

Abstract
Glioblastoma (GBM) represents the most common and most malignant type of primary brain tumor and significantly contributes to cancer morbidity and mortality. Invasion into the healthy brain parenchyma is a major feature of glioblastoma aggressiveness. Reelin (RELN) is a large secreted extracellular matrix glycoprotein that regulates neuronal migration and positioning in the developing brain and sustains functionality in the adult brain. We here show that both RELN and its main downstream effector DAB1 are silenced in glioblastoma as compared to non‐neoplastic tissue and mRNA expression is inversely correlated with malignancy grade. Furthermore, RELN expression is positively correlated with patient survival in two large, independent clinically annotated datasets. RELN silencing occurs via promoter hypermethylation as shown by both database mining and bisulfite sequencing of the RELN promoter. Consequently, treatment with 5′‐Azacytidine and trichostatin A induced RELN expression in vitro. On the functional level, we found RELN to regulate glioblastoma cell migration both in a DAB1 (tyrosine phosphorylation)‐dependent and ‐independent fashion, depending on the substrate provided. Moreover, stimulation of RELN signaling strongly reduced proliferation in glioblastoma cells. This phenotype depends on DAB1 stimulation by RELN, as a mutant that lacks all RELN induced tyrosine phosphorylation sites (DAB1‐5F) failed to induce a growth arrest. Proteomic analyzes revealed that these effects are mediated by a reduction in E2F targets and dephosphorylation of ERK1/2. Taken together, our data establish a relevance of RELN signaling in glioblastoma pathology and thereby might unearth novel, yet unrecognized treatment options.
Keywords: DAB1, glioma, invasion, proliferation, reelin
Introduction
Glioblastoma (GBM) represents the most common and most malignant form of primary brain tumor. Despite an aggressive combination of surgery followed by radiochemotherapy and numerous clinical trials on innovative treatment options, the overall improvements achieved are moderate 45. Thus, the identification of novel molecules and signaling pathways involved in glioblastoma pathology are urgently needed. A hallmark of glioblastoma malignancy is the infiltration of tumor cells into the healthy brain parenchyma that makes complete surgical resection of the tumor impossible. Among others, integrins are believed to play a major role in this context as they mediate the contact of the cells to the extracellular matrix (ECM) 37. However, there are still many unanswered questions regarding the development and regulation of the invasive glioma phenotype.
The large secreted protein reelin (RELN) was discovered as a regulator of neuronal migration in reeler mice. In these mice, cortical neurons fail to reach their normal position in the cortex, resulting in an inside out layering 19. RELN also has been shown to regulate neural precursor cell cycle exit 25 and differentiation 23. It inhibits signaling via integrins by directly disrupting the binding of α3β1 integrin to laminin 14 and the binding of α5β1 integrin to fibronectin in a DAB1‐dependent manner 41. In addition, RELN mediates cofilin phosphorylation in neuronal cells 11 with alterations in cofilin activity exerting strong effects on glioma cell invasion 34. In the canonical signaling cascade, APOER2 and VLDLR mediate the tyrosine phosphorylation of the intracellular adaptor protein DAB1 through RELN binding. This phosphorylation event is catalyzed by SRC family kinases and reinforced by phosphorylated DAB1 in a positive feedback loop 19. Importantly, kinases of the SRC family are well known as important players in tumorigenesis and attractive drug targets in glioblastoma 1, connecting RELN signaling and tumorigenesis mechanistically.
Although primarily studied in post‐mitotic neurons in the central nervous system (CNS), both RELN and DAB1 are also expressed in a variety of tissues outside of the CNS 43. In a cancer context, it has been shown that RELN is involved in the regulation of invasion and proliferation of breast cancer 36, 44. Moreover, RELN was found to inhibit both migration and invasion of pancreatic cancer cells 39. Silencing of reelin in these tumors (breast, pancreatic and gastric cancer) occurred via hypermethylation of the CpG island containing the RELN promoter 13, 39, 44. Of note, also the DAB1 gene maps to a site frequently deleted in different cancers 32.
In contrast to epithelial tumors, a role for RELN signaling has not yet been implicated in brain tumors. In a single investigation on gangliogliomas, transcriptional downregulation of DAB1 was observed but the tumors lacked DAB1 mutations 21. Also, the data on neuroblastoma, a peripheral tumor, are controversial 4, 16. Another study reported decreased RELN expression in various tumor types (including glioblastoma) by analyzing TCGA data 9. This prompted us to follow up on the potential tumor suppressive functions of DAB1 and RELN in gliomas. Interestingly, it has been recently suggested that malignant gliomas can also originate from neural progenitors and/or differentiated neurons 2, 17 and DAB1 as well as RELN have been implicated in the regulation of neuronal differentiation 23, 35. Therefore, it seemed likely that crossconnections between RELN signaling and glioma pathology might exist.
Materials and Methods
In silico analysis
Prenormalized expression datasets from glioblastoma samples and matched clinical data were obtained from the cancer genome atlas 7 data portal (URL: https://tcga-data.nci.nih.gov). In addition, a second set of gene expression data was obtained from Rembrandt 31 and downloaded via the array express database (URL: http://www.ebi.ac.uk/arrayexpress, accession E‐MTAB‐3073). The Rembrandt microarrays were RMA normalized and analyzed with R ver. 3.2.1 and the affylmGUI package. Genome wide methylation 22, 46 data as well as the manufacturer's annotation for each CpG probe were obtained from the Gene Expression Omnibus (GEO) Database (URL: http://www.ncbi.nlm.nih.gov/geo, accession GSE36278 and GSE60274, platform GEO accession GPL13534). The GSEAPreranked module v5 for Gene set enrichment analysis 47 was accessed from the gene pattern server (URL: http://genepattern.broadinstitute.org) and GSEA was performed with the fold changes as calculated by DESeq2.
Clinical samples
Thirty‐five snap‐frozen tumor samples were selected from the tumor tissue collection of the Department of Neuropathology, Heinrich‐Heine‐University, Duesseldorf, Germany and studied according to protocols approved by the institutional review board. It was confirmed via standard histology that all samples had a tumor content of at least 80%. Included were 12 primary glioblastomas WHO grade IV (pGBM IV), 4 secondary glioblastomas WHO grade IV (sGBM IV), 7 anaplastic astrocytomas WHO grade III (AA III) and 12 diffuse astrocytomas WHO grade II (A II). Classification as secondary glioblastoma was based on the presence of a histologically verified preceding lower grade astrocytic lesion. Non‐neoplastic brain control tissues were derived from four different individuals: one from the temporal lobe of a 66‐year‐old male, one from the occipital lobe of a 82‐year‐old female and two from cortex of a 76‐year‐old male and a 72‐year‐old female, respectively. All non‐neoplastic tissues were surgical specimens (snap‐frozen), three with the clinical history of epileptic surgery, one with traumatic brain injury. Extraction of DNA and RNA from frozen tumor tissue was performed as has been described previously 38. The non‐neoplastic brain controls used for analysis of the mRNA expression levels in glioma cell lines were commercially available total RNA preparations derived from a 24‐year‐old male (BioChain, Newark, CA, USA) and a 66‐year‐old female (Agilent Technologies, Santa Clara, CA, USA).
cDNA synthesis and real‐time PCR
cDNA was synthetized from 3 µg total RNA with the super script II reverse transcriptase (Life Technologies, Carlsbad, CA) in a reaction volume of 50 µl. cDNA of 12 ng was then used in a real‐time PCR performed with the Platinum SYBR green Kit (Life Technologies) in a StepOne Plus real‐time PCR thermocycler (Applied Biosystems, Foster City, CA). Initial incubation was performed at 95°C for 10 minutes followed by 40 cycles of 95°C for 10 s and 60°C for one minute. Relative expression was calculated with the ΔΔCt method 29. All primer sequences used are provided in Supporting Information Table 1.
Bisulfite sequencing
Bisulfite treatment was performed as previously described 38. Bisulfite converted DNA of 100 ng was then amplified using a HotStarTaq (Qiagen, Hilden, Germany) and the primer pairs indicated in Supporting Information Table 1. PCR conditions are available on request. The amplified fragment is provided in Supporting Information Fig. 11 Amplified DNA was purified with the Jet quick kit (Genomed/Thermo Fisher, Waltham, MA) and processed for Sanger sequencing on an ABI PRISM® 377 machine (Applied Biosystems).
Cell culture
Cell lines were cultured, obtained and characterized as has been described previously 18 and cell line identity was confirmed by short tandem repeat analysis of 16 loci by CLS Cell Lines Services (Eppelheim, Germany). After thawing, cells were passaged twelve times or cultured for six weeks, respectively, at maximum. The cells were all tested negatively for mycoplasma contamination by PCR including positive and negative controls as has been described by others 33. HEK293T cells expressing GFP or murine RELN were kindly provided by Prof. Eckart Förster (Ruhr University, Bochum, Germany). Cell culture and 5‐Aza‐2′‐deoxycytidine and trichostatin A treatment of glioblastoma cell lines were performed as previously described 3, 18.
