

Abstract
Although the critical role of hypoxia inducible factor‐1α (HIF‐1α) in cerebral neovascularization after stroke has been well characterized, the details regarding the regulation of endothelial progenitor cell (EPC)‐dependent neovascularization by HIF‐1α are not completely understood. Using lentiviral shRNA to knockdown HIF‐1α, we showed that HIF‐1α plays a central role in bone marrow‐derived EPC (bmEPC) homing and sprouting in the post‐acute stage of ischemic Sprague Dawley (SD) rat brains. First, knockdown of HIF‐1α decreased the homing of both endogenous and exogenous bmEPCs to the ischemic brain. Additionally, the knockdown impaired the incorporation and sprouting of bmEPCs in the ischemic brain. In vitro, knockdown of HIF‐1α inhibited the spheroid sprouting and tube formation of bmEPCs. Mechanically, the HIF‐1α‐dependent recruitment of bmEPCs to the ischemic brain was relative to the CXCL12/CXCR4 axis and HMGB1, which were relative to astrocytes. In addition, the loss of HIF‐1α resulted in deficient expression levels of VEGF‐A, Flk‐1, NRP1, and Dll4 in the ischemic brains, bmEPCs, and astrocytes. These findings suggested that HIF‐1α implicates in bmEPC homing via CXCL12/CXCR4 and HMGB1 and that it promotes bmEPC sprouting via VEGF‐A/flk1‐NRP1/Dll4.
Keywords: cell homing, cerebral ischemia, endothelial progenitor cells, HIF‐1α, neovascularization, vascular sprouting
Introduction
Neovascularization promotes neural recovery from ischemic damage in the post‐acute stage of cerebral ischemia 30. Vasculogenesis is one of the most important patterns of neovascularization and depends on endothelial progenitor cells (EPCs) 16. This type of neovascularization is considered promising for cerebral ischemia treatment 6. EPCs comprise multiple different types of cells that are involved in neovascularization. Bone marrow is regarded as one of the most important sources of EPCs. Currently, there are no perfect markers for EPCs, but CD34, CD133, and flk1 are usually used to identify certain subpopulations of bone marrow‐derived EPCs (bmEPCs) 4, 19, 32. After being mobilized from the bone marrow to the peripheral blood in response to a stroke 7, bmEPCs are recruited to the ischemic brain, where CXCL12 (SDF‐1) and HMGB1 are highly expressed 19, 24 Sequentially, EPCs attach to the damaged vessels and repair the vessels, or they migrate out to initiate vasculogenesis 35. Sprouting angiogenesis is another crucial pattern of neovascularization, that is, guided by tip cells 12, 17. Tip cells are typically mobile and are located at the top of a vascular sprout 12, 17. Morphologically, tip cells are rich in filopodia, which help vascular tube formation 25. Endothelial cells (ECs) are the pool of tip cells, and the differentiation of ECs into tip cells is controlled by the VEGF‐A/flk1/Dll4 pathway 12, 17. In detail, when flk1‐positive ECs are stimulated by VEGF‐A, Dll4 levels are elevated in these cells 12, 17. As a result, the differentiation of these cells into tip cells is initiated 12, 17. In addition, NRP1, which is the co‐receptor of flk1, is required for tip cell formation 5, 13. Given these criteria, flk1, NRP1, and Dll4 are the suggested markers for tip cells. Notably, there might be a potential relationship between EPCs and tip cells, as both types of cells share the mutual marker flk1 12, 17, 19, 34.
Hypoxia inducible factor‐1 (HIF‐1) is a transcription factor that regulates neovascularization. HIF‐1α is the α subunit of HIF‐1 and serves as a hypoxia sensor that controls the transcription activity of HIF‐1 20. Under hypoxia, the ubiquitination of HIF‐1α is inhibited due to the inactivity of prolyl hydroxylase domain (PHD) enzyme, resulting in the activation of HIF‐1 20. Indeed, overactivated HIF‐1 damages the blood–brain barrier (BBB) in the acute stage of cerebral ischemia 8, 26. However, its promotion of neovascularization, as well as of neurogenesis, benefits neural recovery from ischemic damage 10, 23. However, how it functions in bmEPC homing and vascular sprouting is still unclear.
In the present study, we speculated that HIF‐1 plays an essential role in bmEPCs‐dependent post‐ischemic cerebral neovascularization in terms of bmEPC homing and sprouting. To test this speculation, we generated middle cerebral artery occlusion (MCAO) rat models via electrocoagulation and knocked down cerebral HIF‐1α using lentiviral shRNA. As CXCL12/CXCR4 and HMGB1 are crucial for EPC homing 19, 23, we tested if the deficient expression of HIF‐1α affects these pivotal factors in brains and in bmEPCs. In terms of sprouting, we investigated the role of HIF‐1α in sprouting angiogenesis and in the VEGF‐A/flk1‐NRP1/Dll4 axis in vivo and in vitro.
Methods
Antibodies and reagents
The commercial antibodies used for the immunofluorescence staining were as follows: CD34 (1:200, Abcam, England), CD133 (20 μg/mL, ThermoFisher, US), flk1 (5 μg/mL, Abcam, England), vWF (1:50, Santa Cruz Biotechnology), CD31 (1:50, Abcam), pERK (1:200, CST, US), BrdU (1:250, Abcam), Dll4 (1:200, Santa Cruz Biotechnology), HMGB1 (1:500; Abcam), GFAP (1:200, CST), CD105 (1:400, Abcam), and NRP1 (1:300, Abcam). For the immunohistochemistry staining, HIF‐1α (1:50, Novus, US) was used. For the western blotting, the following were used: HIF‐1α (1:500, Novus NB100‐105), pERK (1:2000, CST 4370), ERK (1:500, Abcam ab196883), HMGB1 (1:1000; Abcam ab79823), CXCR4 (1:100 Abcam ab124824), CXCL12 (0.1 μg/mL, Abcam ab9797), and beta‐actin, Dll4 (1:500, Santa Cruz Biotechnology sc18641). The commercial reagents DMEM/F12 and fetal bovine serum (FBS) were purchased from Gibco (US). The complete growth factor and basal EGM‐2 medium were purchased from Lonza (US).
Lentiviral shRNA constructs
The short hairpin RNAs (shRNA), targeting rat HIF‐1α mRNA, were designed and constructed by a company (GenePharm). A scrambled non‐silencing (NS) shRNA that does not degrade any known mammalian mRNA was designed as the control shRNA. The sequences were as follows: HIF‐1α‐rat‐1893 (5′‐GATCCGCAGTGACGAAGGACAATATATTCAAGAGATATATTGTCCTTCGTCACTGCTTTTTT‐3′), and NS shRNA (5′GATCCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACG‐3′). The shRNA sequences were integrated into the pGPU6/GFP/Neo vector. The plasmids were packaged into 293T cells using the ViraPowerTM Lentiviral Expression Systems (Invitrogen), according to the manufacturer's protocol. After an incubation for 48 h, the viral supernatants were harvested and ultracentrifuged (100 000 × g for 3 h at 4°C) for concentration. Viral titers were determined by a fluorescence‐activated cell sorting (FACS) analysis. Lentiviruses expressing the HIF‐1α shRNA and NS shRNA were generated, yielding LVshHIF‐1α and LVshNS, respectively (no less than 5 × 108 transducing units).
Animals and animal groups
Two‐month‐old male Sprague–Dawley (SD) rats, 200–250 g, were purchased from the Experimental Animal Center, Chongqing Research Institute of Chinese Medicine (Chongqing, China) and were housed in controlled conditions (22 ± 2°C, 12 h light/dark cycle, free access to food and water) in the Experimental Animal Center, the College of Pharmaceutical Sciences, Southwest University (Chongqing, China). The animal protocols were approved by the Southwest University Animal Use and Care Committee (animal approval number is SYXK (Yu) 2014‐0002). All animal experiments obeyed the ARRIVE guidelines and were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978). All mice used in the animal experiments were randomized. The operator or investigator was blinded for the respective animal treatments throughout the study. Evaluation of all read‐out parameters were done independently and in a blinded fashion.
To test the knockdown efficiency of HIF‐1α in the ischemic brain via lentiviral shRNA, the SD rats were divided into different groups using a random number table. The experimental groups included the following: the sham group (n = 9), MCAO group (n = 9), MCAO+control shRNA group (n = 18) and MCAO+HIF‐1α shRNA group (n = 18). At each time point, 3 rats in each group were sacrificed to check the HIF‐1α knockdown efficiency by western blot. To confirm the knockdown efficiency, 3 rats in the MCAO+control shRNA group and the MCAO+HIF‐1α shRNA group were sacrificed for immunohistochemistry Table S1). To observe the effect of deficient HIF‐1α on the biological process of neovascularization in the post‐ischemic brain, 80 MCAO rats in total in the MCAO+control shRNA group (n = 40) and the MCAO+HIF‐1α shRNA group (n = 40) were used. Among the 40 rats in each group, 7 rats were used to observe several parameters of neural recovery, including mNSS (modified neurological severity scores), rCBF, and infarct volume. The rest of the rats were used to perform immunohistochemistry (n = 5–7 for each index) and immunoblotting (n = 3) at different time points Table S2). To study the influence of deficient HIF‐1α on exogenous bmEPC homing to ischemic brains, the MCAO rats that received control or HIF‐1α shRNA were transplanted with primary bmEPCs in which the HIF‐1α was either knocked down or not (n = 10–12). At different time points, 5–6 rats from each group were sacrificed for immunohistochemistry Table S3).
