

Abstract
Ganglioglioma (GG) is a grade I tumor characterized by alterations in the MAPK pathway, including BRAF V600E mutation. Recently, diffuse midline glioma with an H3 K27M mutation was added to the WHO 2016 classification as a new grade IV entity. As co‐occurrence of H3 K27M and BRAF V600E mutations has been reported in midline tumors and anaplastic GG, we searched for BRAF V600E and H3 K27M mutations in a series of 54 paediatric midline grade I GG (midline GG) to determine the frequency of double mutations and its relevance for prognosis. Twenty‐seven patients (50%) possessed the BRAF V600E mutation. The frequency of the co‐occurrence of H3F3A/BRAF mutations at diagnosis was 9.3%. No H3 K27M mutation was detected in the absence of the BRAF V600E mutation. Double‐immunostaining revealed that BRAF V600E and H3 K27M mutant proteins were present in both the glial and neuronal components. Immunopositivity for the BRAF V600E mutant protein correlated with BRAF mutation status as detected by massARRAY or digital droplet PCR. The median follow‐up of patients with double mutation was 4 years. One patient died of progressive disease 8 years after diagnosis, whereas the four other patients were all alive with stable disease at the last clinical follow‐up (at 9 months, 1 year and 7 years) without adjuvant therapy. We demonstrate in this first series of midline GGs that the H3 K27M mutation can occur in association with the BRAF V600E mutation in grade I glioneuronal tumors. Despite the presence of H3 K27M mutations, these cases should not be graded and treated as grade IV tumors because they have a better spontaneous outcome than classic diffuse midline H3 K27M‐mutant glioma. These data suggest that H3 K27M cannot be considered a specific hallmark of grade IV diffuse gliomas and highlight the importance of integrated histomolecular diagnosis in paediatric brain tumors.
Keywords: ganglioglioma, midline, BRAF V600E, H3 K27M
Introduction
Ganglioglioma (GG) is a rare mixed glioneuronal tumor composed of dysplastic ganglion cells and neoplastic glial cells that occurs mostly in children and young adults. GG is considered a grade I indolent tumor in the WHO 2016 classification 24, and long‐term survival is excellent if the entire tumor can be removed 4, 25. Grade I GG accounts for 1%–5% of all central nervous system neoplasms in children; most are found in the cortex, generally in the temporal lobe (>70%) 1, 21. However, they can occur in the posterior fossa or spinal cord, and other midline locations have also been reported 8, 10, 12, 31. GGs in the brainstem or spinal cord exhibit a poorer prognosis. However, it remains unclear whether this poor prognosis is caused by their location per se or to their molecular profile and/or more frequently incomplete resection 5, 8, 21, 23, 32.
A high frequency of BRAF V600E mutation has been described in GGs and identified as a targetable protein for therapy 3, 6, 7, 35. This mutation results in a glutamic acid to valine substitution within the active site of the encoded serine/threonine kinase domain, resulting in constitutive activation of the mutated protein and driving oncogenic transformation. Whereas alterations in BRAF have been described in paediatric low‐grade gliomas (LGG), recurrent mutations in genes encoding histone H3.1 (HIST1H3B) and H3.3 (H3F3A) are considered a hallmark of midline paediatric high‐grade gliomas (HGG) 11, 38. K27M mutations in the H3.1 or H3.3 genes inhibit Polycomb Repressor Complex 2, resulting in of the loss of lysine 27 trimethylation 22, 41. The frequency of the H3 K27M mutation in midline GGs (midline GGs) is unknown. Until now such mutations were previously reported to be absent from grade I GGs and pilocytic astrocytomas 11, 41. However, rare cases of H3 K27M mutant grade I GG and pilocytic astrocytomas were recently described, suggesting that H3 K27M mutations may be present in a spectrum of glial tumors. H3 K27M mutations should not be used as the sole criterion for the diagnosis of WHO Grade IV midline gliomas 19, 29. In one of these cases, the H3 K27M mutation was associated with a BRAF V600E mutation. This combination of mutations was previously reported in a midline paediatric case 27. Otherwise, only nine other various paediatric tumors (low or high grade) harboring a combination of H3F3A and BRAF mutations [seven gliomas (one grade II astrocytoma, two LGG NOS, three HGG, one diffuse glioma with focal PXA‐like features) and one GG with anaplastic features] have been reported 19, 27, 28, 34, 36, 43, 46. All these tumors were located along the midline in the supratentorial and infratentorial areas. The frequency of H3F3A and BRAF double mutations in paediatric grade I GG and the biological significance of the double mutation are unknown.
In this study, we searched for BRAF V600E and H3 K27M mutations in a series of 54 newly diagnosed paediatric midline grade I GGs using a combination of immunohistochemistry (IHC) and molecular biology.