RELN production
HEK‐293T cells expressing murine (m)RELN (or GFP as a control) 11 were seeded at a density of ∼26000 cells/cm2 in complete medium. After overnight incubation, the medium was exchanged to fresh complete medium or starving medium (no FCS, 0.1% BSA). To remove serum remnants, the cells were washed once with the starving medium. Then, the cells were allowed to grow and secrete RELN for another 48 h. Afterwards, the supernatant was collected and residual cells were removed by centrifugation at 200 × g for 5 minutes. For experiments with the function blocking CR‐50 antibody, a mouse isotype control IgG (#5415, clone G3A1; Cell signaling technology, Danver, CO) or the CR‐50 antibody (MBL, Nagoya, Japan) were added at a final concentration of 20 µg/ml.
Cloning and generation of stably transfected cells
The ORF of human full length DAB1 in pCMV‐Sport6 (Thermo Fisher) was subcloned into the pIRESneo2 vector (Clontech, Palo Alto, CA). As human and mouse DAB1 are identical on the protein level within large parts of their N‐terminus, the N‐terminal portion containing the murine 5F sequence 20 was cloned into the pIRESneo2 vector together with the C‐terminus of human DAB1. This resulted in a construct that is identical to a human DAB1‐5F mutant on protein level. For the establishment of stable cell lines, U87 and U251 cells were seeded at a density of ∼7000 cells per cm2 on Ø10 cm dishes (Sarstedt, Nuembrecht, Germany) and allowed to grow for 48 h. Then, the cells were transfected with 1.5 µg pIRESneo2 empty vector (EV), hDAB1‐WT//pIRES‐Neo2 or hDAB1‐5F//pIRES‐Neo2 with 12 µl of the transfection reagent Lipofectamine 2000 (Life Technologies). For the establishment of stably transfected cells, G418 (Carl Roth, Karlsruhe, Germany) was added to the medium at a concentration of 200 µg/ml (U87) to 400 µg/ml (U251) and the cells were cultured for two weeks until resistant cells emerged.
Plate coating for live cell imaging
Eight well µ‐slides (ibiTreat; Ibidi, Munich, Germany) were coated with 5 µg/ml fibronectin or 2 µg/ml laminin (all from Sigma‐Aldrich, St. Louis, Missouri) for 60 minutes. Then, the coating solution was removed and the slides washed with sterile H2O. The laminin coated plates were allowed to dry for 45 minutes at RT. Finally, the plates were blocked with DPBS containing 0.5% bovine serum album (BSA) for 30 minutes.
Two‐dimensional‐migration assay
Cells were seeded at a density of 6000 cells per cm2 on laminin or fibronectin coated 8 well µ‐slide dishes in CO2‐independent medium (Life Technologies) supplemented with 5% FCS, 2 mM l‐glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies). Cells were allowed to adhere for 6 h, two volumes of RELN or GFP conditioned CO2‐independent medium were added and afterwards cell motility was analyzed using a Leica AF‐6000 (Leica Microsystems, Wetzlar, Germany) microscope and a microscope stage incubator to keep the temperature at 37°C. Pictures were taken every 20 minutes over a period of 12 h. At least 50 randomly chosen cells were tracked using ImageJ 1.47 40 and the manual tracking plugin. Immotile cells and cells that divided during the observation period were excluded from tracking. To calculate velocity and directionality, we imported the tracking files to the chemotaxis and migration tool (Ibidi).
Proliferation assay
Cells per well of 1500 (U87) or 1000 (U251) were seeded on a 96‐well plate in triplicates After 6 h, the medium was changed to starving medium, another 100 µl GFP or RELN conditioned supernatant was added and the cells were incubated at 37°C and 5% CO2. Every 24 h, three wells per condition were labeled with 1 µM (final concentration) Hoechst 33342 for 1 h. Then, four pictures were taken with a low magnification (4×) objective from randomly chosen areas of the triplicate wells and analyzed with an automated cell profiler pipeline (ver. 2.1.1) 8 to count the number of nuclei present.
Cell cycle analysis
Cell cycle analysis was performed via propidium iodide staining. After O/N starvation, cells were trypsinized and counted. Cells of 1 × 106 were fixed in 70% precooled ethanol for 1 h on ice. The cells were washed twice in DPBS, resuspended in staining mix (final concentration 50 µg/ml RNAseA, 40 µg/ml propidium iodide and 0.05% saponin in DPBS) and incubated for 30 minutes at 37°C. Then, the cells were analyzed on a FACS CantoII flow cytometer (Becton Dickinson) and gating as well as quantification was performed with ModFit LT v3.2 (Verity Software House, Topsham, ME, USA).
Western blotting and detection of proteins
Western blotting was performed according to standard protocols. For western blot analysis, 150 000 cells were seeded on Ø1.5 cm tissue culture dishes (Sarstedt) and allowed to grow for 48 h. Primary antibodies used were DAB1 (1:1000, #3328; Cell signaling technology), p(Y220)‐DAB1 (1:1000, #3327; Cell signaling technology), caspase‐3 and cleaved caspase‐3 (1:1000, #14220, clone D3R6Y; Cell signaling technology), p(S491)‐DAB1 (ab5776, Abcam, Cambridge, UK) and tubulin as a loading control (1:10 000, clone DM1A; Sigma‐Aldrich, St. Louis, MO). Anti‐rabbit or anti‐mouse IgG linked to horseradish peroxidase (Santa Cruz, Dallas, Texas) were used as secondary antibodies (1:10 000 dilution). After development with picoluminescence substrate (Thermo Fisher) quantification was performed on a LAS4000 imaging system (GE healthcare, Munich, Germany) using the ImageQuant TL ver. 7.0 software (GE healthcare). For Reln immunohistochemistry, we used the antibody clone E‐5 (#sc‐25346, Santa Cruz Biotechnology, Dallas, Texas) following a standard protocol (1:50 dilution, overnight incubation, 5 minutes heat‐induced antigen retrieval).
Mass spectrometry
Total cellular protein lysates were prepared and subjected to label‐free quantification by SWATH‐MS as published previously 42. Briefly, 100 µg of protein was subjected to tryptic digestion using the FASP‐protocol 50. Six aliquots (1 µg each) of the pooled protein digests were used for generation of the SWATH‐library using an 88 minutes‐LC‐gradient and a TOP40 IDA‐method; database searches were conducted with ProteinPilot 4.5 against the Uniprot database (human). SWATH‐MS‐runs were accomplished with 1 µg of the individual protein digests using the same LC‐gradient and SWATH‐windows of 15 Da width (total m/z range 400–1000). In total, 1950 proteins were quantified using PeakView 2.1. Differential abundance of proteins was calculated based on non‐normalized protein counts with DESeq2 30.
Proteome profiler assay
The phosphorylation of 43 different kinases and the amount of two related proteins was detected with the proteome profiler assay (R&D Systems, Minneapolis, MN). Cells were grown and stimulated with GFP or mRELN as described above. The assay was performed according to the manufacturer's instructions. The chemiluminescent signal was detected with a LAS4000 imaging system (GE healthcare).
DuoSet IC ELISA
Commercially available ELISA assays (R&D Systems) for STAT3(Y705) and ERK1(T202/Y204)/ERK2(T185/Y187) were performed according to the manufacturer's instructions. Briefly, 96‐well ELISA plates (R&D Systems) were coated with capture antibody (diluted 1:180) in PBS. On the next day, the plates were blocked with BSA in PBS, washed with washing buffer (0.05% Tween‐20 in PBS) and cell lysates were incubated for 2 h at RT on the plates. Afterwards, the plates were washed again and detection antibody was added (1:36 diluted). Then, streptavidin‐HRP was added for 20 minutes at RT. After washing, a commercially available substrate solution (R&D Systems) was added and the reaction was stopped after 20 minutes with 2N H2SO4.
Data analysis
All analyzes were performed with GraphPad Prism 4.0 or higher and R ver. 3.2.1. If not otherwise indicated, two‐tailed t‐tests were used for statistical analysis. We only proceeded with post‐tests if the ANOVA or Kruskal‐Wallis test indicated significant differences between groups (P < 0.05).
Results
RELN pathway gene expression is correlated with glioblastoma subtypes and shorter overall survival
Analysis of agilent gene expression microarrays from the TCGA database 7 revealed that RELN is significantly upregulated in the G‐CIMP and neural as compared to the classical and mesenchymal glioblastoma gene expression subtype 49. In line with this finding, RELN was significantly related to IDH1 mutational status in the TCGA dataset (Bonferroni corrected two‐sided Mann‐Whitney test, P = 0.036) but not to mutations in EGFR, TP53, PTEN, PIK3CA, PIK3R1, RB1 or NF1 (Supporting Information Fig. 1). Of note, RELN mutations themselves were also detectable in a significant number of glioblastoma (7.89%), underscoring that RELN is altered in this tumor type. For DAB1, upregulation was only present in the neural subtype (Kruskall‐Wallis test followed by Dunn's multiple comparison test, significant changes are indicated; Figure 1A,B). The upregulation of RELN and DAB1 in the neural subtype was confirmed by analysis of another large glioblastoma cohort from Rembrandt (Figure 1E,F) 31.
Figure 1.