To test if the MCAO model was successful and if a bias was induced by animal allocation, the mNSS and rCBF values were examined after the MCAO surgery. The statistical analyses showed no significant differences between the groups using MCAO rats in terms of mNSS scores or rCBF (Figure S1–S3).
Lentiviral shRNA transfecting rat brains
HIF‐1α‐rat‐1893 was selected for the in vivo transfection. For the in vivo transfection, the vector used to carry the target plasmid was without GFP. One to two days prior to the surgery, the rats received an intracerebroventricular injection of 3 μL of the lentiviral particles once a day. The location coordinates of the right lateral ventricle were 0.8 mm caudal to the bregma, 1.1 mm to the right of the midline, and 4.5‐mm ventral to the skull surface. Rats were sacrificed to check the expression of HIF‐1α via immunohistochemistry and immunoblotting on the 3rd, 5th, and 14th days after the surgery. Brain sections were prepared, each with a 4‐μm thickness. The specific steps for the immunohistochemistry and immunoblotting followed a previously reported protocol 32.
Rat focal cerebral ischemia models, neurological deficiency, and rCBF
Male Sprague–Dawley rats, 200–250 g, were used to establish the MCAO models via electrocoagulation according to a reported protocol 15, 22. Briefly, an incision was made above the right eye socket after each rat was anesthetized with 1% isoflurane. The temporalis was then isolated from the harnpan to expose the temporal fossa, and a small hole was made in the temporal fossa. The hole was extended until the middle cerebral artery was revealed. Then, the artery was blocked by a bipolar coagulator.
The rCBF and modified neurological severity scores (mNSSs) were used to screen for successful MCAO rats. The mNSS scores were measured as previously reported 9. The rCBF in the ischemic core region (2‐mm caudal to bregma and 6‐mm lateral to midline) and in the peripheral region (2‐mm caudal to bregma and 3‐mm lateral to midline) was detected via laser‐Doppler flowmetry (LDF) (ML191, Australia ADInstruments) 30 min before and after the middle cerebral artery was occluded 18. The rCBF in the core region 2–3 h prior to the MCAO surgery was regarded as the baseline for quantifying the decrease in rCBF in the core region after MCAO surgery. In the same way, the rCBF in the peripheral region 2–3 h prior to the MCAO surgery was regarded as the baseline for evaluating the rCBF recovery. The MCAO was considered successful in the rats when the CBF in the ischemic core was reduced by more than 80%, and the mNSS score was between 3 and 8.
Vessel density and vascular tube observations in vivo
On the 14th day after the MCAO surgery, the brains were isolated for sectioning. Brain sections (20 μm) were prepared and fixed for measuring the vessel density via CD31. On the 7th day after the MCAO surgery, 30‐μm brain sections were prepared for observing the vascular tubes via vWF. The vessel density was quantified by counting the isolated CD31‐positive cells and capillaries in the AOIs under a 20‐fold magnification. All samples were blinded and manually counted. The immunofluorescent staining protocol is described in the immunofluorescence staining section.
Tissue immunofluorescence staining
On the seventh day after the MCAO surgery, the brains were removed for immunofluorescence staining. Brain sections (20 μm) were prepared and fixed for the immunofluorescence staining. Tip cells, homing endogenous or exogenous EPCs, incorporated endogenous or exogenous EPCs, Dll4‐expressing EPCs, and reactive astrocytes were manually counted in the AOIs under a 20‐fold magnification. HMGB1, pERK, and VEGF‐A expression levels were analyzed by averaging the gray values in the AOIs under a 20‐fold magnification.
The brain sections were washed with 1 × PBS (3 × 10 minutes) and blocked in a blocking solution (0.3% Triton X‐100 in 1× PBS + 10% serum, which was generated from the species of the secondary antibodies) at room temperature for 1 h. The sections were then incubated in the primary antibody solution at 4°C for 48–72 h. After washing with 1× PBS (3 × 10 minutes), the sections were incubated in a fluorescent secondary antibody solution at 4°C overnight. After washing with 1× PBS, the brain sections were covered with an antifade mounting medium for capture.
Western blot analysis
The proteins in the tissue samples were collected according to the instructions of the manufacturer. The NS‐shRNA‐treated and HIF‐1α‐shRNA‐treated bmEPCs and astrocytes were incubated in hypoxic conditions (2% O2, 5% O2, and 10% O2) for 12 h, and then, the proteins were harvested for western blotting. Notably, all processes were performed on ice. The western blots were performed on SDS‐PAGE gels as previously reported 32. Samples from at least three independent experiments were harvested and analyzed in each group.
BrdU labeling and intra‐arterial delivery of EPCs
EPCs were labeled with BrdU (30 μg/mL; Sigma‐Aldrich) in vitro for 3 days before the delivery. In total, 24 qualified MCAO rats were randomly divided into the control snRNA group and the HIF‐1α shRNA group. Before receiving any shRNA, the MCAO rats in the two groups showed no significant differences in terms of either mNSS scores or rCBF. The bmEPC transplantation was carried out immediately after the occlusion surgery or 24 h after the surgery via an external carotid artery, at a dose of 2 × 106/800 μL. Each time, the intra‐arterial (IA) injection was performed for a duration longer than 5 minutes via a Hamilton syringe. Once the injection was performed, the external carotid artery was immediately ligated. On the 7th day of occlusion, 6–7 brains from each group were removed. Brain sections (20 μm) were prepared and fixed for the immunofluorescence staining. The enrichment of BrdU‐labeled exogenous bmEPCs was quantified by counting BrdU‐positive cells. On the 10th day of occlusion, the incorporation of exogenous bmEPCs was evaluated by counting the BrdU‐positive bmEPCs incorporated into the local vessel. Sprouting exogenous bmEPCs were quantified by counting BrdU/Dll4‐dual‐positive cells. In total, 24–28 AOIs under a 20‐fold magnification were analyzed with those indexes. All samples were blinded when manually analyzed.
Primary cell culture and immunophenotyping
Primary astrocytes were isolated from the cortex of 3‐day‐old rats, as previously report 31. The primary astrocytes were cultured in DMEM/F12 containing 15% FBS. The culture media were changed every other day. After culturing for 7–10 days, astrocytes were identified using GFAP.
Bone marrows from the tibias and femurs of 11 to 12‐week‐old Sprague–Dawley rats were used to isolate bmEPCs. The bone marrow cells were collected using PBS to wash the marrow cavity. Single‐cell suspensions were prepared by tipping the collected bone marrow cells up and down. Mononuclear cells (MNCs) were obtained using density‐gradient centrifugation with HistoPaque (Sigma). After being washed softly, the MNCs were resuspended in complete growth factor media EGM‐2 and were cultured in a 5% CO2 incubator at 37°C. After 14–21 days, lectin binding in the cells was detected with FITC‐labeled Ulex europaeus agglutinin (UEA)‐1, and the expression levels of CD133 and flk1 were identified using a flow cytometer. Furthermore, the phagocytotic activity of EPCs was identified using DiI‐ac‐LDL.
Transfecting primary bmEPCs
For the cell transfections, 1 mL of the unconcentrated lentiviral supernatants and 1 mL of the complete medium with 6 μg/μL of Polybrene were gently added to the cells. After 24 h of incubation, the culture media was changed with fresh complete media for another 48 h, and then, the GFP expression in the transduced cells was evaluated under a fluorescence microscope. The control and HIF‐1α shRNA‐treated bmEPCs were cultured under hypoxic conditions for 4 h and then harvested for western blotting to assess HIF‐1α expression.
Vascular sprouting in vitro
The in vitro vascular sprouting of bmEPCs assessment included tube formations on Matrigel and sphere sprouting in Matrigel under 2% O2. The cells were pretreated with NS shRNA or HIF‐1α shRNA. Matrigel (10 μL) was added carefully to the bottom of the wells of μ‐slide angiogenesis (Ibidi). Then, the bmEPCs (1 × 104) were seeded on Matrigel and cultured under hypoxic conditions for h to observe the endothelial contacts and tube formations. The number of spheroid sprouts was estimated as described in a previous report 27.
