

Abstract
CDKL5 deficiency disorder (CDD) is a rare encephalopathy characterized by early onset epilepsy and severe intellectual disability. CDD is caused by mutations in the X‐linked cyclin‐dependent kinase‐like 5 (CDKL5) gene, a member of a highly conserved family of serine‐threonine kinases. Only a few physiological substrates of CDKL5 are currently known, which hampers the discovery of therapeutic strategies for CDD. Here, we show that SMAD3, a primary mediator of TGF‐β action, is a direct phosphorylation target of CDKL5 and that CDKL5‐dependent phosphorylation promotes SMAD3 protein stability. Importantly, we found that restoration of the SMAD3 signaling through TGF‐β1 treatment normalized defective neuronal survival and maturation in Cdkl5 knockout (KO) neurons. Moreover, we demonstrate that Cdkl5 KO neurons are more vulnerable to neurotoxic/excitotoxic stimuli. In vivo treatment with TGF‐β1 prevents increased NMDA‐induced cell death in hippocampal neurons from Cdkl5 KO mice, suggesting an involvement of the SMAD3 signaling deregulation in the neuronal susceptibility to excitotoxic injury of Cdkl5 KO mice. Our finding reveals a new function for CDKL5 in maintaining neuronal survival that could have important implications for susceptibility to neurodegeneration in patients with CDD.
Keywords: CDKL5, hippocampal neurons, neuronal survival, neuronal maturation, SMAD3, TGF‐β signaling
Introduction
The cyclin‐dependent kinase‐like 5 (CDKL5) gene is a member of the serine/threonine protein kinase family and encodes a protein with kinase activity 39, 49. Mutations in this gene have been associated with X‐linked neurodevelopmental disorder, also known as CDKL5 deficiency disorder (CDD; OMIM #300203). To date, several different mutations have been described in the CDKL5 gene, mainly located within the CDKL5 catalytic domain 13, 20, 42. This strongly suggests that impaired CDKL5 kinase activity plays an important role in the pathogenesis of this encephalopathy 2, 60. Patients with CDD show early onset epileptic seizures, severe intellectual disability, autistic‐like features, and gross motor impairments 2, 20, 39, 72, all clinical features related to central nervous system dysfunctions. In fact, even if CDKL5 is ubiquitously expressed, it is highly expressed in the brain 31, in particular in the forebrain structures (ie, cortex and hippocampus) 56, 70, reaching a peak during postnatal brain development 42, 56.
Existing mouse models of CDD, Cdkl5 knockout (KO) mice 1, 52, 70, have helped to clarify the role of CDKL5 in brain development and function, and therefore in the etiology of CDD. Using a Cdkl5 KO mouse model, we recently showed that CDKL5 plays a fundamental role in postnatal hippocampal development, affecting neural precursor proliferation as well as the survival of newborn granule cells 27. In addition, Cdkl5 deficiency impairs morphogenesis and dendritic arborization of hippocampal neurons and their synaptic connectivity 25, 26, 64. Spontaneous synaptic activity is impaired in hippocampal slices of a forebrain excitatory neuron specific to Cdkl5 KO mice [Nex‐cKO; 59], and excitatory synaptic transmission is reduced in mouse hippocampal cultures silenced for Cdkl5 55. Altered neuronal survival, dendritic development and synaptic function may underlie the severe hippocampus‐dependent cognitive impairment that characterizes Cdkl5 KO mice 24, 26, 64.
The way in which CDKL5 deficiency affects neuronal maturation is starting to be defined progressively with the identification of some of the molecular mechanisms underlying CDKL5 functions. CDKL5 forms a complex with PSD‐95 and NGL‐1 at the spine level 55, 78 to regulate spine morphology and maintenance 14, and with IQ domain‐containing GTPase‐activating protein 1 (IQGAP1) and MAP1S to regulate microtubule dynamics and stability 3, 4, 50. Although data clearly suggest that CDKL5 is important for correct neuronal maturation 14, 24, 27, 51, 56, 64, 66, the role of CDKL5 in the maintenance of neuron survival has been poorly investigated. In addition, the mechanisms by which CDKL5 supports neuronal survival remain unknown. We thus aimed to identify CDKL5 kinase substrates in order to elucidate the neuronal functions of CDKL5 and characterize the role of CDKL5 in the life/death decision of hippocampal neurons.
Methods and Materials
Colony
Mice were handled according to protocols approved by the Italian Ministry for Health (approval number DGSAF 114/2018). The mice used in this work derive from the Cdkl5 KO strain in the C57BL/6N background developed in 1 and backcrossed in C57BL/6J for three generations. Mice for testing were produced as previously described 64. Age‐matched littermates were used for all experiments. The day of birth was designated as postnatal day (P) zero and animals with 24 h of age were considered as 1‐day‐old animals (P1). Mice were housed 3–5 per cage on a 12 h light/dark cycle in a temperature‐controlled environment with food and water provided ad libitum.
Antibody microarray
Six phospho explorer antibody microarrays (PEX100; Full Moon BioSystems, Inc., Sunnyvale, CA, USA) were labeled according to the manufacturer’s protocol, starting from cortex protein extracts from 3 Cdkl5 +/Y and 3 Cdkl5 −/Y mice aged P20. Briefly, proteins were extracted with non‐denaturing lysis buffer, and biotinylation of protein samples was performed with the antibody array assay kit (Full Moon BioSystems). The antibody microarray slides were first blocked in a blocking solution for 30 minutes at room temperature, then rinsed 10 times with Milli‐Q grade water for 3–5 minutes. The slides were then incubated with the biotin‐labeled cell lysates (100 μg of protein) in coupling solution at room temperature for 2 h. The array slides were washed 4–5 times with 1× Wash Solution and rinsed extensively with Milli‐Q grade water before detection of bound biotinylated proteins using Cy3‐conjugated streptavidin. Fluorescence intensity of each array spot was quantified and the average signal intensity of replicate spots was calculated. Raw data were normalized as follows: (Average Signal Intensity – Average Background intensity)/Median Signal. Median Signal is the median value of Average Signal Intensity for all antibodies on the array. For total protein, the fold change was calculated as a ratio between the Cdkl5 −/Y and Cdkl5 +/Y mean signals. Phospho site‐specific signals were first normalized on the corresponding site‐specific signals of the same protein and then the fold change was calculated based on the following equation: Phosphorylation ratio = (phosphoA/unphosphoA)/(phosphoB/unphosphoB) where phosphoA or phosphoB and unphosphoA or unphosphoB referred to signals of the phosphorylated and unphosphorylated proteins, respectively, from the experimental samples: (i) Cdkl5 −/Y (ii) Cdkl5 +/Y samples.