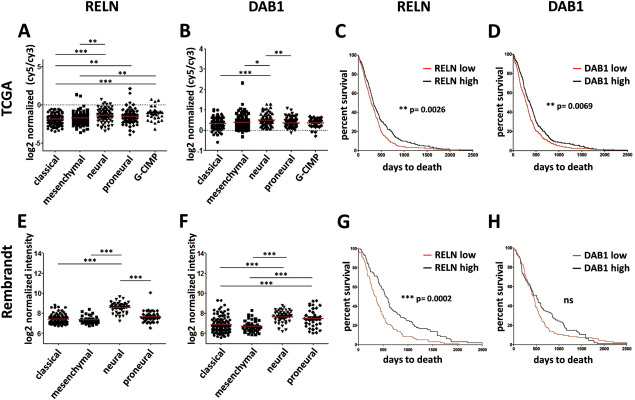
RELN and DAB1 mRNA expression is correlated with transcriptional subtypes and survival. A, B, C, D. Statistical comparison of expression subtypes was performed by the non‐parametric Kruskal‐Wallis test followed by Dunn's multiple comparison test. Red lines indicate means. C, D, G, H. Survival curves were compared with a log‐rank (Mantel‐Cox) test. For survival analyzes, samples were divided by the median expression into the low (red line) and high expression (black line) subgroup. Significant changes are indicated (* = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001). A. RELN expression is low in the mesenchymal and classical subtype and highest in the neural and G‐CIMP subtype in the TCGA dataset (Agilent platform). B. DAB1 expression is significantly lower in the mesenchymal and classical subtype than in the neural subtype in the TCGA dataset. C. High RELN expression is correlated with longer overall survival in the TCGA dataset. D. High DAB1 expression is correlated with longer overall survival in the TCGA dataset. E. RELN expression is significantly lower in the mesenchymal and classical subtype than in the neural subtype in the Rembrandt dataset (Affymetrix platform). F. DAB1 expression is low in the mesenchymal and classical subtype and highest in the neural subtype in the Rembrandt dataset (probe set 228329_at). G. High RELN expression is correlated with longer overall survival in the Rembrandt dataset. H. High DAB1 expression (probe set 228329_at) is correlated with longer overall survival in the Rembrandt dataset. Although not significant, there is a strong trend toward longer overall survival in the DAB1 high expression subgroup.
When the gene expression data were combined with clinical data and patients divided by the median into a low and a high expression subgroup, both high DAB1 and high RELN expression were significantly correlated with increased overall survival in glioblastoma patients in the TCGA dataset (Log‐rank (Mantel‐Cox) test, P = 0.0026 (RELN) and P = 0.0069 (DAB1); Figure 1C,D). Importantly, RELN remained significantly (P < 0.05) associated with survival in a Cox proportional‐hazards regression model that contained G‐CIMP status, DAB1 expression as well as age (below or above 50) as variables. Survival correlations could be confirmed in the Rembrandt dataset for RELN. High RELN expression was significantly correlated with longer overall survival (Log‐rank (Mantel‐Cox) test, P = 0.0002; Figure 1G). For DAB1, there was only a trend toward longer overall survival in the high expression subgroup (Figure 1H).
RELN expression in glioblastomas is silenced by promoter methylation
The genomic region around the RELN transcription startsite (± 2kb) contains a large ∼2kb CpG island as predicted by the CpG island finder software (URL: http://dbcat.cgm.ntu.edu.tw). We designed primers that amplify the part of the CpG island spanning the transcription start site (TSS) (Supporting Information Fig. 2). We analyzed two publicly available datasets that used the Illumina 450k bead array for methylation analysis and additionally contained non‐neoplastic brain tissue. We found that the CpGs that are annotated to the RELN gene and that lie within the CpG island (according to the Illumina annotation) are significantly hypermethylated in glioblastoma samples as compared to non‐neoplastic brain. In contrast, several CpGs in the gene body (including the first exon and 3‐prime UTR according to Illumina annotation) of RELN were hypomethylated (Figure 2A). Own bisulfite sequencing of the region around the transcription start site revealed that RELN is heavily hypermethylated in glioblastoma and that the methylation status is positively correlated with malignancy grade (Supporting Information Table 2). Hypermethylation of the RELN promoter was significantly more often found in glioblastomas as compared to WHO grade II and WHO grade III astrocytomas (P < 0.001 and P < 0.05, respectively, Fisher's exact test; Figure 2B). The RELN promoter was also nearly completely methylated in glioblastoma cell lines in vitro and treatment with the demethylating agent 5′‐Azacytidine (in combination with trichostatin A) led to a reduction in promoter methylation (Supporting Information Table 3). Consequently, the same treatment regimen led to a strong reexpression of RELN in glioblastoma cell lines (Figure 2C). As such, there is compelling evidence that RELN in glioblastoma tissues and cell lines is silenced epigenetically by promoter hypermethylation.
Figure 2.
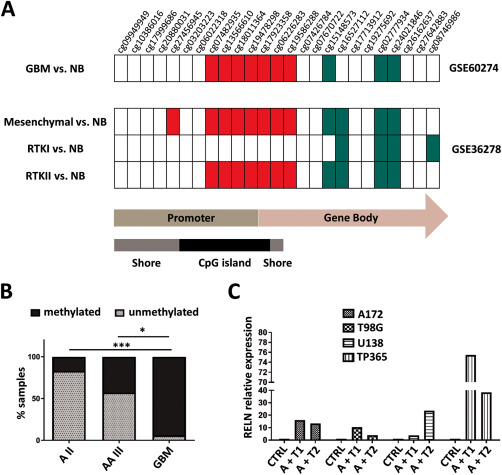
RELN is silenced by promoter hypermethylation in glioblastoma tissues and cell lines. A. Both the array‐based dataset from GSE36278 (46) and GSE60274 (22) show significant hypermethylation in the CpG island spanning the transcription start site (TSS). Significantly hypermethylated CpGs in glioblastoma (GBM) in comparison to non‐neoplastic brain tissue (NB) (adj. P < 0.05) are indicated in red, significantly hypomethylated CpGs in green (adj. P < 0.05) and CpGs with no change in white. Hypermethylation is more pronounced in the receptor tyrosine kinase (RTK) II (corresponding to the classical subtype from the TCGA) and mesenchymal subtype that also show reduced RELN mRNA levels. B. Bisulfite sequencing of the RELN promoter reveals frequent hypermethylation in glioblastoma (also compare Supporting Information Table 2). Hypermethylation is positively correlated with malignancy grade. Significant changes between WHO grades (A II = diffuse astrocytoma, AA III = anaplastic astrocytoma, GBM = glioblastoma) are indicated (* = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001; Fisher's exact test). C. Treatment with 5′‐Azacytidine and trichostatin A leads to reexpression of RELN as measured by real‐time PCR. The RELN mRNA level is increased in the glioblastoma cell lines A172, T98G, U138 and TP365 up to 75‐fold. Cells were treated with DMSO as control (CTRL), 500 nM 5‐Aza‐2′‐deoxycytidine for 48 h + trichostatin A for 24 h (A + T1) or 1 µM 5‐Aza‐2′‐deoxycytidine for 72 h + 1 µM trichostatin A for 24 h (A + T2).
RELN and DAB1 expression is correlated with malignancy grade in gliomas and tissue expression levels are retained in vitro
Real‐time PCR analysis of an own panel of astrocytic tumors of different WHO grades showed that both RELN and DAB1 are strongly downregulated on the mRNA level and that the RELN receptors, VLDLR and APOER2, are upregulated as compared to non‐neoplastic brain tissue. In addition, both RELN and DAB1 expression were inversely correlated with tumor grade and significantly lower in glioblastomas as compared to WHO grade II astrocytomas (Kruskal‐Wallis test followed by Dunn's multiple comparison test, significant changes are indicated; Figure 3A). Loss of Reln expression in tumor cells was also confirmed on the protein level by immunohistochemistry although significant intra‐ as well as intertumoral heterogeneity could be observed (Supporting Information Fig. 3).
Figure 3.

Classical glioblastoma cell lines recapitulate the changes found in glioblastoma tumor tissue. A. Real‐time PCR of RELN, DAB1, VLDLR and APOER2 in astrocytic tumors of different grades (A II: n = 11; AA III: n = 6; GBM IV: n = 9) relative to four non‐neoplastic brain samples (set to 1). Shown are means + SD. RELN expression is inversely correlated with tumor grade and significantly higher in WHO grade II astrocytomas than in glioblastomas. All significant changes between WHO grades are indicated (* = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001; Kruskal‐Wallis test followed by Dunn's multiple comparison test). B. GSEA prediction of the glioblastoma subtype of the cell lines based on next generation sequencing data. All cell lines tested belong to the mesenchymal or classical subtype. C–F. Real‐time PCR of RELN (C), DAB1 (D), VLDLR (E) and APOER2 (F) expression in different glioblastoma cell lines as compared to non‐neoplastic brain tissue (NB). ARF1 was used as an internal control. G. Expression of different APOER2 and VLDLR ligands based on next generation sequencing data. While clusterin (CLU) and Thrombospondin1 (THBS1) are expressed in different glioblastoma cell lines, RELN and APOE are not expressed in vitro.