Image acquisition and analysis
The brain sections were captured under a 20‐fold magnification using a microscope equipped with a PL FLUOTAR 20x/0.4 objective and a Leica DFC310 FX Digital camera. Three‐dimensional reconstructions of confocal z‐stacks were generated by a confocal laser‐scanning microscope (LSM 710, Carl Zeiss) utilizing a 63***x objective (0.3‐μm step size) and assembled in the Zen software. All parameters, such as exposure time, gain, brightness, and contrast, of each image were maintained the same for each fluorescence channel. The cells or averaged gray values in the AOIs (areas of interest) were analyzed using ImageJ and Leica Microsystems LAS AF (AF6000 Modular Systems).
Statistical analysis
Statistical differences between two groups were analyzed with two‐tailed, unpaired Student's t‐tests. ANOVA combined with Holm–Sidak or Tukey's multiple comparison test was used when data from multiple groups were analyzed. P‐values < 0.05 were considered statistically significant.
Results
Lentiviral HIF‐1α shRNA decreased HIF‐1α expression in the ischemic rat brains
HIF‐1 is crucial for post‐ischemic neovascularization in the brain. Once ischemia occurs, HIF‐1α is up‐regulated and maintained in the ischemic area 10. To investigate the role of HIF‐1α in recovery from cerebral ischemia, the expression of HIF‐1α in an ischemic brain was inhibited by lentiviral shRNA. Two days prior to MCAO surgery, rats were administered control shRNA or HIF‐1α shRNA treatment twice (once per day). The MCAO surgery was performed using electrocoagulation as previously reported 15, 22. After the surgery, only MCAO rats exhibiting an rCBF reduction greater than 80% in the ischemic core and mNSS test scores of 3–8 were considered successful cerebral ischemia models (Figure S1–S3). In the whole study, 5 out of 71 of the control shRNA rats and 8 out of 77 of the MCAO rats in the HIF‐1α shRNA group died Table S4). Compared to the sham animals, the MCAO rats presented higher levels of HIF‐1α and maintained these levels until day 14, confirming that HIF‐1α is increased in ischemic brains (Figure S4A,B). Compared to the MCAO group, the control group, who received control shRNA, showed no differences in the levels of HIF‐1α throughout the whole observation, while the MCAO rats in the HIF‐1α shRNA group exhibited lower levels of HIF‐1α in the brain from day 5 to day 14, indicating the inhibition of HIF‐1α in the ischemic brains by the lentiviral HIF‐1α shRNA (Figure S4A,B). Further, immunohistochemistry was used to confirm the knockdown efficiency. In line with the immunoblot data, the MCAO brains presented more HIF‐1α‐positive cells than the sham group (Figure S4C–E), and the HIF‐1α shRNA group presented less HIF‐1α‐positive cells than the control group from day 5 to day 14 (Figure S4D,E). In the meantime, the efficiency of the knockdown was identified in the areas of the ipsilateral cortex, the infarct, and the SVZ (Figure S4F), wherein the neovascularization was investigated in the following studies.
Knockdown of HIF‐1α impaired the recovery of cerebral ischemia
In the acute stage of cerebral ischemia, HIF‐1 increases the risk of hemorrhage due to its damage to the BBB 8. Later, however, HIF‐1 promotes neural recovery from ischemia by promoting angiogenesis and neurogenesis 10. To confirm the beneficial role of HIF‐1 in neural recovery, we examined the neurological deficiency, infarct volume, and rCBF of MCAO rats with treatment of HIF‐1α shRNA or control shRNA (Figure 1A). The mNSS scores of the control group were decreased from 5.59 ± 1.46 (Figure S1A) to 3.14 ± 1.069 (Figure 1B), from day 1 to day 14. The decreased mNSS scores indicated a self‐recovery of neurological function in the ischemic brains. Meanwhile, the scores decreased from 5.73 ± 1.53 to 4.43 ± 0.98 in the HIF‐1α shRNA group, which was higher than that of the control group, indicating that self‐recovery was impaired when HIF‐1α was inhibited (Figure 1B). Consistently, the infarct volumes in the HIF‐1α shRNA group were larger than those in the control shRNA group (23.09 ± 4.13 vs. 16.53 ± 5.20, Figure 1C,D). The infarct areas in both groups were relative to the motor cortex, the corpus callosum, and the striatum, where the blood is supplied by the MCA (Figure 1C). Of note, the infarct volumes in those regions in the HIF‐1α shRNA group were much larger than those in the control group (Figure 1C). Simultaneously, the rCBF in the peripheral ischemic core in the HIF‐1α shRNA group was much lower than that in the control group (26% ± 9.42% vs. 53.71% ± 19.57%, Figure 1E). Collectively, these data highly suggested that HIF‐1α is beneficial for neural recovery, particularly for vessel recovery.
Figure 1.
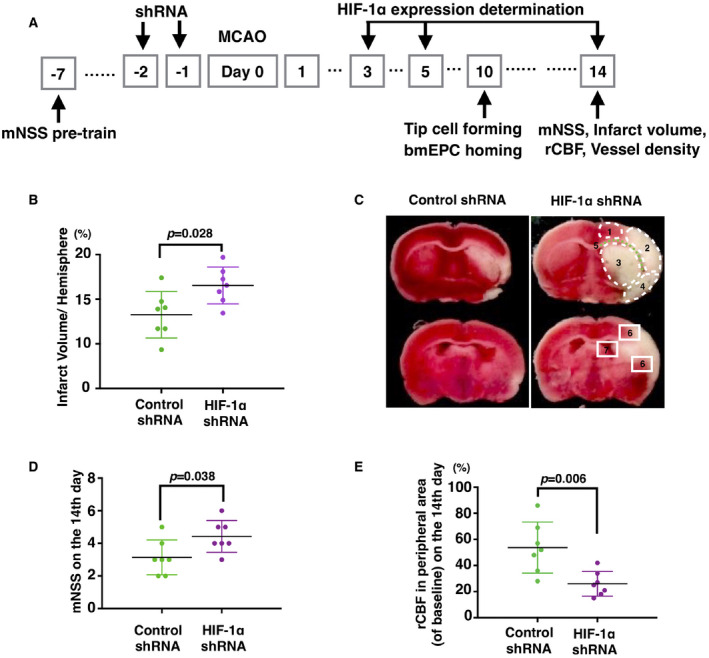
Knockdown of HIF‐1α in post‐acute ischemic brains impaired the recovery of neural deficiency. A. Shows the scheme of the present research. B. Indicates that on the 14th day of MCAO, the infarct volumes in the HIF‐1α shRNA‐treated rats were much larger than those in the control shRNA rats (n = 7). C shows representative images of a cerebral infarct. On day 14, the infarct tissues were related to the following: 1 primary motor cortex, 2 somatosensory cortex, 3 striatum, 4 insular‐piriform cortex, and 5 corpus callosum. According to these damaged areas, the neovascularization focused on ROIs, which were areas 6 (cortex) and 7 (SVZ). D and E show an increase in mNSS scores and a decrease in rCBF in the MCAO rats that were given HIF‐1α shRNA (n = 7). The initial value of rCBF in the peripheral region (2‐mm caudal to bregma and 3‐mm lateral to midline) of the brain before MCAO surgery was regarded as the baseline. Data are presented as the mean ± S.D.
Knockdown of HIF‐1α impaired post‐ischemic neovascularization in rat brains
As HIF‐1 promotes neovascularization under hypoxia, we carefully investigated the effect of HIF‐1 on the neovascularization in the ischemic rat brains. As shown in Figure 2A, a new cerebrovascular architecture was reconstructed around the ischemic areas in the control group (noted by the white arrow). However, the vascular reconstruction was impaired greatly in the HIF‐1α shRNA group. Since the cortex, the corpus callosum, and the striatum were typically damaged by the MCAO surgery (Figure 1C), we evaluated the vessel densities in the cortex and SVZ by CD31 staining. In the cortex, the HIF‐1α shRNA group showed a much lower vessel density than the control group (Figure 2B–D). Simultaneously, a similar result was observed in the SVZ (Figure 2B–D). To further confirm the impaired angiogenesis upon the deduction of HIF‐1α, we did dual immunostaining of CD105 and CD31 to marker the active microvessels in the ischemic area of cerebral cortex 29. Compare to control shRNA, HIF‐1α shRNA group presented much less CD105 positive vessels (Figure S5A,B), indicating an impairment in active angiogenesis upon the decreased HIF‐1α. Next, we observed the vascular branches in the regions where the neovascularization occurred. Consistent with the vessel density findings, the HIF‐1α shRNA group presented many fewer vascular tubes with diameters smaller than 20 μm than the control shRNA group did (Figure S5C,D), suggesting that HIF‐1α might play a critical role in vascular branching.
Figure 2.
