporin (AQP4) and GFAP in CADASIL subjects. (A and B) Localization of AQP4 (red) in profiles of astrocytic cells and GFAP (green) in cell bodies of mostly clasmatodendritic astrocytes in CADASIL. (C) Merged imaged of A and B shows co‐localization of AQP4 and GFAP in the processes of normal appearing astrocytes but mostly nearer cell bodies in clasmatodendrocytes (closed arrow). (D) Normal cells shown at a higher magnification than A–C from an age matched control subject. Open arrow shows an astrocytic process around a blood vessel. Closed arrow shows AQP4 at the end‐feet of an astrocyte juxtaposed a blood vessel. E, Degenerating type B and abnormal clasmatodendritic type C cells (green) with AQP4 dislocation (red) (closed arrows). Sections were counterstained with DAPI (blue) to show nuclei. Magnification bar = 20 μm (A–C) and 10 μm (D and E).
Focusing on the frontal and anterior temporal pole WM, we first observed that the SI was increased in both CADASIL and SVD compared to their relative control age groups (P < 0.001 and P < 0.05, respectively). The mean SI value for frontal WM was 0.43 in CADASIL subjects versus 0.30 in similar age controls. Whereas, more relevant to the astrocytic pathology, in the anterior temporal pole the SI value was 0.46 in CADASIL compared to 0.31 in controls. In SVD, the SI value for the anterior temporal pole was 0.35 versus 0.31 in controls indicating on average a 46% increment in CADASIL. The density of COL4 per area and ratio of GLUT1 to COL4 within microvessels were also increased indicating increased basement membranes in CADASIL and SVD but reduced endothelium. This was demonstrated by the decreased ICAM‐1 immunoreactivity in the order CADASIL <SVD <young controls <old controls (P=0.001). CADASIL subjects showed 1.5‐fold lower ICAM‐1 immunoreactivity per area compared to young controls. Consistent with the data above on total GFAP reactive cells, we found ratios of GFAP to ICAM‐1 immunoreactivity was increased in CADASIL and SVD cases compared to young and older controls (P=0.001)
Mechanisms of astrocytic cell death—Markers of autophagy
In subsequent experiments, we explored mechanisms of astrocytic degeneration particularly focusing on the WM of the anterior temporal pole. We found that p62 immunoreactivities were localized in 90 ± 10% of the clamatodendritic astrocytes (Figure 5). Immunostaining with other markers of the dysfunctional autophagy pathway suggested that LC3 was similarly highly co‐localized in p62 immunoreactive cells. Thus, remarkably immunoreactivities of both LC3 and p62 were identified in clasmatodendritic astrocytes in CADASIL subjects but these were seldom evident in the similar age controls. Quantification of the co‐labeled LC3 marker and GFAP‐positive clasmatodendrocytes again showed 90 ± 9% co‐localization compared to 0% in controls (P=0.000). We also noted Beclin‐1 immunoreactivity in type C cells in the perivascular regions in SVD but not in controls (not shown). Furthermore, we reasoned that since astrocytes were essentially undergoing cell death that Caspase‐3 or Apoptain might be activated. Caspase‐3 is a critical for cell death, as it is either partially or totally responsible for the proteolytic cleavage of many key proteins including the nuclear enzyme poly (ADP‐ribose) polymerase. We found that the hypertrophic appearing astrocytes undergoing clasmatodendrosis were strikingly reactive for activated Caspase‐3, whereas few if any Caspase‐3‐positive cells were observed in similar age controls (Figure 6). In contrast, there were no apparent Caspase‐3‐positive cells in the neocortex in either CADASIL subjects or controls. Caspase‐3 expression in CADASIL was specific to the WM in the damaged astrocytes and end‐feet (Figure 6).
Figure 5.