Material and Methods
Tumor samples
We studied 54 paediatric cases of grade I GG, as defined by the WHO 2016 classification criteria and with a midline location (supratentorial, brainstem or spinal), from a large cohort of 471 GGs (including 323 paediatric cases) diagnosed between January 2005 and December 2015. All cases were retrieved from the pathology archives of Sainte‐Anne‐Necker Hospital and were subjected to local histological (PV) and radiological (NB) review. All the selected cases presented a classic mixed glioneuronal immunophenotype and were analyzed preferentially in solid tissue areas (without persisting axonal networks). Ganglion cells were identified as large pleomorphic neurons displaying positive staining for chromogranin A and/or synaptophysin and NF70 in an environment devoid of residual NF70‐positive axonal networks. The absence of ganglion cells morphologically or by IHC analyses was an exclusion criterion. No patient with neurofibromatosis type 1 was included. Sections for genetic analyses and IHC were prepared from zinc formalin‐fixed paraffin‐embedded tissue specimens (5% formalin, 3 g/L zinc, 8 g/L sodium chloride) or frozen tissues. Two cases displaying anaplastic transformation were described in previous studies 19, 45.
Immunohistochemistry
Representative zinc formalin‐fixed sections were deparaffinized and processed with a Ventana autostainer (BenchMark XT, Ventana Medical Systems or Discovery XT, Ventana Medical Systems) according to a standard protocol. Antibodies against the following proteins were used: glial fibrillary acidic protein (GFAP) (1:200, 6F2, Dako Denmark A/S, Glostrup, Denmark), CD34 (1:40, QBEnd‐10, Dako, Denmark A/S, Glostrup, Denmark), chromogranin A (1:200, LK2H10, Diagnostic BioSystems, Pleasanton, USA), p53 (1:5000, DO‐1, Santa Cruz Biotechnology, Dallas, USA), ATRX (1:200, polyclonal, Sigma Aldrich, St. Louis, MO), BRAF V600E (1:100, VE1, Spring Bioscience, Pleasanton, USA), H3K27M (1:1000, ABE419, EMD Millipore, Billerica, USA) and H3K27me3 (1:1250, C15410195, Diagenode, Seraing, Belgium). Antibody binding was detected by incubation with the chromogen diaminobenzidine. The slides were then scanned in a NanoZoomer 2.ORS (Hamamatsu Photonics, Hamamatsu, Japan).
Double‐immunostaining for the BRAF V600E and H3 K27M mutations
Double chromogenic immunostaining
Representative zinc formalin‐fixed sections were deparaffinized and processed with a Ventana autostainer (BenchMark XT, Ventana Medical Systems or Discovery XT, Ventana Medical Systems) according to a standard protocol. The samples were first incubated with the primary antibody against the K27M histone H3.3 mutation (1:3000, ABE419, EMD Millipore, Billerica, USA) followed by a secondary antibody (Ultra View Universal AP red, Ventana Medical Systems). A second primary antibody, BRAF V600E (1:200, VE1, Spring Bioscience, Pleasanton, USA), was then applied, and the samples were subsequently treated with a second secondary antibody (Optiview kit, Ventana Medical Systems).
Double immunofluorescence staining
The samples were first incubated with the primary antibody against the K27M histone H3.3 mutation (1:1000, ABE419, EMD Millipore, Billerica, USA) and then with the fluorophore (DyLight 550 conjugate, ThermoFisher Scientific, Rockford, USA) ‐conjugated secondary antibody. A second primary antibody, BRAF V600E (1:200, VE1, Spring Bioscience, Pleasanton, USA), was then applied, and the samples were subsequently treated with a fluorophore‐conjugated secondary antibody (Discovery FITC kit, Ventana Medical Systems). Cells on coverslips were counterstained with Hoechst (Sigma Aldrich, MO, USA). Hoechst (blue) was excited by a two‐photon laser at 690 nm. DyLight 550 (red) was excited at 534 nm, and FITC (green) was excited at 488 nm. DyLight 550 and FITC were detected by confocal microscopy (Zeiss LSM 710 NLO). Images were sequentially acquired using a 2‐pixel window (software ZEN 2010).
Mutations analyses of BRAF and H3F3A
DNA extraction
Total DNA was extracted with the use of the QIAampDNA mini‐kit® (Qiagen Inc., Courtaboeuf, France) according to manufacturer's protocols. Briefly, tissues were disrupted in lysis buffer. After removing paraffin, the DNA was purified via sequential centrifugation through membrane spin columns. The purity and quantity of DNA was assessed by measuring the absorbance ratio at 260/280 nm with a NanoDrop® Spectrophotometer (LabTech, Palaiseau, France).
MassARRAY assay
A brain tumor gene mutation panel was developed using the MassARRAY iPlex technology and MassARRAY online design tools (Agena Bioscience), including H3F3A mutations (codon 27–34), HIST1H3B mutations (codon 27) and BRAF mutations (V600E). The MassARRAY iPlex procedure involves a three‐step process consisting of the initial PCR reaction, inactivation of unincorporated nucleotides by shrimp alkaline phosphatase and a single‐base primer extension. Then, the products are nano‐dispensed onto a matrix‐loaded silicon chip (SpectroChipII, Ageno Bioscience). Finally, the mutations are detected by MALDI–TOF (matrix‐assisted laser desorption‐ionization–time of flight) mass spectrometry. Data analysis was performed using MassARRAY Typer Analyzer software 4.0.4.20 (Ageno Bioscience), which facilitates visualization of data patterns as well as the raw spectra.