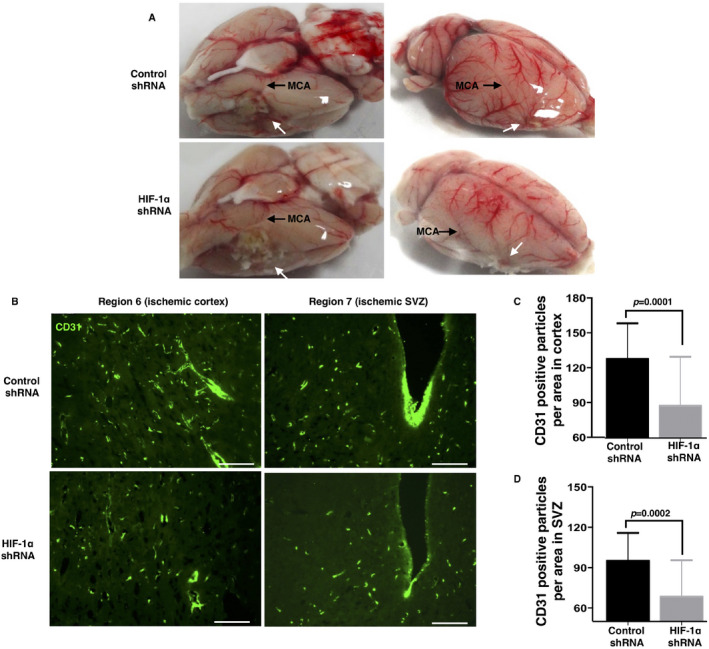
Knockdown of HIF‐1α resulted in an impaired neovascularization in the peripheral area of the ischemic core. Fourteen days after the occlusion, the brains were removed to observe the infarct volume and vessel density. Panel A. shows the impaired reconstruction of the cerebrovascular architecture in the HIF‐1α‐shRNA1‐treated MCAO rats. B. Thirty‐micrometer brain sections were used to observe the vessel density by labeling CD31. Single CD31 particles were counted in the AOIs under a 20‐fold magnification. The results show a significantly reduced vessel density in both the studied areas in the HIF‐1α‐shRNA1‐treated MCAO rats. C and D are the statistical analyses of the vessel densities in the regions of interest, indicating a significant reduction in the vessel density in the MCAO HIF‐1α shRNA brains (n = 29–32 fields from 5 to 6 brains per group). Bar, 200 μm. Data are presented as the mean ± S.D.
Knockdown of HIF‐1α inhibited bmEPC homing in ischemic rat brains
Given that bmEPCs are recruited to ischemic brains 7, 33, we inquired as to whether the inhibition of HIF‐1α in the ischemic brains affects this recruitment. We used flk1/CD34 and flk1/CD133 to label the endogenous bmEPCs in ischemic brains 8. On the seventh day after the occlusion, flk1/CD34‐ or flk1/CD133‐positive cells were observed in the control group. However, these homing endogenous bmEPCs were obviously decreased in the HIF‐1α shRNA group (Figure 3A–D). Simultaneously, the incorporation of flk1/CD34‐positive bmEPCs into the local vessels was studied using the three‐dimensional reconstructions of the confocal z‐stacks. As previously reported, the flk1/CD34‐positive bmEPCs incorporated into native vessels in the control group (Figure 3A), and this incorporation was distinguished from that of pericytes using the triple‐immunostaining of CD34/flk1/α‐SMA (Figure 3E). These incorporations, however, were absent in the HIF‐1α shRNA group (Figure 3A).
Figure 3.
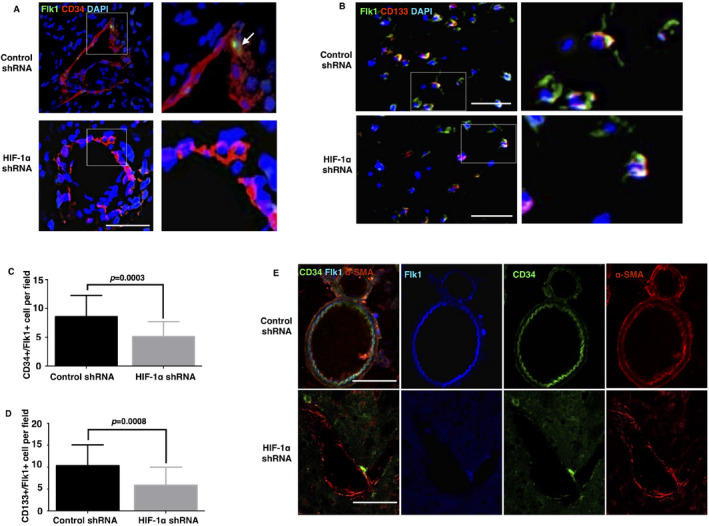
Knockdown of HIF‐1α impaired the homing and incorporation of endogenous bmEPCs in the ischemic brains. On the 7th day after the MCAO surgery, the ischemic brains were isolated. Sections were prepared with thicknesses of 20 μm and 40 μm for immunostaining. The regions of interest were the areas peripheral to the infarct (2‐mm caudal to bregma and 3‐mm lateral to midline). Homing bmEPCs marked by flk1/CD34 (A) or CD133/flk1 (B) were counted in each area at a 20‐fold magnification. Bar, 50 μm. A. The incorporations of homing EPCs are shown by the three‐dimensional reconstructions of the confocal z‐stacks. C and D are the columns of pictures A and B, respectively (n = 21–25 fields from 5–6 brains per group). E. Confocal images of triple‐immunostaining with CD34/flk1/α‐SMA were used to identify the bmEPCs and pericytes in the ischemic area. Data are presented as the mean ± S.D.
To further confirm that HIF‐1α is crucial for the recruitment of bmEPCs to ischemic brains, we implanted bmEPCs (with HIF‐1α shRNA or control shRNA) into the ischemic brains (with HIF‐1α shRNA or control shRNA) and traced them using BrdU (Figure 4A). Primary bmEPCs were isolated from bone marrow and expanded with EGM‐2 medium for 14–21 days (Figure S6A). The bmEPCs were identified by the phagocytotic activity and the binding ability of lectin to UEA‐1 (Figure S6B). A flow cytometry assay indicated a high ratio of CD34/flk1‐positive cells in the cultured bone marrow‐derived cells, approximately 80% (Figure S6C), confirming the identity of the bmEPCs. The bmEPC injections were carried out twice on the MCAO rats via the external carotid artery. The first injection was given immediately after the occlusion, and the second injection was 24 h later. We found that several of the MCAO rats that were given the bmEPC injections died Table S5), potentially due to an embolism and anesthesia. Forty‐eight hours after the last injection, the accumulations of BrdU‐positive bmEPCs in the ischemic brains were detected via BrdU staining. In the control group, in which the brains and bmEPCs were treated with control shRNA, BrdU‐positive cells were abundant in the ischemic brains on the 3rd day of occlusion (Figure 4B,C). The number of these cells, however, was significantly reduced when HIF‐1α was knocked down either from the ischemic brains or from transplanted bmEPCs (Figure 4B,C), and they were further reduced when HIF‐1α was inhibited in both brains and bmEPCs, suggesting that HIF‐1α is crucial for exogenous bmEPC homing.
Figure 4.
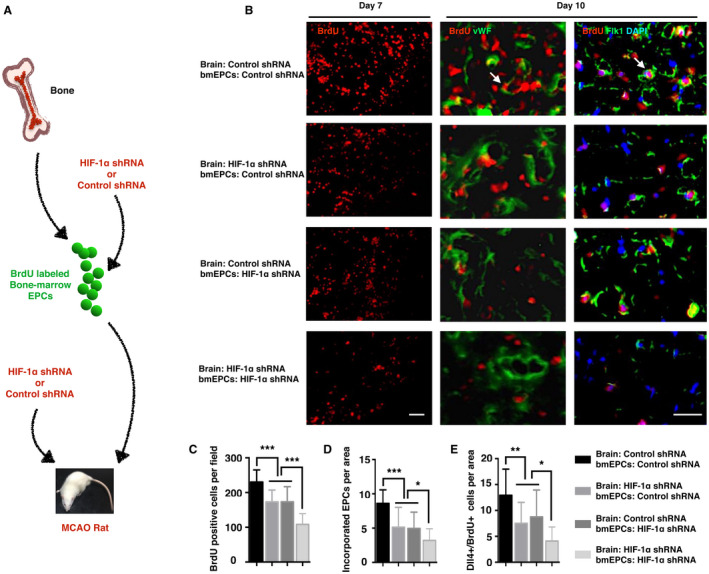
Knockdown of HIF‐1α impaired the homing, incorporation, and sprouting of transplanted bmEPCs in the ischemic brains. A. is the scheme of the experiment. bmEPCs were isolated from the bone marrow and expanded in vitro for 14 days. BrdU‐labeled bmEPCs were treated with control shRNA or HIF‐1α shRNA and injected into the MCAO rat brains, which were given the administration of control shRNA or HIF‐1α shRNA. B. Seven days after the MCAO surgery, the brains were removed for BrdU immunostaining. Ten days after the occlusion, the brains were isolated to observe the incorporation and the Dll4 expression levels of the exogenous bmEPCs. White arrows indicate the incorporated or Dll4‐positive bmEPCs. C–E are the statistical analyses of (B) (n = 22–29 fields from 5‐6 brains per group). Bar, 50 μm. Data are presented as the mean ± S.D.
