

Abstract
Alexander Disease (AxD) is a degenerative disorder caused by mutations in the GFAP gene, which encodes the major intermediate filament of astrocytes. As other cells in the CNS do not express GFAP, AxD is a primary astrocyte disease. Astrocytes acquire a large number of pathological features, including changes in morphology, the loss or diminution of a number of critical astrocyte functions and the activation of cell stress and inflammatory pathways. AxD is also characterized by white matter degeneration, a pathology that has led it to be included in the “leukodystrophies.” Furthermore, variable degrees of neuronal loss take place. Thus, the astrocyte pathology triggers alterations in other cell types. Here, we will review the neuropathology of AxD and discuss how a disease of astrocytes can lead to severe pathologies in non‐astrocytic cells. Our knowledge of the pathophysiology of AxD will also lead to a better understanding of how astrocytes interact with other CNS cells and how astrocytes in the gliosis that accompanies many neurological disorders can damage the function and survival of other cells.
Keywords: Alexander disease, astrocytes, GFAP, leukodystrophy
Introduction
The first autopsy report of a child with Alexander Disease in 1949 [“Progressive Fibrinoid Degeneration of Fibrillary Astrocytes Associated with Mental Retardation in a Hydrocephalic Patient” 1] described a child who did not acquire normal developmental milestones after 7 months, regressed in cognitive and motor skills, developed megalencephaly, seizures and finally died at 16 months of age. This report highlighted two major pathological characteristics of the CNS. The first was the lack of or loss of myelin. In the cerebral hemispheres, there was a gradient of myelin loss, progressing from severe in periventricular locations to more moderate in deep white matter, to well‐preserved arcuate fiber myelin. The second was an enormous number of “hyaline” bodies, which were eventually identified as Rosenthal fibers (RFs), intracellular protein aggregates within astrocytes. Subsequent reports of the neuropathology of AxD also focused on myelin abnormalities, terming the disease, for example, “Dysmyelinogenic Leukodystrophy” 68. Thus, AxD has been historically considered a leukodystrophy, and for good reason. In these early reports, neuropathologists noted a general correlation between the numbers of RFs in white matter and the loss of myelin. However, a mechanistic relationship between the abnormal white matter and the astrocytes with RFs was not at all clear at the time.
The clinical onset of AxD spans a wide range, from infancy to adulthood and produces a range of phenotypes, generally correlating with age of onset. Based on this variability Prust et al 45 have grouped AxD into two major subtypes. Type I, an infantile form, is characterized by macrocephaly, seizures, inability to acquire early motor and cognitive milestones and failure to thrive. Type II occurs in older children and adults and is characterized by bulbar and cerebellar signs and symptoms, eye movement abnormalities, autonomic dysfunction and later onset cognitive changes. Each of the two Types has characteristic MRI patterns.
The identification of GFAP mutations underlying AxD 5 immediately and definitively shifted the focus of pathophysiology to astrocytes and raised the question of how astrocytes could produce such pathological changes. How does a primary astrocyte disease result in myelin pathology? The most logical way to approach this question is to try to understand how the massive accumulation of GFAP, both normal and mutant, changes astrocyte morphology, physiology and metabolism, and then to understand how astrocyte abnormalities cause the pathological phenotype of the disease.
The Degree of White Matter Pathology Varies and Predominates in Type I AxD
The degree of white matter pathology varies considerably in AxD. In general, the earlier the onset, the more pronounced is the pathology and the later the onset, the milder is the pathology and the more prolonged the clinical course. Thus, infants show severe cerebral hemispheric white matter degeneration, with many RFs (Figure 1A) and in some cases even cavitation (see for eg, 10, 15, 29). Many astrocytes are enlarged, bizarre in morphology and often contain eosinophilic granules in cell bodies, representing small RFs (Figure 1B). It is common to see multinucleated astrocytes (Figure 1C), representing mitotic activity without cytokinesis. Indeed, in areas of severe degeneration, one can find mitotic figures (Figure 1D), resembling “Creutzfeldt” astrocytes. The white matter contains many activated microglia and macrophages. Hemispheric cerebral white matter pathology is accompanied by similar changes in the cerebellar white matter (Figure 1E) and in the white matter tracts of the brain stem and spinal cord.
Figure 1.
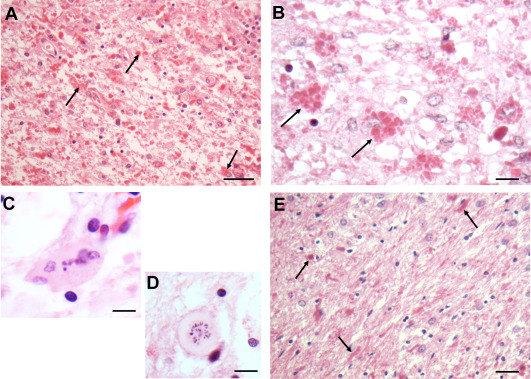
Hemispheric white matter from a patient with Type I AxD. A. The white matter contains innumerous RFs (bright red profiles, arrows). There is a generally pale background, reflecting myelin loss. B. RFs inside astrocyte cell bodies (two marked with arrows) in this AxD individual. C. A multinucleated astrocyte. D. An astrocyte with a mitotic figure. This cell resembles the “Creutzfeldt” astrocyte – mitotic figures in reactive astrocytes, described in inflammatory diseases such as multiple sclerosis. E. Deep cerebellar white matter from this patient. There are many RFs (arrows) and many enlarged astrocytes (astrocyte nuclei are large and round to oval). All Figures H&E. Scale Bars (A, E) 200 μm, (B) 100 μm, (C) 50 μm, (D) 50 μm.
AxD patients who develop clinical signs and symptoms later in life do not display the same severe degree of cerebral white matter degeneration, which is mainly confined to the periventricular regions (Figure 2A), where there is loss of myelin and axons and the astrocytes contain many RFs. Interestingly, in several patients with mild white matter degeneration, the astrocytes in deep white matter, which appears well myelinated, are also moderately enlarged and contain RFs (Figure 2B). In contrast, astrocytes in subcortical white matter (U fibers) appear normal with no RFs (Figure 2C). The major areas of the CNS affected in late onset AxD are the brain stem and cerebellum, correlating well with the clinical picture of ataxias and brain stem motor functional abnormalities such as dysarthria and dysphagia. Why the stem is so affected is not clear, but GFAP levels in brain stem and spinal cord white matter astrocytes are high in comparison to levels in other areas.