Droplet digital polymerase‐chain reaction
Using numerical PCR 42 of a droplet 17 (ddPCR, Bio‐Rad), a sample is distributed such that each sample of nucleic acid molecules is located and concentrated in separate reaction chambers. The conventional PCR and ddPCR assays are performed for each sample. However, the sample is separated into a large number of partitions, and the reaction is performed in each partition individually. The nucleic acid limiting dilution is accompanied by dilution of the factors influencing the kinetics of PCR. Furthermore, the quantification is obtained by counting the positive and negative chambers at the end point of the PCR reaction, thus eliminating any kinetic problem. The absolute quantification of a PCR amplicon is obtained by a count of 15 000 to 18 000 positive and negative chambers/droplets. In the PCR assay, absolute quantification is obtained by transformation of positive/negative chambers with Poisson law for DNA copies. By ddPCR, we measured the proportion of BRAF p.Val600Glu c.1799 T > A, p.Val600Lys c.1798_1799delinsAA and p.Lys601Glu c.1801 A > G mutations in FFPE DNA samples following the supplier's recommendations (Bio Rad).
Sanger sequencing
Histone H3F3A gene was analyzed by direct sequencing of PCR‐amplified products from tumor DNA using the following primers (Forward: 5′‐TCAATGCTGGTAGGTAAGTAAGGA‐3′; Reverse: 5′‐GGTTTCTTCACCCCTCCAGT‐3′) as previously described 2. The amplified products were studied by direct sequencing after clean‐up exonuclease ExoSAP‐IT (Affymetrix, Santa Clara, CA) using the Big Dye Terminator Cycle Sequencing Kit and capillary electrophoresis on the automated sequencer ABI3730 (Applied Biosystems, Carlsbad, CA). Sense and antisense sequences were screened for exonic alterations using SeqScape v2.5 software (Applied Biosystems) and compared with the NCBI reference sequences: H3F3A (NM_002107.4).
Results
The characteristics of the patients are summarized in Figure 1. The study cohort comprised 30 (55%) boys and 24 (45%) girls, with a median age at surgery of 8 years (range: 2–17 years). At diagnosis, 52% (28/54) were between 2‐ and 8‐year old, 28% (15/54) were nine to 12‐year old and 20% (11/54) were older than 13‐year old (Supporting Information Figure S1). Twenty‐seven (50%) tumors were supratentorial (ST) (median age: 9.5 years, range 3–15 years), 22 (41%) were located in the brainstem (BS) (median age: 6.5 years, range 2–17 years) and five (9%) had a spinal (SP) location (median age: 13 years, range 11–15 years). The distribution of tumor locations differed between age groups. Between the ages of 2 and 8 years, the BS was the predominant tumor location (57%), with no tumors detected in the spinal cord. By contrast, in the 9 to 12 years age group, 73% of tumors were ST, with the BS and SP sites accounting for 20% and 7% of cases, respectively. In children aged 13 years and older, 35.5% of tumors were ST, 27% were located in the BS and 36.5% had a SP location (Supporting Information Figure S1).
Figure 1.
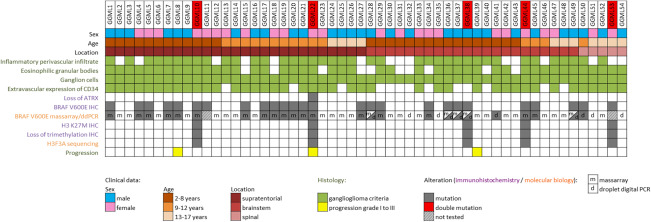
Clinical characteristics, histological features and genetic alterations in 54 pediatric grade I midline GGs.
The histological characteristics of the tumors are summarized in Figure 1. An inflammatory perivascular infiltrate and eosinophilic granular bodies were present in 72% (39/54) and 81% (44/54) of cases, respectively. The mitotic index remained low in all cases, and the MIB index was less than 5% in all cases. Forty‐one tumors (76%) displayed extravascular CD34 expression. A loss of ATRX immunostaining was observed in one case, and three cases (7%) displayed a histologically confirmed secondary malignant transformation.