To establish a suitable model system for in vitro experiments, we screened seven classical glioblastoma cell lines as well as four stem‐like cell lines for RELN and DAB1 expression via real‐time PCR (Figure 3C,D). Non‐neoplastic brain tissues were used as controls. Consistent with the data from the tissue samples (Figure 3A), expression of both genes was absent or low in all glioblastoma cell lines (classical and stem‐like cells). We thus decided to make use the classical cell lines for our further experiments. As classified by ssGSEA, all classical cell lines tested corresponded to either the classical or mesenchymal expression subtype (Figure 3B). Also congruent with the tissue data, the expression of APOER2 was high in the glioblastoma cell lines as compared to non‐neoplastic brain tissue (Figure 3E). The expression of VLDLR was highly variable in glioblastoma cell lines (Figure 3F). We also reanalyzed previously obtained next generation sequencing data of our cell lines 18 to check if any other ligands for the receptors VLDLR and APOER2 were expressed. Indeed, THBS1 and Clusterin were highly expressed (RPKM > 10, Figure 3G).
We then decided to construct an expression system in which DAB1 overexpression was driven from a pCMV promoter followed by an IRES sequence. DAB1 overexpressing cells were selected by adding G418 to the cell culture medium. A DAB1 mutant was used as an additional control. This mutant (5F) has all five tyrosine residues mutated to phenylalanine that are phosphorylated following RELN stimulation 20. Interestingly, we observed varying abundance of DAB1‐WT proteins of different length in U251 and U87. While in U87 the predominant band was visible at 60 kDa for both DAB1‐WT and 5F, in U251 the predominant band of the DAB1‐WT protein was visible at 45 kDa. We checked that the different cell lines had integrated full length DAB1‐WT and DAB1‐5F at equal levels by semi‐quantitative PCR as integration artifacts could have potentially caused the length differences. For this assay, we designed the primers in a way that one pair covers the five‐prime and one pair covers the three‐prime border of the insert. We quantified if the five‐prime to three‐prime ratio is altered in one of the cell lines what was not the case. The integration of both full‐length constructs was equally efficient in U251 and U87. In addition, we amplified the full‐length DAB1‐WT and DAB1‐5F inserts from genomic DNA and verified that they did not contain any mutations (Supporting Information Fig. 4). Thus, it appears most likely that phosphorylation‐dependent processing of DAB1—as already described in neuronal cells 6—might account for the observed differences in western blotting.
Proteomics identifies E2F targets as well as ERK1/2 and STAT3 phosphorylation as downstream mediators of RELN signaling
To identify possible effects of RELN and DAB1 deregulation, we subjected U87 cells expressing DAB1‐WT or DAB1‐5F to RELN stimulation for 1 h and quantified both protein abundance and phosphorylation status of proteins. The abundance of few individual proteins changed significantly (P‐adj.<0.1) comparing DAB1‐WT expressing with non‐WT (empty vector and 5F) expressing cells (Supporting Information Table 4). No additional changes were observed on RELN stimulation. Interestingly, on a pathway level, E2F target gene sets were significantly downregulated in the U87‐DAB1‐WT group as compared to empty vector or 5F cells (GSEA; Figure 4A,B).
Figure 4.
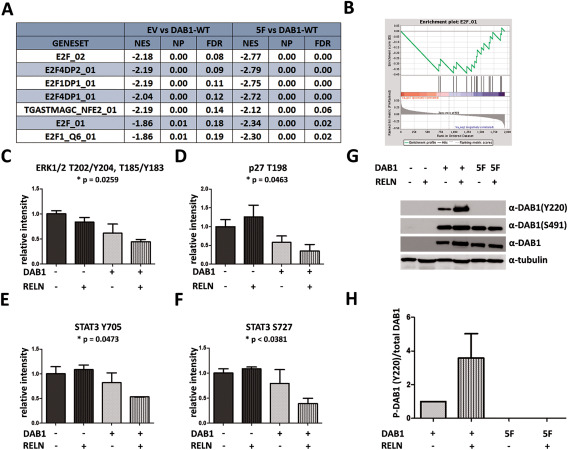
Proteomic analysis reveals enrichment for E2F pathway components in control cells compared to DAB overexpressing cells. A. GSEA of proteomic data based on fold changes as calculated by DESeq2. N = 5 in each group. E2F signaling is the main deregulated pathway after DAB1‐WT expression, represented by several significant gene sets at P < 0.05 and FDR < 0.2. B. Enrichment plot for the E2F gene set for DAB1‐WT vs. empty vector cells. C–F. Quantification of the proteome profiler analysis. Phosphorylation of ERK1/2 on T202/Y204 and T185/Y183 (C), of STAT3 on Y705 (D) and S727 (F) as well as phosphorylation of p27 on T198 are markedly decreased after DAB1 expression and RELN stimulation. One‐way ANOVA indicates significant differences between groups for all four phosphorylation sites. Shown are means + SD, n = 2. G, H. Representative western blot and quantification of n = 3 western blots probed with P‐DAB1 Y220, P‐DAB1 S491 and total DAB1 antibody, shown are means + SEM.
In addition, the proteome profiler array that measures the phosphorylation level of a number of kinases, indicated that phosphorylation of STAT3, MAPK and p27 in U87 cells were all lower in DAB1 expressing cells and that this effect was potentiated by RELN stimulation and statistical analysis indicated significant differences between groups for all four phosphoproteins (one‐way ANOVA, Figure 4C‐F). The expression of DAB1‐WT and ‐5F protein and the effectiveness of RELN stimulation was controlled by western blotting (Figure 4G). Results of the proteome profiler were then confirmed by ELISA in all stable cell lines (Figure 5). In U87, there was a highly significant reduction of both ERK1(T202/Y204)/ERK2(T185/Y187) and STAT3(Y705) phosphorylation in DAB1‐WT cells (both RELN and GFP‐stimulated) as compared to empty vector cells treated with GFP (P < 0.001 or <0.01, one‐way ANOVA followed by Dunnett's post hoc test of all conditions vs. empty vector + GFP). Also, in U87 and U251 cells, ERK1/2 activity was significantly reduced in empty vector and DAB1‐5F transfected cells stimulated with RELN as compared to empty vector cells treated with GFP (P < 0.01 and P < 0.001, one‐way ANOVA followed by Dunnett's post hoc test of all conditions vs. empty vector + GFP). For STAT3 in U251, there was no difference in protein phosphorylation after RELN stimulation, but even a slight increase in WT‐GFP cells as compared to EV‐GFP cells. Thus, overlappingly between both cell lines we found a reduction in ERK1/2 phosphorylation following RELN stimulation. Importantly, this effect could be rescued by the function blocking CR‐50 antibody, indicating its RELN‐specificity (Supporting Information Fig. 5).
Figure 5.
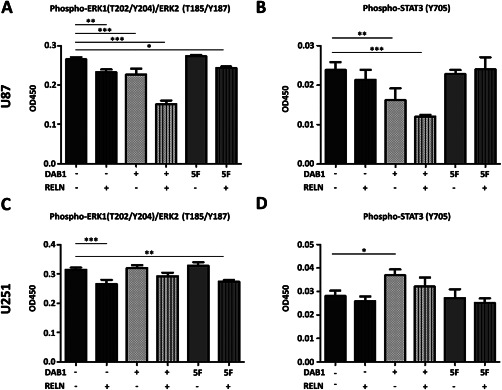
ELISA assays corroborate influences of RELN signaling on ERK1/2 and STAT3 phosphorylation. Phospho‐STAT3(Y705) and Phospho‐ERK1(T202/Y204)/ERK2 (T185/Y187) DuoSet IC ELISAs in U87–A, B and U251–C, D. Groups were compared with one‐way ANOVA followed by Dunnett's post hoc test. Significant changes are indicated (* = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001). Shown are means + SD, n = 3. DAB1‐WT expressing U87 cells show reduced Phospho‐STAT3 and Phospho‐ERK1/2 levels under control (GFP stimulation) conditions. In DAB1‐WT cells, RELN treatment reduces the phoshorylation further. In U251 cells, there is a RELN‐induced reduction of ERK1/2 phosphorylation that is DAB1 (tyrosine phosphorylation)‐independent. In contrast to U87 cells, in U251 RELN does not alter the phosphorylation levels of STAT3(Y705), suggesting cell line‐specific effects of RELN on STAT3 activity.
Activation of RELN signaling leads to a reduction in proliferation but does not affect chemosensitivity
Activation of the DAB1/RELN signaling axis led to significantly decreased cell numbers on day four and five in both U251 and U87 cells expressing DAB1‐WT with or without RELN stimulation (repeated measures, two‐way ANOVA with Bonferroni corrected post‐test, P < 0.001, Figure 6A,B). The DAB1‐5F mutant did not achieve comparable effects to DAB1‐WT overexpression, strongly arguing for the fact that RELN stimulated tyrosine phosphorylation is important to convey DAB1 inhibitory effects on proliferation. Only in U87 cells, there was a significant decrease in cell numbers on day five when EV + GFP cells were compared to 5F + RELN cells (repeated measures, two‐way ANOVA with Bonferroni corrected post‐test, P < 0.05). Together with the observation that RELN stimulation itself in DAB1‐WT cells was not able to reduce the proliferation further, it thus may not be excluded that other ligands contribute to influence this pathway in vitro, too. We analyzed if this change in proliferation is because of senescence, apoptosis or alterations in the cell cycle in U251 cells. There was no sign of senescence in our stable cell cultures as analyzed by ß‐galactosidase staining (data not shown). In addition, the cleaved form of caspase‐3 was undetectable. In contrast, the amount of cells in G2/M was increased significantly in DAB1‐WT as compared to empty vector transfected cells (two‐way ANOVA followed by Bonferroni corrected posttests, P < 0.001, Supporting Information Fig. 6). There was no significant difference between DAB1‐5F and empty vector transfected cells.