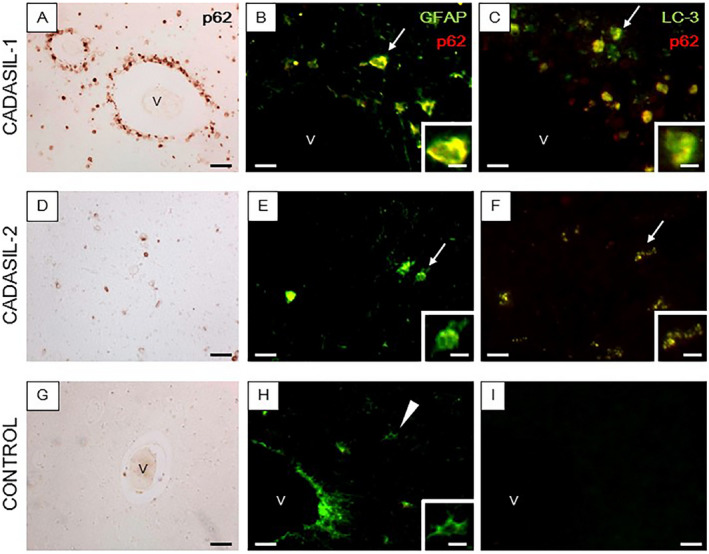
Co‐localization of LC3, p62 and GFAP immunoreactivities in the WM of CADASIL subjects. (A–I) Sections from the anterior temporal lobe from a 61‐year‐old male with CADASIL carrying the R133C mutation (A–C), 55‐year‐old male with CADASIL carrying the R558C mutation (D–F) and age matched control cases (G–I) were immunostained for p62 (red) and GFAP (green) or LC3 (green). (A, D and G) Perivascular immunoreactivity of p62 was observed around >90% of the vessel profiles in CADASIL with R133C mutation (A), whereas less perivascular p62 immunoreactivity was observed in CADASIL with R133C mutation (D) and no apparent p62 immunoreactivity was observed in control (G). (B, C, E, F, H, and I) Compared to control (H and I) the merged immunofluorescent images from CADASIL subjects show strong co‐immunolocalization of p62 antigen in degenerating GFAP‐labeled astrocytes (arrows in B and E) which are also immunopositive for LC3 (arrows in C and F). (H–I) Normal astrocytes stained for GFAP with no apparent immunopositivity to p62 (H) (arrow head) and negative immunoreactivitiy for LC3 (I). Magnification bar = 50 μm (A, D and G), 25 μm (B, C, E, F and H) and in insets B, C, E, F and H =10μm. ‘v’ indicates a blood vessel (B, C, G, H and I).
Figure 6.
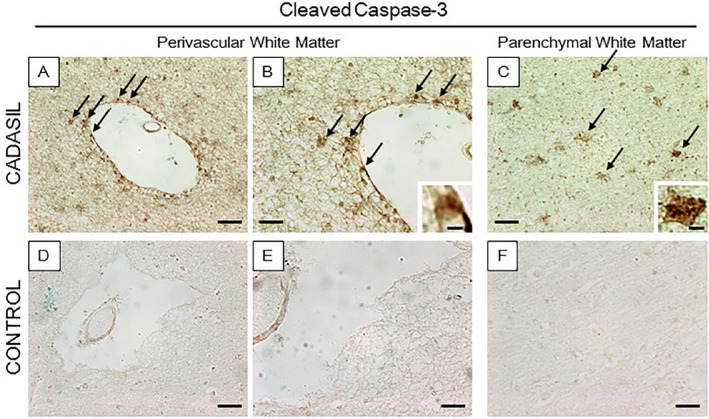
Caspase‐3 immunoreactivity associated with abnormal astrocytes in CADASIL. Caspase‐3 was frequently found in abnormal type B cells with large cell bodies and some retracted processes but not in normal astrocytes in either CADASIL or control subjects. (A–B) Perivascular immunoreactivity of Caspase‐3 was noted in deep WM of a CADASIL (R133C mutation) subject. Some astrocytes within the WM parenchyma were also observed in CADASIL (C) but there was no Caspase‐3 immunoreactivity in controls (D–F). Magnification bar = 50 μm in A and D, 25 μm in B, C, E, and F, and in insets = 5 μm (B and C).