Plasmids
The following plasmids were used: pHA‐hCDKL5 45 and phCDKL5‐3xFLAG 64, carrying the first characterized CDKL5 isoform that generates a protein of 1030 amino acids [115 kDa; 45]; pCS2‐FLAG‐SMAD2 (Addgene, Watertown, MA, USA), pCS2‐FLAG‐SMAD3 36, pCS2‐FLAG‐SMAD3‐MH1, pCS2‐FLAG‐SMAD3‐MH2, and pCS2‐FLAG‐SMAD3‐L‐MH2. SMAD3 deletion mutant plasmids were produced by whole around PCR using pCS2‐FLAG‐SMAD3 plasmid as a template and the primers indicated in Supporting Table 1. pCS2‐FLAG‐SMAD3 plasmid was provided by Prof. Stefano Piccolo (University of Padova, Italy) 36, (CAGA)12‐luc plasmid, reporter gene for SMAD3 activity, was provided by Caroline Hill (Lincoln’s Inn Fields Laboratories, London, UK) 44, and pTKRL plasmid was obtained from Promega, Madison, WI, USA.
Co‐immunoprecipitation assays
HEK293T cells were transfected with HA‐CDKL5 alone or co‐transfected with HA‐CDKL5 and SMAD3‐FLAG or SMAD3 mutants using Metafectene Easy Plus (Biontex Laboratories GMBH, Munich, Germany). Twenty‐four hours after transfection, cell lysates were processed for co‐immunoprecipitation as described in the Supporting Methods section. For endogenous SMAD3 co‐immunoprecipitation, SH‐SY5Y cells were infected with CDKL5‐FLAG adenovirus particles (Ad‐CDKL5) or GFP adenovirus particles as a control (Ad‐GFP; Vector BioLabs, Malver, PA, USA) at 100 multiplicities of infection (MOI) and cell lysates were processed as described in the Supporting Methods.
In vitro phosphorylation assay
Two milligram of protein lysate from HEK293T cells transfected with SMAD3‐FLAG or SMAD3 mutants was added to 20 μL of EZview™ Red ANTI‐FLAG® M2 Affinity Gel (Sigma‐Aldrich, St. Louis, MO, USA) and incubated overnight at 4°C. 3xFLAG peptide was used to elute purified protein according to the manufacturer’s instructions (Sigma‐Aldrich). For recombinant CDKL5 kinase assay, CDKL5ΔN protein (rCDKL5 1‐498aa, Aurogene s.r.l.; Rome, Italy) was incubated with 2 mM cold ATP, 1× kinase buffer (20 mM HEPES, pH 7.4, 10 mM MgCl2, 10 mM NaCl), 10 μCi of [γ‐ 32P]‐ATP (Perkin Elmer; Waltham, MA, USA), and SMAD3 purified proteins at 32°C for 60 minutes. The reaction was terminated by the addition of 15 μL loading buffer and boiled at 95°C for 10 minutes. For the wild‐type CDKL5 kinase assay, 2 mg of protein lysate from HEK293T cells transfected with hCDKL5‐FLAG was immunoprecipitated on an EZview™ Red ANTI‐FLAG® M2 Affinity Gel (Sigma‐Aldrich). The kinase assay was performed with SMAD3 or SMAD2 purified proteins as previously described 64. Western blotting was performed as described below and membrane was exposed to an autoradiographic film. After the decay of radioactivity, membranes were stained with PonceauS or immunostained with anti‐ SMAD3 and SMAD2 antibodies.
Primary hippocampal cultures
Primary hippocampal neuronal cultures were prepared from P1 Cdkl5 +/Y and Cdkl5 −/Y mice as previously described 64. Treatments were performed as described in the Supporting Methods.
Immunocytochemistry
Hippocampal cultures were fixed in 4% paraformaldehyde + 4% sucrose in 100 mM phosphate buffer, pH 7.4. Fluorescent images were acquired using a Nikon Eclipse TE600 microscope equipped with a Nikon Digital Camera DXM1200 ATI System (Nikon Instruments, Inc., Melville, NY, USA).
SMAD3 and P‐SMAD3 (Ser213) nuclear intensity
Primary hippocampal neurons were fixed after 10 days in culture DIV10 and immunostaining was performed using a primary anti‐SMAD3 antibody (rabbit polyclonal anti‐SMAD3 Ab, 1:200, Cell Signaling Technology, Inc., Danvers, MA, USA) or a primary anti‐Phospho‐SMAD3 antibody (Ser213) (rabbit polyclonal anti‐phospho‐SMAD3 Ab, 1:200; Full Moon BioSystems, Inc., Sunnyvale, CA, USA) and a Cy3‐conjugated anti‐rabbit IgG (1:200, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) secondary fluorescent antibody. Nuclei were counterstained with Hoechst‐33342 (Sigma‐Aldrich), and fluorescence images were acquired at the same intensity. To assess SMAD3 or P‐SMAD3 nuclear intensity Hoechst and SMAD3 or P‐SMAD3 images of the same cell were processed. The perimeter of the nucleus was traced using Hoechst counterstaining as a guide to define the nuclear area of each cell, and the intensity of Cy3‐staining corresponding to the SMAD3 or P‐SMAD3 signal was quantified by determining the number of positive (bright) pixels of the cell and within the nucleus. A total of 200 cells for each condition were quantified for SMAD3 signal intensity. A total of 50 cells for each condition were quantified for P‐SMAD3 signal intensity.
Apoptotic cell death
To assess apoptotic cell death primary hippocampal cultures were fixed on DIV4 or DIV10‐12 (after exposure to different neurotoxic stimuli as above described) and double‐stained with the following primary antibodies: anti‐α‐tubulin (mouse monoclonal anti‐α‐tubulin Ab, 1:500, Sigma‐Aldrich) and anti‐cleaved caspase‐3 antibody (rabbit polyclonal anti‐cleaved caspase‐3 Ab, 1:200, Cell Signaling). Detection was performed with a FITC‐conjugated anti‐mouse IgG (1:200, Jackson Immunoresearch) and a Cy3‐conjugated anti‐rabbit IgG (1:200, Jackson Immunoresearch) antibody. The number of cleaved caspase‐3 positive neurons (α‐tubulin positive cells with a neuronal phenotype) was counted manually and expressed as a percentage of the total number of neurons. A total of 100 cells for each condition were counted.
Neuronal maturation
In order to assess axon elongation and neuritic outgrowth primary hippocampal neurons were fixed on DIV10 and stained with the following primary antibodies: anti‐TAU1 (mouse monoclonal anti‐TAU1 Ab, 1:200, Merck Millipore, Burlington, MA, USA) and anti‐MAP2 (rabbit polyclonal anti‐MAP2 Ab, 1:100, Merck Millipore). Detection was performed with a Cy3‐conjugated anti‐mouse IgG (1:200, Jackson Immunoresearch) and a FITC‐conjugated anti‐rabbit IgG (1:200, Jackson Immunoresearch) antibody. Neurites with a significant intensity of TAU1 staining increasing along the proximal to distal axis were counted as axons. Axon and dendritic length was analyzed using the image analysis system Image Pro Plus as previously described 64. A total of 50 neurons for each condition were evaluated for axon elongation and neuritic outgrowth.