Knockdown of HIF‐1α reduced the number of vascular tip cells in ischemic rat brains
Since we observed a decrease in vascular branches in the HIF‐1α shRNA group Figure S5C,D), we wondered if knockdown of HIF‐1α impairs the event of vascular sprouting. Vascular ECs, those that are Dll4‐positive, are widely considered tip cells, which initiate the event of vascular sprouting 12, 17. Therefore, we investigated rat brain cross sections using Dll4/vWF co‐immunofluorescence. The HIF‐1α shRNA group showed many fewer Dll4/vWF‐positive cells in the brain than the control group on the seventh day after the occlusion (Figure 5A, 5C). Flk1, also known as VEGFR2, is a suggested marker for tip cells since it induces a high expression of Dll4 in the candidate tip cells with the stimulation of VEGF‐A 12, 17. Therefore, we used flk1 and Dll4 to identify tip cells in the ischemic brains. Consistent with the Dll4/vWF immunostaining, the HIF‐1α shRNA group presented many fewer flk1/Dll4‐positive cells than the control group (Figure 5B,D). In addition to Dll4 and flk1, NRP1, the co‐receptor of flk1, plays a critically positive role in vascular sprouting 5, 13. Given this, we inferred that the knockdown of HIF‐1α might affect NRP1/Dll4‐positive cells. As expected, the number of NRP1/Dll4‐positive cells was significantly reduced in the HIF‐1α shRNA group (Figure 5E,F). Altogether, these data suggested that deleting HIF‐1α expression during the post‐acute stage of cerebral ischemia impairs the formation of tip cells.
Figure 5.
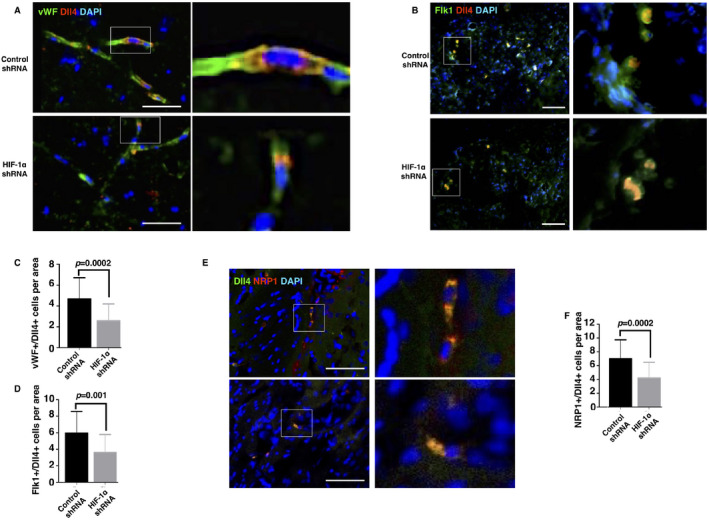
Knockdown of HIF‐1α reduced the number of tip cells in the ischemic brains. On the 7th day after the MCAO surgery, the brains were removed. Sections were prepared with a thickness of 30 μm and fixed. The regions of interest were the areas peripheral to the infarct (2 mm caudal to bregma and 3 mm lateral to midline). Tip cells were labeled with vWF/Dll4 (A), flk1/Dll4 (B), and NRP1/Dll4 (E). Dual‐positive cells were counted in each area at a 20‐fold magnification. C, D, and F are the statistical analyses for pictures A, B, and E, respectively (n = 24–28 fields from 5 to 6 brains per group). Bar, 60 μm. Data are presented as the mean ± S.D.
Knockdown of HIF‐1α decreased the expression of CXCR4 in bmEPCs and astrocytic CXCL12 and HMGB1
Studies have indicated that activated astrocytes recruit bmEPCs by secreting HMGB1 and CXCL12, the ligands of RAGE and CXCR4, respectively, both of which are expressed on the membranes of EPCs 11, 19. We speculated that HIF‐1α might regulate bmEPC homing via these signals. We first checked astrocytes using GFAP to label reactive astrocytes. The immunostaining images indicated that inhibiting HIF‐1α significantly reduced the GFAP‐positive cells in the ischemic brains (Figure 6A,B), suggesting that HIF‐1α is required for maintaining the appropriate reactive astrocytes in ischemic brains.
Figure 6.
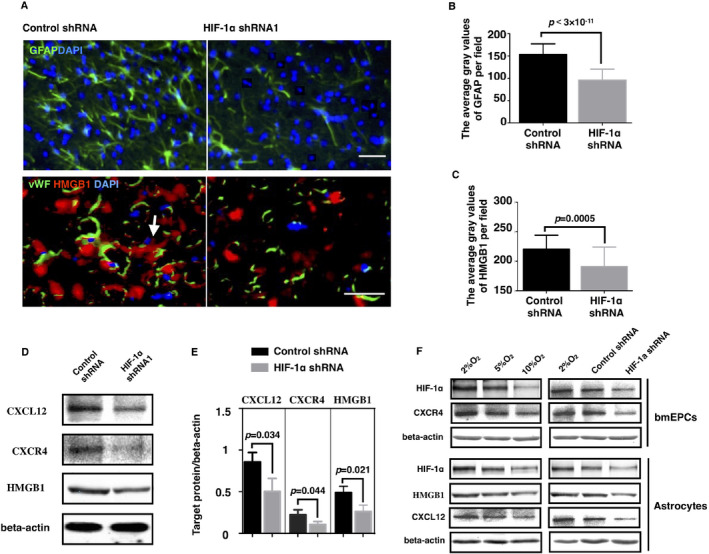
Knockdown of HIF‐1α impaired the microenvironment that recruited bmEPCs. A. On the 7th day of MCAO, the brains were removed, and 20‐μm brain sections were prepared and fixed for immunostaining. The average gray value of the AOIs was used to quantify the HMGB1 expression and GFAP expression. B and C are the statistical analyses of panel (A) (n = 3). D, E Brains were isolated for western blotting on the 7th day after the MCAO surgery. F. Primary bone marrow‐derived EPCs and astrocytes were cultured under a gradient oxygen situation for 8 h. Then, the cells were harvested for the protein expression analysis of CXCR4, HMGB1, and CXCL12 by western blotting. Data were generated from at least 3 independent experiments. (n = 23–28 fields from 5 to 6 brains per group). Bar, 60 μm. Data are presented as the mean ± S.D.
Next, we examined the effect of HIF‐1α on the CXCL12/CXCR4 axis in vivo. Compared to the control group, the HIF‐1α shRNA group showed much lower levels of CXCL12 and CXCR4 in the brain (Figure 6D,E), suggesting a positive role of HIF‐1α in the CXCL12/CXCR4 axis. To confirm that HIF‐1α regulates the CXCL12/CXCR4 axis between bmEPCs and astrocytes, we tested the protein levels of CXCL12 and CXCR4 in primary astrocytes and bmEPCs under gradual hypoxia (Figure 6F). As previously reported 31, primary astrocytes were identified by GFAP staining (Figure S6D,E). Under hypoxic conditions, the levels of CXCL12 in astrocytes and of CXCR4 in bmEPCs were increased with the decreases in oxygen (10%, 5%, and 2%), suggesting the activation of the CXCL12/CXCR4 axis under hypoxia. To assess if HIF‐1α underlies the activation, we checked the protein levels of CXCL12 and CXCR4 in both types of cells with the knockdown of HIF‐1α under hypoxia (2% O2). In line with our hypothesis, knockdown of HIF‐1α significantly inhibited the hypoxia‐induced CXCL12 in astrocytes and the CXCR4 in bmEPCs (Figure 6F), suggesting the implication of HIF‐1α in bmEPC homing via the CXCL12/CXCR4 axis between astrocytes and bmEPCs.
Next, we asked if knockdown of HIF‐1α affects HMGB1, which is generated by astrocytes in ischemic brains 19. First, we examined whether HMGB1 is regulated by HIF‐1α in ischemic brains. Using immunofluorescence, HMGB1 expression was shown around the vessels in ischemic brains (Figure 6A). However, when HIF‐1α was deleted, the HMGB1 levels were sharply decreased (Figure 6A,C), which was confirmed with the western blot assay (Figure 6D,E). Similar to CXCL12, we observed a hypoxia‐dependent increase in HMGB1 in primary astrocytes in vitro, and knockdown of HIF‐1α completely abolished the hypoxia‐dependent increase (Figure 6F). Altogether, these in vivo and in vitro data highly suggested that HIF‐1α is involved in the astrocyte‐driven bmEPCs homing via CXCL12/CXCR4 and HMGB1.
