

Abstract
VWM is one of the most prevalent leukodystrophies with unique clinical, pathological and molecular features. It mostly affects children, but may develop at all ages, from birth to senescence. It is dominated by cerebellar ataxia and susceptible to stresses that act as factors provoking disease onset or episodes of rapid neurological deterioration possibly leading to death. VWM is caused by mutations in any of the genes encoding the five subunits of the eukaryotic translation initiation factor 2B (eIF2B). Although eIF2B is ubiquitously expressed, VWM primarily manifests as a leukodystrophy with increasing white matter rarefaction and cystic degeneration, meager astrogliosis with no glial scarring and dysmorphic immature astrocytes and increased numbers of oligodendrocyte progenitor cells that are restrained from maturing into myelin‐forming cells. Recent findings point to a central role for astrocytes in driving the brain pathology, with secondary effects on both oligodendroglia and axons. In this, VWM belongs to the growing group of astrocytopathies, in which loss of essential astrocytic functions and gain of detrimental functions drive degeneration of the white matter. Additional disease mechanisms include activation of the unfolded protein response with constitutive predisposition to cellular stress, failure of astrocyte‐microglia crosstalk and possibly secondary effects on the oxidative phosphorylation. VWM involves a translation initiation factor. The group of leukodystrophies due to defects in mRNA translation is also growing, suggesting that this may be a common disease mechanism. The combination of all these features makes VWM an intriguing natural model to understand the biology and pathology of the white matter.
Keywords: astrocytes, leukodystrophy, vanishing white matter
Introduction and Historical Perspective
Vanishing white matter (VWM, OMIM number 603896), also referred to as childhood ataxia with CNS hypomyelination, is one of the most prevalent leukodystrophies 102. Its exact incidence has however not been determined. Although it had already been reported in single cases, VWM was recognized as a single disease entity only in the 1990s 39, 91, 101.
The first description of VWM dates back to 1962 with the clinical–pathological report of a female patient with and adult‐onset chronic‐progressive “atypical diffuse sclerosis” characterized by gait difficulties and secondary amenorrhea 26. The patient was susceptible to minor physical trauma inducing superimposed episodes of rapid neurological deterioration. Pathology showed cystic destruction of the cerebral white matter with mild fibrillary astrogliosis and increased number of oligodendrocytes in relatively spared white matter areas. In the following 20 years similar reports followed 2, 17, 35, 37, 38, 65, 115, all describing cavitatory degeneration of the white matter and increased numbers of oligodendrocytes. Many patients displayed fast clinical deterioration after minor traumas and febrile infections.
VWM re‐entered the literature in the 1990s, this time described as a single leukodystrophy with childhood‐onset, chronic‐progressive course and autosomal recessive inheritance. Minor head trauma and febrile infections were confirmed as provoking factors for the disease 39, 91, 101. A typical proton magnetic resonance spectroscopy profile was identified with disappearance of all major white matter metabolites that are replaced by signals representing lactate and glucose. At the light of autopsy findings, this profile was interpreted as indicative of progressive cystic degeneration 101.
Clinical Features and Imaging: Classic Phenotype and Phenotypic Variation
VWM affects individuals of all ages, from prenatal ages to senescence, but is most prevalent in young children. At this age, the disease features cerebellar ataxia and, to a lesser degree, spasticity; cognition is usually better preserved and epilepsy and optic atrophy may ensue, but are less prominent and later signs 39, 91, 101. The peripheral nerve is usually not involved 101. Stresses, including febrile infections, acute fright and minor head trauma act as factors provoking the onset of the disease or episodes of rapid neurological deterioration 39, 49, 101, 104, 114. These episodes are clinically characterized by appearance or increase of irritability, vomiting, epilepsy, muscle hypotonia and impairment of consciousness varying from somnolence to coma. During the episodes death may occur 101, 104. Besides these, the disease is slowly progressive, but nonetheless fatal 39, 91, 101, 104.
VWM however has a broader clinical spectrum in which, as normally occurs in neurological diseases, the clinical severity is inversely related to age at onset 31, 104, 108. The disease may show an early‐infantile or even antenatal onset 32, 108. A typical early‐onset form of VWM is described amongst Cree and Chippewayan indigenous populations in Manitoba and North Quebec. This form, named Cree encephalopathy, has onset in the first months of life and is fatal within the second year 6, 7, 32. Prenatal‐onset VWM 108 is clinically characterized by oligohydramnios and reduced fetal movements in the third trimester of pregnancy. Microcephaly and variable extraneurologic signs are apparent since birth, including congenital arthrogryposis, growth failure, hepatosplenomegaly, renal hypoplasia, pancreatitis and cataracts. The involvement of the white matter is clear and accompanied in females by ovarian dysgenesis 8, 108. Milder clinical variants of VWM affect adolescents and adults, including older individuals. At these later ages the episodes of major neurological deterioration are usually less prominent, and the clinical course progresses slowly and is more protracted 58, 60, 104, 106. In adults, VWM features epilepsy, migraine, cognitive deterioration and psychiatric symptoms 18, 68, 73, 78, 81, 92, 106. Many women with VWM leukodystrophy are also affected by premature ovarian failure or primary amenorrhea, a combination known as ovarioleukodystrophy 30, 67, 92. It should be noted that the association of leukodystrophy and ovarian failure is not exclusively typical of VWM, but also occurs in other defects of translation 16. Notably, the ovarian failure may precede the neurological symptoms 5, 30, 101.
Magnetic resonance imaging (MRI) of the brain is diagnostic in VWM, especially in the classic early‐childhood disease (Figure 1). It shows a bilateral, diffuse and symmetric involvement of the cerebral white matter, with low signal on T1‐weighted, proton density and fluid‐attenuated inversion recovery (FLAIR) images and high signal on T2‐weighted images and no enhancement after contrast administration. The U‐fibers, internal capsules, anterior commissure and outer rim of the corpus callosum are usually largely spared. This pattern of signal abnormalities is also detected in presymptomatic patients 33, 84, 101, 104, 106, 118. Over the clinical course, proton density and FLAIR sequences reveal a progressive rarefaction and cystic degeneration of the affected white matter, that is eventually replaced by fluid with signal features very similar to those of cerebrospinal fluid (CSF) 101, 104. This spectrum is diagnostic of VWM. Inside the cystic regions, T1‐weighted, proton density and FLAIR images commonly show a pattern of radiating stripes stretching from the ventricular walls to the subcortical areas 101, 104. Proton spectroscopy accordingly demonstrates a progressive reduction to disappearance of all major white matter metabolites, with accumulation of lactate and glucose at concentrations similar to that of CSF 23, 39, 91, 99, 101, 104. Diffusion‐weighted images show that diffusivity is increased in cystic areas 76, compatible with loss of tissue and replacement by fluid, and decreased in relatively spared areas, correlating with increased cellularity 76. The cerebellar white matter may also be involved in VWM, however, without cystic degeneration 101, 104. In the brainstem, signal changes may appear in the central tegmental tracts 101, 104. Over time, cerebellar and brainstem atrophy ensue. The cerebral and cerebellar cortices have normal signals. Minor and sometimes temporary signal abnormalities may be detected in the globus pallidus, thalamus and pons 104.
Figure 1.