To assess the degree of synaptic innervation primary hippocampal neurons were fixed on DIV10 and stained with the following antibodies: anti‐synaptophysin (mouse monoclonal anti‐SYN Ab, 1:500, clone SY38, Merck Millipore) and anti‐MAP2 (rabbit polyclonal anti‐MAP2 Ab, 1:100, Merck Millipore). Detection was performed with a Cy3‐conjugated anti‐mouse IgG (1:200, Jackson Immunoresearch) and a FITC‐conjugated anti‐rabbit IgG (1:200, Jackson Immunoresearch) antibody. The degree of synaptic innervation was evaluated by counting the number of synaptic puncta (SYN‐positive) along the proximal dendrites and expressed as the number of SYN puncta per 10 µm of dendritic length. In order to evaluate spine density fluorescence images (MAP2‐postitive protrusions) were acquired using a Leica TCS confocal (Leica Microsystems, Wetzlar, Germany) 63× oil immersion lens at a 0.6 mm intervals at 1.024 × 1.024 pixels resolution with a 1× zoom. Spine density was measured by counting the number of dendritic protrusions (spines) on proximal dendrites and expressed as the number of spines per 10 µm of dendritic length. A total of 50 neurons for each condition were evaluated for the number of synaptic puncta and number of spines.
Luciferase assay
SMAD3 activity in SH‐SY5Y cells and primary hippocampal cultures was monitored through luciferase assays using a p(CAGA)12‐luc reporter plasmid as described in the Supporting Methods.
Western blot analysis
Total proteins from SKNBE cells transfected with wild‐type CDKL5 or CDKL5ΔN, SH‐SY5Y cells infected with CDKL5‐FLAG (Ad‐CDKL5) or GFP (Ad‐GFP) adenovirus particles, and primary hippocampal cultures at DIV10 were lysed in ice‐cold RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Triton‐X100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 1mM PMSF, and with 1% protease and phosphatase inhibitor cocktail (Sigma‐Aldrich). Total proteins from the hippocampus and cortex of 9–12‐week‐old Cdkl5 −/Y and Cdkl5 +/Y male mice were homogenized in ice‐cold RIPA buffer. Protein concentration for both cell and tissue extracts was determined using the Lowry method 46 and equivalent amounts (50 μg) of protein were subjected to electrophoresis on a 4%–12% Mini‐PROTEAN® TGX™ Gel (Bio‐Rad, Hercules, CA, USA) and transferred to a Hybond‐ECL nitrocellulose membrane (GE Healthcare Life Sciences, Amersham, UK). The following primary antibodies were used: anti‐HA antibody (rabbit polyclonal anti‐HA Ab, 1:1000, Cell Signaling Technology), anti‐FLAG M2 antibody (mouse monoclonal anti‐FLAG M2 Ab, 1:1000, Sigma‐Aldrich), anti‐SMAD3 antibody (rabbit polyclonal anti‐SMAD3 Ab, 1:1000; Cell Signaling Technology), anti‐SMAD2 antibody (rabbit polyclonal anti‐SMAD2 Ab, 1:1000; Cell Signaling Technology), anti‐Phospho‐SMAD3 antibody (Ser423/425) (rabbit polyclonal anti‐phospho‐SMAD3 Ab, 1:1000; Cell Signaling Technology), anti‐Phospho‐SMAD3 antibody (Ser425) (rabbit polyclonal anti‐phospho‐SMAD3 Ab, 1:1000; Full Moon Biosystem Inc), anti‐Phospho‐SMAD3 antibody (Ser213) (rabbit polyclonal anti‐phospho‐SMAD3 Ab, 1:1000; Full Moon Biosystem Inc), anti‐GFP antibody (rabbit polyclonal anti‐green fluorescent protein Ab, 1:1000; Thermo Fisher Scientific, Waltham, MA, USA), anti‐CDKL5 antibody (rabbit polyclonal anti‐green fluorescent protein Ab, 1:500, Sigma‐Aldrich), and anti‐GAPDH antibody (rabbit polyclonal anti‐GAPDH Ab, 1:5000; Sigma‐Aldrich). Densitometric analysis of digitized images was performed using Chemidoc XRS Imaging Systems and Image Lab™ Software (Bio‐Rad).
In vivo experiments
Experiments were carried out on a total of 38 Cdkl5 −/Y and 30 Cdkl5 +/Y mice. Seizures were induced in 10–12‐week‐old mice by intraperitoneal administration of 60 mg/kg NMDA (Sigma‐Aldrich) in phosphate‐buffered saline (PBS). Seizure grades were scored according to 74 and recorded in a 35‐minutes observation period. Rescue experiments with TGF‐β1 (Relia Tech GMBH, Wolfenbüttel, Germany) were performed 60 minutes after NMDA injection as described in the Supporting Methods. Animals were preceded for immunohistochemical and histological procedures as described in the Supporting Methods.
Statistical analysis
Values are expressed as means ± standard error (SE). The significance of results was obtained using Student’s t test and one‐way or two‐way ANOVA followed by Fisher’s LSD post hoc test. A probability level of P < 0.05 was considered to be statistically significant.
Results
Reduced SMAD3 protein levels in the brains of Cdkl5 KO mice
To search for CDKL5 target proteins, brain protein extracts from Cdkl5 −/Y and wild‐type (+/Y) mice were applied onto Phospho Explorer antibody microarrays 30, 64. These microarrays consist of 1318 site‐specific and phospho site‐specific antibodies against proteins related to multiple signaling pathways and biological processes. Among the antibodies against the SMAD family proteins, array data obtained with the site‐specific antibody against SMAD2 (Smad2 Ab‐245) and that against SMAD3 (Smad3 Ab‐204) suggested reduced SMAD2 and SMAD3 protein levels in the absence of Cdkl5 (Table 1). No difference in SMAD protein phosphorylation levels between Cdkl5 −/Y and +/Y mice was highlighted by the SMAD phospho site‐specific antibodies on the array (Table 1).
Table 1.
A summary of phospho explorer antibody microarray results for the SMAD family of proteins is presented with corresponding antibodies. Cdkl5 −/Y (KO) vs. Cdkl5 +/Y (WT) ratio of total SMAD proteins and their phospho‐isoforms are indicated with fold change, error, and P‐value.
| PhosphoExplorer Antibody Microarray—SMAD family signals | |||||
|---|---|---|---|---|---|
| KO/WT ratio | P‐value | KO/WT ratio | P‐value | ||
| Smad1 (Ab‐187) | 0.94 ± 0.10 | 0.560 | Smad1 | ||
| Smad1 (Ab‐465) | 1.17 ± 0.11 | 0.202 | P‐Ser187/Smad1 (Ab‐187) | 1.16 ± 0.13 | 0.238 |
| Smad1‐mean | 1.05 ± 0.12 | P‐Ser465/Smad1 (Ab‐465) | 0.70 ± 0.15 | 0.183 | |
| Smad2 (Ab‐220) | 1.13 ± 0.22 | 0.555 | Smad2 | ||
| Smad2 (Ab‐245) | 0.86 ± 0.05 | 0.047 ** | P‐Ser250/Smad2 (Ab‐250) | 0.94 ± 0.09 | 0.619 |
| Smad2 (Ab‐250) | 0.89 ± 0.05 | 0.173 | P‐Ser467/Smad2 (Ab‐467) | 1.33 ± 0.29 | 0.286 |
| Smad2 (Ab‐255) | 0.96 ± 0.11 | 0.732 | P‐Thr220/Smad2 (Ab‐220) | 0.91 ± 0.13 | 0.544 |
| Smad2 (Ab‐467) | 0.83 ± 0.08 | 0.120 | |||
| Smad2‐mean | 0.93 ± 0.05 | Smad2/3 | |||
| P‐Thr8/Smad2/3 (Ab‐8) | 1.13 ± 0.15 | 0.385 | |||
| Smad2/3 (Ab‐8) | 0.83 ± 0.13 | 0.320 | |||
| Smad3 (Ab‐179) | 0.96 ± 0.02 | 0.117 | Smad3 | ||
| Smad3 (Ab‐204) | 0.75 ± 0.07 | 0.054 * | P‐Ser204/Smad3 (Ab‐204) | 1.08 ± 0.08 | 0.309 |
| Smad3 (Ab‐213) | 0.88 ± 0.13 | 0.480 | P‐Thr213/Smad3 (Ab‐213) | 1.00 ± 0.17 | 0.972 |
| Smad3 (Ab‐425) | 1.02 ± 0.08 | 0.819 | P‐Ser425/Smad3 (Ab‐425) | 0.93 ± 0.12 | 0.645 |
| Smad3‐mean | 0.90 ± 0.06 | P‐Thr179/Smad3 (Ab‐179) | 0.92 ± 0.06 | 0.315 | |
Values are represented as means ± SE.