BRAF status was determined by IHC and massARRAY and/or ddPCR (Figure 1). BRAF V600E status by IHC was available for all cases. Twenty‐six cases exhibited positivity by IHC (48%). In two cases, BRAF V600E status was available only by IHC. By massARRAY, 46 cases were tested (85%), including 19 positive cases (19/46, 41%). Five immunopositive cases were not confirmed by this technique but were confirmed by ddPCR. Among six cases only tested by ddPCR (few tumoral sample), one case was immunopositive (midline GG50) and one did not show any staining on repeated IHC (midline GG41). The discordant results are summarized in Supporting Information Table 1. In total, 27 patients (50%) presented a BRAF V600E mutation (14 boys and 13 girls). Specifically, 15 patients had ST (15/26%–58%), 10 had BS (10/22%–45%) and 2 had SP (2/5%–40%) tumors. The BRAF V600E mutation was observed in 50% (14/28) of 2‐ to 8‐year‐old patients, 67% (10/15) of 9‐ to 12‐year‐old patients and 27% (3/11) in 13‐ to 17‐year‐old patients. Positive staining for glial and neuronal components was clearly observed by IHC (Figure 2A–F). In one case [described by Joyon et al 19], the BRAF V600E mutation was detected at diagnosis but lost at spontaneous malignant transformation, as shown by IHC and confirmed by sequencing. In this peculiar case, no BRAF V600E positive clone was detected by IHC and no CMMRD was found by IHC. Immunostatus (H3 K27me3 and H3.3 K27M mutations) and sequencing were available for all cases. Five cases (9.3%) displayed both a loss of H3 K27 trimethylation and an H3F3A K27M mutation in both glial and neuronal components. No single H3.3 K27M mutation was detected, and all such mutations were associated with the BRAF V600E mutation. No other mutation in H3F3A or in HIST1H3B genes was found.
Figure 2.
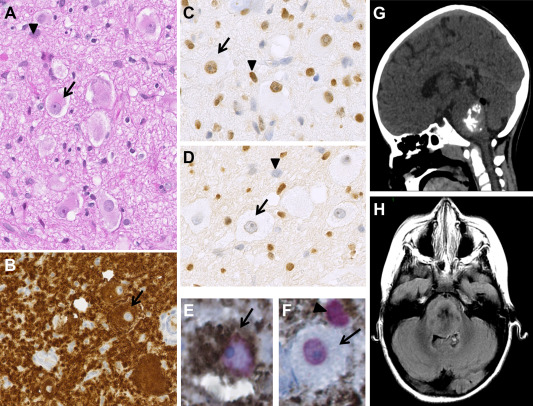
Radiological, histological and immunohistochemical findings of midline GG38. Serial sections showing mixed glial (arrowhead) and neuronal ganglion cell elements (arrow) (H&E) (A) with strong, diffuse immunostaining for BRAF V600E in both the glial and neuronal components (B), positive immunostaining for H3 K27M in both the glial and neuronal components (C) and a loss of H3K27me3 trimethylation in both the glial and neuronal components (D). Double‐immunostaining for BRAF V600E (diaminobenzidine in brown)/H3 K27M (AP Red in red) depicting colocalization in the glial and ganglion cell components (E and F). Very high magnification depicting a double‐mutated ganglion cell (E) and glial cell (arrowhead, F). Note that the ganglion cell could only be positive for H3 K27M (F). A–D. Magnification ×400, E and F magnification ×600. Coronal plane tomography (G) and axial plane MRI T2‐FLAIR (H). Images depict a well‐circumscribed solid mass arising from the pontine area. The computed tomography images show hyperdensity and calcification. MR images depict a heterogeneous but well‐circumscribed lesion.
The clinical and radiological characteristics of the patients with both BRAF V600 and H3.3 K27M mutations are summarized in Table 1 and Figure 2G,H,B and Supporting Information Figure S2. These patients had a median age of 10 years (range: 6–14 years). Four patients were girls, and one was a boy. The tumors were all located in the thalamus, pons, peduncle or spinal area (T9‐T10). No gross total resection was performed. The median follow‐up period was 4 years (range: 0.8–8 years). None of the patients received adjuvant treatment. One patient died from spontaneous malignant in situ transformation 8 years after diagnosis. Three patients are still alive, with no evidence of progression of the residual tumor based on MRI. One patient was lost to follow‐up four months after diagnosis. Interestingly, on detailed radiological review, 4 out 5 double‐mutated cases presented a hyperdensity on tomography, which is uncharacteristic for a grade I GG. These tumors all enhanced heterogeneously with gadolinium and could be calcified (Figure 2G and Supporting Information Figure S2). Histopathologically, a ganglion cell component was present in all cases as confirmed immunohistochemically (Figure 2A–F and Supporting Information Figure S3). This component was always associated with eosinophilic granular bodies and/or inflammatory perivascular infiltrate (Figure 1). None of the cases presented necrosis. Microvascular proliferation was present in one case. The mitotic index was low and MIB‐1 labeling was weak. Extravascular CD34 staining was observed in all but one case. IHC demonstrated the presence of both the BRAF V600E and H3.3 K27M proteins in the glial and neuronal components (Figures 2B–F, 3 and Supporting Information Figure S3). The colocalization of BRAF V600E and H3.3 K27M was noted in both glial and neuronal components, but some neurons were only immunostained for the H3.3 K27M mutation (Figures 2E,F and 3).
Table 1.