Figure 2.
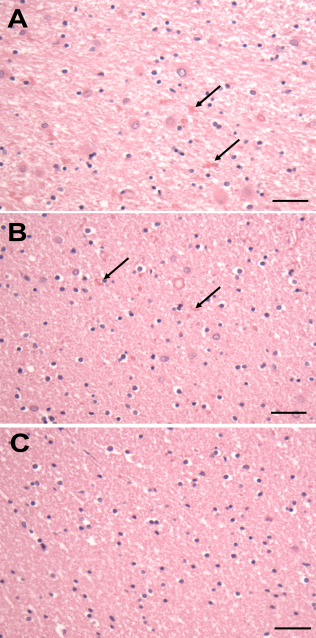
Temporal white matter from a patient with Type II AxD. A. The periventricular white matter is pale and contains RFs (arrows) and enlarged astrocytes. B. The deep white matter shows scattered RFs (arrows), but the degree of myelination appears normal. C. The astrocytes and the degree of myelination in U fibers appears normal. All Figures H&E. Scale Bars (A, B, C) 200 μm.
The astrocyte changes in AxD largely spare isocortex, but involve subpial astrocytes
Astrocytes in the AxD isocortex rarely enlarge and rarely accumulate RFs and GFAP (Figure 3A). The exception is astrocytes in the subpial region, which do enlarge and do accumulate GFAP and RFs. RFs in the subpial endfeet of these astrocytes appear as a layer of RFs against the pial surface (Figure 3B). The obvious explanation for this difference is that subpial astrocytes have far higher GFAP levels than astrocytes in other isocortical layers. Indeed, the protoplasmic astrocytes of the cortex have low levels of GFAP, much lower than that of astrocytes in subpial locations or in white matter.
Figure 3.
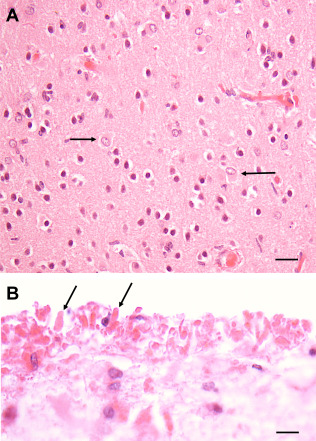
Isocortex of a Type I AxD patient. A. Isocortex does not show RFs, although some astrocytes are enlarged (arrows). B. RFs (2 marked by arrows) accumulate at the pial surface (top of image) in the endfeet of astrocytes. All Figures H&E. Scale Bars (A) 50 μm, (B) 150 μm.
AxD can present clinically as a mass lesion
AxD has presented in rare patients as a mass lesion, involving, for example, the brainstem or optic chiasm 38, 46, 59, 64, suggesting the diagnosis of an astrocytoma. The Ki67 indices are low but raise above the lower levels present in the normal CNS. These lesions contain large numbers of RFs. The AxD diagnosis is made by GFAP sequencing.
Neuronal loss in AxD
Only a few reports describe neuronal loss in AxD brains 4. Neuronal loss was found usually in the CA1 pyramidal layer of the hippocampus 60 and also occurred in the striatum. Neuronal loss is not a general and widespread phenomenon, however. For example, in the brain stem, one can find enlarged astrocytes with RFs interspersed with normal‐appearing neuronal cell bodies (Figure 4A). The neuronal loss seems to appear in areas that have large glutamatergic input, suggesting that the loss of glutamate buffering by compromised astrocytes may be deleterious for the neurons. Seizures may also add to the glutamate‐induced excitotoxic neuronal death. In cultures, astrocytes that contain mutant GFAP do not prevent glutamate‐induced neuronal death at glutamate concentrations at which normal astrocytes preserve neurons 60. In a Drosophila model for AxD, in which mutant GFAP is expressed by glia, there is substantial neuronal loss via glutamate toxicity 65.
Figure 4.
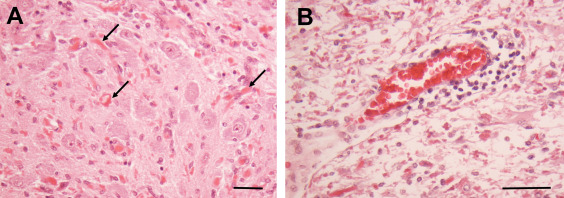
A. Inferior olive of a Type I AxD patient. There are many RFs (arrows) interspersed among olivary neurons, which themselves appear morphologically unremarkable. B. Pons of a Type I AxD patient showing a perivascular lymphocytic infiltrate. Many of the surrounding astrocytes contain RFs. All Figures H&E. Scale Bars (A) 100 μm, (B) 50 μm.
Does AxD have an inflammatory component?
Lymphocytic accumulations around vessels have been noted in several reports of human AxD, primarily in the brain stem (18, 48, 61; Figure 4B). Little attention, however, has been paid to microglia. We have examined microglia in several AxD specimens and regularly found enlarged microglia with thickened processes, suggesting activation in different brain areas 43. How AxD astrocytes might promote such an inflammatory environment is considered below.
Murine Models of AxD
Murine models of AxD include both transgenic (TG), with insertion of several copies of human GFAP 36, and knockin (KI), with substitution of one GFAP allele with mutant mouse GFAP containing a missense mutation homologous to a common one found in the human disorder 20. These mice survive for over a year. They develop subclinical seizures and are more susceptible to kainic acid 20. Mating the two mutants (TGxKI) produces a mouse that develops seizures and does not live more than 5–6 weeks. However, despite severe alterations in astrocytes that accumulate large amounts of GFAP and RFs, the pathology of white matter and of oligodendrocytes is not as pronounced and does not show the severe changes found in the Type I human AxD. Although prominent changes in white matter have not been reported in AxD mice 20, 36, 56, we have observed mild myelin abnormalities in KI and TG AxD mice 6 months of age and older. Most widespread is the appearance of swollen profiles immunopositive for typical myelin markers MBP (myelin basic protein), CNP (cyclic nucleotide phosphohydrolase) and PLP (proteolipid protein) (Figure 5A‐C). These unusual structures may correspond to axonal spheroids although this suggestion needs further validation.