* P = 0.054.
** P < 0.05 (Unpaired t‐test).
Western blot and immunohistochemistry analyses were used to confirm array data. While we did not find reduced SMAD2 levels in the cortex of Cdkl5 −/Y mice in comparison with +/Y mice (Supporting Figure 1A), we confirmed that SMAD3 levels are reduced both in the cortex (Figure 1A–D) and hippocampus (Figure 1E–F, Supporting Figure 1B) of Cdkl5 −/Y mice. In particular, we observed that Cdkl5 −/Y mice showed, in all cortical layers, a reduced number of SMAD3 positive cells compared to wild‐type (+/Y) mice (Figure 1B,D). These latter mice exhibited a strong SMAD3 immunopositivity, whereas Cdkl5 −/Y mice showed a moderate SMAD3 immunopositivity (Figure 1C). Similarly, hippocampal CA1 pyramidal neurons showed a reduced SMAD3 immunopositivity in Cdkl5 −/Y mice in comparison with wild‐type (+/Y) mice (Figure 1F, Supporting Figure 1B).
Figure 1.
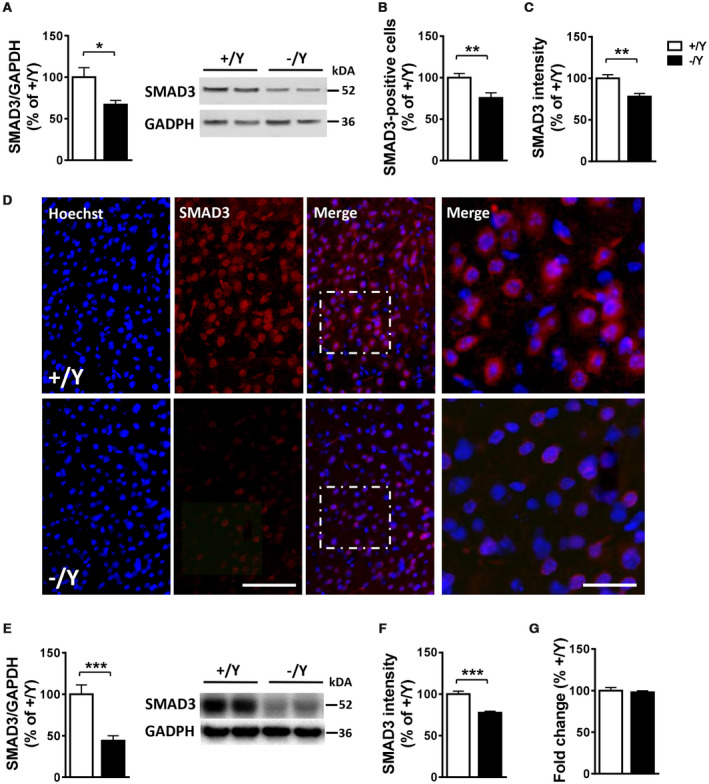
Reduced SMAD3 levels in the cortex and hippocampus of Cdkl5 KO mice. A. Western blot analysis of SMAD3 levels normalized to GAPDH levels in the somatosensory cortex of wild‐type (+/Y; n = 3) and Cdkl5 −/Y (n = 4) adult mice. Immunoblots are examples from two animals of each experimental group. B, C. Number of SMAD3 positive cells (B) and SMAD3 nuclear signal intensity (C) in the somatosensory cortex of wild‐type (+/Y; n = 10, n = 4, respectively) and Cdkl5 −/Y (n = 7, n = 4, respectively) adult mice. D. Representative images of cortical sections processed for fluorescent SMAD3 immunostaining of wild‐type (+/Y) and Cdkl5 −/Y mice. The dotted boxes indicate the regions shown at a higher magnification. Scale bar = 50 μm lower magnification, 15 μm higher magnification. E. Western blot analysis of SMAD3 levels normalized to GAPDH levels in the hippocampus of wild‐type (+/Y; n = 7) and Cdkl5 −/Y (n = 8) adult mice. Immunoblots are examples from two animals of each experimental group. F. SMAD3 nuclear signal intensity in the hippocampus of wild‐type (+/Y; n = 8) and Cdkl5 −/Y (n = 8) mice. G. Quantification by RT‐qPCR of SMAD3 expression in the hippocampus of wild‐type (+/Y; n = 8) and Cdkl5 −/Y (n = 8) mice. Data are expressed as a percentage of the values of Cdkl5 +/Y mice. Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Unpaired t‐test).
As suggested by the array data (Table 1), we did not observe a difference in SMAD3 phosphorylation levels at Ser213 and Ser425 in the cortex of Cdkl5 −/Y mice in comparison with +/Y mice (Supporting Figure 1C).
SMAD3 is regulated at the mRNA level and at the level of protein stability 12, 53. No differences in SMAD3 mRNA levels were observed between Cdkl5 −/Y and +/Y mice in either the hippocampus (Figure 1G) or cortex (Supporting Figure 1D), suggesting a CDKL5‐dependent post‐transcriptional regulation of SMAD3.
SMAD3 is a phosphorylation target of CDKL5
SMAD3, together with SMAD2, is one of the primary mediators of TGF‐β action 21, 32, 47. Upon phosphorylation by the TGF‐β receptors, the SMAD proteins translocate into the nucleus, where they regulate transcription 34.