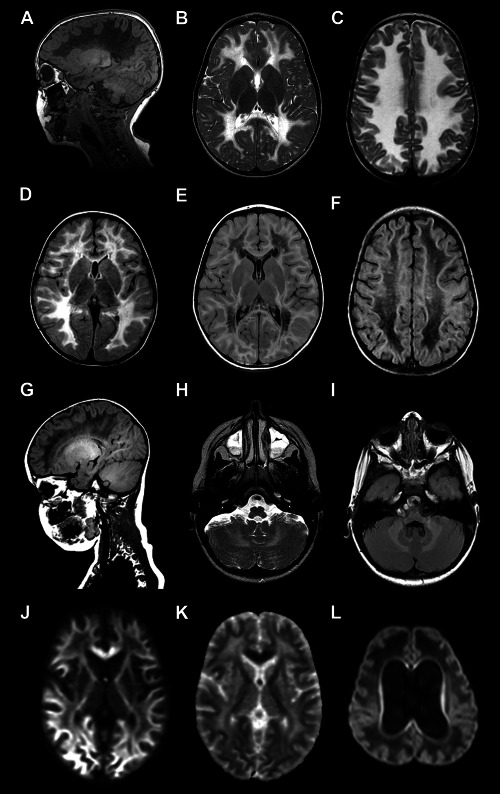
MRI features of VWM. A. Sagittal T1‐weighted image of a 3‐year‐old child shows diffuse low signal involving all cerebral hemispheric white matter. Only in the occipital white matter the U‐fibers are partially preserved. The cerebral cortex and thalamus are not affected. B, C. Axial T2‐weighted images of a 2.5‐ and 2‐year‐old child, respectively, show a diffuse, bilateral and symmetric signal hyperintensity in the cerebral white matter extending to the posterior limb of the internal capsules. The anterior limb of the internal capsule, and external capsule are preserved. D–F. Axial fluid‐attenuated inversion recovery (FLAIR) images of two children both aged 3 years shows variable rarefaction and cystic degeneration of the white matter, that is limited in one child (D) whereas is more accentuated in another child (E, F). In the second child the affected white matter shows a signal intensity approaching that of the cerebrospinal fluid. G. Sagittal T1‐weighted image of a 3‐year‐old child shows fine radiating stripes extending from the ventricular wall to the cerebral cortex. Also note that the white matter of the cerebellum has a normal signal intensity. H, I. Axial images through the posterior fossa of a 3‐ and a 2.5‐year old child, respectively, show that the cerebellar white matter has a mildly increased signal intensity on the T2‐weighted image (H), but is not cavitated on the FLAIR image (I). J. Sagittal T1‐weighted image of an 8.5‐month‐old infant shows massive involvement of the cerebral white matter that shows a low signal. Note the dilation of the ventricles, thinning of the corpus callosum and atrophy of the cerebellar vermis. K, L. Axial T2‐weighted images of the same child at 7 and 8.5 months, respectively, show diffuse signal hyperintensity of the white matter extending into the capsules and U‐fibers. At 8.5 months severe atrophy of the white matter with also thinning of the cerebral cortex is seen. M, N. Diffusion‐weighted image of a 1.5‐year‐old child (M) shows a low signal in the deep white matter, indicating increased diffusion, and high signal in the subcortical white matter, indicating restricted diffusion, confirmed by high and low apparent diffusion coefficient (ADC) values on the ADC map (N) (high signal in the deep white matter, low signal in the subcortical white matter). O. Diffusion‐weighted image of an 8.5‐month‐old infant shows diffuse low signal in the white matter, compatible with increased diffusion, related to the severe white matter tissue loss.
In early‐infantile and adult VWM, the MRI picture is more difficult to interpret. In neonates, there is swelling and immaturity of the white matter with little, if any rarefaction. Subcortical atrophy occurs over time, leading to collapse of the cortex onto the ventricular walls over dilated lateral ventricles 8, 108. In Cree encephalopathy, signal changes in the thalami and basal ganglia accompany the white matter involvement 1. In adults with VWM, white matter cystic degeneration may be limited or even absent, whereas some degree of white matter atrophy is the rule 92, 104, 106.
Neuropathology
External examination of the brain of VWM children shows no pathological features 10, 82, 104. Only in infants, there is some degree of swelling with flattening of the gyri 101. The brain of adults is usually mildly to moderately atrophic, with enlargement of the lateral ventricles and subarachnoid spaces. Atrophy is uncommon in children 82, 101. On cut, the cerebral white matter appears greyish and partly gelatinous to frankly cystic 9, 34, 82, 101, 104, 116. Cystic degeneration is more prominent in the deep brain regions 104 and, as revealed by MRI, the U‐fibers, anterior commissure and capsules are better preserved. All grey matter structures appear normal, including the cortex, basal nuclei, thalami and brainstem nuclei 104.
In the macroscopically affected white matter (Figure 2), histopathology shows paucity of myelin and thin to vacuolated myelin sheaths 9, 82, 101, 104, 116, which ultrastructurally correspond to uncompacted myelin close to the axonal membrane 82. Macrophages containing myelin products may be present, but are disproportionally scarce compared with the severity of the myelin involvement 9, 82, 101, 104. In line with this, biochemistry revealed markedly reduced myelin yields 34, 82, 91. Axonal numbers are decreased commensurate to the degree of lack of myelin 9, 104, 116. Axons are completely lost in cavitated areas, but preserved although thinned in the relatively spared white matter where axonal swellings may also be appreciated 9, 29, 48, 82, 104, 116. The ultrastructure of axons is normal 82, 116.
Figure 2.
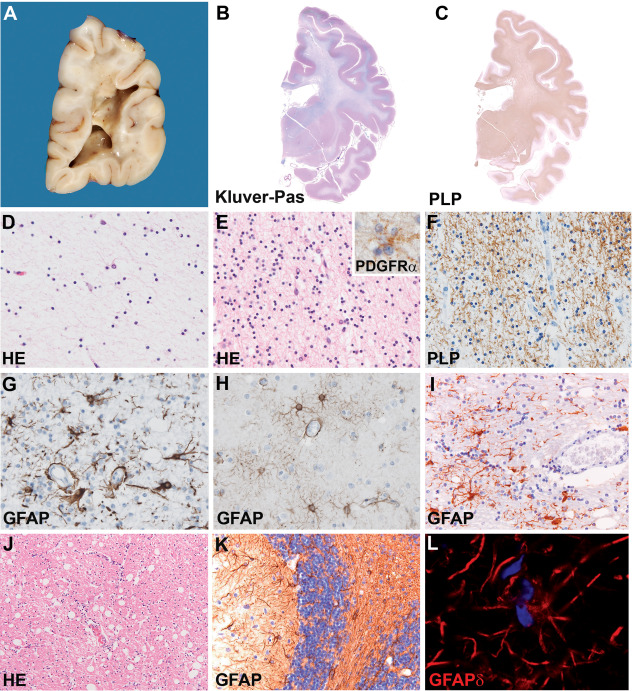
Neuropathology of classic childhood‐onset VWM. A. Formalin‐fixed coronal section of the brain of a 5‐year‐old child taken just behind the pulvinar shows gelatinous appearance of the white matter that is rarefied to frankly cavitated. The U‐fibers are relatively spared. The cerebral cortex appears normal. B, C. Whole mounts of a coronal brain section of a 10‐year‐old child stained with Klüver stain form myelin (blue) and periodic acid‐Schiff (pink) (B) or stained against the major myelin protein proteolipid protein (PLP, C) show diffuse lack of myelin in de hemispheric white matter extended in places into the U‐fibers. The internal capsule appears slightly better myelinated. D. HE stained tissue section from the deep white matter shows global degeneration with decrease in the number of all cell types and cystic degeneration of the neuropil. E. In better preserved areas as the U‐fibers the cellular density is highly increased and most cells have the morphology [small round compact nuclei surrounded by a clear halo, Hematoxylin‐eosin (HE)] and immunohistochemical properties (platelet‐derived growth factor receptor α, PDGFRα, inset) of oligodendrocyte progenitor cells. F. The scarcity of mature myelin‐forming cells correlates with lack of mature myelin, as shown by this PLP stained section of the parietal white matter. G. Stain against the astrocyte‐specific intermediate filament glial fibrillary acidic protein (GFAP) shows that cerebral white matter astrocytes have an aberrant morphology with short, blunt processes. H. Stain against GFAP of a cortical section shows that grey matter astrocytes have a normal morphology with elongated, thin cell processes. I. In the cerebellar white matter, stain against GFAP reveals that astrocytes are morphologically less affected. J. HE E stain of the cerebellar white matter shows cystic vacuolization of the neuropil without cavitation. K. Stain against GFAP of the cerebellar cortex shows numerous immunopositive Bergmann glia, the cell body of which is translocated from the Purkinje cell to the molecular layer and with disorganized cell processes. L. Stain against the GFAP isoform GFAPδ shows that white matter astrocytes are intensely GFAPδ‐immunopositive. Original magnifications: (D–I) 200×; (J, K) 100×; (inset in E and L) 400×.
The degree of reactive astrogliosis is typically meager in VWM and absolutely disproportional to the degree of tissue involvement 34, 82, 101, 104, 116. Astrocytes are only found surrounding blood vessels in cystic to cavitated white matter areas, corresponding to the radiating pattern of stripes seen on MRI 82, 101, 104. Additionally, white matter astrocytes are dysmorphic with multiple nuclei and blunt, coarse cell processes instead of the fine processes seen in normal controls and other leukodystrophies 29, 78, 82, 90, 110, 116. A particular class of astrocytic cells, the Bergmann glia in the cerebellum, shows mislocalization of the cell body and nucleus from the Purkinje cell to the molecular layer 21. Inflammation is typically scarce, with low numbers of activated microglia/macrophages and absent T‐ and B‐lymphocytes 9, 57, 82.