Figure 6.
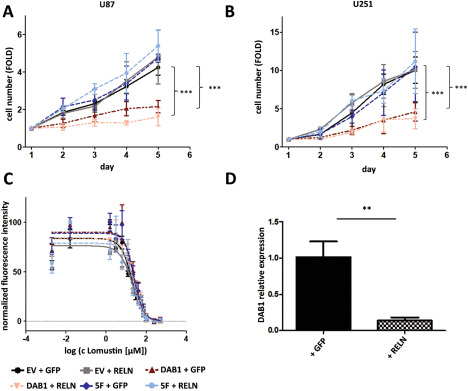
Activation of RELN signaling inhibits cellular proliferation. A, B. Proliferation assay in U87 (A) and U251 (B) cells stably expressing DAB1‐WT or DAB1‐5F stimulated with supernatants from HEK293T cells expressing mRELN or GFP as a control. The influence of DAB1‐WT expression on the cell number with or without RELN stimulation was highly significant (repeated measures, two‐way ANOVA followed by Bonferroni corrected post‐tests, P < 0.001 on day 4 and 5). Shown are means ± SD, n = 3. C. DAB1 expression and RELN treatment do not alter resistance toward the chemotherapeutic agent lomustine. Neither RELN treatment nor DAB1 expression did influence the IC50 of U87 cells toward lomustine, n = 2. D. DAB1 expression is strongly decreased in U118 cells after long‐term treatment with mRELN. The expression was measured via real‐time PCR. Shown are means + SD, n = 3.
The antiproliferative effects of RELN could also be stated in a different fashion. We speculated that adding recombinant RELN to the supernatant of a cell line that expresses at least some DAB1 should select for cells with lower DAB1 expression. We used the U118 cell line for this experiment as it showed the highest endogenous DAB1 expression (Figure 3D). Indeed, after long‐term stimulation with RELN‐conditioned medium for three weeks, U118 cells showed a significantly lower DAB1 expression as compared to U118 cells cultured in control (GFP‐) conditioned medium (Figure 6D).
As DNA damage signaling is known to be influenced by E2F targets 12 and others have reported an influence of RELN on chemoresistance 28, we also assessed chemoresistance following RELN/DAB1 manipulation. However, we could not observe any change in chemoresistance of U87 cells to lomustine (Figure 6C). DAB1 expression and effectiveness of RELN stimulation were controlled by routine western blotting.
RELN has both DAB1‐dependent and DAB1 tyrosine phosphorylation independent effects on cell migration
We studied tumor cell migration following DAB1‐WT and ‐5F overexpression in our two cell lines (U251 and U87) under two different matrix conditions (fibronection and laminin) (Figure 7A–D). Across both cell lines and matrix conditions RELN‐stimulated DAB1‐WT cells had markedly reduced migration in comparison to empty vector (GFP) and 5F (GFP) cells. These comparisons were concordantly significant in U251 cells on fibronectin and laminin and on U87 cells on laminin. U87 RELN‐stimulated DAB1‐WT cells on fibronectin showed significantly lower migration in comparison to 5F (GFP) cells and still a trend toward reduced migration in comparison to empty vector (GFP) cells (one‐way ANOVA, followed by Tukey's multiple comparison test, P < 0.05 for all significant comparisons). Thus, these findings argue for strong DAB1 tyrosine phosphorylation dependent suppressive RELN effects on glioma cell migration.
Figure 7.
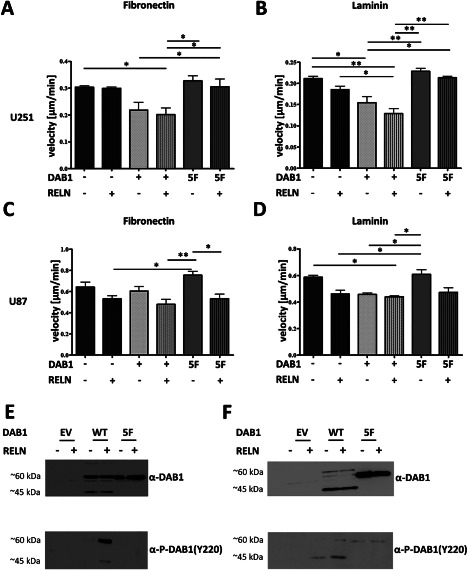
Activation of RELN signaling alters migration in a DAB1 (tyrosine phosphorylation)‐dependent and ‐independent fashion. Groups were compared with one‐way ANOVA followed by Tukey's multiple comparison test. Significant changes are indicated (* = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001). Shown are means + SEM, n = 2–3. A, B. Velocity of U251 cells on fibronectin (A) and laminin (B). DAB1‐WT expressing cells that are stimulated with RELN migrate markedly slower than cells expressing the inactive 5F mutant or EV transfected cells. There is a trend toward slower overall migration after RELN stimulation on laminin also in EV and 5F cells, indicative of DAB1 (tyrosine phosphorylation)‐independent effects on cell migration. C, D. Velocity of U87 cells on fibronectin (C) and laminin (D). On fibronectin, DAB1‐WT expressing cells that are stimulated with RELN migrate markedly slower than GFP‐stimulated cells expressing the inactive 5F mutant or EV‐transfected cells. On laminin, RELN stimulated DAB1‐WT cells show a significantly slower migration in comparison to 5F (GFP) cells and still a trend toward reduced migration in comparison to empty vector (GFP) cells. In addition, RELN stimulated 5F cells migrate significantly slower on fibronectin than GFP‐stimulated cells and there is a trend toward slower overall migration after RELN stimulation on laminin and fibronectin for 5F and EV cells, indicative of DAB1 (tyrosine phosphorylation)‐independent effects on cell migration. E, F. Western blot of DAB1 protein and P‐DAB1 Y220 in U87 (E) and U251. (F). While in U87 the predominant band is visible at 60 kDa for both DAB1‐WT and 5F, in U251 the predominant band of the DAB1‐WT protein is visible at 45 kDa, indicating higher basal activity of RELN signaling in U251 cells.
Interestingly, we also observed DAB1 (tyrosine phosphorylation)‐independent RELN effects: There was a significant decrease in migration comparing U87 5F (RELN) to U87 5F (GFP) cells on fibronection (one‐way ANOVA, followed by Tukey's multiple comparison test, P < 0.05). A similar (although non‐significant) trend was observed‐ independently of the matrix condition used‐ in both cell lines not only for the comparison 5F (RELN vs. GFP‐stimulated) but also for the comparison empty vector (RELN vs. GFP‐stimulated). Overall, particularly when considering the U251 experiments on fibronection, it appeared that the DAB1 (tyrosine phosphorylation)‐independent RELN effects were lower in U251 than in U87. Importantly, the reduction of velocity could be rescued by addition of CR‐50, again indicating that the observed effect is RELN‐specific. DAB1 expression and effectiveness of RELN stimulation were controlled by routine western blotting (Figure 7E,F; representative example).
In addition, we checked if DAB1 or RELN regulate the expression of the adhesion molecules ITGA3, ITGA5, ITGB1, FAK or PXN (paxillin). However, there was no consistent significant difference between the empty vector cells and (RELN‐treated) DAB1‐transfected cell lines (one‐way ANOVA followed by Dunnett's post hoc test, Supporting Information Fig. 7).
Discussion
The reelin signaling pathway plays a pivotal role in brain development by regulating neuronal migration and cortical layering 19. In contrast to these physiological functions, the role of reelin signaling in glioblastoma had not yet been investigated. We here can show that the RELN gene is silenced via promoter hypermethylation in glioblastoma tumors and cell lines, a mechanism that had also been observed in breast, pancreatic and gastric cancers 13, 44. Importantly, we found that DAB1 expression was also strongly reduced in high‐grade brain tumors. This suggests that downregulation of RELN in brain tumor cells alone is not sufficient to inactivate the pathway. It is well possible that either RELN from the surrounding healthy brain parenchyma and/or other ligands can bind to the RELN receptors APOER2 and VLDLR, activate DAB1 and thereby exert effects on the tumor cells. Indeed, it had been shown that thrombospondin 1, clusterin or ApoE can all bind to APOER2 and VLDLR and interfere with RELN in vitro and in vivo 5, 27. Our in vitro findings suggest that particularly thrombospondin 1 is expressed by glioma cells.
We next assessed the correlation of RELN and DAB1 expression with WHO grade, glioblastoma expression subtype and overall survival. RELN and DAB1 expression were significantly inversely associated with WHO malignancy grade. Moreover, high RELN transcriptional levels were associated with significantly longer overall survival in large glioblastoma patient cohorts. This suggests the RELN/DAB1 signaling axis as a novel candidate diagnostic and prognostic marker and potential therapeutic target. Interestingly, when comparing the expression of RELN and DAB1 between the different transcriptional glioblastoma subtypes 49 highest expression levels were observed in the neural subtype. This is in accord with the physiological function of RELN signaling as the neural expression subtype is suggestive of cells with a differentiated phenotype and typified by the expression of neuron markers, such as NEFL, GABRA1, SYT1 and SLC12A5 49.