HIF‐1α promoted the sprouting angiogenesis of bmEPCs via VEGF‐a/flk1‐NRP1/Dll4
To investigate if HIF‐1α regulates bmEPC incorporation and sprouting, we labeled exogenous bmEPCs with BrdU and traced them. On the seventh day after occlusion, abundant BrdU‐positive cells incorporated into the native vessels in the control shRNA group (Figure 4D), confirming the incorporation of EPCs into the damaged vessels 35. However, the incorporated cells were reduced from HIF‐1α knockdown either in the ischemic brains or in the exogenous bmEPCs, and these cells were further decreased when HIF‐1α was knocked down in both the brains and bmEPCs (Figure 4B,D).
In the meantime, we found a high and selective expression of Dll4 in the exogenous bmEPCs in the ischemic brains, suggesting that bmEPCs may participate in the biological process of vascular sprouting (Figure 4B). These Dll4‐positive bmEPCs were significantly decreased when HIF‐1α was inhibited either in the ischemic brains or in exogenous bmEPCs, and they were further decreased when HIF‐1α was inhibited in both the brains and the bmEPCs (Figure 4B,E). To further confirm the essentiality of HIF‐1α for bmEPC sprouting, we investigated if knockdown of HIF‐1α impairs tube formation and spheroid sprouting in the primary bmEPCs under hypoxia. In contrast to the control group, the HIF‐1α shRNA group indeed presented much less tube formation (Figures 7A,C) and spheroid branches (Figures 7B,D). Collectively, these data highly indicated a critical role of HIF‐1α in the specification of bmEPCs into tip cells.
Figure 7.

Knockdown of HIF‐1α resulted in deficient in vitro angiogenesis of bmEPCs. A. bmEPCs were cultured on Matrigel under 2% O2 to observe tube formation. The cell bodies of the bmEPCs treated with the control shRNA stretched (noted by a white arrow) and connected with each other to form tubes. However, in the HIF‐1α shRNA‐treated group, tube formation was inhibited. B. Spheroid bmEPCs were cultured in Matrigel under 2% O2 to observe bmEPCs sprouting. Spheroid sprouts in each bmEPC sphere of the groups were counted. C and D are the statistical analyses of A and B, respectively (n = 5–7). (E) is the western blot of VEGF‐A and flk1 in ischemic brains treated with the control shRNA or HIF‐1α shRNA. (F) Primary bmEPCs and astrocytes were cultured under gradient oxygen; primary bmEPCs and astrocytes treated with HIF‐1α shRNA or control shRNA were cultured under 2% O2. White bar, 30 μm. Black bar, 200 μm. Data are presented as the mean ± S.D.
The VEGF‐A/flk1 axis controls tip cell formation by activating Dll4 12, 17. Given this, we first checked the axis in ischemic brains. We found that the loss of HIF‐1α decreased the levels of VEGF‐A and flk1 protein in ischemic brains (Figure 7E), suggesting a positive role of HIF‐1α in the VEGF‐A/flk1 axis. As VEGF‐A can be generated from astrocytes and bmEPCs 3, we speculated that HIF‐1α regulates the expression of VEGF‐A in both cells. We observed a hypoxia‐dependent expression of VEGF‐A in primary astrocytes and bmEPCs, and the manner in both cells was shown to be HIF‐1α‐dependent (Figure 7F). Next, we determined if the receptor of VEGF‐A, flk1, in bmEPCs is regulated by HIF‐1α. Similar to VEGF‐A, the expression of flk1 in the bmEPCs under hypoxia was dependent on HIF‐1α (Figure 7F). In addition to the VEGF‐A/flk1 axis, NRP1 and Dll4 have been reported to be critical for tip cell formation 5, 12, 13, 17. We, therefore, studied the expression patterns of these two proteins in primary bmEPCs under hypoxic situations. Similar to the VEGF‐A/flk1 axis, Dll4 and NRP1 were positively regulated by HIF‐1α, as the two receptors increased with the decrease in hypoxia, specifically from 10% O2 to 2% O2 (Figure 7F), and these increases were abolished when HIF‐1α was inhibited by shRNA (Figure 7F). Altogether, these results indicated that HIF‐1α promotes the sprouting of bmEPCs in ischemic brains via VEGF‐A/flk1‐NRP1/Dll4 between bmEPCs and astrocytes.
Discussion
In the present study, we identified that HIF‐1α is required for bmEPC‐dependent sprouting angiogenesis in the ischemic brain (Figure 8. Specifically, bmEPCs may engage in vascular sprouting by via specifying into tip cells. During the biological process, HIF‐1α is required for the recruitment of bmEPCs to ischemic brains and the specification of these cells into tip cells. Mechanistically, the recruitment of bmEPCs by HIF‐1α may be relative to CXCL12/CXCR4 and HMGB1, and it may promote the specification of bmEPCs into tip cells via the VEGF‐A/flk1‐NRP1/Dll4. Notably, astrocytes play a critical role in both biological processes.
Figure 8.

The model of HIF‐1 regulation of bmEPC‐dependent neovascularization. When cerebral ischemia occurs, HIF‐1 is stabilized due to the stabilization of its hypoxic sensor subunit HIF‐1α. Increased HIF‐1 helps bone marrow‐derived EPCs (bmEPCs) home to the ischemic brain by activating the CXCL12/CXCR4 axis and HMGB1 between reactive astrocytes and bmEPCs. In addition, HIF‐1 promotes sprouting angiogenesis by promoting the specification of bmEPCs into tip cells, the process of which is relative to the VEGF‐A/flk1‐NRP1/Dll4 axis. This working model provides cellular and molecular understandings of neovascularization for the pathology and the potential treatment targets of cerebral ischemia.
Once cerebral ischemia occurs, bmEPCs are mobilized into the peripheral blood from the bone marrow 7. Then, the circulating bmEPCs are recruited to the ischemic brain, particularly to the penumbra zone 2. Here, we verified that HIF‐1α either in the ischemic brain or in the bmEPCs is required for the bmEPCs homing (Figures 3A–D, Figures 4B,C). In response to ischemic damage, astrocytes expand and help the EPCs home to ischemic brains 34. In vivo, we showed that knockdown of HIF‐1α reduced the number of reactive astrocytes in ischemic brains (Figures 6A,B), consistent with a previous report showing an increase in reactive astrocytes in ischemic mouse brains with deficient PHD, which the deficiency leads to the stabilization of HIF‐1α 21. These data indicated that astrocytes serve as the effector cells of HIF‐1α to recruit bmEPCs. Studies have suggested that astrocytes recruit EPCs by releasing CXCL12 and HMGB1, the two ligands of CXCR4 and RAGE, respectively 11, 19, 33. Beyond this, we showed that HIF‐1α positively regulated CXCL12 and HMGB1 in astrocytes and CXCR4 in bmEPCs (Figures 6,F), indicating that HIF‐1α may recruit bmEPCs via the CXCL12/CXCR4 axis and the HMGB1/RAGE axis between astrocytes and bmEPCs.
After homing, bmEPCs perform their ability to form new vascular networks, which first requires the incorporation of bmEPCs into local vessels. We found that HIF‐1α is essential for this incorporation (Figures 3A, 4B,D). We considered several reasons for this. The first one is the significantly decreased homing of bmEPCs in the ischemic brain upon knockdown of HIF‐1α (Figure 3 and Figures 4B,C). Then, the decreases in chemokines, such as CXCL12 and HMGB1 (Figure 6, may impair the adherence of bmEPCs. Third, as several MMPs locate downstream of HIF‐1 28, loss of HIF‐1α should decrease MMPs, thus facilitating the incorporation of EPCs into vessels.
ECs have been regarded as the main source of tip cells, which stand on the top of a sprout and guide the direction of the sprout. Interestingly, EPCs and tip cells share a mutual maker, namely, flk1 12, 17, 19, suggesting the close relationship between these two types of cells. Moreover, the other two tip cell markers, Dll4 and NRP1 5, 12, 13, 17, were expressed in the recruited bmEPCs (Figures 4B, 7F). Consistent with the potential relationship between bmEPCs and tip cells, there has been indicative evidence showing the involvement of bmEPCs in sprouting angiogenesis 1. Therefore, this highly suggested that bmEPCs might be the pool for tip cells. Of interest, we further found that HIF‐1α is essential for the EPC‐dependent sprouting angiogenesis, as knockdown of HIF‐1α clearly reduced both the homing bmEPCs and the tip cells in the ischemic brains (Figures 3A–D). More encouragingly, the loss of HIF‐1α reduced Dll4 levels, the typical marker for tip cells, in bmEPCs both in vivo and in vitro (Figures 4B,E, 7F).