In less affected white matter regions, the absolute cellularity is remarkably increased 34, 82, 104, 116. These cells show the morphology and immunohistochemical properties of oligodendrocyte progenitor cells 11. Mature oligodendrocytes are scarce, explaining the lack of myelin. A variable portion of these oligodendrocytes display an abundant and finely vacuolated cytoplasm that ultrastructurally corresponds to membranous structures associated with mitochondrial membranes and myelin lamellae 82, 116. These so‐called “foamy oligodendrocytes” have been advocated to be typical of VWM, but are scarce and difficult to identify. The cerebral cortex is normal, whereas neuronal loss may occur in the neostriatum 82, 101.
The neuropathology of VWM in infants is somehow different (Figure 3) (34, unpublished data). White matter rarefaction occurs, but frank cavitation is absent. Compared with older children, adolescents and adults, astrocytes are present in higher numbers and their morphology is normal, although reactive gliosis and glial scarring do not take place. The innate immunity appears to be better activated, with presence of microglia/macrophages throughout the affected white matter. Oligodendrocyte numbers are within normal limits and foamy oligodendrocytes have been described 7, 32. Variable expression of proliferation and pro‐and anti‐apoptotic markers in brains of patients of different ages and in different white matter regions suggests that oligodendrocytes are lost by apoptosis in young children and early white matter lesion formation 9, 34, whereas in older patients with longer standing disease these cells actively proliferate accounting for the striking increase in their numbers 111, 116. Loss of Purkinje and granular neurons may be seen in the cerebellum of infants and younger children 29, 82, 97, suggesting some degree of trans‐synaptic degeneration. Infants with Cree encephalopathy may also lose neurons in the hippocampus, possibly due to epilepsy 32.
Figure 3.
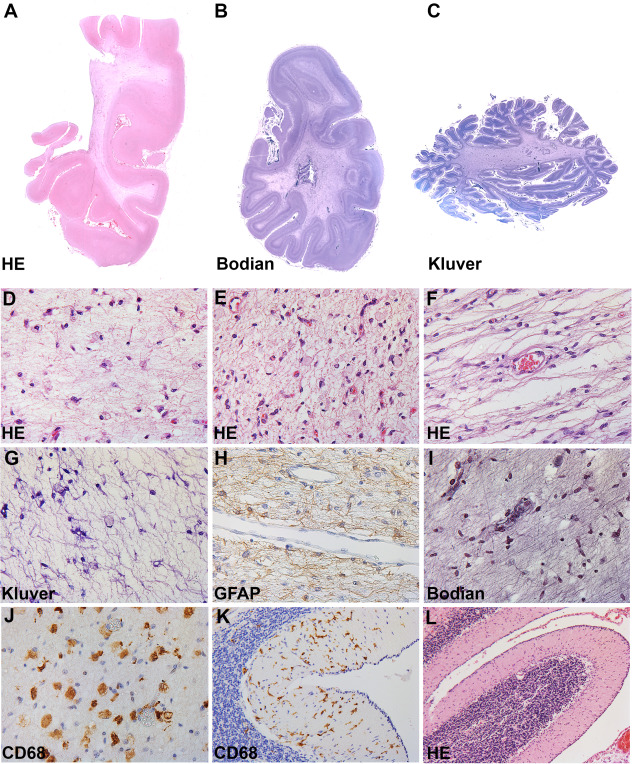
Neuropathology of infantile‐onset VWM. A, B. Whole mounts of coronal sections of the parietal and occipital lobe of a 5‐month‐old infant (A, HE; B, Bodian stain) show diffuse lack of myelin in the white matter without tissue cavitation. C. Whole mount section of the cerebellum stained with Klüver stain for myelin (blue) shows atrophy of the cortex and white matter pallor both deep in the hemisphere and in the folia. D–F. HE stain of the deep, subcortical and periventricular white matter show variable degrees of cystic degeneration of the tissue without tissue cavitation. The number of oligodendrocytes is reduced (D) to slightly increased (E). In the white matter macrophages are present. The blood vessels are normal. G. Klüver stain of the frontal white matter shows absolute lack of myelin. The macrophages do not contain myelin debris. H. Stain against GFAP shows that white matter astrocytes in the parietal lobe have normal morphology with thin, elongated cell processes. There is mild clustering of astrocytes around the blood vessel. I. Bodian stain of the frontal white matter shows thin, naked axons without swellings or varicosities. J. Stain against the microglia/macrophage marker CD68 shows abundance of macrophages or ameboid microglia in the white matter of the occipital lobe. K. Numerous activated CD68‐immunopositive microglia are also observed in the cerebellar cortex. L. HE stain of a cerebellar cortical folium shows loss of Purkinje cells. Note the presence of an external granular layer, compatible with the young age of this infant. Original magnifications: (D–J) 200×; (K) 100×; (L) 50×.
Disease Mechanisms
Genetics and genotype–phenotype correlation
VWM is due to mutations in either of the genes (EIF2B1, EIF2B2, EIF2B3, EIF2B4 and EIF2B5) coding the five non‐identical subunits (α through ɛ) of the eukaryotic translation initiation factor eIF2B. Mutations in any of these genes can independently cause the disease 60, 105. Most mutations are missense mutations involving non‐conserved amino acid residues. Nonsense and frameshift mutations preventing the expression of full‐length eIF2B subunits have only been found in a compound‐heterozygous state with a missense mutation as the second mutation 60, 105, 109.
There are a few known founder effects. Dutch patients share two common ancestors and have the same mutation in EIF2B5 (271A > G) or EIF2B2 (638A > G) 59, 60. Cree leukoencephalopathy is due to a specific mutation in EIF2B5 (584 G > H) 32. There is a clear genotype–phenotype correlation 109. Some mutations are consistently related to a mild phenotype and others to a severe phenotype. There is, however, a wide phenotypic variability among patients with the same mutations, including siblings, suggesting that other genetic and/or environmental factors significantly influence the phenotype.
Astrocytes play a central role in vanishing white matter
The paucity of astrogliosis despite severe global white matter degeneration and the abnormal morphology of astrocytes in affected white matter areas are exquisitely typical of VWM 10 (Figure 2) and suggested a deficiency in normal astrocytic functions and acquisition of pathologic functions 20. This brought forward the hypothesis that astrocytes are the white matter cell type bearing the largest functional brunt and, as such, driving the other aspects of VWM neuropathology.
One affected function of VWM white matter astrocytes is failure to reach full maturity 11. They consistently show the immunohistochemical profile of astrocyte progenitors, with high expression of immature markers as nestin and CD44. Especially surrounding the cavitated areas, a portion of white matter astrocytes also display the bipolar morphology typical of precursor cells 12. Consistent with this finding, astrocytic immaturity is the first histopathological sign of disease in two VWM mouse models and correlates with disease severity and progression 21. Another aberrant feature of VWM white matter astrocytes pertains the composition of their intermediate filament cytoskeletal network 11. Besides overexpressing the intermediate filament nestin, VWM astrocytes also show an abnormal alternative splicing of the intermediate filament glial fibrillary acidic protein (GFAP) 11, 46. GFAP has different isoforms. In the brain, GFAPα is the most abundantly expressed isoform 50. Alternative isoforms, including GFAPδ, differ from GFAPα in their carboxy‐terminus 70. GFAPα has the best intrinsic capacity to form normal filaments, whereas increased expression of other isoforms, including GFAPδ, yields condensed cytoskeletal networks 50. VWM white matter astrocytes and Bergmann glia show an absolute overexpression of GFAPδ (Figure 2), but not total GFAP and GFAPα, resulting in increased GFAPδ/α ratio 11, 21. This finding, typical of VWM, may bear various functional consequences. In the developing human brain, astrocytic differentiation and maturation coincides with a decrease in GFAPδ/α ratio, suggesting that GFAP alternative splicing plays a role in the maturation of astrocytes 69, 83. This does not occur in VWM astrocytes, possibly contributing to their persistent immaturity. Increase in total GFAP and GFAPα expressions are also a feature of reactive astrocytes 95. In VWM astrocytes such expression is not increased 11, which may be a cause of the failed process of reactive gliosis and glial scarring leading to the formation of cavitated lesions. Consistent with an incorporation of excessive GFAPδ in the cytoskeletal network, VWM astrocytes also accumulate αB‐crystallin 11, a small heat shock protein that binds and stabilizes unstable conformers of other proteins 66. It has been of particular interest for astrocytes, since it was found to be a major component of Rosenthal fibers 47, the pathologic hallmark of the leukodystrophy Alexander disease caused by mutations in GFAP. The αB‐crystallin promotes disassembly of preformed GFAP filaments and disaggregates Rosenthal fibers 55. This suggests that, in VWM as in Alexander disease, overexpression of αB‐crystallin is a part of a stress response aiming at protecting astrocytes from the accumulation of abnormal GFAP or from the aberrant GFAP isoform composition. Failure of such protective stress response could have further functional consequences and also contribute to the aberrant morphology of VWM astrocytes. In mouse models of VWM, only immature astrocytes overexpressing GFAPδ have blunt cell processes 21, confirming that these aspects of astrocytic pathology are related. Furthermore, hippocampal astrocytes forced to overexpress mutant GFAP in a cross between two Alexander disease mouse models show morphologic changes similar to VWM astrocytes 96.