Figure 5.
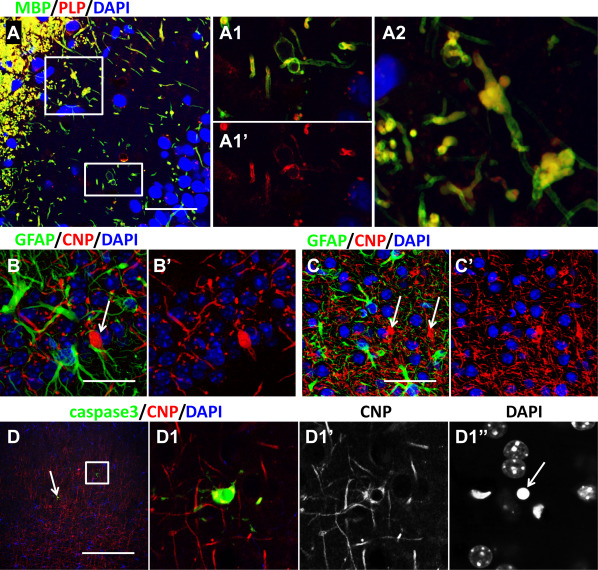
Oligodendrocyte changes in AxD mice. A–C. Bulbous‐like enlargements of myelin in TG mice identified with MBP and PLP (A, enlarged boxed areas shown in A1 and A2) and with CNP (B, C, arrows) immunostaining. A, B – hippocampus; C – isocortex. D. Death‐related changes in TG/KI mouse oligodendrocytes. Cleaved caspase 3 (caspase 3) immunopositive oligodendrocytes are indicated with boxed area and arrow in D. D1 – enlarged boxed area in (D). Note shrunken nucleus of caspase3 immunopositive oligodendrocyte in D1″. Confocal microscopy. Scale bars: 45 μm in (A, C); 15 μm in (B); 85 µm in (D).
With increasing age of the mice, some oligodendrocyte (2–4 cells in isocortex and hippocampus per 40 micron‐thick coronal section at the level of the hippocampus) display positive immunostaining for cleaved caspase 3 and nuclear fragmentation, indicative of cell death. Earlier, in 1‐month‐old KI and TG AxD mice, similar dying oligodendrocytes were not observed. Thus, it seems that pathology of white matter in the TG and KI AxD mice develops with age and probably correlates with aggravation of astrocytic changes. Such dynamics and intensity of pathological alterations correspond to Type II human AxD. In contrast, we have observed more severe pathology in the TGxKI AxD mice, more typical for Type I human AxD disease. These mice have many caspase 3 immunopositive cells, including oligodendrocytes (Figure 5D). In the AxD TGxKI mice, Western blots of MBP from the spinal cord show lower levels than do controls (Olabarria and Goldman, unpublished observations). Myelin pathology and oligodendrocyte death in the AxD mice needs to be investigated further.
As noted above, patients with Type II AxD often present with bulbar and cerebellar signs and symptoms. It is not known why this might be, although an analysis of AxD model mice revealed that transcript levels of KCNJ10 and SLC1A2, which encode Kir4.1 (the main astrocytic channel responsible for K+ uptake) and GLT‐1 (glutamate transporter responsible for glutamate uptake), respectively are significantly reduced in stem and spinal cord early in the disease 40.
There are inflammatory changes in the mouse models, including, microglial activation and an immune response, judging by transcriptional changes, immunocytochemistry and ELISA assays 19, 43.
Physiological and morphological changes in AxD astrocytes
Accumulation of GFAP and RFs
The most notable change in astrocytes is the massive accumulation of GFAP with formation of RFs. RFs are intracellular, cytoplasmic protein aggregates that in the electron microscope look like osmiophilic, unstructured masses directly linked to many intermediate filaments. In H&E staining they have the canonical appearance of bright pink, homogeneous spots. Their size varies from very small, seen only in the electron microscope, to large, oval/elongated shapes up to many µm. They contain, as major proteins, GFAP and small heat shock proteins predominantly alphaB‐crystallin and hsp27, as well as several other proteins in varying amounts 22. RFs can appear in astrocyte cell bodies, but often aggregate in astrocyte processes and endfeet, in subpial zones and around blood vessels.
RFs are not specific to AxD, occurring in old glial scars 55 and in pilocytic astrocytomas 67, indicating that GFAP mutations are not required for RF formation, although mutations may accelerate the formation of RFs. However, the numbers of RFs and their widespread distribution in AxD are markedly higher than in any other pathologies. GFAP filaments appear to be scaffolds onto which RFs form, in association with alphaB‐crystallin 51.
Accumulation of GFAP is a pronounced and prominent feature in AxD, but the mechanisms responsible for abnormal proteostasis remain elusive. In all of the AxD mouse models, GFAP protein levels and transcript levels are increased 20, 36, 43, 50, 56.
Changes in protein turnover/inhibition of proteasome activity
Studies of astrocyte cell lines that express mutant GFAP show a slowed protein turnover and the accumulation of ubiquitinated GFAP and other proteins, and studies in vitro show that mutant GFAP inhibits proteasome activity and the mutant protein is a far stronger inhibitor of proteasome function than is normal GFAP 58. Proteasome inhibition may represent an early stage in the pathogenesis of AxD and contribute substantially to the cellular stress. For example, the inhibition of proteasome activity in cultured CHO cells produces an oxidized state in the cytoplasm and in mitochondria 34. The membrane potential of mitochondria decreased, and the amount of reactive oxygen species increased.
Although it would sound intuitively correct that the inhibition of proteasomes increases protein levels, several studies show that the inhibition of proteasome activity can actually decrease levels of proteins. For example, studies in cell cultures (glioblastoma cell lines) show that the inhibition of proteasomes causes a significant decrease in the levels of GFAP mRNA and the subsequent decline of GFAP protein levels 37. In primary neuronal cultures from rat E18 cortices, proteasome inhibition results in a decrease of global protein synthesis 14. These experiments were not performed with cells that expressed mutant GFAP, however.