Clinical features of cases with H3 K27M/BRAF V600E double mutations.
| Sex | Age at diagnosis (y) | Location | Gross total resection | Adjuvant treatment | Follow‐up period | Follow‐up | Status at last follow‐up | |
|---|---|---|---|---|---|---|---|---|
| Midline GG10 | F | 8 | Thalamus | No | No | 7 years | Stability of the residual tumor | Alive |
| Midline GG22 | F | 12 | Thalamus | No | No | 8 years | Spontaneous malignant in situ transformation 7 years after diagnosis | Dead |
| Midline GG38 | M | 6 | Pons | No | No | 9 months | Stability of the residual tumor | Alive |
| Midline GG44 | F | 10 | Peduncle | No (metastatic) | No | NA | NA | NA |
| Midline GG54 | F | 14 | Spinal (T9‐T10) | No | No | 1 year | Stability of the residual tumor | Alive |
Abbreviations: F = female; M = male; NA = not available; y = year.
Figure 3.
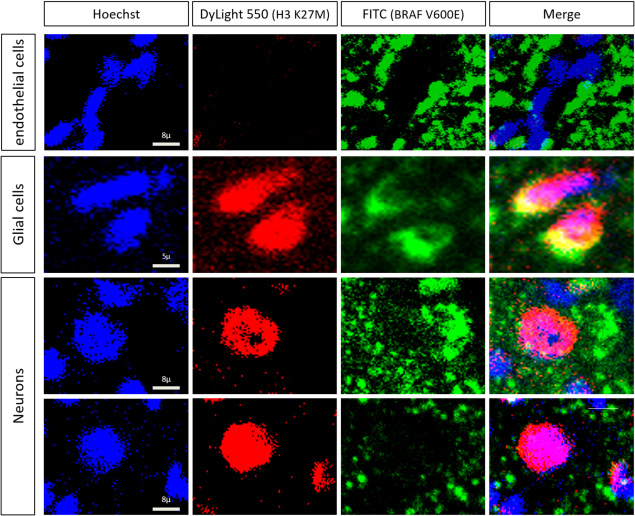
Double immunofluorescence staining of BRAF V600E and H3 K27M in midline GG38. The upper row depicts endothelial cells without labeling for H3 K27M (red) and BRAF V600E (green). Imaging revealed evidence of H3 K27M and BRAF V600E colocalization in glial cells (second row) and neurons (third row) in nuclear and cytoplasmic areas, respectively. Note that neurons could only be positive for H3 K27M (lower row).
Discussion
Grade I GGs display considerable glial and neuronal structural and morphological heterogeneity. We therefore carefully selected classical tumors with a clear neuronal component by morphological and/or IHC analysis of solid tumor tissue areas without residual NF70‐positive axonal networks. In our cohort, the infratentorial tumors corresponded to classic GGs, or group I according to Gupta et al 12. The positive BRAF V600E and H3.3 K27M staining in both glial and neuronal components in all cases with the double mutation confirmed the mixed glioneuronal nature of these tumors. Until recently, it was unclear whether the neoplastic compartment of GGs was glial, neuronal or both 16, 20, 47.
Few tissues were available for molecular analyses caused by the midline location. Consequently, BRAF V600E and histone H3.3 K27M IHC were prioritized, allowing a better characterization and comparison of the distinct neuronal and glial components of these biphasic tumors. Inter‐assay reproducibility for the detection of BRAF V600E mutation was very good. Furthermore, IHC and ddPCR were more effective than massARRAY to detect this mutation. Numerous studies on melanomas and neuro‐oncopathology have demonstrated a strong correlation between BRAF V600E IHC and sequencing data, and previous studies have also demonstrated a high level of concordance between ddPCR and exome sequencing 13, 33. Similarly, high sensitivity and specificity were reported for detection of the H3 K27M mutation by targeted sequencing‐based methods 2, 41.
The five cases of double mutations studied here were grade I GGs without malignant features. The detection of the H3.3 K27M mutation within a grade I tumor entity is intriguing, as histone mutations are generally considered to be the major driver of grade IV midline diffuse gliomas. The size of the cohort does not allow performing any statistical analysis but some cases showed a long survival without adjuvant treatment (7 years, GG10). In the same way, for double‐mutant tumors previously reported (Table 2), OS (known for five cases) was significantly increased compared with H3 K27M diffuse midline glioma 28. This natural indolent course strongly contrasts with the rapid tumor progression of H3 K27M diffuse midline gliomas as the median overall survival (OS) for patients with H3.3 K27M supratentorial midline gliomas is less than 12 months, and only 9.2 months for patients with diffuse infiltrating pontine gliomas (DIPG) 2, 38. However, some rare atypical brainstem gliomas progress more slowly 18. These findings suggest that H3.3 K27M mutation is not specific to malignant grade IV glial tumors. In addition, the H3 K27M mutation has recently been described in posterior fossa ependymoma 9.
Table 2.