Astrocytic pathology impacts on the normal functions of oligodendrocytes in the VWM white matter. Immature astrocytes appear to not be functionally competent to support the maturation of oligodendrocyte progenitors into myelin‐forming cells 12, 21. VWM astrocytes overexpress the astrocyte‐restricted progenitor marker CD44 12, 63. CD44 is the membrane receptor of the glycosaminoglycan hyaluronan, a major component of the brain extracellular matrix predominantly produced by astrocytes 94. Hyaluronan inhibits oligodendrocyte progenitor maturation in different white matter pathologies, including multiple sclerosis and ischemic injury 3, 4, 13. In the VWM white matter, there is massive overabundance of hyaluronan, and hyaluronan amounts co‐vary with the astrocytic immaturity, the numbers of oligodendrocyte progenitors and the degree of lack of myelin in differently affected white matter areas 12. Co‐culture systems of astrocytes and oligodendrocyte progenitors from wild‐type and VWM mouse models in variable combinations demonstrated that VWM astrocytes inhibit oligodendrocyte maturation via secreted factors, one of which at least at end‐stage disease is hyaluronan 21. These findings conclusively demonstrate that the progenitor maturation defect is not intrinsic to VWM oligodendrocytes, but rather results from astrocytic pathology. VWM astrocytes also have an impact on axons. In VWM patients, neuropathology at end‐stage disease shows axonal loss and reduced axonal caliber in the affected white matter 10, 104, 107. In the white matter of VWM mouse models, axons are thinner and the percentage of thin axons is higher than in controls 21, 36. Myelin sheath thickness, axonal diameter and G‐ratio are initially normal, but later axons become thinner and the G‐ratio is abnormally low 54. These changes co‐vary with disease severity. Additionally, an increased percentage of thin, unmyelinated axons and increased axonal density are found. Co‐cultures of neurons with astrocytes show that mutant astrocytes induce increased axonal density, also of control neurons, whereas control astrocytes induce normal axonal density, also of mutant neurons 54. In mutant mice and patients, signs of axonal transport defects and cytoskeletal abnormalities are notably minimal 54.
The central role of astrocytes in VWM pathophysiology is further supported by the finding that other types of astrocytes are affected in addition to white matter astrocytes 21. The involvement of cerebellar Bergmann glia is discussed above. Mouse models of VWM also pointed to the involvement of Müller glia, the retinal astrocytes 21. These animals develop signs of laminar retinal disorganization that parallel the severity of the neurologic phenotype. The histopathological features are those of a retinal dysplasia with ectopia of inner nuclear and granule cells and disruption of the photoreceptor layer. There is loss of normal glutamine synthetase immunoreactivity in Müller glia, which also parallels the degree of clinical severity. In the more severely affected mouse model, Müller glia cells have an abnormal morphology with thick cell processes spanning through the disorganized retinal layers. Electroretinographic (ERG) data of VWM patients reveals preservation of the a‐wave with loss of the b‐wave of the trace. The ERG a‐wave reflects the activity of photoreceptors and is largely independent of Müller cell activity. The b‐wave is mostly generated by outer nuclear (ON)‐bipolar cells. ON‐bipolar cells are driven by photoreceptors via a glutamatergic pathway and glutamate released by photoreceptors is mostly taken up by Müller glia. In case of malfunctioning of the glutamate re‐uptake transporters, including when Müller cells are not functional 25, glutamate levels increase in the outer retina and saturate the glutamate receptors of bipolar cells. As a result, bipolar neurons cease responding and the ERG b‐wave is decreased 25. These combined mouse and human data indicate that retinopathy is also part of the human VWM phenotype and that it can be ascribed to an involvement of the retinal Müller glia.
Translation initiation and the unfolded protein response
eIF2B is an evolutionary conserved protein complex involved in the first steps of protein synthesis (Figure 4) 79, 88, 89. In eukaryotes initiation of translation is a multistep process, in which the start of translation at the AUG codon of the mRNA is ensured by the interplay of mRNA, initiator methionyl‐tRNA (), the ribosomal subunits and different translation initiation factors (eIFs) 79, 89. The process begins with formation of a ternary complex composed of , eIF2 and GTP that binds to the ribosome. When the start codon of the mRNA is recognized, the eIF2‐bound GTP is hydrolyzed and eIF2 is then released in its GDP‐bound inactive form. This starts the translation of the new protein, followed by dissociation of the ribosome from the mRNA and then release of the newly synthesized protein. To allow the formation of another ternary complex and the synthesis of another protein, active eIF2 must be regenerated by exchange of GDP to GTP. Such GDP/GTP exchange is necessary for every round of translation and is catalyzed by eIF2B 19. The eIF2B‐catalyzed step is rate limiting in the process of translation initiation and thus regulates the rate of global protein synthesis 79. eIF2B activity is regulated by numerous factors under different conditions 44. One of the most important mechanisms is phosphorylation of eIF2α subunit, which then acts as a competitive inhibitor of eIF2B, inhibiting mRNA translation.
Figure 4.
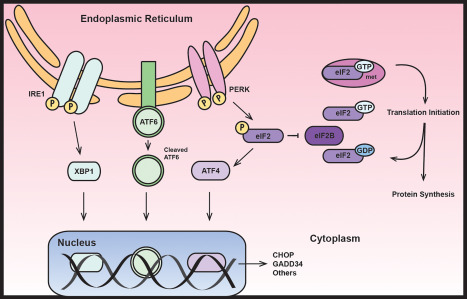
The normal role of eIF2B in translation initiation (right) and in the unfolded protein response (left). ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; CHOP, C/EBP homologous protein; eIF2B, eukaryotic translation initiation factor 2B; GADD34, growth arrest and DNA damage protein 34; IRE1, serine/threonine‐protein kinase/endoribonuclease inositol‐requiring enzyme 1; PERK, pancreatic endoplasmic reticulum kinase; XBP1, X‐box binding protein 1.
Inhibition of protein synthesis is a central mechanism in a cellular stress response known as unfolded protein response (Figure 4) 61, 75, 93. This process aims at limiting the accumulation and promoting the degradation of unfolded or denaturated proteins and preserving cellular energy to enhance cell survival under stress. Different cellular stresses lead to eIF2α phosphorylation, including viral infection, oxidative and endoplasmic reticulum stress and thermal trauma 15, 24, 40, 77, 87. In VWM patients, neurological deterioration usually follows stress 101, which has led to the hypothesis that the unfolded protein response plays a role in the pathophysiology. Activation of the pancreatic endoplasmic reticulum kinase (PERK) is the first step of the unfolded protein response leading to phosphorylation of eIF2α, its inhibition of eIF2B and thus inhibition of translation 56, 85. Some mRNAs however escape this mechanism because of specific features in their 5′ untranslated regions 42. One such mRNA encodes activating transcription factor 4 (ATF4), a factor that leads to activation of different target genes. Amongst these are the growth arrest and DNA damage protein 34 (GADD34) and the C/EBP homologous protein (CHOP) 41, 64. Under stress conditions the balance between the unfolded protein response downstream effectors determines the fate of the cell, either CHOP‐mediated cycle arrest and apoptosis or GADD34‐related survival and growth 72, 74.
The functional consequences of VWM mutations are diverse, including defects in eIF2B complex integrity, substrate binding and GDP/GTP exchange activity. However, some mutations also causing severe disease do not affect these functions at all 62. Decreased eIF2B activity does not affect the rate of global protein synthesis, the regulation of protein synthesis under stress, or patients' cells ability to survive and proliferate 51, 56, 112. Decreased eIF2B activity theoretically activates the unfolded protein response by leading to upregulation of ATF4 and its transcriptome. Indeed, activation of the unfolded protein response with enhanced activity of PERK, phosphorylated eIF2α, ATF4 and CHOP was found in the white matter of VWM patients in vivo and in cells bearing VWM mutations in vitro 52, 110, 113. Thus, VWM appears to be related to a constitutive predisposition to stress 52, 110.
It is still unclear why the brain white matter is selectively affected, while other organs with high rate of protein synthesis are spared, including bone marrow and liver. In VWM patients, the unfolded protein response was found to be selectively activated in the white matter astrocytes and oligodendrocytes 113. Such selective activation in glial cells remains to date unexplained.