Another contributing factor to GFAP protein levels is the turnover rate of the protein. In an interesting study of protein turnover, that of GFAP in the AxD KI mice in vivo and in primary astrocyte cultures from these mice was compared to GFAP turnover in control mice 41. The half‐life of GFAP protein in vitro was much shorter than in vivo and did not differ between KI and control astrocytes. In contrast, the half‐life of GFAP in mutant mice in vivo was much shorter than in control mice (∼ 15 days vs. 28 days, respectively). The faster turnover of GFAP in mutant mice in vivo, as the authors propose, may be because of an acceleration of both protein synthesis and protein degradation 41. It should be noted that the analysis of proteins in vivo was performed at 2 months of age, when astrocytes have already accumulated high levels of GFAP and many RFs. Notably, levels of GFAP in the KI mice did not change between 2 and 4 months, indicating that at this young adult age the dynamics of pathological changes is not prominent and possibly has reached a stable plateau. The findings also may indicate that at earlier times the synthesis of GFAP predominated over the degradation to produce high protein levels. The study was performed on AxD KI mice containing both mutant and normal copies of GFAP, but whether both proteins turn over at the same rate was not determined.
Activation of cell stress pathways
AxD astrocytes show an activation of MAPK stress pathways and also show constitutive activation of both p38 and JNK kinases 57. If constitutively activated in astrocytes, MLK2 or MLK3 activate JNK and result in impairment of proteasome activity 57. Thus, the activation of JNK may increase GFAP levels by further inhibiting proteasomes.
A further interaction between proteasome inhibition and stress pathways occurs because polyubiquitinated proteins interact with histone deacetylase 6 (HDAC6) through its ubiquitin‐binding domain. This binding causes a dissociation of the protein complex composed of p97/VCP (valosin‐containing protein), Hsp90, HSF‐1 (heat shock factor‐1) and HDAC6 (histone deactylase 6) 27. p38, which is acetylated in resting cells, is deacetylated by HDAC6, allowing for p38 phosphorylation and the subsequent downstream changes, including the activation of Nrf‐2. HSF‐1 is a transcriptional factor responsible, as is Nrf‐2, for the transcriptional activation of many stress proteins.
It is worth noting that p38 MAPK may also be, in part, responsible for the accumulation of GFAP, because in mice and in astrocyte culture knockout of p38 diminishes expression of GFAP 47. However, there appears to be a balance in the effects of cell stress pathways and GFAP accumulation. Activation of p38 in astrocytes promotes autophagy by negatively regulating mTOR activity, and thus may serve to decrease GFAP levels.
Oxidative stress
Astrocytes in AxD mice display markers of oxidative stress 20 and a transcriptional analysis reveals that a number of genes that respond to oxidative stress are upregulated 19. Similarly, a Drosophila model of AxD also shows significant oxidative stress 66. The cause of the oxidative stress is not completely clear. A recent report on giant axonal neuropathy (GAN), characterized by large accumulations of neurofilaments in neuronal cell bodies and axons, describes significant oxidative stress associated with mitochondrial dysmotility 23. A similar situation may occur in AxD, as the filament aggregates and RFs isolate areas of cytoplasm containing mitochondria and other organelles in the astrocyte cell body and processes 51. We have not directly measured mitochondria motility or mitochondrial function in these cells, however.
Endoplasmic reticulum (ER) stress in AxD astrocytes
Many astrocytes in AxD brains and in murine models of AxD display markers of ER stress (BIP/Grp78 and CHOP/GADD153) (Guilfoyle, Sosunov and Goldman unpublished data). ER stress activates an IRE1‐TRAF2‐ASK1‐JNK pathway 35, 63 as well as MAP kinases, including JNK and p38 MAP kinases 49. Simultaneously ER stress activates NF‐κB 13, 24. As noted above, the activation of these pathways may cause the appearance of a reactive astrocyte phenotype. What may be the cause of ER stress in AxD astrocytes needs further investigations.
Nitic oxide (NO) as a pathogenic factor in AxD
The Drosophila model of AxD is a most useful strategy to study basic mechanisms of the disease. Drosophila glia with the GFAP R49H mutation produce a lot of NO, toxic to neighboring neurons and astrocytes 66. The activation of NOS (nitric oxide synthase, enzyme responsible for production of NO; flies have only one isoform in contrast to the mammalian three) is a direct effect of mutant GFAP, although the molecular mechanisms are not determined. The iNOS pathway is activated in the KI and TG AxD mice 66. High concentrations of NO may generate reactive oxidant species that could result in oxidative stress and DNA damage in neurons and oligodendrocytes.
Activation of Nf‐kB
Transcript levels for Nf‐kB are elevated, as are protein levels and the Nf‐kB is found in astrocyte nuclei (Twardus, Sosunov and Goldman, unpublished observations). Upregulation of many inflammatory genes that are regulated by Nf‐kB is also observed 43. In AxD mice, two of the genes that are most increased are CXCL10 and CCL2 (43). These changes are part of an inflammatory environment that includes the activation of microglia and a modest influx of T cells into the CNS 43. Notably, several neuropathologists have described lymphocyte cuffs around blood vessels in AxD brains, as noted above. What the effects of T cells in the tissue might be is not known.
Loss of critical astrocyte functions
Astrocytes are responsible for the homeostasis of extraneuronal milieu, particularly the levels of K+ and glutamate below the thresholds of neuronal overexcitation and seizure initiation 44. Diminution of the major glutamate transporter (GLT‐1 in rodents, EAAT2 in humans), and the diminution of levels of Kir4.1 (40, 50, 60) occur. These losses profoundly affect the astrocyte's ability to buffer glutamate and potassium. The loss of glutamate buffering could lead to the seizures commonly seen in human AxD, and in the TGxKI AxD mouse 20, 50, 60. Furthermore, in the context of seizures, glutamate toxicity may play a role in the death of neuronal populations (hippocampal CA1 pyramids). In cell cultures, astrocytes that carry an AxD mutation do not prevent glutamate‐induced neuronal death at glutamate concentrations at which normal astrocytes preserve neurons 60. In the Drosophila, AxD model there is substantial neuronal loss via glutamate toxicity 65.
Simultaneously with excitatory abnormalities, inhibitory inputs may also be altered. Mouse AxD astrocytes reveal diminished levels of GAT2, a major transporter for GABA and GABA levels in astrocytes appear to increase (Olabarria, unpublished observations).