Clinical and histopathological features of H3 K27M/BRAF V600E double‐mutated gliomas and glioneuronal tumors reported in the literature.
| Cases | Age (y), sex | Midline location | Histological diagnosis | OS (years) | Status at last follow‐up |
|---|---|---|---|---|---|
| Zhang 2013 (27) | 10, M | Supratentorial | Grade II astrocytoma | NA | NA |
| Wu 2014 (26) | 10, F | Supratentorial | HGG (grade IV) | NA | NA |
| Nguyen 2015 (24) | 2, F | Pons | Diffuse LGG NOS | 5.2 | Alive |
| Nguyen 2015 (24) | 16, F | Pineal | Grade III GG/PXA with aggressive features vs. grade III GG | 3.1 | Dead |
| Nguyen 2015 (24) | 10, F | Brainstem | PA with aggressive features vs. diffuse HGG NOS | 1.0 | Dead |
| Mistry 2015 (23) | NA, M | Supratentorial (midline) | Secondary grade III GG | NA | NA |
| Solomon 2015 (25) | 5, F | Thalamus | Diffuse glioma with focal PXA‐like features | NA | NA |
| Ryall 2016 (28) | NA, M | Thalamus | High grade glioma | 1.4 | Dead |
| Ryall 2016 (28) | NA, F | Thalamus | Low grade glioma | NA | NA |
| Joyon 2016 (21)* | 12, F | Thalamus | Malignant transformation of grade I GG | 8.0 | Dead |
Abbreviations: F = female; M = male; NA = not available; y = year.
*Corresponding to our midline GG22.
Midline GG22 displayed spontaneous malignant transformation 7 years after diagnosis, with subsequent loss of the BRAF V600E mutation. Interestingly, by double immunostaining for BRAF V600E‐H3.3 K27M mutations, double positivity was observed in both glial and neuronal components. However, some cells exhibited positive immunostaining only for the H3.3 K27M mutation. These cells may have lost the BRAF V600E mutation during tumoral progression. These data suggest that the BRAF V600E mutation may influence long‐term survival if associated with the H3.3 K27M mutation. Anaplastic GGs (grade III), a controversial and poorly characterized entity with a poor prognosis 24, 40, 45, can arise de novo or secondary to the malignant transformation of WHO grade I GG, but the rate of malignant transformation is unclear, with reported values of 0.6% to 14.5% depending on the study 25, 26, 44. Three patients in our series presented secondary malignant transformation, whatever the mutational status. Hochart et al described a H3 K27M pilocytic astrocytoma that underwent spontaneous malignant transformation into glioblastoma 10 years after diagnosis 15.
All together these data suggest that glial tumors with H3.3 K27M mutation but no malignant features should not be systematically treated as grade IV. The histological features associated with GGs (ie, granular bodies, lymphocytic infiltration, extravascular CD34 expression and MRI findings) and BRAF status should be assessed. These findings highlight the importance of integrated characterization, including imaging, histology and molecular biology, for these CNS tumors. It must be stressed that the integration of histopathological and molecular data must remain the rule when establishing a diagnosis 24. For example, SMARCB1‐deficient tumors in the CNS mainly comprise very aggressive ATRT but also a benign intraventricular tumor called cribriform neuroepithelial tumor (CRINET) 14. Nevertheless, given the rapid progression classically associated with midline paediatric HGG with H3.3 mutation and DIPG, the comparison with the evolution of these double‐mutated cases is easy but the comparison of these cases with GG without H3 K27M mutation (BRAF V660E mutated or not) remains to be investigated.
Consistent with published findings, the sex distribution of cases with BRAF V600E mutation showed a female predominance. Thereby, combining our series with previous reports (Table 2), 71% of all patients with double mutations are female (10/14). A similar sex ratio has been reported for other recently described paediatric tumors, such as YAP1 ependymomas, high‐grade neuroepithelial tumors with MN1 alteration (HGNET‐MN1) and DIPG with ACVR1 mutation. However, the reasons for this female preponderance remain unclear 30, 37, 39.
In conclusion, in this first series of midline grade I GGs, we demonstrate that the H3.3 K27M mutation occurs always in association with BRAF V600E mutation. Despite the presence of the H3.3 K27M mutation, these cases should not be graded and treated as grade IV tumors. Further investigations, including long‐term survival data and treatment information, will clarify the prognosis of these grade I tumors and determine the role of BRAF, especially in the context of targeted therapy.
Conflict of interest
The authors declare that they have no conflict of interest except Pascale Varlet (Hoffmann La roche, Novartis and Boehringer‐Ingelheim) and Jacques Grill (Novartis, Roche, Bristol‐Myers Squibb).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Distribution of age categories and locations by age at diagnosis.
Figure S2. Radiological findings of cases with H3 K27M/BRAF V600E double mutation.
Figure S3. Histological and immunohistochemical analyses of cases with the H3 K27M/BRAF V600E double mutation. Serial sections (H&E) exhibiting mixed glial and neuronal ganglion cell elements with strong diffuse immunostaining for BRAF V600E in both the glial and neuronal components, loss of H3K27me3 trimethylation in both the glial and neuronal components and positive immunostaining for H3 K27M in both components. Magnification x400.
Table 1. Discordant cases for BRAF V600E analysis.
Table 2. Characteristics of the cohort.