Other possible disease mechanisms
Neuroinflammation is typically scarce in VWM, at least in children, adolescents and adults 10. This prompted the hypothesis that such asthenic inflammatory response contributes to the pathophysiology of the disease 14. After treatment with lipopolysaccharide, a mouse model of VWM failed to adequately synthesize and secrete cytokines as IL6 and IL1β. The same was observed in primary cultures of astrocytes and microglia, accompanied by a failed overexpression of TNFα and MCP1. This suggests that the reduction in eIF2B activity prevents the appropriate increase in translation rates upon exposure to inflammatory stressors. It also suggests a failure of restoration of brain homeostasis in VWM via the microglia‐astrocyte crosstalk.
Another hypothetic disease mechanism pertains the failure of oxidative phosphorylation and mitochondrial function in VWM. Mass‐spectroscopy measurements of total proteomes of whole brains and cultured fibroblasts isolated from a mutant VWM mouse revealed abnormally increased or decreased abundances of multiple proteins and unbalanced stoichiometry of subunits within large protein complexes, including mitochondrial proteins. A major influence was found on the composition of the respiratory chain complexes leading to defective mitochondrial performance and adenosine triphosphate production 80. All in all, these findings led to the hypothesis that eIF2B mutations can cause a defective mitochondrial function due to unbalanced abundance of respiratory chain subunits having a deleterious effect on the global mitochondrial translation efficacy and leading to defective oxidative phosphorylation 27.
Extra‐Cerebral Involvement
Irrespective of their age, females with VWM are also often affected by primary or premature ovarian failure 30, 108. In infants that have involvement of multiple organs, pathology showed streak ovaries 108. Analysis of the ovaries of two children with VWM revealed that the ovarian failure is due to accelerated follicular atresia (Figure 5). This is a hormonally regulated physiologic process by which follicles are eliminated by apoptosis in a coordinated manner 45. By onset of puberty, this process ensures that only 20% of the original follicles are left 100. Evaluation of follicle numbers on autopsy material showed that the total number of follicles in VWM ovaries is significantly lower than in age‐matched controls (unpublished data). Analysis of pathologic features of atresia also revealed that the percentage of atretic follicles is increased to double in VWM ovaries (unpublished data). VWM follicles displayed multiple morphologic features of atresia, including pyknosis of the oocyte nucleus consistent with loss by apoptosis and presence of inflammatory cells beyond the zona pellucida 43, 117. Interestingly, the degree of follicular atresia was higher in a 6‐year‐old than in a 10‐year‐old VWM child (unpublished data). This finding mirrors the clinical observation of a more severe neurological phenotype in younger children with earlier‐onset disease variants, suggesting that the severity of ovarian failure in VWM is age‐dependent and related to the global phenotypic severity.
Figure 5.
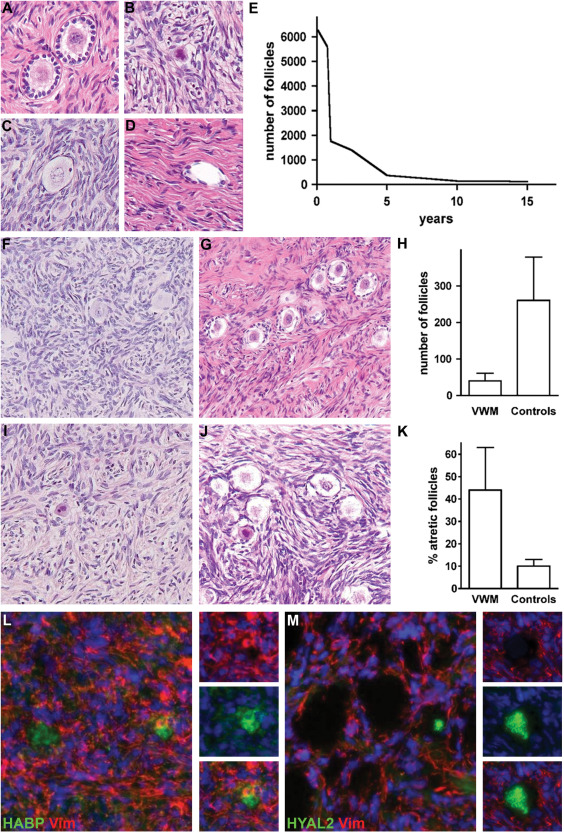
Ovarian involvement in VWM. A–D. Morphology of normal and atretic human primary follicles. Normal follicles are composed of a central oocyte surrounded by the thin membrane of the zona pellucida and by a single row of granulosa cells (A). Follicles in different stages of atresia (B–D) are characterized by pyknotic oocyte nuclei (B), infiltration of mononuclear inflammatory cells beyond the zona pellucida (C) and, eventually, disappearance of the oocyte (D). E. The number of follicles counted on a single central longitudinal 4 µm‐thick human ovary tissue section progressively decreases from neonatal age through adolescence. F–K. Accelerated follicular atresia in VWM ovaries. At 10 years of age, the density of follicles is much lower in a VWM ovary (F) than in a control ovary (G, H). Most residual follicles in a VWM ovary are atretic (I), whereas in the control ovary (J, K) atretic follicles are sporadic. L. Fluorescence stain against the hyaluronan‐binding protein (HABP, green) and vimentin (Vim, red) shows that only a portion of the residual follicles contains hyaluronan in the antrum. M. Fluorescence stain against the hyaluronan‐catalyzing enzyme hyaluronidase 2 (HYAL2, green) and vimentin (Vim, red) shows that VWM empty follicles are filled with HYAL2. Original magnifications: 200×.
We investigated the possibility that leukodystrophy and ovarian failure may share one or more disease mechanisms in VWM. In particular, we focused on hyaluronan as its metabolism plays a central role in follicular development (Figure 5). It is synthetized in granulosa cells and secreted into the follicular fluid where it maintains folliculogenesis and prevents oocyte degeneration 86, 98. Hyaluronan degradation may be associated with follicular atresia 71. In all controls analyzed, ranging from 1 month to 45 years of age, hyaluronan was detected in growing preantral cells and mature Graafian follicles and surrounded by granulosa cells expressing the hyaluronan receptor CD44. In VWM ovaries, by contrast, we detected hyaluronan only in a few follicles and in those follicles where no oocyte could be visualized the antrum and surrounding stroma were consistently devoid of hyluronan (unpublished data). Consistent with this, we detected a stronger immunoreactivity for the hyaluronan‐degrading enzyme hyaluronidase‐2 in VWM ovaries compared with controls. Additionally, VWM granulosa cells showed loss of CD44 expression. These combined findings of decreased hyaluronan in the follicular antrum and abolished CD44 immunoreactivity in the granulosa cells suggest that hyaluronan metabolism plays a role in the early degeneration of all follicular components in VWM.
With the exception of a single case in whom the density of large diameter myelinated fibers was moderately reduced in absence of axonal degeneration and storage material in Schwann cells 28, the peripheral nerve is not affected 101.
The few available reports on other organs only showed nonspecific findings in the liver 108.
Therapeutic Perspectives and Conclusions
VWM still lacks a curative treatment. Current management includes avoidance of stresses known to provoke the episodes of rapid neurological deterioration 107. Recently, an attempt at modulating eIF2B activity was conducted in VWM mouse models 22. VWM mutant mice received long‐term treatment with Guanabenz, an FDA‐approved α2‐adrenergic receptor agonist used as anti‐hypertensive agent. Bergmann glia pathology and myelin pathology both improved upon treatment. This finding opens to the possibility that compounds regulating eIF2 phosphorylation may also benefit VWM patients.
VWM is an intriguing disease with unique clinical, pathological and molecular features. Although eIF2b is ubiquitously expressed, VWM primarily manifests as a white matter disorder. Recent findings point to a central role for astrocytes in driving the brain pathology. In this, VWM belongs to the growing group of astrocytopathies, in which loss of essential astrocytic functions and gain of detrimental functions lead to degeneration of the white matter 103. VWM involves a translation initiation factor, eIF2B. The group of leukodystrophies due to defects in mRNA translation is also growing, suggesting that this may be a common disease mechanism 53. The combination of these features makes VWM a fascinating model to understand the biology and pathology of the white matter.