Changes in astrocyte shape
Changes in astrocyte shape are prominent in AxD, due in part to the cellular hypertrophy and the accumulation of GFAP and RFs, usually in cell bodies and in endfeet. In the TGxKI AxD mouse hippocampus, a profound reconstruction of morphology involves changes in the shapes and sizes of the main branches. Some astrocytes displayed unusual shapes of processes that were thickened and simultaneously shortened (normal astrocytes have tapered processes). The consistent feature of such astrocytes was an abundance of RFs that filled unusual truncated processes (Figure 6). Astrocytes also lose their fine distal processes, which normally enwrap synapses 50. These fine distal processes are responsible for astrocyte‐neuronal interactions and their disappearance presumably severely compromises neuronal function and may contribute to excitotoxic damage and seizures. These morphological changes are correlated with the new expression of CD44, which encodes a cell receptor that interacts with several extracellular matrix molecules, including hyuronan and osteopontin. Note that in mouse brain CD44 is normally found on subpial and white matter astrocytes, which do not elaborate fine distal processes. Protoplasmic astrocytes in the gray matter are devoid of CD44 (50).
Figure 6.
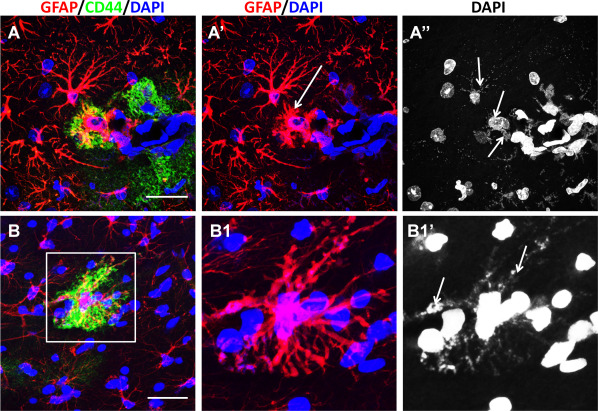
Aberrant forms of astrocytes with thick, short, “truncated” processes in AxD mice. A. An astrocyte with abnormal processes (arrow in A′) is located near an astrocyte with longer, more branched processes, A, B. These abnormal astrocytes show the presence of CD44. A″, B′. Abnormal astrocytes contain many RFs (arrows in A″, B1′). RFs are delineated with DAPI stain, as shown in 51. Confocal microscopy. Scale bars: 55 μm.
Astrocyte proliferation
In the normal CNS, there is little proliferation of astrocytes. Following damage, such as ischemia or trauma, astrocytes will enter cell cycle, judging by thymidine or BrdU incorporation or Ki67 labeling index, but such cycling does not last long. The mouse TGxKI model shows an increased Ki67 labeling index of astrocytes at 2 weeks of age, which then settles to a normal degree by 4 weeks 50. In infantile AxD in the severe white matter degeneration, one can find mitotic figures in astrocytes (Figure 1D). Furthermore, it is common to find bi‐ and multi‐nucleated astrocytes in AxD (Figure 1C), indicating that the cells have gone through S‐phase and split nuclei but are unable to undergo cytokinesis. Thus, many astrocytes in AxD appear to be polyploid. Astrocytes cultured from AxD model mice do not proliferate as much as those from their wild type littermates 8.
Astrocytes degeneration in AxD
Although several studies in culture have shown that cells expressing mutant GFAP or overexpressing normal GFAP have a decreased viability 7, 8, data on astrocytes damage in situ in AxD pathology are elusive. In AxD mice, there are cleaved caspase 3 immunopositive astrocytes with small, round nuclei that we considered as apoptotic changes (Figure 7A). Examination of the microglial reaction in AxD mice showed groups of microglial cells surrounding astrocytes that had lost their regular shape but could be recognized by the presence of RFs and GFAP (Figure 7B). In the electron microscope, “dark” astrocytes were a typical finding in AxD mice, albeit rare (Figure 7C, D). These cells contain many RFs, a high electron density of cytosol and nucleoplasm, but do not show typical features of apoptotic cells. Similar changes in neurons were described in an experimental model of Huntington's disease and were considered as an example of non‐apoptotic death 62.
Figure 7.
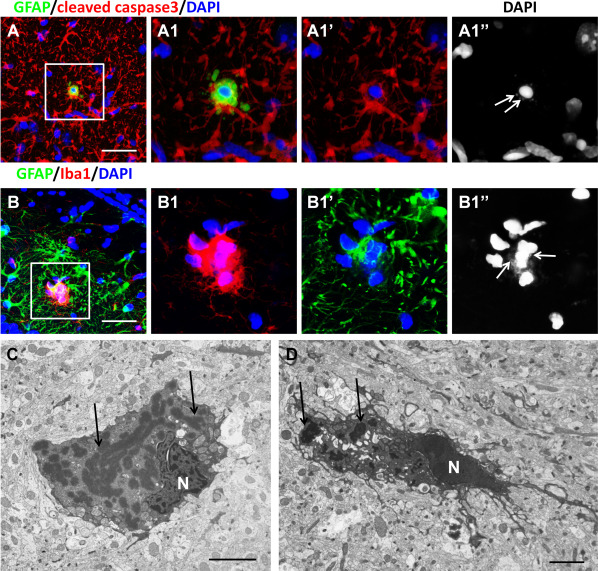
Degenerative features of astrocytes in TG AxD mice. A. Death of astrocyte with shrunken nucleus. RFs are marked with arrows in A1″. B. Activated microglial cells (Iba1+) encircle astrocyte (GFAP+). RFs are marked with arrows in B1″. Confocal microscopy. (C, D) “Dark” forms of astrocytes filled with RFs (arrows, indicating some RFs). Electron microscopy. N‐nucleus. Scale bars; 50 µm in (A, B); 3 μm in (C, D).
AxD astrocytes promote an inflammatory environment
Recent studies of the mouse models show that the AxD astrocytes promote significant inflammation in the CNS. Transcription analysis of AxD mice shows a significant upregulation of a number of inflammatory genes 19, 43. In fact, a gene ontology analysis showed that inflammatory or immune events are the most numerous ones and the cytokines and chemokines in particular represent a third of the total of immune‐related functions. CXCL10 and CCL2 are the most increased chemokines both at the transcript and protein levels, and both chemokines are localized in astrocytes, while CCL2 is also in microglia 43. Pro‐inflammatory cytokines such as TNFα, IL‐1, IL‐6 and IFNγ or anti‐inflammatory ones like IL‐4 and IL‐10 did not change in mouse AxD 43. It will be important to analyze cytokine and chemokine levels in human AxD, as mouse and human inflammatory reactions are not always similar.