To establish whether SMAD3 and CDKL5 physically interact in vivo, we performed co‐immunoprecipitation assays from cell lysates of HEK293T cells transfected with both HA‐CDKL5 and SMAD3‐FLAG. Using an anti‐FLAG antibody we found co‐immunoprecipitation of SMAD3 and CDKL5, indicating their interaction (Figure 2B; lane 4 arrow). SMAD3 consists of two highly‐conserved MAD homology domains, in the amino (MH1) and carboxyl (MH2) termini that are connected by a proline‐rich nonconserved linker region (Figure 2A). To identify the SMAD3 domain that interacts with CDKL5, FLAG‐tagged SMAD3 deletion constructs (Figure 2A), FLAG‐MH1 (amino acids 1–136) and FLAG‐L‐MH2 (amino acids 136–425), were co‐transfected with HA‐CDKL5 into HEK293T cells. As shown in Figure 2B, CDKL5 interacted with the SMAD3 deletion construct MH1 (Figure 2B; lane 6 arrow), but not with the L‐MH2 construct (Figure 2B; lane 8), indicating that the association of CDKL5 and SMAD3 in cells is mediated via the N‐terminal MH1 domain of SMAD3. To confirm the interaction of CDKL5 with SMAD3 in cells, we also performed co‐immunoprecipitation experiments with endogenous SMAD3 protein in SH‐SY5Y cells, a neuroblastoma cell line that exhibits relatively high basal levels of SMAD3 (Figure 2C, lane 1). We infected SH‐SY5Y cells with CDKL5‐FLAG adenovirus particles and precipitated the overexpressed protein from the cell extracts with anti‐FLAG antibodies (Figure 2C). Subsequent immunoblotting with an anti‐SMAD3 antibody revealed that endogenous SMAD3 co‐precipitated with overexpressed CDKL5‐FLAG (Figure 2C).
Figure 2.
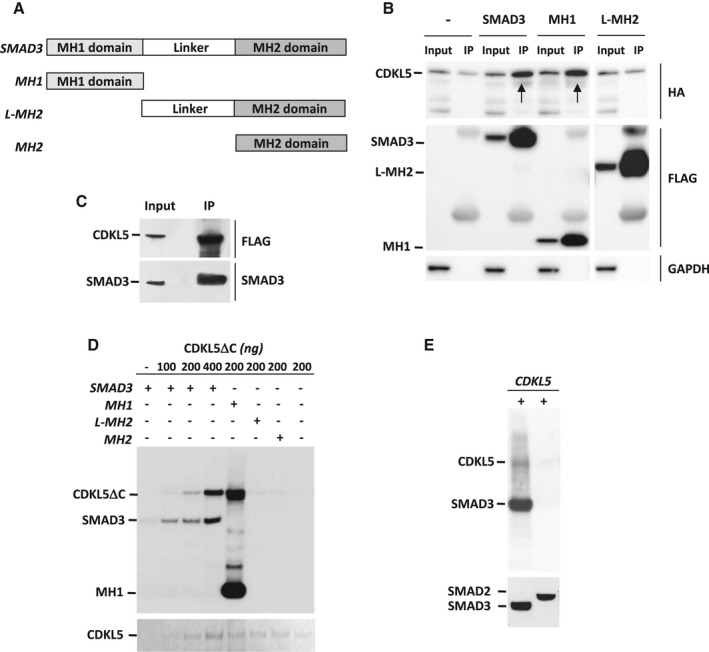
CDKL5 interacts with, and phosphorylates, SMAD3 protein. A. Schematic representation of SMAD3 and mutant SMAD3 domains. The locations of MH1 domain (light gray), linker region, and MH2 domain (dark gray) are shown. B. Interaction between CDKL5 and SMAD3. HEK293T cells were co‐transfected with HA‐CDKL5 and wild‐type SMAD3‐FLAG or the indicated SMAD3 mutant‐FLAG plasmids, and cell lysates (Input) were immunoprecipitated with anti‐FLAG antibodies (IP). GAPDH was used as an internal control for Input. Immunoprecipitated proteins were detected by anti‐HA (CDKL5) and anti‐FLAG antibodies (SMAD3 and SMAD3 mutants). Arrows indicate co‐immunoprecipitated CDKL5. Lysates of cells overexpressing only HA‐CDKL5 (Input; lane 1) were immunoprecipitated with anti‐FLAG antibodies as a control (IP; lane 2). Irrelevant lanes were spliced out with a white space. C. SH‐SY5Y cells, infected with CDKL5‐FLAG adenoviral particles or GFP adenoviral particles as control, were lysed (Input) and immunoprecipitated with anti‐FLAG antibodies (IP). Immunoprecipitated CDKL5, SMAD3 and GFP were detected by anti‐CDKL5, anti‐SMAD3 and anti‐GFP antibodies, respectively. D. CDKL5 phosphorylates SMAD3 at the MH1 domain. Kinase assays were conducted with purified CDKL5DC (1‐498aa) and SMAD3 or SMAD3 mutants. Samples were resolved by SDS‐PAGE, transferred onto nitrocellulose membrane and exposed to film by autoradiography. CDKL5DC was detected with PonceauS staining (lower panel). E. Immunoprecipitated FLAG‐tagged wild‐type CDKL5 was subjected to an in vitro kinase assay to test its ability to phosphorylate purified SMAD3 and SMAD2. Samples were resolved by SDS‐PAGE, transferred onto nitrocellulose membrane and exposed to film by autoradiography. The same membrane was subjected to immunoblot analyses using anti‐ SMAD3 and SMAD2 antibodies. [Corrections added on 10 February 2020, after first online publication: Figure 2 and the legend have been corrected in this version.]
To determine whether SMAD3 is a direct phosphorylation substrate for CDKL5, we immunoprecipitated overexpressed SMAD3 from transfected HEK293T cells and incorporated it into a reaction mixture containing [γ‐ 32P]ATP, in the presence of increasing concentrations of the CDKL5 kinase domain (amino acids 1‐498; CDKL5ΔC). We observed a CDKL5ΔC dose‐dependent increase in SMAD3 phosphorylation (Figure 2D), indicating that SMAD3 is a direct CDKL5 phosphorylation target. To determine the critical domain in SMAD3 phosphorylated by CDKL5, the SMAD3 deletion constructs MH1, L‐MH2, and MH2 were incubated with CDKL5ΔC. While L‐MH2 and MH2 were not phosphorylated in the presence of CDKL5, MH1 was highly phosphorylated (Figure 2D). Confirming previous evidence 5, 45, we found that CDKL5ΔC exhibits autophosphorylation activity, which increased in the presence of a target protein (Figure 2D).
SMAD3 and SMAD2 are closely related TGF‐β downstream effectors with 92% amino acid sequence similarity 7. To investigate whether CDKL5 specifically phosphorylates SMAD3 and not SMAD2, we compared the effect of wild‐type CDKL5 kinase activity on SMAD3 and SMAD2. Full‐length CDKL5 phosphorylated SMAD3, similarly to CDKL5ΔC, but did not phosphorylate SMAD2 (Figure 2E), indicating a specific CDKL5/SMAD3 interaction.
CDKL5‐mediated phosphorylation of SMAD3 is required for SMAD3 protein stability
Various types of phosphorylation of SMAD3, mediated by protein kinases and phosphatases, have been reported to affect its activity, stability, and localization in cells 63, 73, 75. SMAD3 regulates transcription of genes by binding to specific sequences within the promoter of target genes and by interacting with other proteins 57. To explore whether CDKL5‐dependent phosphorylation affects the transcriptional activity of SMAD3, we performed assays with a luciferase reporter that is sensitive to SMAD3 (Figure 3A). CAGA(12)‐luc reporter, containing CAGA elements in the promoter which bind activated SMAD3 16, was transfected into SH‐SY5Y cells. Expression of CDKL5 did not modify luciferase activity in SH‐SY5Y cells (Figure 3A), indicating that CDKL5 does not directly affect SMAD3 transcriptional activity. Treatment with TGF‐β1 strongly increased SMAD3 transcriptional activity (Figure 3A), while treatment with SB431542 (SB), a potent and specific inhibitor of TGF‐β receptor, decreased SMAD3 transcriptional activity (Figure 3A), indicating the presence of a functional TGF‐β/SMAD3 signaling in SH‐SY5Y cells.