References
- 1. Alorainy IA, Patenaude YG, O'Gorman AM, Black DN, Meagher‐Villemure K (1999) Cree leukoencephalopathy: neuroimaging findings. Radiology 213:400–406. [DOI] [PubMed] [Google Scholar]
- 2. Anzil AP, Gessaga E (1972) Late‐life cavitating dystrophy of the cerebral and cerebellar white matter. A form of sudanophil leucodystrophy. European Neurology 7:79–94. [DOI] [PubMed] [Google Scholar]
- 3. Back SA, Kroenke CD, Sherman LS, Lawrence G, Gong X, Taber EN et al (2011) White matter lesions defined by diffusion tensor imaging in older adults. Annals of Neurology 70:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J et al (2005) Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med 11:966–972. [DOI] [PubMed] [Google Scholar]
- 5. Biancheri R, Rossi A, Di Rocco M, Filocamo M, Pronk JC, van der Knaap MS, Tortori‐Donati P (2003) Leukoencephalopathy with vanishing white matter: an adult onset case. Neurology 61:1818–1819. [DOI] [PubMed] [Google Scholar]
- 6. Black DN, Booth F, Watters GV, Andermann E, Dumont C, Halliday WC et al (1988) Leukoencephalopathy among native Indian infants in northern Quebec and Manitoba. Ann Neurol 24:490–496. [DOI] [PubMed] [Google Scholar]
- 7. Black DN, Harris R, Schiffmann R, Wong K (2002) Fatal infantile leukodystrophy: a severe variant of CACH/VWM syndrome, allelic to chromosome 3q27. Neurology 58:161–162. [DOI] [PubMed] [Google Scholar]
- 8. Boltshauser E, Barth PG, Troost D, Martin E, Stallmach T (2002) “Vanishing white matter” and ovarian dysgenesis in an infant with cerebro‐oculo‐facio‐skeletal phenotype. Neuropediatrics 33:57–62. [DOI] [PubMed] [Google Scholar]
- 9. Bruck W, Herms J, Brockmann K, Schulz‐Schaeffer W, Hanefeld F (2001) Myelinopathia centralis diffusa (vanishing white matter disease): evidence of apoptotic oligodendrocyte degeneration in early lesion development. Ann Neurol 50:532–536. [DOI] [PubMed] [Google Scholar]
- 10. Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS (2010) Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol 69:987–996. [DOI] [PubMed] [Google Scholar]
- 11. Bugiani M, Boor I, van Kollenburg B, Postma N, Polder E, van Berkel C et al (2011) Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol 70:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bugiani M, Postma N, Polder E, Dieleman N, Scheffer PG, Sim FJ et al (2013) Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain: J Neurol 136:209–222. [DOI] [PubMed] [Google Scholar]
- 13. Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K et al (2012) Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol 71:93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cabilly Y, Barbi M, Geva M, Marom L, Chetrit D, Ehrlich M, Elroy‐Stein O (2012) Poor cerebral inflammatory response in eIF2B knock‐in mice: implications for the aetiology of vanishing white matter disease. PloS One 7:e46715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clemens MJ (2001) Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol 27:57–89. [DOI] [PubMed] [Google Scholar]
- 16. Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ, Salomons GS et al (2014) Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 82:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deisenhammer E, Jellinger K (1976) Cavitating neutral fat leukodystrophy with recurrent course. Neuropadiatrie 7:111–121. [DOI] [PubMed] [Google Scholar]
- 18. Denier C, Orgibet A, Roffi F, Jouvent E, Buhl C, Niel F et al (2007) Adult‐onset vanishing white matter leukoencephalopathy presenting as psychosis. Neurology 68:1538–1539. [DOI] [PubMed] [Google Scholar]
- 19. Dever TE (2002) Gene‐specific regulation by general translation factors. Cell 108:545–556. [DOI] [PubMed] [Google Scholar]
- 20. Dietrich J, Lacagnina M, Gass D, Richfield E, Mayer‐Pröschel M, Noble M et al (2005) EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat Med 11:277. [DOI] [PubMed] [Google Scholar]
- 21. Dooves S, Bugiani M, Postma NL, Polder E, Land N, Horan ST et al (2016) Astrocytes are central in the pathomechanisms of vanishing white matter. J Clin Investig 126:1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dooves S, Bugiani M, Wisse LE, Abbink TEM, van der Knaap MS, Heine VM (2017) Bergmann glia translocation: a new disease marker for vanishing white matter identifies therapeutic effects of Guanabenz treatment. Neuropathol Appl Neurobiol. doi: 10.1111/nan.12411. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 23. Dreha‐Kulaczewski SF, Dechent P, Finsterbusch J, Brockmann K, Gärtner J, Frahm J, Hanefeld FA (2008) Early reduction of total N‐acetyl‐aspartate‐compounds in patients with classical vanishing white matter disease. A long‐term follow‐up MRS study. Pediatric Res 63:444–449. [DOI] [PubMed] [Google Scholar]
- 24. Duncan RF, Hershey JW (1989) Protein synthesis and protein phosphorylation during heat stress, recovery, and adaptation. J Cell Biol 109:1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eckstein AK, Reichenbach A, Jacobi P, Weber P, Gregor M, Zrenner E (1997) Hepatic retinopathia. Changes in retinal function. Vis Res 37:1699–1706. [DOI] [PubMed] [Google Scholar]
- 26. Eicke W (1962) Polycystische umwandlung des marklagers mit progredientem verlauf. Atypische diffuse sklerose?. Arch Psychiat Nervenkr 599–602. [Google Scholar]
- 27. Elroy‐Stein O (2017) Mitochondrial malfunction in vanishing white matter disease: a disease of the cytosolic translation machinery. Neural Regener Res 12:1610–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Federico A, Scali O, Stromillo ML, Di Perri C, Bianchi S, Sicurelli F et al (2006) Peripheral neuropathy in vanishing white matter disease with a novel EIF2B5 mutation. Neurology 67:353–355. [DOI] [PubMed] [Google Scholar]
- 29. Fogli A, Dionisi‐Vici C, Deodato F, Bartuli A, Boespflug‐Tanguy O, Bertini E (2002) A severe variant of childhood ataxia with central hypomyelination/vanishing white matter leukoencephalopathy related to EIF21B5 mutation. Neurology 59:1966–1968. [DOI] [PubMed] [Google Scholar]
- 30. Fogli A, Rodriguez D, Eymard‐Pierre E, Bouhour F, Labauge P, Meaney BF et al (2003) Ovarian failure related to eukaryotic initiation factor 2b mutations. Am J Hum Genet 72:1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fogli A, Schiffmann R, Bertini E, Ughetto S, Combes P, Eymard‐Pierre E et al (2004) The effect of genotype on the natural history of eIF2B‐related leukodystrophies. Neurology 62:1509–1517. [DOI] [PubMed] [Google Scholar]
- 32. Fogli A, Wong K, Eymard‐Pierre E, Wenger J, Bouffard JP, Goldin E et al (2002) Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann Neurol 52:506–510. [DOI] [PubMed] [Google Scholar]
- 33. Fontenelle LM, Scheper GC, Brandao L, van der Knaap MS (2008) Atypical presentation of vanishing white matter disease. Arq Neuro‐Psiquiatr 66:549–551. [DOI] [PubMed] [Google Scholar]
- 34. Francalanci P, Eymard‐Pierre E, Dionisi‐Vici C, Boldrini R, Piemonte F, Virgili R et al (2001) Fatal infantile leukodystrophy: a severe variant of CACH/VWM syndrome, allelic to chromosome 3q27. Neurology 57:265–270. [DOI] [PubMed] [Google Scholar]
- 35. Gautier JC, Gray F, Awada A, Escourolle R (1984) Cavitary orthochromatic leukodystrophy in the adult. Oligodendroglial proliferation and inclusions. Rev Neurol 140:493–501. [PubMed] [Google Scholar]
- 36. Geva M, Cabilly Y, Assaf Y, Mindroul N, Marom L, Raini G et al (2010) A mouse model for eukaryotic translation initiation factor 2B‐leucodystrophy reveals abnormal development of brain white matter. Brain: J Neurol 133:2448–2461. [DOI] [PubMed] [Google Scholar]
- 37. Girard PF, Tommasi M, Rochet M, Boucher M (1968) Leukoencephalopathy with large bilateral symmetrical cavitation. Post‐traumatic decortication syndrome. Presse Med 76:163–166. [PubMed] [Google Scholar]