The VEGF‐A/flk1/Dll4 axis underlies the specification of ECs into tip cells 12, 17. In addition, the co‐receptor of flk1, NRP1, plays an equivalent role, that is, even more important in EC transformation into tip cells 5, 13. Given that HIF‐1α is a typical positive transcription factor for VEGF‐A 14 and that knockdown of HIF‐1α reduced the expression levels of flk1, NRP1 and Dll4 in the bmEPCs both in vivo and in vitro (Figures 7E,7F), HIF‐1α may promote the specification of bmEPCs into tip cells via the VEGF‐A/flk1‐NRP1/Dll4 axis.
Notably, several limitations should be addressed for the present study. First, the immune response induced by the lentiviral particles was one of the potential side effects that may have influenced the experimental results to some extent, as the immune response may cause the expansion of monocytes and the activation of inflammation, both of which affect the biological process of neovascularization 4, 33. Second, bmEPCs are a type of poorly characterized cells due to the absence of specific markers. Due to this shortage, the definition of true EPCs is controversial and studies of EPCs are limited. Therefore, more attentions and efforts to find a specific marker for EPCs are needed. Finally, although two markers of tip cells, NRP1 and Dll4, were applied to identify the sprouting bmEPCs in the present study, more direct evidence showing the morphology of sprouting bmEPCs is needed. In addition to these concerns, questions regarding the direction in which the sprouting bmEPCs guide vascular sprouting, whether in native vessels or in de novo vascular‐trees, or both, need to be investigated in future work.
Conclusion
In the present study, we showed that inhibiting HIF‐1α reduced neural recovery form ischemic damage and impaired the bmEPC‐dependent neovascularization in ischemic brains, including bmEPC homing and sprouting. Maintaining the appropriate reactive astrocytes in the ischemic brain was relative to the regulation of bmEPC homing and sprouting by HIF‐1α. Molecularly, the signals from the CXCL12/CXCR4 axis, HMGB1, and the VEGF‐A/flk1‐NRP1/Dll4 axis between the astrocytes and the bmEPCs underlie the regulation of bmEPCs homing and sprouting by HIF‐1α.
Conflict of Interest
All authors declare that there are no conflicts of interest.
Authors’ Contributions
Yang L. designed and performed the experiments, analyzed the data, and wrote and revised the article. Ran H. performed the experiments and analyzed the data. Yaping X. performed the in vitro experiments and analyzed the data. Hongjin Wang analyzed the data. Yi C. and Weihai C. funded the study and reviewed the article. Xiaoyu X designed the study, funded the study, and reviewed the article.
Supporting information
Figure S1. SD rats were randomly divided into the sham group (n = 9), the MCAO group (n = 9), MCAO + control shRNA group (n = 18) and the MCAO + HIF‐1a shRNA group (n = 18). Compared to the sham animals, the rats that received the MCAO treatment showed an increase in mNSS scores and a decrease in rCBF. Among the MCAO group, the MCAO+control shRNA group, and the MCAO+HIF‐1a shRNA group, the MCAO rats showed no significant differences in terms of the mNSS scores or the rCBF at the beginning of the study, indicating that no significant difference was induced by the grouping. The rCBF data were collected immediately after the occlusion. The mNSS scores were assessed after the MCAO rats awakened. Figure S2. MCAO rats used for the bmEPC transplantation experiment in all three groups showed no significant differences in terms of the mNSS scores or rCBF (n = 10–12), indicating that there was no significant difference induced by the grouping. The rCBF data were collected immediately after the occlusion. The mNSS scores were assessed after the MCAO rats awakened. Figure S3. MCAO rats were used to estimate the mNSS scores, infarct volumes, rCBF, and vessel densities on the 14th day, and the tip cells and homing EPCs were observed on the 10th day in the control shRNA group and the HIF‐1a shRNA group. The two groups showed no significant differences in terms of the mNSS scores or rCBF on the day of the surgery (n = 40), indicating that there was no significant difference between the groups at the beginning of the experiment. The rCBF data were collected immediately after the occlusion. The mNSS test was performed after the MCAO rats awakened. Figure S4. HIF‐1a protein in the ischemic rat brains was inhibited by the shRNA. Rats received the lentivirus 2 days before the surgery (once per day). A shows the HIF‐1a expression level detected by western blotting, showing that HIF‐1a was knocked down in the HIF‐1a shRNA group from day 5 to day 14 compared to the control shRNA group (n = 3). B shows the statistical analysis of the western blot results. C shows a very slight expression of HIF‐1a in the brains of the sham rats. D and E show no significant difference in HIF‐1a expression between the MCAO rats treated with the HIF‐1a shRNA or the control shRNA on the 3rd day after the surgery. However, the HIF‐1a expression in the MCAO rats treated with the HIF‐1a shRNA was rapidly reduced between the 5th day and the 14th day (n = 9 field from 3 rat brains/group). F shows that HIF‐1a was knocked down in areas of the cortex, the infarct, and the SVZ of the HIF‐1a shRNA group on the 5th day, compared to the control shRNA group. Black bar, 50 μm; white bar, 25 μm; red bar, 150 μm. Data are presented as the mean ± S.D. Figure S5. Loss of HIF‐1a impaired active microvessels and vessel branches in the ischemic brains. (A) On day 14, brains were isolated and sliced for detecting active microvessels using dual immunostaining of CD105 and CD31. Column B is the quantification of CD105 (n = 9). (C) Brains were removed on day 10. To observe the vascular tubes, 30‐μm brain sections were stained with vWF. Panel C shows the subbranches with diameters less than 20 μm (noted by white arrow) in the control shRNA group and HIF‐1a shRNA group. Colum D is the statistical analysis of C (n = 15–16). Green bar, 100 μm. White bar, 200 μm. Black bar, 20 μm. Figure S6. Identification of primary bmEPCs and astrocytes. A shows the pictures of the bmEPCs cultured in vitro for 3 days, 7 days, 14 days, and 21 days. B shows that the cultured bmEPCs possessed a phagocytic function, as assessed by UEA1 and acLDL. C indicates that approximately 80% of the cultured cells were Flk1/CD34 dual‐positive as determined by flow cytometry. D is the primary astrocytes cultured for 5 days and 2 days after the purification. E is the immunophenotyping of the astrocytes via GFAP. Table S1. Rat distribution for checking the HIF‐1α knockdown effeciency Table S2. Rat distribution for observing the effect of deficient HIF‐1α on neovascularization Table S3. Rat distribution for the exogenous bmEPC transplantation Table S4. Mortality rates of the rats with shRNA administration Table S5. Mortality rates of the rats with bmEPC transplantation
Acknowledgments
This research was funded by the Fundamental Research Funds for the Central Universities (No. XDJK2015D016), the National Natural Science Foundation of China (No. 81473549, No. 81402441, NO. 81302757) and the China Postdoctoral Science Foundation funded project (No.2014 M562272).