Figure 3.
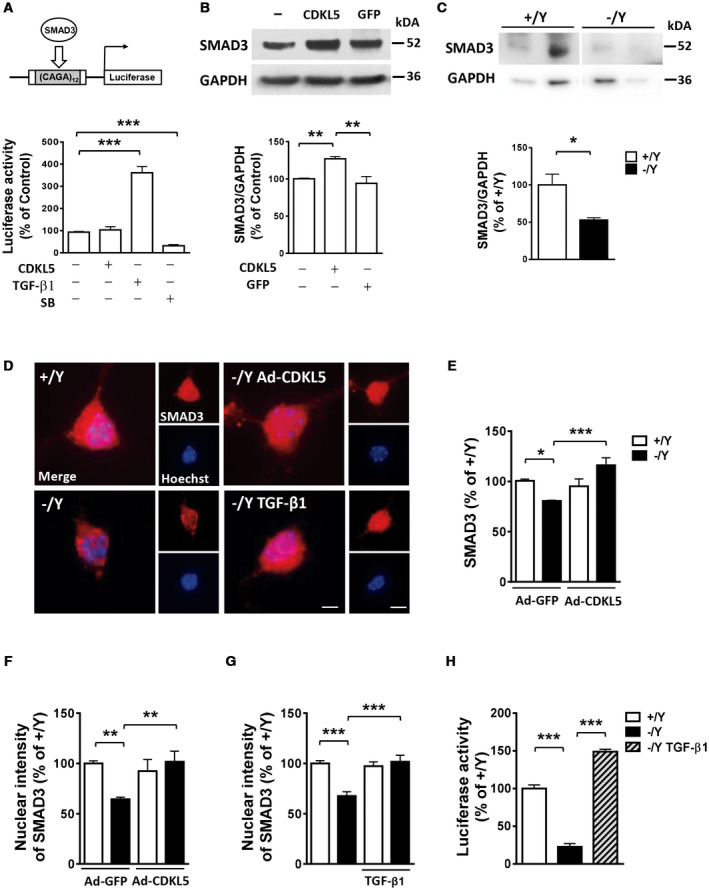
CDKL5 phosphorylation regulates SMAD3 protein levels. A. Luciferase reporter analysis of SMAD3‐dependent promoter (CAGA12‐luc reporter; schematic representation in the upper panel) in SH‐SY5Y cells transfected with CDKL5 or treated with TGF‐β1 (5 ng/ml) or SB431542 (SB; 10 µM). B. Western blot analysis of SMAD3 levels normalized to GAPDH levels in SH‐SY5Y cells infected with CDKL5 adenoviral particles (Ad‐CDKL5; n = 3), with GFP adenoviral particles (Ad‐GFP; n = 3), or not infected (n = 3). Immunoblots (upper panel) are examples from each experimental group. C. Western blot analysis of SMAD3 levels in 10‐day (DIV10) differentiated hippocampal neurons from wild‐type (+/Y, n = 5) and Cdkl5 −/Y (n = 6) mice. Immunoblots (upper panel) are two examples from each experimental group. D. Representative fluorescent images of 10‐day (DIV10) differentiated hippocampal neurons from wild‐type (+/Y) and Cdkl5 −/Y mice immunopositive for SMAD3 and counterstained with Hoechst. SMAD3 localizes both in the nucleus and in the cytoplasm. Cdkl5 −/Y hippocampal cultures were infected with adenoviral particle for CDKL5 (Ad‐CDKL5) or GFP as a control (Ad‐GFP) on DIV3, or treated with TGF‐β1 (1 ng/ml) administered on alternate days starting from DIV2. Scale bar = 1.5 µm higher magnification, 6 μm lower magnification. E. Quantification of SMAD3 signal intensity in hippocampal neurons infected with adenoviral particle for GFP (Ad‐GFP; +/Y n = 5, −/Y n = 4) or CDKL5 (Ad‐CDKL5; +/Y n = 5, −/Y n = 4). F, G. Quantification of SMAD3 nuclear signal intensity in hippocampal neurons infected with adenoviral particles for GFP (Ad‐GFP ; +/Y n = 5, −/Y n = 4) or CDKL5 (Ad‐CDKL5; +/Y n = 5, −/Y n = 4) in F and untreated (+/Y n = 5, −/Y n = 5) or treated with TGF‐β1 (+/Y n = 4, −/Y n = 5) in G. H. Luciferase reporter analysis of SMAD3‐dependent promoter in primary hippocampal neurons from wild‐type (+/Y, n = 5) and Cdkl5 −/Y (n = 5) mice and in Cdkl5 −/Y cultures treated with TGF‐β1 (5 ng/ml; n = 5). Data are expressed as a percentage of the values of control samples. Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Unpaired t‐test in B, C; Fisher’s LSD after ANOVA in A, E–H).
Since phosphorylation levels could affect SMAD3 basal turnover and protein stability 29, 35, 43, 68 we hypothesized that CDKL5‐dependent phosphorylation of SMAD3 might affect SMAD3 protein levels. To support this hypothesis we over‐expressed CDKL5 or GFP as a control in SH‐SY5Y neuroblastoma cells. We found higher SMAD3 protein levels in cells expressing CDKL5, compared to SH‐SY5Y cells that were not infected or expressing GFP (Figure 3B). Similarly, in a neuroblastoma cell line, SKNBE, that does not express endogenous CDKL5 66, we found that the stability of co‐expressed SMAD3 protein was affected by whether it was CDKL5 or CDKL5 lacking the kinase domain (CDKL5ΔN) that was expressed (Supporting Figure 2A). We found higher SMAD3 protein levels in SKNBE cells expressing CDKL5, compared to cells expressing CDKL5ΔN (Supporting Figure 2A). Moreover, as observed in vivo in Cdkl5 −/Y mice (Figure 1E), we found reduced SMAD3 levels in hippocampal cultures from Cdkl5 −/Y mice compared to wild‐type mice (Figure 3C,D), which were paralleled by a reduced SMAD3 nuclear intensity (Figure 3F–G). Re‐expression of CDKL5 in Cdkl5 −/Y neurons restored SMAD3 protein levels (Figure 3D,E) and, consequently, its nuclear intensity (Figure 3D,F), suggesting a CDKL5‐dependent regulation of SMAD3 protein levels. In a similar way, treatment with TGF‐β1 was able to restore SMAD3 nuclear intensity in Cdkl5 −/Y hippocampal neurons (Figure 3D,G).
In agreement with reduced SMAD3 nuclear levels, we found a decreased SMAD3‐dependent transcriptional activity in hippocampal cultures from Cdkl5 −/Y mice compared to wild‐type mice (Figure 3H). Treatment with TGF‐β1 in Cdkl5 −/Y neurons restored SMAD3 activity to control levels (Figure 3H).