Discussion
We report severe albeit variable astrocytopathy in CADASIL. Examination of CADASIL brains revealed essentially three morphologically distinct GFAP‐positive cell profiles. Higher percentage of abnormal and lower percentage of normal cells was indicative of greater clasmatodendrosis in CADASIL brains compared to controls. The abnormal type C largely comprised clasmatodendrocytes with retracted processes and dislocated end‐feet containing AQP4, similar to what we recently reported in post‐stroke survivors 6. As increased astrocytopathy was evident, our findings implicate profound effects on disease pathophysiology, white matter lesions, vascular pathology, and BBB damage. Astrocytes are continually being broken down and new cells being formed but this may slow down with age and duration of disease 34. Consistent with previous findings that large perivascular spaces were found in the WM of the temporal pole and greater pericyte and vascular smooth muscle cell degeneration 40, we suggest there is high turnover of astrocytes. This is likely increased also due to strategic location of subcortical infarcts 11.
Our observations foremost provide evidence that astrocytic cell death may involve deregulation of autophagy. We found the co‐localization of key markers in the autophagy pathway. In light of the current knowledge of the autophagy pathway, p62 likely binds to ubiquinated astrocytic proteins and these complexes are then recruited into LC3‐labeled autophagosomes within dying cells. Accumulation of LC3‐positive structures may suggest either autophagy upregulation at the level of autophagosome biogenesis or a block in autophagy at the later stages of the pathway, for example, impairment of autophagosome–lysosome fusion 30. However, simultaneous accumulation of LC3 and the autophagic substrate p62 strongly argues that the changes we observe are likely to be due to the autophagy blockade, which has been implicated in a range of neurological pathologies and could result in the loss of cell viability 22. We propose that autophagy dysfunction or an insufficient autophagy in stress conditions may be the cause of apoptosis in CADASIL astrocytes and result in greater turnover of astrocytes. Mechanisms leading to autophagy dysfunction as well as those linking defective autophagy to death of astrocytes in CADASIL require further investigation.
Besides a consequence of the vascular pathology, is it plausible that astrocytes are directly affected by the mutant NOTCH3 receptor? Related to this proposal, Tang et al 35 recently showed that demyelination occurs secondary to vascular pathology. They showed that compared to cells overexpressing wild‐type NOTCH3, oligodendrocytes overexpressing mutant NOTCH3 (R90C) were less viable and had a higher rate of apoptosis. Cells with mutant NOTCH3 also had higher levels of intrinsic mitochondrial apoptosis, extrinsic death receptor path‐related apoptosis, and autophagy. Thus, early defects in glia influenced by NOTCH3 mutants may directly contribute to WM pathology in addition to secondary vascular defects. Consistent with our findings, in vitro model of ischemia induced by oxygen and glucose deprivation there was increased formation of autophagosomes and autolysosomes and monodansylcadaverine (MDC)‐labeled vesicles in cultured astrocytes 25. This was accompanied by increased production of microtubule‐associated protein 1 light chain 3‐II (LC3‐II) and upregulation of Beclin 1, lysosome‐associated membrane protein 2 (LAMP2), and lysosomal cathepsin B expression in primary astrocytes.