There is also a dramatic activation of microglia in AxD, both in mouse models and in humans 43. Morphology, the microglial cells were enlarged, with a few ameboid cells 19, 43. However, we have found occasional microglia that appear to have phagocytosed astrocytes, judging by GFAP in the cytoplasm (Figure 7B).
How does astrocyte pathology lead to oligodendrocyte/myelin disease?
There are a number of possible mechanisms by which the astrocyte pathology in AxD could lead to oligodendrocyte and/or myelin pathology, thus producing a “leukodystrophic” phenotype.
Glutamate toxicity
Neuronal activity has a positive effect on OPC proliferation and differentiation 16, 17. Glutamate released by axons directly affects oligodendrocytes and OPCs through glutamate receptors and the synapses that glutamatergic neurons form with OPCs 25, 26. However, it is unclear to what extent glutamate promotes remyelination or contributes to excitotoxic demyelination 53. Both AMPA and NMDA receptors are found in OPCs but diminish in mature oligodendrocytes 12, 30. Glutamate can induce a Ca2+‐dependent damage/death of oligodendrocytes, not through NMDA receptors (as in neurons), but rather through H+‐gated, transient receptor potential channels that are similar to TRPA1 channels and allow massive Ca2+ and Mg2+ influx into oligodendrocytes 21. Activation of these channels finalizes several critical steps beginning with the release of glutamate and activation of axons that produces a release of K+. The increase in extracellular K+ with simultaneous K+ uptake by oligodendrocytes causes a rise in H+ concentration in the cells. This acidification opens TRPA1 channels and lets Ca2+ and Mg2+ into the cell with subsequent myelin damage 21. Pathological astrocytes may thus play an important role in oligodendrocyte damage if astrocytes do not support glutamate and K+ buffering. Astrocytes may even aggravate conditions because of a reverse functioning of their glutamate transporters that begin to release glutamate.
Conversely, astrocytes may help to protect oligodendrocytes in excitotoxicity by releasing transforming growth factor β (TGF‐β). This may not be the case with pathological astrocytes, however. The activation of metabotrophic glutamate receptor, mGluR4, rather typical for astrocytes, is a necessary step in the protective effect 52. It worth noting that our transcriptome analysis of AxD mice did not reveal upregulation of mGluR4.
The young AxD patients almost always experience seizures. Several studies have examined the influences of neuronal activity on OPCs and resulted in variable observations, ranging from proliferation to no effect; the differences may be explained by different types/intensity of seizures and different contexts of experiments 17, 32.
Hyaluronan inhibition of myelination
The deposition of the glycosaminoglycan hyaluronan in the extracellular space accompanies astrocyte gliosis of various etiologies and occurs in the AxD mice (Guilfoyle and Goldman, unpublished observations). Hyaluronan inhibits the maturation of OPCs 2. The significance of hyaluronan for myelination is well documented in another leukodystrophy, vanishing white matter disease, in which the deposition of hyaluronan directly correlates with an inhibition of oligodendrocyte maturation 6.
Lipocalin (LCN2) as a possible pathogenetic factor in AxD pathology
The expression of LCN2 is a characteristic feature of reactive astrocytes 54. Astrocytes in AxD and in mouse models of AxD express high levels of LCN2 (Guilfoyle and Goldman, manuscript in preparation). LCN2 belongs to the family of acute response genes and is involved in CNS immune responses. Thus, LCN2 is a powerful inducer of chemokines and causes an upregulation of CXCL10 through JAK2/STAT3 and IKK/NF‐κB pathways 31. The significance of increased levels of LCN2 for white matter damage of optic nerves was demonstrated with LCN2 KO mice, which have much more limited damage in experimental autoimmune optic neuritis 9.
Inflammation
Studies of inflammatory and demyelinating diseases are consistent with significant but highly complex effects of chemokines and cytokines on the severity of the pathology. The secretion of CXCL10 and CCL2 from astrocytes has significant effects on the progression of demyelinating diseases. For example, knocking out astrocyte CXCL10 delays the onset of EAE 39. Another study using the cuprizone demyelination model found that microglial activation by CXCL10 promoted oligodendrocyte survival and remyelination 11. An in vitro study showed that reactive astrocyte‐derived CXCL10 decreases human OPC differentiation, reflected by a reduced number of O4+ cells 42. Astrocyte‐conditional CCL2 knockout EAE mice showed decreased disease severity with reduced macrophage and T cell infiltration, less astrocyte and microglial activation and less axonal loss and demyelination 28. Note that these studies have been performed in mouse models, so it is not known how the interference of chemokine or cytokine activity could affect the course of human AxD.
Loss of astrocyte‐to‐astrocyte coupling
Astrocytes in the normal CNS are coupled by gap junctions, containing two of the members of the connexin family, Cx30 and Cx43. The junctions allow the passage of water and ions, particularly potassium and other small molecules, thus distributing these elements from an area of focal increase to widespread areas (spatial buffering) 3. The loss of both connexins, Cx43 and Cx30, in astrocytes of a mouse model leads to dysmyelination 33. These mice show edema within myelin sheaths and vacuolated oligodendrocytes. The cause of the dysmyelination is not clear, although Lutz et al 33 note that a loss of coupling prevents spatial buffering of water, ions and small molecules, and postulate that this could cause edema formation and subsequent damage to the myelin sheath. However, vacuolation of oligodendrocytes and intramyelinenic edema have not been noted in AxD.
Conclusions
Astrocytes in AxD acquire a large number of pathological features. These include the accumulation of GFAP with the formation of RFs, proteasome inhibition, NO and chemokine production and oxidative stress, activation of cell stress pathways, changes in morphology and the diminution of important astrocyte functions. Furthermore, AxD astrocytes promote a significant inflammatory environment in the CNS. A number of these changes are likely to be responsible for the white matter degeneration and the variable degrees of neuronal loss. Even in the absence of significant neuronal death, the clinical features of AxD, such as seizures and cognitive and motor problems, must reflect neuronal dysfunction. Our knowledge of AxD will illuminate how pathological astrocytes interact with other CNS cells in many other neurological disorders.