References
- 1. André H, Tunik S, Aronsson M, Kvanta A (2015) Hypoxia‐inducible factor‐1α is associated with sprouting angiogenesis in the murine laser‐induced choroidal neovascularization model. Investigative Ophthalmol Visual Sci 56:6591–6604. [DOI] [PubMed] [Google Scholar]
- 2. Arai K, Jin G, Navaratna D, Lo EH (2009) Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J 276:4644–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN et al (2012) Astrocyte‐derived VEGF‐A drives blood‐brain barrier disruption in CNS inflammatory disease. J Clin Invest 122:2454–2468. 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–966. [DOI] [PubMed] [Google Scholar]
- 5. Aspalter IM, Gordon E, Dubrac A, Ragab A, Narloch J, Vizán P et al (2015) Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nature Communications 6:7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beck H, Plate KH (2009) Angiogenesis after cerebral ischemia. Acta Neuropathological 117:481–496. [DOI] [PubMed] [Google Scholar]
- 7. Borlongan CV, Glover LE, Tajiri N, Kaneko Y, Freeman TB (2011) The great migration of bone marrow‐derived stem cells toward the ischemic brain: Therapeutic implications for stroke and other neurological disorders. Prog. Neurobiol 95:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen C, Hu Q, Yan J, Yang X, Shi X, Lei J et al (2009) Early inhibition of HIF‐1α with small interfering RNA reduces ischemic–reperfused brain injury in rats. Neurobiol. Dis 33:509–517. [DOI] [PubMed] [Google Scholar]
- 9. Chen J, Zhang C, Jiang H, Li Y, Zhang L, Robin A et al (2005) Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb. Blood Flow Metab 25:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cunningham LA, Candelario K, Li L (2012) Roles for HIF‐1α in neural stem cell function and the regenerative response to stroke. Behav Brain Res 227:410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Domanska UM, Kruizinga RC, Nagengast WB, Timmer‐Bosscha H, Huls G, de Vries EGE, Walenkamp AME (2013) A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer 49:219–230. [DOI] [PubMed] [Google Scholar]
- 12. Eilken HM, Adams RH (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22:617–625. [DOI] [PubMed] [Google Scholar]
- 13. Fantin A, Vieira JM, Plein A, Denti L, Fruttiger M, Pollard JW, Ruhrberg C (2013) NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood 121:2352–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL (Mol Cellular Biol) Activation of vascular endothelial growth factor gene transcription by hypoxia‐inducible factor 1. 1996;16:4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haipeng L, Yongbo L, Xiaodong Z, Na W, Peng X (2011) Improved electrocoagulation method for establishing rat cerebral apoplexy model. Third Mil Med Univ 33:1798–2011. [Google Scholar]
- 16. Ishikawa H, Tajiri N, Shinozuka K, Vasconcellos J, Kaneko Y, Lee HJ et al (2013) Vasculogenesis in experimental stroke after human cerebral endothelial cell transplantation. Stroke; a J Cereb Circ 44:3473–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12:943–953. [DOI] [PubMed] [Google Scholar]
- 18. Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y (2012) Suppressing inflammation by inhibiting the NF‐kappaB pathway contributes to the neuroprotective effect of angiotensin‐(1–7) in rats with permanent cerebral ischaemia. British J Pharmacol 167:1520–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayakawa K, Pham L‐DD, Katusic ZS, Arai K, Lo EH (2012) Astrocytic high‐mobility group box 1 promotes endothelial progenitor cell‐mediated neurovascular remodeling during stroke recovery. PNAS 109:7505–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koh MY, Spivak‐Kroizman TR, Powis G (2008) HIF‐1 regulation: not so easy come, easy go. Trends Biochem Sci 33:526–534. [DOI] [PubMed] [Google Scholar]
- 21. Li L, Saliba P, Reischl S, Marti HH, Kunze R (2016) Neuronal deficiency of HIF prolyl 4‐hydroxylase 2 in mice improves ischemic stroke recovery in an HIF dependent manner. Neurobiol Dis 91:221–235. [DOI] [PubMed] [Google Scholar]
- 22. Llovera G, Roth S, Plesnila N, Veltkamp R, Liesz A (2014) Modeling stroke in mice: permanent coagulation of the distal middle cerebral artery. J Vis Exp e51729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuda T, Abe T, Wu JL, Fujiki M, Kobayashi H (2005) Hypoxia‐inducible factor‐1α DNA induced angiogenesis in a rat cerebral ischemia model. Neurol Res 27:503–508. [DOI] [PubMed] [Google Scholar]
- 24. Miller JT, Bartley JH, Wimborne HJ, Walker AL, Hess DC, Hill WD, Carroll JE (2005) The neuroblast and angioblast chemotaxic factor SDF‐1 (CXCL12) expression is briefly up regulated by reactive astrocytes in brain following neonatal hypoxic‐ischemic injury. BMC Neurosci 6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phng L‐K, Stanchi F, Gerhardt H (2013) Filopodia are dispensable for endothelial tip cell guidance. Development 140:4031–4040. [DOI] [PubMed] [Google Scholar]
- 26. Singh N, Sharma G, Mishra V, Raghubir R (2012) Hypoxia inducible factor‐1: its potential role in cerebral ischemia. Cell Mol Neurobiol 32:491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Korff T, Augustin HG (1998) Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J Cell Biol 143:1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakamoto T, Weng JS, Hara T, Yoshino S, Kozuka‐Hata H, Oyama M, Seiki M (2014) Hypoxia‐inducible factor 1 regulation through cross talk between mTOR and MT1‐MMP. Mol Cellular Biol 34:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Laake LW, van den Driesche S, Post S, Feijen A, Jansen MA, Driessens MH et al (2006) Endoglin has a crucial role in blood cell‐mediated vascular repair. Circulation 114:2288–2297. [DOI] [PubMed] [Google Scholar]
- 30. Xiong Y, Mahmood A, Chopp M (2010) Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Invest Drugs 11:298–308. [PMC free article] [PubMed] [Google Scholar]
- 31. Xue Q, Liu Y, Qi H, Ma Q, Xu L, Chen W et al (2013) A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int J Biol Sci 9:174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang L, Qiang X, Xu L, Jifen Z, Zhifeng F, Binbin F et al (2014) Amelioration of stroke‐induced neurological deficiency by lyophilized powder of catapol and puerarin. Int J Biol Sci 10:448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yip H, Chang L, Chang W, Lu C, Liou C, Lan M et al (2008) Level and value of circulating endothelial progenitor cells in patients after acute ischemic stroke. Stroke 39:69–74. [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Huang J, Li Y, Yang G‐Y (2012) Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr Drug Targets 13:166–172. [DOI] [PubMed] [Google Scholar]
- 35. Zhang ZG, Zhang L, Jiang Q, Chopp M (2002) Bone marrow‐derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res 90:284–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SD rats were randomly divided into the sham group (n = 9), the MCAO group (n = 9), MCAO + control shRNA group (n = 18) and the MCAO + HIF‐1a shRNA group (n = 18). Compared to the sham animals, the rats that received the MCAO treatment showed an increase in mNSS scores and a decrease in rCBF. Among the MCAO group, the MCAO+control shRNA group, and the MCAO+HIF‐1a shRNA group, the MCAO rats showed no significant differences in terms of the mNSS scores or the rCBF at the beginning of the study, indicating that no significant difference was induced by the grouping. The rCBF data were collected immediately after the occlusion. The mNSS scores were assessed after the MCAO rats awakened. Figure S2. MCAO rats used for the bmEPC transplantation experiment in all three groups showed no significant differences in terms of the mNSS scores or rCBF (n = 10–12), indicating that there was no significant difference induced by the grouping. The rCBF data were collected immediately after the occlusion. The mNSS scores were assessed after the MCAO rats awakened. Figure S3. MCAO rats were used to estimate the mNSS scores, infarct volumes, rCBF, and vessel densities on the 14th day, and the tip cells and homing EPCs were observed on the 10th day in the control shRNA group and the HIF‐1a shRNA group. The two groups showed no significant differences in terms of the mNSS scores or rCBF on the day of the surgery (n = 40), indicating that there was no significant difference between the groups at the beginning of the experiment. The rCBF data were collected immediately after the occlusion. The mNSS test was performed after the MCAO rats awakened. Figure S4. HIF‐1a protein in the ischemic rat brains was inhibited by the shRNA. Rats received the lentivirus 2 days before the surgery (once per day). A shows the HIF‐1a expression level detected by western blotting, showing that HIF‐1a was knocked down in the HIF‐1a shRNA group from day 5 to day 14 compared to the control shRNA group (n = 3). B shows the statistical analysis of the western blot results. C shows a very slight expression of HIF‐1a in the brains of the sham rats. D and E show no significant difference in HIF‐1a expression between the MCAO rats treated with the HIF‐1a shRNA or the control shRNA on the 3rd day after the surgery. However, the HIF‐1a expression in the MCAO rats treated with the HIF‐1a shRNA was rapidly reduced between the 5th day and the 14th day (n = 9 field from 3 rat brains/group). F shows that HIF‐1a was knocked down in areas of the cortex, the infarct, and the SVZ of the HIF‐1a shRNA group on the 5th day, compared to the control shRNA group. Black bar, 50 μm; white bar, 25 μm; red bar, 150 μm. Data are presented as the mean ± S.D. Figure S5. Loss of HIF‐1a impaired active microvessels and vessel branches in the ischemic brains. (A) On day 14, brains were isolated and sliced for detecting active microvessels using dual immunostaining of CD105 and CD31. Column B is the quantification of CD105 (n = 9). (C) Brains were removed on day 10. To observe the vascular tubes, 30‐μm brain sections were stained with vWF. Panel C shows the subbranches with diameters less than 20 μm (noted by white arrow) in the control shRNA group and HIF‐1a shRNA group. Colum D is the statistical analysis of C (n = 15–16). Green bar, 100 μm. White bar, 200 μm. Black bar, 20 μm. Figure S6. Identification of primary bmEPCs and astrocytes. A shows the pictures of the bmEPCs cultured in vitro for 3 days, 7 days, 14 days, and 21 days. B shows that the cultured bmEPCs possessed a phagocytic function, as assessed by UEA1 and acLDL. C indicates that approximately 80% of the cultured cells were Flk1/CD34 dual‐positive as determined by flow cytometry. D is the primary astrocytes cultured for 5 days and 2 days after the purification. E is the immunophenotyping of the astrocytes via GFAP. Table S1. Rat distribution for checking the HIF‐1α knockdown effeciency Table S2. Rat distribution for observing the effect of deficient HIF‐1α on neovascularization Table S3. Rat distribution for the exogenous bmEPC transplantation Table S4. Mortality rates of the rats with shRNA administration Table S5. Mortality rates of the rats with bmEPC transplantation