In light of Reelin's physiological function, it is somewhat astonishing that the potential association between RELN signaling and brain cancer pathology had not yet been systematically assessed. This might be caused by hindrances in modelling this pathway, that is, the cumbersome size of the RELN protein, the low expression of RELN (and DAB1) in most glioma cell lines and the lack of vector constructs to overexpress human reelin in glioma cells. On account of these limitations and the simultaneous downregulation of DAB1 in glioblastomas described above, for our functional analyzes we decided to take an approach in which DAB1 was stably overexpressed in two different qualities: (i) as DAB1‐WT with the full functionality of the DAB1 protein and (ii) DAB1‐5F mutant with point mutations at all five Dab1 tyrosine phosphorylation sites (Tyr185, tyr198, Tyr200, Tyr220 and Tyr232). This DAB1‐5F mutant has been previously well characterized and is devoid of all RELN‐mediated tyrosine phosphorylation‐dependent downstream signaling effects 20. Empty vector cells served as an additional control. As such, we were able to assess the common final signaling pathway with and without reelin stimulation but also completely free of endogenous RELN effects (5F‐mutant and CR‐50 blocking antibody experiments).
In this experimental setting, we observed major effects on RELN/DAB1 signaling on glioma cell proliferation and migration. DAB1‐WT overexpression led to a significant decrease in proliferation and this effect was RELN‐dependent as it could not be observed in cells expressing the DAB1‐5F mutant. Consistently with this finding, E2F signaling was the major pathway altered in our proteomic screen comparing DAB1‐WT and DAB1‐5F expressing U87 cell lines. E2F transcription factors are important regulators of cell cycle progression 15. They are also known to be frequently altered in glioblastoma, as for example mediated by other well‐known molecular alterations like RB1 mutations, CDK4 amplification or CDKN2A deletion 7, 49. Our findings therefore describe a new mechanism of E2F control, previously probably not observed because of the fact that RELN signaling via DAB1 was examined mostly in post‐mitotic neurons.
In addition, we described cell line‐specific effects of RELN signaling on ERK1/2 as well as on STAT3 activity. In U87, DAB1 expression and RELN stimulation were able to reduce ERK1/2 and STAT3 activity. Moreover, short term RELN stimulation after medium exchange was able to reduce ERK1/2 activity in both U251 and U87 cells. As U251 DAB1‐WT cells in the proliferation assays grew nonetheless much slower after longer (3–5 days) growth factor removal and ERK and STAT3 phosphorylation were not reduced in these cells, we speculate that alternative ligands, secreted factors or adaptive responses to growth factor removal may trigger this behavior. In contrast to neuronal migration, where alterations in p‐cofilin levels after RELN stimulation have been reported 10, 11, we could not find any change in p‐cofilin to the total cofilin ratio or a qualitative change in the actin cytoskeleton (data not shown).
Nevertheless, functionally we observed a relevant impact of RELN/DAB1 signaling on glioma cell migration. This suggests that RELN acts only in small areas of the cell (precluding the analysis of a quantitative phenotype) or by a cofilin‐independent mechanism, for example by regulation of N‐WASP 48. In our rather elaborated setting of migration assays involving different DAB1‐overexpressing clones under different matrix conditions with and without RELN stimulation, the most consistent finding was a significant negative impact of RELN‐dependent DAB1 activation on tumor cell migration. However, as outlined above, we also observed DAB1 (tyrosine phosphorylation)‐independent RELN effects and these appeared to be gradually different between the cell lines (less pronounced in U251 on fibronectin than in U87). Although both lines are PTEN‐deficient 26, they differ in their expression subtype and p53 status what might account for the cell‐line specific differences. More importantly, U251 (in comparison to U87) also contains a much higher content of thrombospondin (Figure 3G). Thus, alternative ligand binding might mitigate the DAB1 (tyrosine phosphorylation)‐independent RELN effects in U251 cells. As thrombospondin has also been described to specifically bind to fibronectin, this interaction might additionally account for the slightly different migrational behavior of U251 cells on this specific matrix 24. We highlight this example here to underline the advantage of our approach studying RELN signaling by mainly comparing DAB1‐WT and DAB1‐5F cells over an approach solely relying on stimulation with recombinant RELN. The latter approach may be more prone to uncontrolled interference with endogenous alternative ligand binding and might thereby obscure distinct results.
In summary, we show that the canonical RELN signaling cascade via DAB1 exerts tumor suppressive functions and is a yet unknown barrier to cellular transformation in gliomagenesis. These effects are mediated via suppression of E2F activity by the DAB1‐WT protein and harbor strong prognostic implications. Our findings might thereby unearth novel, yet unrecognized treatment options in glioblastomas.
Author Contributions
Conception and design: MJ Riemenschneider, M Schulze
Development of methodology: MJ Riemenschneider, C Violonchi, M Schulze, S Swoboda
Acquisition of data: M Schulze, C Violonchi, S Swoboda, J Simbürger, T Welz, S Hoja
Analysis and interpretation of data: M Schulze, MJ Riemenschneider, J Reinders, E Kerkhoff
Writing, review and/or revision of the manuscript: M Schulze, MJ Riemenschneider
Administrative, technical or material support: MJ Riemenschneider, J Reinders, E Kerkhoff
Study supervision: MJ Riemenschneider
Conflict of Interest
The authors declare no potential conflicts of interest.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Figure 1. Analysis of RELN expression and recurrent mutations in GBM. While there is no significant difference between EGFR‐, NF1‐, TP53‐, PIK3CA‐, PIK3R1‐, PTEN‐ and RB1‐mutant vs. wildtype (WT) tumors, there is a small but significant difference between IDH1‐mutant and WT‐tumors (Bonferroni corrected two‐sided Mann‐Whitney test, P < 0.05).
Figure 2. Depiction of the region of the RELN promoter analyzed by bisulfite sequencing. CpG dinucleotides (red) are numbered according to their position relative to the transcription start site (TSS). Primers are marked with grey arrows.
Figure 3. Reln protein expression as assessed by immunohistochemistry. Reln is expressed in non‐neoplastic brain tissue with strongest staining in neurons of the grey matter (A, x400) but positivity also in the white matter (B, x200, showing the transition zone between grey and white matter). Reln is also expressed in tumor cells but in a very heterogeneous fashion C (×400) and D (×200) show regions of differential Reln expression within a single case of IDH‐wildtype glioblastoma. (E, x200 and F, x200) Two further cases of glioblastoma with one (E) completely devoid of Reln protein expression and the other (F) exhibiting only interspersed Reln positive cells with some of the cells most likely corresponding to intermixed reactive astrocytes. (G, x200) A case of IDH‐mutant anaplastic astrocytic with very low to absent Reln protein expression. (H, x400) A case of IDH‐mutant WHO grade II diffuse astrocytoma with uniform Reln expression close to the levels observed in non‐neoplastic brain tissue.
Figure 4. A) Outline of the strategy for semiquantitative PCR. The five‐prime PCR product is located in the CMV promoter, followed by the DAB1 full length ORF. The three‐prime product is amplified from a site between the ORF and the IRES site. B) Semiquantitative PCR of the regions flanking the DAB1‐WT and DAB1‐5F expression construct on the three‐prime and five‐prime end. All cell lines tested show integration of the full‐length construct at an equal level. M, Marker (100 bp DNA ladder plus); +, positive control (20 ng DAB1‐WT DNA construct); −, negative control (ultrapure water). A‐F, Five‐prime region PCR of stably transfected U87MG and U251 cell lines: A, U87‐MG + EV; B, U87‐MG + DAB1‐WT; C, U87‐MG + DAB1‐5F; D, U251 + EV; E, U251 + DAB1‐WT; F, U251 + DAB1‐5F. G‐L, PCR of the three‐prime region from stably transfected U87‐MG and U251 cell lines: G, U87‐MG + EV; H, U87‐MG + DAB1‐WT; I, U87‐MG + DAB1‐5F; J, U251+ EV; K, U251 + DAB1‐WT; L, U251 + DAB1‐5F. C) Quantification of the PCR as shown in ‐B). The fluorescence intensities were calculated as ratio of the five‐prime PCR intensity divided by the three‐prime PCR intensity. +, positive control A, U87‐MG + EV; B, U87‐MG + DAB1‐WT; C, U87‐MG + DAB1‐5F; D, U251 + EV; E, U251 + DAB1‐WT; F, U251 + DAB1‐5F. D) PCR of full length DAB1 inserts. M, Marker (100 bp DNA ladder plus); A, U87‐MG + DAB1‐WT; C, U87‐MG + DAB1‐5F; D, U251 + DAB1‐WT; E, U251 + DAB1‐5F.
Figure 5. Both reduction of ERK1/2 phosphorylation (A and B) and migration (C and D) can be rescued by addition of the CR‐50 antibody that blocks the function of mRELN. Shown are means + SD. Groups were compared by one‐way ANOVA followed by Tukey's post hoc test.