Acknowledgment
The study was funded in part by the Tuberous Sclerosis Alliance and NIH PO1NS042803.
Re‐use of this article is permitted in accordance with the Terms and Conditions set out at http://wileyonlinelibrary.com/onlineopen#OnlineOpen_Terms
References
- 1. Alexander WS (1949) Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 72:373–381. [DOI] [PubMed] [Google Scholar]
- 2. Back SA, Tuohy TMF, Chen H, Wallingford N, Craig A, Struve J et al (2005) Hyaluronan accumulates in de‐ myelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med 11:966–972. [DOI] [PubMed] [Google Scholar]
- 3. Bellot‐Saez A, Kékesi O, Morley JW, Buskila Y (2017) Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci Biobehav Rev 77:87–97. [DOI] [PubMed] [Google Scholar]
- 4. Borrett D, Becker LE (1985) Alexander's disease. A disease of astrocytes. Brain 108:367–385. [DOI] [PubMed] [Google Scholar]
- 5. Brenner M, Johnson AB, Boespflug‐Tanguy O, Rodriguez D, Goldman JE, Messing A (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 27:117–120. [DOI] [PubMed] [Google Scholar]
- 6. Bugiani M, Postma N, Polder E, Dieleman N, Scheffer PG, Sim FJ et al (2013) Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain 136:209–222. [DOI] [PubMed] [Google Scholar]
- 7. Chen YS, Lim SC, Chen MH, Quinlan RA, Perng MD (2011) Alexander disease causing mutations in the C‐terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability. Exp Cell Res 317:2252–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cho W, Messing A (2009) Properties of astrocytes cultured from GFAP over‐expressing and GFAP mutant mice. Exp Cell Res 315:1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chun BY, Kim JH, Nam Y, Huh MI, Han S, Suk K (2015) Pathological Involvement of Astrocyte‐Derived Lipocalin‐2 in the Demyelinating Optic Neuritis. Invest Ophthalmol Vis Sci 56:3691–3698. [DOI] [PubMed] [Google Scholar]
- 10. Crome L (1953) Megalencephaly associated with hyaline pan‐neuropathy. Brain 76:215–228. [DOI] [PubMed] [Google Scholar]
- 11. Clarner T, Janssen K, Nellessen L, Stangel M, Skripuletz T, Krauspe B et al (2015) CXCL10 triggers early microglial activation in the cuprizone model. J Immunol 194:3400–3413. [DOI] [PubMed] [Google Scholar]
- 12. De Biase LM, Nishiyama A, Bergles DE (2010) Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci 30:3600–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N et al (2004) Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24:10161–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding H, Wang X, Wang H, Zhu L, Wang Q, Jia Y et al (2017) Nrf2‐ARE signaling provides neuroprotection in traumatic brain injury via modulation of the ubiquitin proteasome system. Neurochem Int S0197–0186:30078–30075. [DOI] [PubMed] [Google Scholar]
- 15. Escourolle R, de Baecque C, Gray F, Baumann N, Hauw J‐J (1979) Etude en microscopie electronique et neurochimique d'un cas de maladie d'Alexander. Acta Neuropathol 45:133–140. [DOI] [PubMed] [Google Scholar]
- 16. Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I et al (2015) Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun 6:8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS et al (2014) Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goebel HH, Bode G, Caesar R, Kohlschütter A (1981) Bulbar palsy with Rosenthal fiber formation in the medulla of a 15‐year‐old girl. Localized form of Alexander's disease? Neuropediatrics 12:382–391. [DOI] [PubMed] [Google Scholar]
- 19. Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A (2005) Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet 14:2443–2458. [DOI] [PubMed] [Google Scholar]
- 20. Hagemann TL, Connor JX, Messing A (2006) Alexander disease‐associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci 26:11162–11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamilton NB, Kolodziejczyk K, Kougioumtzidou E, Attwell D (2016) Proton‐gated Ca(2+)‐permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 529:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heaven MR, Flint D, Randall SM, Sosunov AA, Wilson L, Barnes S et al (2016) Composition of rosenthal fibers, the protein aggregate hallmark of Alexander disease. J Proteome Res 115:2265–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Israeli E, Dryanovski DI, Schumacker PT, Chandel NS, Singer JD, Julien JP et al (2016) Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum Mol Genet 25:2143–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC (2003) Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF‐kappaB in response to diverse cellular stresses. Mol Cell Biol 23:5651–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Káradóttir R, Cavelier P, Bergersen LH, Attwell D (2005) NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438:1162–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Káradóttir R, Attwell D (2007) Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience 145:1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kästle M, Woschee E, Grune T (2012) Histone deacetylase 6 (HDAC6) plays a crucial role in p38MAPK‐dependent induction of heme oxygenase‐1 (HO‐1) in response to proteasome inhibition. Free Radic Biol Med 53:2092–2101. [DOI] [PubMed] [Google Scholar]
- 28. Kim RY, Hoffman AS, Itoh N, Ao Y, Spence R, Sofroniew MV, Voskuhl RR (2014) Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 274:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klein EA, Anzil AP (1994) Prominent white matter cavitation in an infant with Alexander's disease. Clin. Neuropathol 13:31–38. [PubMed] [Google Scholar]
- 30. Kukley M, Nishiyama A, Dietrich D (2010) The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci 30:8320–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee S, Kim JH, Kim JH, Seo JW, Han HS, Lee WH et al (2011) Lipocalin‐2 Is a chemokine inducer in the central nervous system: role of chemokine ligand 10 (CXCL10) in lipocalin‐2‐induced cell migration. J Biol Chem 286:43855–43870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Q, Brus‐Ramer M, Martin JH, McDonald JW (2010) Electrical stimulation of the medullary pyramid promotes proliferation and differentiation of oligodendrocyte progenitor cells in the corticospinal tract of the adult rat. Neurosci Lett 479:128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF (2009) Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci 29:7743–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maharjan S, Oku M, Tsuda M, Hoseki J, Sakai Y (2014) Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep 4:5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsuzawa A, Nishitoh H, Tobiume K, Takeda K, Ichijo H (2002) Physiological roles of ASK1‐mediated signal transduction in oxidative stress‐ and endoplasmic reticulum stress‐induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal 4:415–425. [DOI] [PubMed] [Google Scholar]