Acknowledgment
We would like to thank Michelle Oliveiro, Joëlle Lacombe and Daniel Fiole for technical assistance. We acknowledge the Nuovo‐Soldati forundation for funding sources (MP).
References
- 1. Blümcke I, Wiestler OD (2002) Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 61:575–584. [DOI] [PubMed] [Google Scholar]
- 2. Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N et al (2015) Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol (Berl) 130:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chappé C, Padovani L, Scavarda D, Forest F, Nanni‐Metellus I, Loundou A et al (2013) Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol Zurich Switz 23:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Compton JJ, Laack NNI, Eckel LJ, Schomas DA, Giannini C, Meyer FB (2012) Long‐term outcomes for low‐grade intracranial ganglioglioma: 30‐year experience from the Mayo Clinic. J Neurosurg 117:825–830. [DOI] [PubMed] [Google Scholar]
- 5. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR (2013) BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol (Berl) 125:901–910. [DOI] [PubMed] [Google Scholar]
- 6. Donson AM, Kleinschmidt‐DeMasters BK, Aisner DL, Bemis LT, Birks DK, Mulcahy Levy JM et al (2014) Pediatric brainstem gangliogliomas show BRAFV600E mutation in a high percentage of cases. Brain Pathol Zurich Switz 24:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ et al (2010) Activating mutations in BRAF characterize a spectrum of pediatric low‐grade gliomas. Neuro‐Oncol 12:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dudley RWR, Torok MR, Gallegos DR, Mulcahy‐Levy JM, Hoffman LM, Liu AK et al (2015) Pediatric Low Grade Ganglioglioma/Gangliocytoma: epidemiology, treatments, and outcome analysis on 348 children from the SEER database. Neurosurgery 76:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gessi M, Capper D, Sahm F, Huang K, von Deimling A, Tippelt S et al (2016) Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol (Berl) 132:635–637. [DOI] [PubMed] [Google Scholar]
- 10. Gessi M, Dörner E, Dreschmann V, Antonelli M, Waha A, Giangaspero F et al (2016) Intramedullary gangliogliomas: histopathologic and molecular features of 25 cases. Hum Pathol 49:107–113. [DOI] [PubMed] [Google Scholar]
- 11. Gielen GH, Gessi M, Hammes J, Kramm CM, Waha A, Pietsch T (2013) H3F3A K27M mutation in pediatric CNS tumors: a marker for diffuse high‐grade astrocytomas. Am J Clin Pathol 139:345–349. [DOI] [PubMed] [Google Scholar]
- 12. Gupta K, Orisme W, Harreld JH, Qaddoumi I, Dalton JD, Punchihewa C et al (2014) Posterior fossa and spinal gangliogliomas form two distinct clinicopathologic and molecular subgroups. Acta Neuropathol Commun 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA (2015) Large multiallelic copy number variations in humans. Nat Genet 47:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hasselblatt M, Oyen F, Gesk S, Kordes U, Wrede B, Bergmann M et al (2009) Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol 68:1249–1255. [DOI] [PubMed] [Google Scholar]
- 15. Hochart A, Escande F, Rocourt N, Grill J, Koubi‐Pick V, Beaujot J et al (2015) Long survival in a child with a mutated K27M‐H3.3 pilocytic astrocytoma. Ann Clin Transl Neurol 2:439–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoischen A, Ehrler M, Fassunke J, Simon M, Baudis M, Landwehr C et al (2008) Comprehensive characterization of genomic aberrations in gangliogliomas by CGH, array‐based CGH and interphase FISH. Brain Pathol Zurich Switz 18:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Närvä E et al (2011) Copy number variation and selection during reprogramming to pluripotency. Nature 471:58–62. [DOI] [PubMed] [Google Scholar]
- 18. Jackson S, Patay Z, Howarth R, Pai Panandiker AS, Onar‐Thomas A, Gajjar A, Broniscer A (2013) Clinico‐radiologic characteristics of long‐term survivors of diffuse intrinsic pontine glioma. J Neurooncol 114:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joyon N, Tauziède‐Espariat A, Alentorn A, Giry M, Castel D, Capelle L et al (2016) K27M mutation in H3F3A in ganglioglioma grade I with spontaneous malignant transformation extends the histopathological spectrum of the histone H3 oncogenic pathway. Neuropathol. Appl Neurobiol [Epub ahead of print; doi: 10.1111/nan.12329]. [DOI] [PubMed] [Google Scholar]
- 20. Koelsche C, Wöhrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M et al (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol (Berl) 125:891–900. [DOI] [PubMed] [Google Scholar]
- 21. Lang FF, Epstein FJ, Ransohoff J, Allen JC, Wisoff J, Abbott IR, Miller DC (1993) Central nervous system gangliogliomas. Part 2: clinical outcome. J Neurosurg 79:867–873. [DOI] [PubMed] [Google Scholar]
- 22. Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA et al (2013) Inhibition of PRC2 activity by a gain‐of‐function H3 mutation found in pediatric glioblastoma. Science 340:857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lindsay AJ, Rush SZ, Fenton LZ (2014) Pediatric posterior fossa ganglioglioma: unique MRI features and correlation with BRAF V600E mutation status. J Neurooncol 118:395–404. [DOI] [PubMed] [Google Scholar]