Figure 6. A) Cell cycle distribution of stably transfected U251 cells. While the fraction of cells in G1 is significantly reduced, the fraction of cells in G2/M is significantly increased in DAB1‐WT expressing cells as compared to empty vector cells (two‐way ANOVA followed by Bonferroni corrected posttests, P < 0.001). In contrast, there are no significant differences between empty vector and DAB1‐5F expressing cells. B) Caspase 3 western blot analysis in U251 cells. Only full‐length caspase 3 can be detected in all stable cell lines, while cleaved caspase 3 is absent.
Figure 7. Real‐time PCR analysis of the adhesion molecules PXN, FAK, ITGA3, ITGA5 and ITGB1. Shown are means + SD, n=2. There was no consistent significant difference between the empty vector cells and (RELN‐treated) DAB1‐transfected cell lines (one‐way ANOVA followed by Dunnett's post hoc test, all conditions vs. empty vector + GFP).
Table 1. Primers used for bisulfite sequencing and real‐time PCR analyses.
Table 2. Overview of the methylation status (+/‐), the methylation score and the methylation pattern of RELN as detected by direct sodium bisulfite sequencing of the RELN promoter region (CpGs sites in Supporting Information Fig. 1). mRNA expression levels are also provided. Allmost all glioblastomas show hypermethylation of the RELN promoter (for % samples hypermethylated in the different WHO grades compare Fig. 2B). For promoter methylation scoring, the CG sites were first semiquantitatively assigned to the categories unmethylated (no cytosine detectable, =0), weakly methylated (peak for cytosine detectable, but smaller that thymine, =1), moderately methylated (C and T at equal amounts detectable, =2) and strongly methylated (thymine peak smaller than cytosine, =3). Then, the scores for each individual sample were summed across the whole investigated CpG site to determine the overall methylation score. Tumors with a score ≥22 were considered as hypermethylated (+), tumors with a score <22 as not hypermethylated (‐) compared to the non‐neoplastic controls.
Table 3. Methylation analysis of the RELN promoter after 5‐Aza‐2′deoxycytidine treatment in different cell lines. Treatment with 5‐Aza‐2′deoxycytidine in combination with trichostatin A reduces the methylation in all cell lines, although significant methylation still remains.
Table 4. Differentially abundant proteins as identified by DESeq2 at P‐adjusted < 0.1. Proteins that are less abundant in DAB1‐WT cells are labeled in green, whereas proteins that are more abundant in DAB1‐WT cells are shown in red.
Acknowledgments
We acknowledge Maria Hirblinger (Neuropathology Regensburg) for expert technical assistance. We thank Prof. Jonathan A. Cooper (Fred Hutchinson Cancer Research Center, Seattle, Washington) for providing the murine DAB1‐5F plasmid and Prof. Eckart Förster (Ruhr University, Bochum, Germany) for providing HEK cells producing GFP or mouse RELN. The HEK cells were originally transfected with the full length reelin clone pCrl that was provided by Prof. Thomas Curran, The Children's Hospital of Philadeliphia (CHOP). The pCRL clone had been generated while Prof. Curran was employed by St. Jude Childrens's Research Hospital (St. Jude), Memphis, TN. We thank Florian Zeman (Centre for Clinical Studies, Regensburg University Hospital, Regensburg, Germany) for a critical discussion of the manuscript. We thank Prof. Christel Herold‐Mende (Experimental Neurosurgery, Department of Neurosurgery, University of Heidelberg, Heidelberg, Germany) for providing stem‐like cell lines. Early parts of this work were performed in the Max Eder‐Junior Research Group (grant to Markus J. Riemenschneider while employed at the Department of Neuropathology, Heinrich Heine University Düsseldorf). We thank Prof. Guido Reifenberger for kindly supporting this work at that time and contributing tissue samples from the Düsseldorf CNS tumor bank.
References
- 1. Ahluwalia MS, de Groot J, Liu WM, Gladson CL (2010) Targeting SRC in glioblastoma tumors and brain metastases: rationale and preclinical studies. Cancer Lett 298:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK et al (2009) Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barski D, Wolter M, Reifenberger G, Riemenschneider MJ (2010) Hypermethylation and transcriptional downregulation of the TIMP3 gene is associated with allelic loss on 22q12.3 and malignancy in meningiomas. Brain Pathol 20:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Becker J, Frohlich J, Perske C, Pavlakovic H, Wilting J (2012) Reelin signalling in neuroblastoma: migratory switch in metastatic stages. Int J Oncol 41:681–689. [DOI] [PubMed] [Google Scholar]
- 5. Blake SM, Strasser V, Andrade N, Duit S, Hofbauer R, Schneider WJ, Nimpf J (2008) Thrombospondin‐1 binds to ApoER2 and VLDL receptor and functions in postnatal neuronal migration. EMBO J 27:3069–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bock HH, Jossin Y, May P, Bergner O, Herz J (2004) Apolipoprotein E receptors are required for reelin‐induced proteasomal degradation of the neuronal adaptor protein Disabled‐1. J Biol Chem 279:33471–33479. [DOI] [PubMed] [Google Scholar]
- 7. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O et al (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castellano E, Molina‐Arcas M, Krygowska AA, East P, Warne P, Nicol A, Downward J (2016) RAS signalling through PI3‐Kinase controls cell migration via modulation of Reelin expression. Nat Commun 7:11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chai X, Forster E, Zhao S, Bock HH, Frotscher M (2009) Reelin acts as a stop signal for radially migrating neurons by inducing phosphorylation of n‐cofilin at the leading edge. Commun Integr Biol 2:375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chai X, Forster E, Zhao S, Bock HH, Frotscher M (2009) Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n‐cofilin phosphorylation at serine3. J Neurosci 29:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dimova DK, Dyson NJ (2005) The E2F transcriptional network: old acquaintances with new faces. Oncogene 24:2810–2826. [DOI] [PubMed] [Google Scholar]
- 13. Dohi O, Takada H, Wakabayashi N, Yasui K, Sakakura C, Mitsufuji S et al (2010) Epigenetic silencing of RELN in gastric cancer. Int J Oncol 36:85–92. [PubMed] [Google Scholar]
- 14. Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA et al (2000) Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27:33–44. [DOI] [PubMed] [Google Scholar]
- 15. Dyson NJ (2016) RB1: a prototype tumor suppressor and an enigma. Genes Dev 30:1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evangelisti C, Florian MC, Massimi I, Dominici C, Giannini G, Galardi S et al (2009) MiR‐128 up‐regulation inhibits Reelin and DCX expression and reduces neuroblastoma cell motility and invasiveness. FASEB J 23:4276–4287. [DOI] [PubMed] [Google Scholar]
- 17. Friedmann‐Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O et al (2012) Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338:1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoja S, Schulze M, Rehli M, Proescholdt M, Herold‐Mende C, Hau P, Riemenschneider MJ (2016) Molecular dissection of the valproic acid effects on glioma cells. Oncotarget 7:62989–63002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Honda T, Kobayashi K, Mikoshiba K, Nakajima K (2011) Regulation of cortical neuron migration by the Reelin signaling pathway. Neurochem Res 36:1270–1279. [DOI] [PubMed] [Google Scholar]
- 20. Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA (2000) Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol 10:877–885. [DOI] [PubMed] [Google Scholar]
- 21. Kam R, Chen J, Blumcke I, Normann S, Fassunke J, Elger CE et al (2004) The reelin pathway components disabled‐1 and p35 in gangliogliomas–a mutation and expression analysis. Neuropathol Appl Neurobiol 30:225–232. [DOI] [PubMed] [Google Scholar]
- 22. Kurscheid S, Bady P, Sciuscio D, Samarzija I, Shay T, Vassallo I et al (2015) Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX‐signature in glioblastoma. Genome Biol 16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon IS, Cho SK, Kim MJ, Tsai MJ, Mitsuda N, Suh‐Kim H, Lee YD (2009) Expression of Disabled 1 suppresses astroglial differentiation in neural stem cells. Mol Cell Neurosci 40:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lahav J, Lawler J, Gimbrone MA (1984) Thrombospondin interactions with fibronectin and fibrinogen. Mutual inhibition in binding. Eur J Biochem 145:151–156. [DOI] [PubMed] [Google Scholar]
- 25. Lakoma J, Garcia‐Alonso L, Luque JM (2011) Reelin sets the pace of neocortical neurogenesis. Development 138:5223–5234. [DOI] [PubMed] [Google Scholar]
- 26. Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL, Lee JS (2011) PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ 18:666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leeb C, Eresheim C, Nimpf J (2014) Clusterin is a ligand for apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and signals via the Reelin‐signaling pathway. J Biol Chem 289:4161–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin L, Yan F, Zhao D, Lv M, Liang X, Dai H et al (2016) Reelin promotes the adhesion and drug resistance of multiple myeloma cells via integrin beta1 signaling and STAT3. Oncotarget 7:9844–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, Buetow K (2009) Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol Cancer Res 7:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McAvoy S, Zhu Y, Perez DS, James CD, Smith DI (2008) Disabled‐1 is a large common fragile site gene, inactivated in multiple cancers. Genes Chromosomes Cancer 47:165–174. [DOI] [PubMed] [Google Scholar]