- 36. Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M (1998) Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 152:391–398. [PMC free article] [PubMed] [Google Scholar]
- 37. Middeldorp J, Kamphuis W, Sluijs JA, Achoui D, Leenaars CH, Feenstra MG et al (2009) Intermediate filament transcription in astrocytes is repressed by proteasome inhibition. FASEB J 23:2710–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mignot C, Desguerre I, Burglen L, Hertz‐Pannier L, Renaldo F, Gadisseux JF et al (2009) Tumor‐like enlargement of the optic chiasm in an infant with Alexander disease. Brain Dev 31:244–247. [DOI] [PubMed] [Google Scholar]
- 39. Mills Ko E, Ma JH, Guo F, Miers L, Lee E, Bannerman P et al (2014) Deletion of astroglial CXCL10 delays clinical onset but does not affect progressive axon loss in a murine autoimmune multiple sclerosis model. J Neuroinflammation 11:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Minkel HR, Anwer TZ, Arps KM, Brenner M, Olsen ML (2015) Elevated GFAP induces astrocyte dysfunction in caudal brain regions: a potential mechanism for hindbrain involved symptoms in type II Alexander disease. Glia 63:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moody LR, Barrett‐Wilt GA, Sussman MR, Messing A (2017) Glial fibrillary acidic protein exhibits altered turnover kinetics in a mouse model of Alexander disease. J Biol Chem 292:5814–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore CS, Cui QL, Warsi NM, Durafourt BA, Zorko N, Owen DR et al (2015) Direct and indirect effects of immune and central nervous system‐resident cells on human oligodendrocyte progenitor cell differentiation. J Immunol 194:761–772. [DOI] [PubMed] [Google Scholar]
- 43. Olabarria M, Putilina M, Riemer EC, Goldman JE (2015) Astrocyte pathology in Alexander disease causes a marked inflammatory environment. Acta Neuropathol 130:469–486. [DOI] [PubMed] [Google Scholar]
- 44. Parpura V, Verkhratsky A (2012) Homeostatic function of astrocytes: Ca(2+) and Na(+) signalling. Transl Neurosci 3:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E et al (2011) GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 77:1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ring S, Lackner H, Urban C, Brunner‐Krainz M, Plecko‐Startinig B (2010) Alexander disease: an important mimicker of focal brainstem glioma. Pediatr Blood Cancer 54:486. [DOI] [PubMed] [Google Scholar]
- 47. Roy Choudhury G, Ryou MG, Poteet E, Wen Y, He R, Sun F et al (2014) Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res 1551:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Russo LS Jr, Aron A, Anderson PJ (1976) Alexander's disease: a report and reappraisal. Neurology 26:607–614. [DOI] [PubMed] [Google Scholar]
- 49. Sekine Y, Takeda K, Ichijo H (2006) The ASK1‐MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med 6:87–97. [DOI] [PubMed] [Google Scholar]
- 50. Sosunov AA, Guilfoyle E, Wu X, McKhann GM II, Goldman JE (2013) Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in Alexander disease. J Neurosci 33:7439–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sosunov AA, McKhann GM II, Goldman JE (2017) The origin of Rosenthal fibers and their contributions to astrocyte pathology in Alexander disease. Acta Neuropathol Commun 5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Spampinato SF, Merlo S, Chisari M, Nicoletti F, Sortino MA (2015) Glial metabotropic glutamate receptor‐4 increases maturation and survival of oligodendrocytes. Front Cell Neurosci 8:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spitzer S, Volbracht K, Lundgaard I, Káradóttir RT (2016) Glutamate signalling: a multifaceted modulator of oligodendrocyte lineage cells in health and disease. Neuropharmacology 110:574–585. [DOI] [PubMed] [Google Scholar]
- 54. Suk K (2016) Lipocalin‐2 as a therapeutic target for brain injury: an astrocentric perspective. Prog Neurobiol 144:158–172. [DOI] [PubMed] [Google Scholar]
- 55. Sun DA, Yu H, Spooner J, Tatsas AD, Davis T, Abel TW et al (2008) Postmortem analysis following 71 months of deep brain stimulation of the subthalamic nucleus for Parkinson disease. J Neurosurg 109:325–329. [DOI] [PubMed] [Google Scholar]
- 56. Tanaka KF, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T et al (2007) Murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. Glia 55:617–631. [DOI] [PubMed] [Google Scholar]
- 57. Tang G, Xu Z, Goldman JE (2006) Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem 281:38634–38643. [DOI] [PubMed] [Google Scholar]
- 58. Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE (2010) Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein αB‐crystallin reverses the inhibition. J Biol Chem 285:10527–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tavasoli A, Armangue T, Ho CY, Whitehead M, Bornhorst M, Rhee J et al (2017) Alexander Disease. J Child Neurol 32:184–187. [DOI] [PubMed] [Google Scholar]
- 60. Tian R, Wu X, Hagemann TL, Sosunov AA, Messing A, McKhann GM et al (2010) Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol 69:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Towfighi J, Young R, Sassani J, Ramer J, Horoupian DS (1983) Alexander's disease: further light‐, and electron‐microscopic observations. Acta Neuropathol 61:36–42. [DOI] [PubMed] [Google Scholar]
- 62. Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW (2000) Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA 97:8093–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666. [DOI] [PubMed] [Google Scholar]
- 64. Van Poppel K, Broniscer A, Patay Z, Morris EB (2009) Alexander disease: an important mimicker of focal brainstem glioma. Pediatr Blood Cancer 53:1355–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang L, Colodner KJ, Feany MB (2011) Protein misfolding and oxidative stress promote glial‐mediated neurodegeneration in an Alexander disease model. J Neurosci 31:2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang L, Hagemann TL, Kalwa H, Michel T, Messing A, Feany MB (2015) Nitric oxide mediates glial‐induced neurodegeneration in Alexander disease. Nat Commun 6:8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2016) WHO Classification of Tumours of the Central Nervous System, 4th edn. Lyon, France: IARC. [Google Scholar]
- 68. Wohlwill FJ, Bernstein J, Yakovlev PI (1959) Dysmyelinogenic leukodystrophy; report of a case of a new, presumably familial type of leukodystrophy with megalobarencephaly. J Neuropathol Exp Neurol 18:359–383. [DOI] [PubMed] [Google Scholar]