- 38. Graveleau P, Gray F, Plas J, Graveleau J, Brion S (1985) Cavitary orthochromatic leukodystrophy with oligodendroglial changes. A sporadic adult case. Rev Neurol 141:713–718. [PubMed] [Google Scholar]
- 39. Hanefeld F, Holzbach U, Kruse B, Wilichowski E, Christen HJ, Frahm J (1993) Diffuse white matter disease in three children: an encephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy. Neuropediatrics 24:244–248. [DOI] [PubMed] [Google Scholar]
- 40. Harding HP, Calfon M, Urano F, Novoa I, Ron D (2002) Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol 18:575–599. [DOI] [PubMed] [Google Scholar]
- 41. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 6:1099–1108. [DOI] [PubMed] [Google Scholar]
- 42. Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397:271–274. [DOI] [PubMed] [Google Scholar]
- 43. Himelstein‐Braw R, Byskov AG, Peters H, Faber M (1976) Follicular atresia in the infant human ovary. J Reprod Fertil 46:55–59. [DOI] [PubMed] [Google Scholar]
- 44. Hinnebusch AG (2000) Mechanism and regulation of initiation methionyl‐tRNA binding to ribosomes. In: Translational Control of Gene Expression. In: Sonenberg N, Hershey JWB, Mathews MB (eds). Cold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 45. Hsueh AJ, Billig H, Tsafriri A (1994) Ovarian follicle atresia: a hormonally controlled apoptotic process. Endocr Rev 15:707–724. [DOI] [PubMed] [Google Scholar]
- 46. Huyghe A, Horzinski L, Henaut A, Gaillard M, Bertini E, Schiffmann R et al (2012) Developmental splicing deregulation in leukodystrophies related to EIF2B mutations. PloS One 7:e38264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iwaki T, Kume‐Iwaki A, Liem RK, Goldman JE (1989) Alpha B‐crystallin is expressed in non‐lenticular tissues and accumulates in Alexander's disease brain. Cell 57:71–78. [DOI] [PubMed] [Google Scholar]
- 48. Jansen AC, Andermann E, Niel F, Creveaux I, Boespflug‐Tanguy O, Andermann F (2008) Leucoencephalopathy with vanishing white matter may cause progressive myoclonus epilepsy. Epilepsia 49:910–913. [DOI] [PubMed] [Google Scholar]
- 49. Kaczorowska M, Kuczynski D, Jurkiewicz E, Scheper GC, van der Knaap MS, Jozwiak S (2006) Acute fright induces onset of symptoms in vanishing white matter disease‐case report. Eur J Paediatr Neurol 10:192–193. [DOI] [PubMed] [Google Scholar]
- 50. Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH et al (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PloS One 7:e42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kantor L, Harding HP, Ron D, Schiffmann R, Kaneski CR, Kimball SR, Elroy‐Stein O (2005) Heightened stress response in primary fibroblasts expressing mutant eIF2B genes from CACH/VWM leukodystrophy patients. Hum Genet 118:99–106. [DOI] [PubMed] [Google Scholar]
- 52. Kantor L, Pinchasi D, Mintz M, Hathout Y, Vanderver A, Elroy‐Stein O (2008) A point mutation in translation initiation factor 2b leads to a continuous hyper stress state in oligodendroglial‐derived cells. PloS One 3:e3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kevelam SH, Steenweg ME, Srivastava S, Helman G, Naidu S, Schiffmann R et al (2016) Update on leukodystrophies: a historical perspective and adapted definition. Neuropediatrics 47:349–354. [DOI] [PubMed] [Google Scholar]
- 54. Klok MD, Bugiani M, de Vries SI, Gerritsen W, Breur M, van der Sluis S et al (2018) Axonal abnormalities in vanishing white matter. Ann Clin Transl Neurol. doi: 10.1002/acn3.540 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koyama Y, Goldman JE (1999) Formation of GFAP cytoplasmic inclusions in astrocytes and their disaggregation by alphaB‐crystallin. Am J Pathol 154:1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG (2001) Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol 21:5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuhlmann T, Lassmann H, Bruck W (2008) Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol 115:275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Labauge P, Horzinski L, Ayrignac X, Blanc P, Vukusic S, Rodriguez D et al (2009) Natural history of adult‐onset eIF2B‐related disorders: a multi‐centric survey of 16 cases. Brain: J Neurol 132:2161–2169. [DOI] [PubMed] [Google Scholar]
- 59. Leegwater PA, Konst AA, Kuyt B, Sandkuijl LA, Naidu S, Oudejans CB et al (1999) The gene for leukoencephalopathy with vanishing white matter is located on chromosome 3q27. Am J Hum Genet 65:728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leegwater PA, Vermeulen G, Konst AA, Naidu S, Mulders J, Visser A et al (2001) Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet 29:383–388. [DOI] [PubMed] [Google Scholar]
- 61. Liu CY, Kaufman RJ (2003) The unfolded protein response. J Cell Sci 116:1861–1862. [DOI] [PubMed] [Google Scholar]
- 62. Liu R, van der Lei HDW, Wang X, Wortham NC, Tang H, van Berkel CGM et al (2011) Severity of vanishing white matter disease does not correlate with deficits in eIF2B activity or the integrity of eIF2B complexes. Hum Mutat 32:1036–1045. [DOI] [PubMed] [Google Scholar]
- 63. Liu Y, Han SS, Wu Y, Tuohy TM, Xue H, Cai J et al (2004) CD44 expression identifies astrocyte‐restricted precursor cells. Dev Biol 276:31–46. [DOI] [PubMed] [Google Scholar]
- 64. Lu PD, Harding HP, Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Macchi G, Taramelli M, Borri P (1968) Su d un caso di leucopatia spongiocavitaria. Acta Neurol 24:565–571. [PubMed] [Google Scholar]
- 66. MacRae TH (2000) Structure and function of small heat shock/alpha‐crystallin proteins: established concepts and emerging ideas. Cell Mol Life Sci: CMLS 57:899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mathis S, Scheper GC, Baumann N, Petit E, Gil R, van der Knaap MS, Neau JP (2008) The ovarioleukodystrophy. Clin Neurol Neurosurg 110:1035–1037. [DOI] [PubMed] [Google Scholar]
- 68. Matsui M, Mizutani K, Ohtake H, Miki Y, Ishizu K, Fukuyama H et al (2007) Novel mutation in EIF2B gene in a case of adult‐onset leukoencephalopathy with vanishing white matter. Eur Neurol 57:57–58. [DOI] [PubMed] [Google Scholar]
- 69. Middeldorp J, Boer K, Sluijs JA, De Filippis L, Encha‐Razavi F, Vescovi AL et al (2010) GFAPdelta in radial glia and subventricular zone progenitors in the developing human cortex. Development (Cambridge, England) 137:313–321. [DOI] [PubMed] [Google Scholar]
- 70. Middeldorp J, Hol EM (2011) GFAP in health and disease. Prog Neurobiol 93:421–443. [DOI] [PubMed] [Google Scholar]
- 71. Miyake Y, Sakurai M, Tanaka S, Tunjung WA, Yokoo M, Matsumoto H et al (2009) Expression of hyaluronan synthase 1 and distribution of hyaluronan during follicular atresia in pig ovaries. Biol Reprod 80:249–257. [DOI] [PubMed] [Google Scholar]
- 72. Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34‐mediated dephosphorylation of eIF2alpha. J Cell Biol 153:1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ohtake H, Shimohata T, Terajima K, Kimura T, Jo R, Kaseda R et al (2004) Adult‐onset leukoencephalopathy with vanishing white matter with a missense mutation in EIF2B5. Neurology 62:1601–1603. [DOI] [PubMed] [Google Scholar]
- 74. Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389. [DOI] [PubMed] [Google Scholar]
- 75. Paschen W (2003) Shutdown of translation: lethal or protective? Unfolded protein response versus apoptosis. J Cerebr Blood Flow Metab 23:773–779. [DOI] [PubMed] [Google Scholar]
- 76. Patay Z (2005) Diffusion‐weighted MR imaging in leukodystrophies. Eur Radiol 15:2284–2303. [DOI] [PubMed] [Google Scholar]
- 77. Petrov T, Underwood BD, Braun B, Alousi SS, Rafols JA (2001) Upregulation of iNOS expression and phosphorylation of eIF‐2alpha are paralleled by suppression of protein synthesis in rat hypothalamus in a closed head trauma model. J Neurotrauma 18:799–812. [DOI] [PubMed] [Google Scholar]
- 78. Prass K, Bruck W, Schroder NW, Bender A, Prass M, Wolf T et al (2001) Adult‐onset Leukoencephalopathy with vanishing white matter presenting with dementia. Ann Neurol 50:665–668. [DOI] [PubMed] [Google Scholar]