- 33. Molla Kazemiha V, Shokrgozar MA, Arabestani MR, Shojaei Moghadam M, Azari S, Maleki S et al (2009) PCR‐based detection and eradication of mycoplasmal infections from various mammalian cell lines: a local experience. Cytotechnology 61:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagai S, Moreno O, Smith CA, Ivanchuk S, Romagnuolo R, Golbourn B et al (2011) Role of the cofilin activity cycle in astrocytoma migration and invasion. Genes Cancer 2:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez‐Martinez FJ, Luque‐Rio A, Sakakibara A, Hattori M, Miyata T, Luque JM (2012) Reelin‐dependent ApoER2 downregulation uncouples newborn neurons from progenitor cells. Biol Open 1:1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramaker RC, Lasseigne BN, Hardigan AA, Palacio L, Gunther DS, Myers RM, Cooper SJ (2017) RNA sequencing‐based cell proliferation analysis across 19 cancers identifies a subset of proliferation‐informative cancers with a common survival signature. Oncotarget 8:38668–38681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Riemenschneider MJ, Mueller W, Betensky RA, Mohapatra G, Louis DN (2005) In situ analysis of integrin and growth factor receptor signaling pathways in human glioblastomas suggests overlapping relationships with focal adhesion kinase activation. Am J Pathol 167:1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riemenschneider MJ, Reifenberger J, Reifenberger G (2008) Frequent biallelic inactivation and transcriptional silencing of the DIRAS3 gene at 1p31 in oligodendroglial tumors with 1p loss. Int J Cancer 122:2503–2510. [DOI] [PubMed] [Google Scholar]
- 39. Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M (2006) Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology 130:548–565. [DOI] [PubMed] [Google Scholar]
- 40. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sekine K, Kawauchi T, Kubo K, Honda T, Herz J, Hattori M et al (2012) Reelin controls neuronal positioning by promoting cell‐matrix adhesion via inside‐out activation of integrin alpha5beta1. Neuron 76:353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simburger JM, Dettmer K, Oefner PJ, Reinders J (2016) Optimizing the SWATH‐MS‐workflow for label‐free proteomics. J Proteomics 145:137–140. [DOI] [PubMed] [Google Scholar]
- 43. Smalheiser NR, Costa E, Guidotti A, Impagnatiello F, Auta J, Lacor P et al (2000) Expression of reelin in adult mammalian blood, liver, pituitary pars intermedia, and adrenal chromaffin cells. Proc Natl Acad Sci USA 97:1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stein T, Cosimo E, Yu X, Smith PR, Simon R, Cottrell L et al (2010) Loss of reelin expression in breast cancer is epigenetically controlled and associated with poor prognosis. Am J Pathol 177:2323–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC et al (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol 10:459–466. [DOI] [PubMed] [Google Scholar]
- 46. Sturm D, Witt H, Hovestadt V, Khuong‐Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. [DOI] [PubMed] [Google Scholar]
- 47. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suetsugu S, Tezuka T, Morimura T, Hattori M, Mikoshiba K, Yamamoto T, Takenawa T (2004) Regulation of actin cytoskeleton by mDab1 through N‐WASP and ubiquitination of mDab1. Biochem J 384:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wiśniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Figure 1. Analysis of RELN expression and recurrent mutations in GBM. While there is no significant difference between EGFR‐, NF1‐, TP53‐, PIK3CA‐, PIK3R1‐, PTEN‐ and RB1‐mutant vs. wildtype (WT) tumors, there is a small but significant difference between IDH1‐mutant and WT‐tumors (Bonferroni corrected two‐sided Mann‐Whitney test, P < 0.05).
Figure 2. Depiction of the region of the RELN promoter analyzed by bisulfite sequencing. CpG dinucleotides (red) are numbered according to their position relative to the transcription start site (TSS). Primers are marked with grey arrows.
Figure 3. Reln protein expression as assessed by immunohistochemistry. Reln is expressed in non‐neoplastic brain tissue with strongest staining in neurons of the grey matter (A, x400) but positivity also in the white matter (B, x200, showing the transition zone between grey and white matter). Reln is also expressed in tumor cells but in a very heterogeneous fashion C (×400) and D (×200) show regions of differential Reln expression within a single case of IDH‐wildtype glioblastoma. (E, x200 and F, x200) Two further cases of glioblastoma with one (E) completely devoid of Reln protein expression and the other (F) exhibiting only interspersed Reln positive cells with some of the cells most likely corresponding to intermixed reactive astrocytes. (G, x200) A case of IDH‐mutant anaplastic astrocytic with very low to absent Reln protein expression. (H, x400) A case of IDH‐mutant WHO grade II diffuse astrocytoma with uniform Reln expression close to the levels observed in non‐neoplastic brain tissue.
Figure 4. A) Outline of the strategy for semiquantitative PCR. The five‐prime PCR product is located in the CMV promoter, followed by the DAB1 full length ORF. The three‐prime product is amplified from a site between the ORF and the IRES site. B) Semiquantitative PCR of the regions flanking the DAB1‐WT and DAB1‐5F expression construct on the three‐prime and five‐prime end. All cell lines tested show integration of the full‐length construct at an equal level. M, Marker (100 bp DNA ladder plus); +, positive control (20 ng DAB1‐WT DNA construct); −, negative control (ultrapure water). A‐F, Five‐prime region PCR of stably transfected U87MG and U251 cell lines: A, U87‐MG + EV; B, U87‐MG + DAB1‐WT; C, U87‐MG + DAB1‐5F; D, U251 + EV; E, U251 + DAB1‐WT; F, U251 + DAB1‐5F. G‐L, PCR of the three‐prime region from stably transfected U87‐MG and U251 cell lines: G, U87‐MG + EV; H, U87‐MG + DAB1‐WT; I, U87‐MG + DAB1‐5F; J, U251+ EV; K, U251 + DAB1‐WT; L, U251 + DAB1‐5F. C) Quantification of the PCR as shown in ‐B). The fluorescence intensities were calculated as ratio of the five‐prime PCR intensity divided by the three‐prime PCR intensity. +, positive control A, U87‐MG + EV; B, U87‐MG + DAB1‐WT; C, U87‐MG + DAB1‐5F; D, U251 + EV; E, U251 + DAB1‐WT; F, U251 + DAB1‐5F. D) PCR of full length DAB1 inserts. M, Marker (100 bp DNA ladder plus); A, U87‐MG + DAB1‐WT; C, U87‐MG + DAB1‐5F; D, U251 + DAB1‐WT; E, U251 + DAB1‐5F.
Figure 5. Both reduction of ERK1/2 phosphorylation (A and B) and migration (C and D) can be rescued by addition of the CR‐50 antibody that blocks the function of mRELN. Shown are means + SD. Groups were compared by one‐way ANOVA followed by Tukey's post hoc test.
Figure 6. A) Cell cycle distribution of stably transfected U251 cells. While the fraction of cells in G1 is significantly reduced, the fraction of cells in G2/M is significantly increased in DAB1‐WT expressing cells as compared to empty vector cells (two‐way ANOVA followed by Bonferroni corrected posttests, P < 0.001). In contrast, there are no significant differences between empty vector and DAB1‐5F expressing cells. B) Caspase 3 western blot analysis in U251 cells. Only full‐length caspase 3 can be detected in all stable cell lines, while cleaved caspase 3 is absent.
Figure 7. Real‐time PCR analysis of the adhesion molecules PXN, FAK, ITGA3, ITGA5 and ITGB1. Shown are means + SD, n=2. There was no consistent significant difference between the empty vector cells and (RELN‐treated) DAB1‐transfected cell lines (one‐way ANOVA followed by Dunnett's post hoc test, all conditions vs. empty vector + GFP).
Table 1. Primers used for bisulfite sequencing and real‐time PCR analyses.
Table 2. Overview of the methylation status (+/‐), the methylation score and the methylation pattern of RELN as detected by direct sodium bisulfite sequencing of the RELN promoter region (CpGs sites in Supporting Information Fig. 1). mRNA expression levels are also provided. Allmost all glioblastomas show hypermethylation of the RELN promoter (for % samples hypermethylated in the different WHO grades compare Fig. 2B). For promoter methylation scoring, the CG sites were first semiquantitatively assigned to the categories unmethylated (no cytosine detectable, =0), weakly methylated (peak for cytosine detectable, but smaller that thymine, =1), moderately methylated (C and T at equal amounts detectable, =2) and strongly methylated (thymine peak smaller than cytosine, =3). Then, the scores for each individual sample were summed across the whole investigated CpG site to determine the overall methylation score. Tumors with a score ≥22 were considered as hypermethylated (+), tumors with a score <22 as not hypermethylated (‐) compared to the non‐neoplastic controls.
Table 3. Methylation analysis of the RELN promoter after 5‐Aza‐2′deoxycytidine treatment in different cell lines. Treatment with 5‐Aza‐2′deoxycytidine in combination with trichostatin A reduces the methylation in all cell lines, although significant methylation still remains.
Table 4. Differentially abundant proteins as identified by DESeq2 at P‐adjusted < 0.1. Proteins that are less abundant in DAB1‐WT cells are labeled in green, whereas proteins that are more abundant in DAB1‐WT cells are shown in red.