- 24. Louis DN, Ohgaki H, Wiestler OD et al., eds. (2016) WHO Classification of Tumours of the Central Nervous System. Lyon: IARC; 144–146. [Google Scholar]
- 25. Luyken C, Blümcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J (2004) Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow‐up of 8 years. Cancer 101:146–155. [DOI] [PubMed] [Google Scholar]
- 26. Majores M, von Lehe M, Fassunke J, Schramm J, Becker AJ, Simon M (2008) Tumor recurrence and malignant progression of gangliogliomas. Cancer 113:3355–3363. [DOI] [PubMed] [Google Scholar]
- 27. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M et al (2015) BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high‐grade glioma. J Clin Oncol off J Am Soc Clin Oncol 33:1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nguyen AT, Colin C, Nanni‐Metellus I, Padovani L, Maurage C‐A, Varlet P, et al (2015) Evidence for BRAF V600E and H3F3A K27M double mutations in paediatric glial and glioneuronal tumours. Neuropathol Appl Neurobiol 41:403–408. [DOI] [PubMed] [Google Scholar]
- 29. Orillac C, Thomas C, Dastagirzada Y, Hidalgo ET, Golfinos JG, Zagzag D et al (2016) Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol Commun 4:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pan C‐C, Chen X, Xu C, Wu W‐H, Zhang P, Wang Y et al (2016) Brainstem gangliogliomas: prognostic factors, surgical indications and functional outcomes. J Neurooncol 128:445–453. [DOI] [PubMed] [Google Scholar]
- 32. Puget S, Alshehri A, Beccaria K, Blauwblomme T, Paternoster G, James S et al (2015) Pediatric infratentorial ganglioglioma. Childs Nerv Syst ChNS off J Int Soc Pediatr Neurosurg 31:1707–1716. [DOI] [PubMed] [Google Scholar]
- 33. Routhier CA, Mochel MC, Lynch K, Dias‐Santagata D, Louis DN, Hoang MP (2013) Comparison of 2 monoclonal antibodies for immunohistochemical detection of BRAF V600E mutation in malignant melanoma, pulmonary carcinoma, gastrointestinal carcinoma, thyroid carcinoma, and gliomas. Hum Pathol 44:2563–2570. [DOI] [PubMed] [Google Scholar]
- 34. Ryall S, Krishnatry R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R et al (2016) Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun 4:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol (Berl) 121:397–405. [DOI] [PubMed] [Google Scholar]
- 36. Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJJ, Perry A (2015) Diffuse midline gliomas with histone H3‐K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol Zurich Switz 26:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 164:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sturm D, Witt H, Hovestadt V, Khuong‐Quang D‐A, Jones DTW, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. [DOI] [PubMed] [Google Scholar]
- 39. Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C et al (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terrier L‐M, Bauchet L, Rigau V, Amelot A, Zouaoui S, Filipiak I, Club de Neuro‐Oncologie of the Société Française de Neurochirurgie et al (2016) Natural course and prognosis of anaplastic gangliogliomas: a multicenter retrospective study of 43 cases from the French Brain Tumor Database. Neuro‐Oncol [Epub ahead of print; doi: 10.1093/neuonc/now186]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Venneti S, Santi M, Felicella MM, Yarilin D, Phillips JJ, Sullivan LM et al (2014) A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol (Berl) 128:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vogelstein B, Kinzler KW (1999) Digital PCR. Proc Natl Acad Sci USA 96:9236–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yust‐Katz S, Anderson MD, Liu D, Wu J, Yuan Y, Olar A et al (2014) Clinical and prognostic features of adult patients with gangliogliomas. Neuro‐Oncol 16:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zanello M, Pages M, Tauziède‐Espariat A, Saffroy R, Puget S, Lacroix L et al (2016) Clinical, imaging, histopathological and molecular characterization of anaplastic ganglioglioma. J Neuropathol Exp Neurol 75:971–980. [DOI] [PubMed] [Google Scholar]
- 46. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al (2013) Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet 45:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu JJ, Leon SP, Folkerth RD, Guo SZ, Wu JK, Black PM (1997) Evidence for clonal origin of neoplastic neuronal and glial cells in gangliogliomas. Am J Pathol 151:565–571. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Distribution of age categories and locations by age at diagnosis.
Figure S2. Radiological findings of cases with H3 K27M/BRAF V600E double mutation.
Figure S3. Histological and immunohistochemical analyses of cases with the H3 K27M/BRAF V600E double mutation. Serial sections (H&E) exhibiting mixed glial and neuronal ganglion cell elements with strong diffuse immunostaining for BRAF V600E in both the glial and neuronal components, loss of H3K27me3 trimethylation in both the glial and neuronal components and positive immunostaining for H3 K27M in both components. Magnification x400.
Table 1. Discordant cases for BRAF V600E analysis.
Table 2. Characteristics of the cohort.