Recent publications have described clasmatodendrosis in various neurodegenerative diseases and brain injury 32, 36. However, it appears that the astrocytopathy largely associated with the WM in CADASIL is a profound, albeit related to the intensity of disease onset. Previous studies have suggested loss of AQP4 is associated with clasmatodendrosis and edema in neuromyelitis optica 23. We recently reported AQP4 dislocation and and astrocytopathy/clasmatodendrosis in post‐stroke survivors and an experimental animal model 6. Such similarities with CADASIL indicate that new information regarding treatment of neuromyelitis optica could also be applicable to CADASIL and SVD. The extent of astrocyte involvement in causing WM degeneration in acute haemorrhagic leukoencephalitis and Binswanger's disease has been hotly discussed. It is argued that clasmatodendrosis is causing demyelination in acute haemorrhagic leukoencephalitis because it occurs before any evidence of demyelination can be detected 29 whereas in Binswanger‐like disease, clasmatodendrosis may occur without any signs of myelin loss 31. The natural history of the changes in these and even neurodegenerative disorders suggests that arteriolosclerosis is one of the first changes 10. The arteriopathy via reduction in the vascular bed could be the direct link in causing damage to astrocytes 33. That this is plausible is because reducing brain perfusion even in subcortical structures and the WM with bilateral carotid artery stenosis non‐invasively to the brain in mice leads to increased percentage of clasmatodendritic astrocytes 15, 16. Damaged astrocytes would then escalate WM changes through progressively altered BBB function. BBB damage causes leaky vessels and fibrinogen, leaked from brain vessels to parenchyma, could be taken up by damaged astrocytes (clasmatodendrocytes), as several clasmatodendrocytes were immuostained by fibrinogen in CADASIL. At this stage, severity of clasmatodendrosis might be more severe and we observed them as Type C cells (severe clasmatodendrocytes). Furthermore, ischemic preconditioning prevents astrocyte injury through upregulating protective protein 14‐3‐3γ in murine models. Preservation of astrocytes could therefore offer a potential therapy in minimizing arteriopathy in CADASIL.
Our study is not without limitations. Despite analysis of a sizeable number of different CADASIL cases, we could not demonstrate the features of astrocytic cell death by ultrastructural morphology. The final proof how the process occurs could be resolved by this means but it was difficult to examine well preserve post‐mortem tissue. Access to a larger cohort of CADASIL cases, there would be the possibility of exploring genotype–phenotype relationships and whether all genotypes have similar effects on astroglial demise. A recent study 13 showed that although specific NOTCH3 genotypes did not relate to WM changes, overrepresentation of the apolipoprotein E ε2 allele was associated with greater WM hyperintensity volumes. Our analysis was also limited to the examination of astrocytes in the WM. It is possible that dysfunctional gray matter astrocytes cause similar effects, particularly BBB disruption.
In summary, we report that there is significant astrocyte pathology in CADASIL, with the temporal lobe being most affected. We also provide clear evidence that astrocyte death in CADASIL involves the autophagy pathway. Our findings have implications for both CADASIL and aging related small vessel disease, where agents, which modulate astrocyte signaling, may provide therapeutic strategies to reduce the morbidity in vascular dementias.
Sources of Funding
Our work is supported by grants from the UK Medical Research Council (MRC, G0500247), Newcastle Centre for Brain Ageing and Vitality (BBSRC, EPSRC, ESRC and MRC, LLHW), and Alzheimer's Research (ARUK). Tissue for this study was collected by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK MRC (G0400074), by the Newcastle NIHR Biomedical Research Centre in Ageing and Age‐Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust, and by a grant from the Alzheimer's Society and ART as part of the Brains for Dementia Research Project. Y.H. was supported by SENSHIN Medical Research Foundation, Osaka, Japan and The Great Britain Sasakawa Foundation, London, UK.
Author Contributions
Yoshiki Hase: analysis and interpretation of data, acquisition of the microvascular results and revising the manuscript at various stages of preparation.
Aiqing Chen: analysis, interpretation, and acquisition of the data.
Letitia Bates: acquisition of the original astrocyte numerical data.
Lucinda Craggs: analysis, interpretation, and acquisition of the data.
Elizabeth Gemmell: analysis, interpretation, and acquisition of the data.
Arthur Oakley: analysis/interpretation/acquisition of data and technical advice on imaging.
Viktor Korolchuk: advising, editing the manuscript, and interpretation of data
Raj N Kalaria: drafting, editing the manuscript, and interpretation of data, diagnosing the cases and obtaining funding.
Disclosures
None related to this article.