- 79. Proud CG (2001) Regulation of eukaryotic initiation factor eIF2B. Prog Mol Subcell Biol 26:95–114. [DOI] [PubMed] [Google Scholar]
- 80. Raini G, Sharet R, Herrero M, Atzmon A, Shenoy A, Geiger T, Elroy‐Stein O (2017) Mutant eIF2B leads to impaired mitochondrial oxidative phosphorylation in vanishing white matter disease. J Neurochem 141:694–707. [DOI] [PubMed] [Google Scholar]
- 81. Riecker A, Nagele T, Henneke M, Schols L (2007) Late onset vanishing white matter disease. J Neurol 254:544–545. [DOI] [PubMed] [Google Scholar]
- 82. Rodriguez D, Gelot A, della Gaspera B, Robain O, Ponsot G, Sarlieve LL et al (1999) Increased density of oligodendrocytes in childhood ataxia with diffuse central hypomyelination (CACH) syndrome: neuropathological and biochemical study of two cases. Acta Neuropathol 97:469–480. [DOI] [PubMed] [Google Scholar]
- 83. Roelofs RF, Fischer DF, Houtman SH, Sluijs JA, Van Haren W, Van Leeuwen FW, Hol EM (2005) Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia 52:289–300. [DOI] [PubMed] [Google Scholar]
- 84. Rosemberg S, Leite C. d C, Arita FN, Kliemann SE, Lacerda MTC (2002) Leukoencephalopathy with vanishing white matter: report of four cases from three unrelated Brazilian families. Brain Dev 24:250–256. [DOI] [PubMed] [Google Scholar]
- 85. Rowlands AG, Panniers R, Henshaw EC (1988) The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biol Chem 263:5526–5533. [PubMed] [Google Scholar]
- 86. Salustri A, Camaioni A, Di Giacomo M, Fulop C, Hascall VC (1999) Hyaluronan and proteoglycans in ovarian follicles. Hum Reprod Update 5:293–301. [DOI] [PubMed] [Google Scholar]
- 87. Scheper GC, Mulder J, Kleijn M, Voorma HO, Thomas AA, van Wijk R (1997) Inactivation of eIF2B and phosphorylation of PHAS‐I in heat‐shocked rat hepatoma cells. J Biol Chem 272:26850–26856. [DOI] [PubMed] [Google Scholar]
- 88. Scheper GC, Proud CG, van der Knaap MS (2006) Defective translation initiation causes vanishing of cerebral white matter. Trends Mol Med 12:159–166. [DOI] [PubMed] [Google Scholar]
- 89. Scheper GC, van der Knaap MS, Proud CG (2007) Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet 8:711–723. [DOI] [PubMed] [Google Scholar]
- 90. Schiffmann R, Elroy‐Stein O (2006) Childhood ataxia with CNS hypomyelination/vanishing white matter disease—A common leukodystrophy caused by abnormal control of protein synthesis. Mol Genet Metab 88:7–15. [DOI] [PubMed] [Google Scholar]
- 91. Schiffmann R, Moller JR, Trapp BD, Shih HH, Farrer RG, Katz DA et al (1994) Childhood ataxia with diffuse central nervous system hypomyelination. Ann Neurol 35:331–340. [DOI] [PubMed] [Google Scholar]
- 92. Schiffmann R, Tedeschi G, Kinkel RP, Trapp BD, Frank JA, Kaneski CR et al (1997) Leukodystrophy in patients with ovarian dysgenesis. Ann Neurol 41:654–661. [DOI] [PubMed] [Google Scholar]
- 93. Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63. [DOI] [PubMed] [Google Scholar]
- 94. Sherman LS, Back SA (2008) A 'GAG' reflex prevents repair of the damaged CNS. Trends Neurosci 31:44–52. [DOI] [PubMed] [Google Scholar]
- 95. Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sosunov AA, Guilfoyle E, Wu X, McKhann GM, 2nd , Goldman JE (2013) Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in Alexander disease. J Neurosci 33:7439–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sugiura C, Miyata H, Oka A, Takashima S, Ohama E, Takeshita K (2001) A Japanese girl with leukoencephalopathy with vanishing white matter. Brain Dev 23:58–61. [DOI] [PubMed] [Google Scholar]
- 98. Takahashi N, Tarumi W, Ishizuka B (2014) Involvement of hyaluronan synthesis in ovarian follicle growth in rats. Reproduction (Cambridge, England) 147:189–197. [DOI] [PubMed] [Google Scholar]
- 99. Tedeschi G, Schiffmann R, Barton NW, Shih HH, Gospe SM, Jr. , Brady RO et al (1995) Proton magnetic resonance spectroscopic imaging in childhood ataxia with diffuse central nervous system hypomyelination. Neurology 45:1526–1532. [DOI] [PubMed] [Google Scholar]
- 100. Tilly JL, Kowalski KI, Johnson AL, Hsueh AJ (1991) Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology 129:2799–2801. [DOI] [PubMed] [Google Scholar]
- 101. van der Knaap MS, Barth PG, Gabreels FJ, Franzoni E, Begeer JH, Stroink H et al (1997) A new leukoencephalopathy with vanishing white matter. Neurology 48:845–855. [DOI] [PubMed] [Google Scholar]
- 102. van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J (1999) Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 213:121–133. [DOI] [PubMed] [Google Scholar]
- 103. van der Knaap MS, Bugiani M (2017) Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 134:351–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. van der Knaap MS, Kamphorst W, Barth PG, Kraaijeveld CL, Gut E, Valk J (1998) Phenotypic variation in leukoencephalopathy with vanishing white matter. Neurology 51:540–547. [DOI] [PubMed] [Google Scholar]
- 105. van der Knaap MS, Leegwater PA, Konst AA, Visser A, Naidu S, Oudejans CB et al (2002) Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol 51:264–270. [DOI] [PubMed] [Google Scholar]
- 106. van der Knaap MS, Leegwater PA, van Berkel CG, Brenner C, Storey E, Di Rocco M et al (2004) Arg113His mutation in eIF2Bepsilon as cause of leukoencephalopathy in adults. Neurology 62:1598–1600. [DOI] [PubMed] [Google Scholar]
- 107. van der Knaap MS, Pronk JC, Scheper GC (2006) Vanishing white matter disease. Lancet Neurol 5:413–423. [DOI] [PubMed] [Google Scholar]
- 108. van der Knaap MS, van Berkel CG, Herms J, van Coster R, Baethmann M, Naidu S et al (2003) eIF2B‐related disorders: antenatal onset and involvement of multiple organs. Am J Hum Genet 73:1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. van der Lei HD, van Berkel CG, van Wieringen WN, Brenner C, Feigenbaum A, Mercimek‐Mahmutoglu S et al (2010) Genotype‐phenotype correlation in vanishing white matter disease. Neurology 75:1555–1559. [DOI] [PubMed] [Google Scholar]
- 110. van der Voorn JP, van Kollenburg B, Bertrand G, Van Haren K, Scheper GC, Powers JM, van der Knaap MS (2005) The unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol 64:770–775. [DOI] [PubMed] [Google Scholar]
- 111. Van Haren K, van der Voorn JP, Peterson DR, van der Knaap MS, Powers JM (2004) The life and death of oligodendrocytes in vanishing white matter disease. J Neuropathol Exp Neurol 63:618–630. [DOI] [PubMed] [Google Scholar]
- 112. van Kollenburg B, Thomas AAM, Vermeulen G, Bertrand GAM, van Berkel CGM, Pronk JC et al (2006) Regulation of protein synthesis in lymphoblasts from vanishing white matter patients. Neurobiol Dis 21:496–504. [DOI] [PubMed] [Google Scholar]
- 113. van Kollenburg B, van Dijk J, Garbern J, Thomas AA, Scheper GC, Powers JM, van der Knaap MS (2006) Glia‐specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol 65:707–715. [DOI] [PubMed] [Google Scholar]
- 114. Vermeulen G, Seidl R, Mercimek‐Mahmutoglu S, Rotteveel JJ, Scheper GC, van der Knaap MS (2005) Fright is a provoking factor in vanishing white matter disease. Ann Neurol 57:560–563. [DOI] [PubMed] [Google Scholar]
- 115. Watanabe I, Muller J (1967) Cavitating “diffuse sclerosis”. J Neuropathol Exp Neurol 26:437–455. [PubMed] [Google Scholar]
- 116. Wong K, Armstrong RC, Gyure KA, Morrison AL, Rodriguez D, Matalon R et al (2000) Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta Neuropathol 100:635–646. [DOI] [PubMed] [Google Scholar]
- 117. Wu J, Zhang L, Wang X (2000) Maturation and apoptosis of human oocytes in vitro are age‐related. Fertil Steril 74:1137–1141. [DOI] [PubMed] [Google Scholar]
- 118. Wu Y, Jiang YW, Qin J, Xiao JX, Wang JM, Yang YL et al (2007) Clinical characteristics of cases with leukoencephalopathy with vanishing white matter. Chin J Pediatr 45:115–120. [PubMed] [Google Scholar]