Increased TGF‐β1‐induced SMAD3 transcriptional activity is mediated by TGF‐β type I receptor‐induced SMAD3 phosphorylation at the Ser213 site in the linker region 8. As expected we found increased SMAD3 phosphorylation at Ser213 in hippocampal cultures from both Cdkl5 KO and wild‐type mice treated with TGF‐β1 (Supporting Figure 2B,C). Differently, as observed in vivo in Cdkl5 −/Y mice (Supporting Figure 1C), a lack of Cdkl5 did not affect SMAD3 phosphorylation at Ser213 in hippocampal cultures (Supporting Figure 2B), suggesting that TGF‐β1 furthers SMAD3 activity through a CDKL5‐independent pathway.
Restoration of TGF‐β/SMAD3 signaling in primary hippocampal neurons from Cdkl5 KO mice recovers neuronal survival and maturation
Based on evidence that TGF‐β/SMAD signaling regulates many physiological processes in the brain, including neuronal survival, development, and activity 17, we sought to investigate whether restoration of TGF‐β/SMAD3 signaling improves the neurodevelopmental alterations that characterize Cdkl5 −/Y hippocampal neurons 25, 26, 64.
The assessment of apoptotic cell death revealed that differentiating hippocampal neurons generated from Cdkl5 −/Y mice had more apoptotic (cleaved caspase‐3 positive) cells compared to control cultures (Figure 4A). We found that treatment with TGF‐β1 restored the number of cleaved caspase‐3 positive cells in hippocampal cultures from Cdkl5 −/Y mice (Figure 4A), suggesting that TGF‐β/SMAD3 signaling plays a role in CDKL5‐dependent neuronal survival. As expected, the re‐expression of CDKL5 restored the number of apoptotic cells in hippocampal cultures from Cdkl5 −/Y mice (Supporting Figure 3A).
Figure 4.
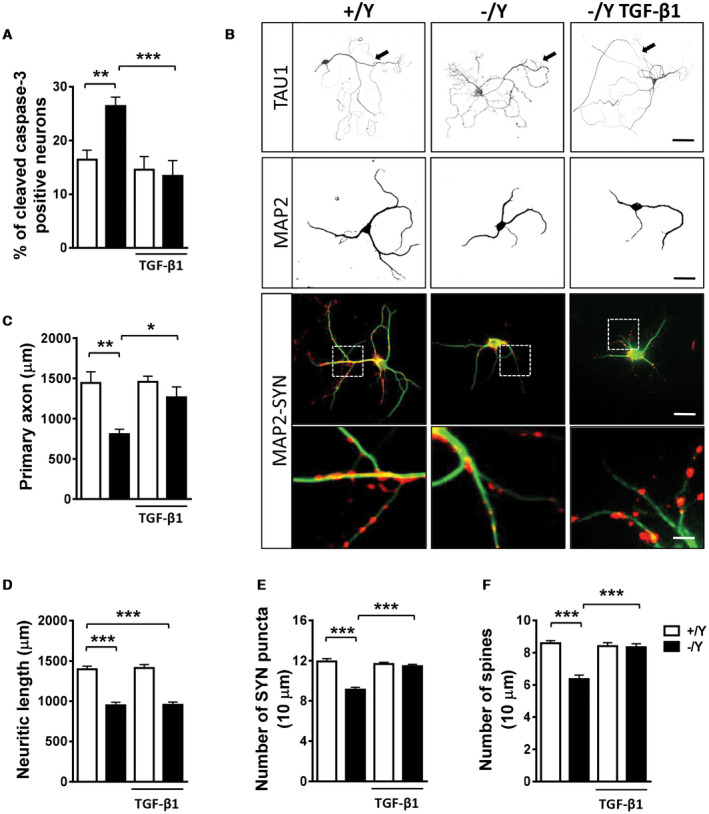
Effect of treatment with TGF‐β1 on survival and maturation of hippocampal neurons from Cdkl5 KO mice. A. Percentage of cleaved caspase‐3 positive neurons in 4‐day differentiated (DIV4) hippocampal neurons from wild‐type (+/Y n = 5) and Cdkl5 −/Y (n = 5) mice. Hippocampal cultures were treated with TGF‐β1 (1 ng/ml) on day 2 postplating (DIV2). B. Representative images of 10‐day (DIV10) differentiated +/Y and −/Y hippocampal neurons and −/Y hippocampal neurons treated with TGF‐β1 (1 ng/ml), administered on alternate days starting from DIV2, immunopositive for the axon marker TAU1 (upper panel; scale bar = 50 µm, arrows indicate the primary axon), microtubule‐associated protein 2 (MAP2; scale bar = 30 µm), or MAP2 (green) plus synaptophysin (SYN, red). The dotted boxes indicate the regions shown at a higher magnification. Scale bar = 30 μm lower magnification, 2.5 μm higher magnification. C–F. Quantification of the length of the primary axon (C, TAU1‐positive; +/Y = 4, −/Y = 4), the total length of MAP2‐positive neurites (D, +/Y = 6, −/Y = 6), the number of SYN‐immunoreactive puncta per 10 μm in proximal dendrites (E, +/Y = 6, −/Y = 6), and the number of MAP2‐positive spines (F, +/Y = 6, −/Y = 6) from differentiated hippocampal cultures from Cdkl5 +/Y and Cdkl5 −/Y mice treated as in (B). Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Fisher’s LSD after ANOVA).
Hippocampal neurons from Cdkl5 −/Y mice are characterized by reduced axon [Figure 4B,C; 51] and neurite [Figure 4B,D; 64] outgrowth. We found that treatment with TGF‐β1 restored primary axon length in hippocampal neurons from Cdkl5 −/Y mice (Figure 4B,C), but did not improve the reduced neurite outgrowth (Figure 4B,D), suggesting that the TGF‐β signal has a specific involvement in axonal development. The re‐expression of CDKL5 restored both primary axon length and neurite outgrowth (Supporting Figure 3B,C).
As previously reported 64, the assessment of synaptophysin (SYN) puncta in neurites revealed that hippocampal neurons from Cdkl5 −/Y mice had a reduction in the number of presynaptic connections (Figure 4B,E). Treatment with TGF‐β1 restored the number of SYN puncta in hippocampal neurons from Cdkl5 −/Y mice (Figure 4B,E). Confirming the reduced number of synaptic connections, we found a reduced spine density in hippocampal neurons from Cdkl5 −/Y mice compared to those of control cultures (Figure 4F, Supporting Figure 3F). Treatment with TGF‐β1 restored the density of dendritic spines in hippocampal neurons from Cdkl5 −/Y mice (Figure 4F, Supporting Figure 3F). As expected, the re‐expression of CDKL5 restored synaptic connections (Supporting Figure 3D,E). In control neurons treatment with TGF‐β1 or increased CDKL5 levels had no effect on neuronal survival (Figure 4A; Supporting Figure 3A), axon and neurite growth (Figure 4D; Supporting Figure 3B,C), or connectivity (Figure 4E,F; Supporting Figure 3D–F).