Acknowledgments
The authors are grateful to the patients and families for their cooperation in the investigation of this study. We thank Janet Y Slade for expert technical assistance. We also thank Carina Tham and Sarah Hewitt for assisting in collating some of the data used in this report. The authors acknowledge the MRC London Brain Bank for Neurodegenerative Diseases (Dr. Claire Troakes), Institute for Stroke and Dementia Research, Ludwig Maximilians University, Munich Germany (Prof Dr Martin Dichgans), and the MRC Sudden Death Brain and Tissue Bank, the University of Edinburgh, for providing one each of the CADASIL cases and two control cases, respectively. We are also grateful to Drs Glyn Nelson and Alison Spilsbury for expert advice on analysis of immunofluorescent staining and technical assistance on the Zeiss Spinning Disk (Invert).
References
- 1. Allan LM, Rowan EN, Firbank MJ, Thomas AJ, Parry SW, Polvikoski TM et al (2011) Long term incidence of dementia, predictors of mortality and pathological diagnosis in older stroke survivors. Brain 134:3716–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arboleda‐Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C, Vanderburg CR et al (2011) Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small‐vessel disease. Proc Natl Acad Sci USA 108:E128–E135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carloni S, Buonocore G, Balduini W (2008) Protective role of autophagy in neonatal hypoxia‐ischemia induced brain injury. Neurobiol Dis 32:329–339. [DOI] [PubMed] [Google Scholar]
- 4. Chabriat H, Joutel A, Dichgans M, Tournier‐Lasserve E, Bousser MG (2009) CADASIL. Lancet Neurol 8:643–653. [DOI] [PubMed] [Google Scholar]
- 5. Chai H, Diaz‐Castro B, Shigetomi E, Monte E, Octeau JC, Yu X et al (2017) Neural circuit‐specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95:e539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen A, Akinyemi RO, Hase Y, Firbank MJ, Ndungu MN, Foster V et al (2016) Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post‐stroke dementia. Brain 139:242–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cherra SJ 3rd, Chu CT (2008) Autophagy in neuroprotection and neurodegeneration: a question of balance. Future Neurol 3:309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Craggs LJ, Hagel C, Kuhlenbaeumer G, Borjesson‐Hanson A, Andersen O, Viitanen M et al (2013) Quantitative vascular pathology and phenotyping familial and sporadic cerebral small vessel diseases. Brain Pathol 23:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Craggs LJ, Yamamoto Y, Ihara M, Fenwick R, Burke M, Oakley AE et al (2014) White matter pathology and disconnection in the frontal lobe in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Neuropathol Appl Neurobiol 40:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deramecourt V, Slade JY, Oakley AE et al (2012) Staging and natural history of cerebrovascular pathology in dementia. Neurology 78:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duering M, Csanadi E, Gesierich B, Jouvent E, Herve D, Seiler S et al (2013) Incident lacunes preferentially localize to the edge of white matter hyperintensities: insights into the pathophysiology of cerebral small vessel disease. Brain 136:2717–2726. [DOI] [PubMed] [Google Scholar]
- 12. Foster V, Oakley AE, Slade JY, Hall R, Polvikoski TM, Burke M et al (2014) Pyramidal neurons of the prefrontal cortex in post‐stroke, vascular and other ageing‐related dementias. Brain 137:2509–2521. [DOI] [PubMed] [Google Scholar]
- 13. Gesierich B, Opherk C, Rosand J, Gonik M, Malik R, Jouvent E et al (2016) APOE varepsilon2 is associated with white matter hyperintensity volume in CADASIL. J Cereb Blood Flow Metab 36:199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hara A, Mori H, Niwa M (2000) Novel apoptotic evidence for delayed neuronal death in the hippocampal CA1 pyramidal cells after transient ischemia. Stroke 31:236–238. [DOI] [PubMed] [Google Scholar]
- 15. Hase Y, Craggs L, Hase M, Stevenson W, Slade J, Lopez D et al (2017) Effects of environmental enrichment on white matter glial responses in a mouse model of chronic cerebral hypoperfusion. J Neuroinflammation 14:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hattori Y, Enmi J, Iguchi S, Saito S, Yamamoto Y, Nagatsuka K et al (2016) Substantial reduction of parenchymal cerebral blood flow in mice with bilateral common carotid artery stenosis. Sci Rep 6:32179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hewitt G, Korolchuk VI (2017) Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol 27:340–351. [DOI] [PubMed] [Google Scholar]
- 18. Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, Oakley AE et al (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer's disease, and dementia with Lewy bodies. Acta Neuropathol 119:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T (2004) Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 226:75–80. [DOI] [PubMed] [Google Scholar]
- 20. Levine B, Kroemer G. Autophagy in the pathogenesis of disease (2008) Cell 132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A et al (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034. [DOI] [PubMed] [Google Scholar]
- 23. Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S et al (2013) Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol 125:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perry RH, Oakley AE (1993) ‘Newcastle Brain Map’. In: Neuropsychiatric Disorders, pp. 1–10, London: Wolfe. [Google Scholar]
- 25. Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L et al (2010) Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 6:738–753. [DOI] [PubMed] [Google Scholar]
- 26. Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N et al (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106:107–120. [DOI] [PubMed] [Google Scholar]
- 27. Rami A, Kogel D (2008) Apoptosis meets autophagy‐like cell death in the ischemic penumbra: two sides of the same coin? Autophagy 4:422–426. [DOI] [PubMed] [Google Scholar]
- 28. Rami A, Langhagen A, Steiger S (2008) Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy‐like cell death. Neurobiol Dis 29:132–141. [DOI] [PubMed] [Google Scholar]
- 29. Robinson CA, Adiele RC, Tham M, Lucchinetti CF, Popescu BF (2014) Early and widespread injury of astrocytes in the absence of demyelination in acute haemorrhagic leukoencephalitis. Acta Neuropathol Commun 2:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, Klionsky DJ (2009) In search of an “autophagomometer”. Autophagy 5:585–589. [DOI] [PubMed] [Google Scholar]
- 31. Sahlas DJ, Bilbao JM, Swartz RH, Black SE (2002) Clasmatodendrosis correlating with periventricular hyperintensity in mixed dementia. Ann Neurol 52:378–381. [DOI] [PubMed] [Google Scholar]
- 32. Sakai K, Fukuda T, Iwadate K (2013) Beading of the astrocytic processes (clasmatodendrosis) following head trauma is associated with protein degradation pathways. Brain Inj 27:1692–1697. [DOI] [PubMed] [Google Scholar]
- 33. Simpson JE, Fernando MS, Clark L, Ince PG, Matthews F, Forster G et al (2007) White matter lesions in an unselected cohort of the elderly: astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol Appl Neurobiol 33:410–419. [DOI] [PubMed] [Google Scholar]
- 34. Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang M, Shi C, Song B, Yang J, Yang T, Mao C et al (2017) CADASIL mutant NOTCH3(R90C) decreases the viability of HS683 oligodendrocytes via apoptosis. Mol Biol Rep 44:273–280. [DOI] [PubMed] [Google Scholar]
- 36. Tomimoto H, Akiguchi I, Wakita H, Suenaga T, Nakamura S, Kimura J (1997) Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer's disease patients. Acta Neuropathol 94:146–152. [DOI] [PubMed] [Google Scholar]
- 37. Xu F, Gu JH, Qin ZH (2012) Neuronal autophagy in cerebral ischemia. Neurosci Bull 28:658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamamoto Y, Craggs L, Baumann M, Kalimo H, Kalaria RN (2011) Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol Appl Neurobiol 37:94–113. [DOI] [PubMed] [Google Scholar]
- 39. Yamamoto Y, Craggs LJ, Watanabe A, Booth T, Attems J, Low RW et al (2013) Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and granular osmiophilic material in CADASIL. J Neuropathol Exp Neurol 72:416–431. [DOI] [PubMed] [Google Scholar]
- 40. Yamamoto Y, Ihara M, Tham C, Low RW, Slade JY, Moss T et al (2009) Neuropathological correlates of temporal pole white matter hyperintensities in CADASIL. Stroke 40:2004–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]