Increased susceptibility to neurotoxic stress in primary hippocampal neurons from Cdkl5 KO mice is rescued by treatment with TGF‐β1
Primary hippocampal neurons are known to be susceptible to excitotoxicity and oxidative stress, which lead to the induction of apoptotic cell death 9, 10, 33, 71. Numerous studies have shown a protective effect of TGF‐β signaling against various toxins and injurious agents in cultured neurons 6, 22, 65. To test the hypothesis that CDKL5, with its function on TGF‐β/SMAD3 signaling regulation, is required for neuronal apoptotic resistance, we exposed hippocampal neuronal cultures from Cdkl5 −/Y mice to an oxidative stress (100 μM H2O2) or an excitotoxic stimulus (100 μM NMDA). Apoptotic cell death was evaluated using cleaved caspase‐3 immunocytochemistry or Hoechst staining to visualize pyknotic nuclei. Interestingly, neuronal vulnerability to H2O2‐ or NMDA‐induced apoptosis was higher in hippocampal neurons from Cdkl5 −/Y mice in comparison with control neurons (Figure 5A–D). Treatment with TGF‐β1 after H2O2 or NMDA exposure prevented apoptosis in hippocampal neuronal cultures from Cdkl5 −/Y mice (Figure 5A–D).
Figure 5.
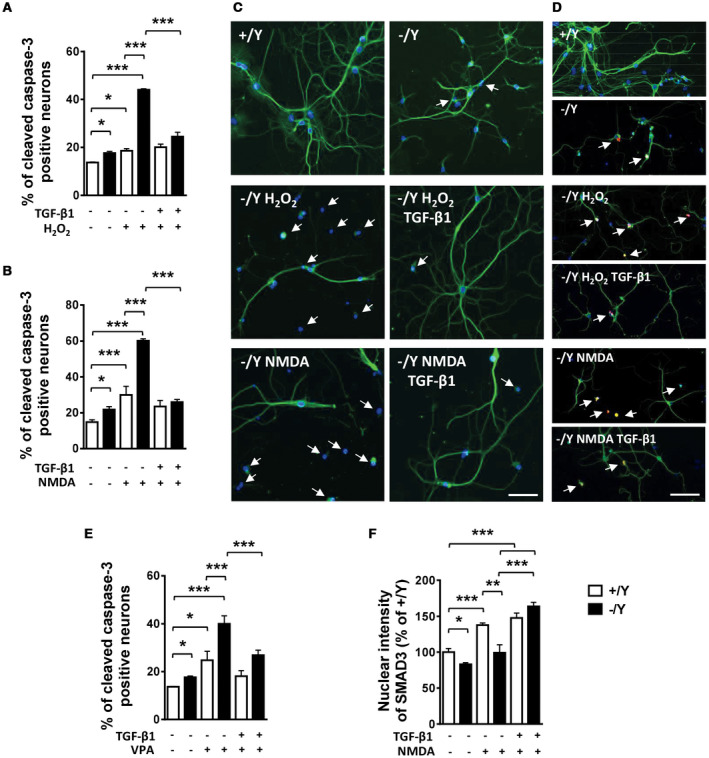
TGF‐β1 treatment rescues the increased susceptibility to neurotoxic stress of hippocampal neurons from Cdkl5 KO mice. A, B. Percentage of cleaved caspase‐3 positive neurons in primary hippocampal neurons from wild‐type and Cdkl5 −/Y mice. Hippocampal cultures were treated on DIV10 with H2O2 (100 μM; +/Y n = 4, −/Y n = 4) or H2O2 + TGF‐β1 (1 ng/ml; +/Y n = 4, −/Y n = 4) in A, and NMDA (100 μM; +/Y n = 4, −/Y n = 4) or NMDA + TGF‐β1 (1 ng/ml; +/Y n = 3, −/Y n = 4) in B, and fixed after 24 h. C. Representative fluorescent images of differentiated hippocampal neurons from wild‐type (+/Y) and Cdkl5 −/Y mice immunopositive for MAP2 (green) and stained with Hoechst (blue). Cultures were treated as in (A, B). White arrows indicate pyknotic nuclei. Scale bar = 30 μm. D. Representative fluorescent images of differentiated hippocampal neurons from wild‐type (+/Y) and Cdkl5 −/Y mice immunopositive for MAP2 (green) and cleaved caspase 3 (red), and stained with Hoechst (blue). Cultures were treated as in (A, B). White arrows indicate apoptotic cells positive for cleaved caspase 3. Scale bar = 40 μm. E. Percentage of cleaved caspase‐3 positive neurons over total neuron number from wild‐type and Cdkl5 −/Y mice. Hippocampal cultures were treated with VPA (1 mM; +/Y n = 4, −/Y n = 4) or VPA + TGF‐β1 (1 ng/ml; +/Y n = 4, −/Y n = 4). F. Quantification of SMAD3 signal intensity in untreated (+/Y n = 3, −/Y n = 3), NMDA‐treated (+/Y n = 4, −/Y n = 3), and NMDA + TGF‐β1‐treated (+/Y n = 3, −/Y n = 3) hippocampal neurons immunostained for SMAD3. Data in E are expressed as a percentage of the values of untreated +/Y. Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Fisher’s LSD after ANOVA).
The developmental neurotoxicity of the commonly used antiepileptic drug valproic acid (VPA) on differentiating hippocampal neurons is well recognized 69. After exposing differentiating hippocampal neurons to VPA (1 mM) for 4 days, we found a higher neuronal vulnerability to VPA in hippocampal neurons from Cdkl5 −/Y mice in comparison with control neurons (Figure 5E); this increased susceptibility was prevented by treatment with TGF‐β1 (Figure 5E).
In order to elucidate the mechanism underlying the higher neuronal vulnerability associated with Cdkl5 loss of function, we investigated the effect of NMDA on TGF‐β/SMAD3 signaling activation by evaluating SMAD3 nuclear‐immunopositivity. NMDA‐induced excitatory stimulation resulted in an increase in SMAD3 nuclear levels in control cultures (Figure 5F). However, Cdkl5 −/Y neurons did not show a significant increase in SMAD3 levels after NMDA exposure (Figure 5F), suggesting an impaired NMDA‐induced SMAD3 activation in the absence of CDKL5. On the contrary, after TGF‐β1 treatment, similarly increased SMAD3 levels were detected in hippocampal neurons from Cdkl5 −/Y and control mice (Figure 5F).
Treatment with TGF‐β1 protects CA1 pyramidal neurons from Cdkl5 KO mice against NMDA‐induced cell death
To determine whether Cdkl5 KO neurons are also more susceptible to excitotoxic stimuli in vivo, we injected Cdkl5 −/Y and wild‐type (+/Y) mice intraperitoneally with NMDA (60 mg/kg; Figure 6A). No difference in seizure intensity was observed between Cdkl5 −/Y and wild‐type (+/Y) mice in the 35 minutes following NMDA administration. Seizure intensity reached a maximum of stage 3 on a 0–5 seizure scale, between 10 and 20 minutes following NMDA administration (Figure 6B). Similarly, a low mortality rate was observed in treated Cdkl5 −/Y (two out of 17 treated mice) and wild‐type (1 out of 13 treated mice) mice.
Figure 6.
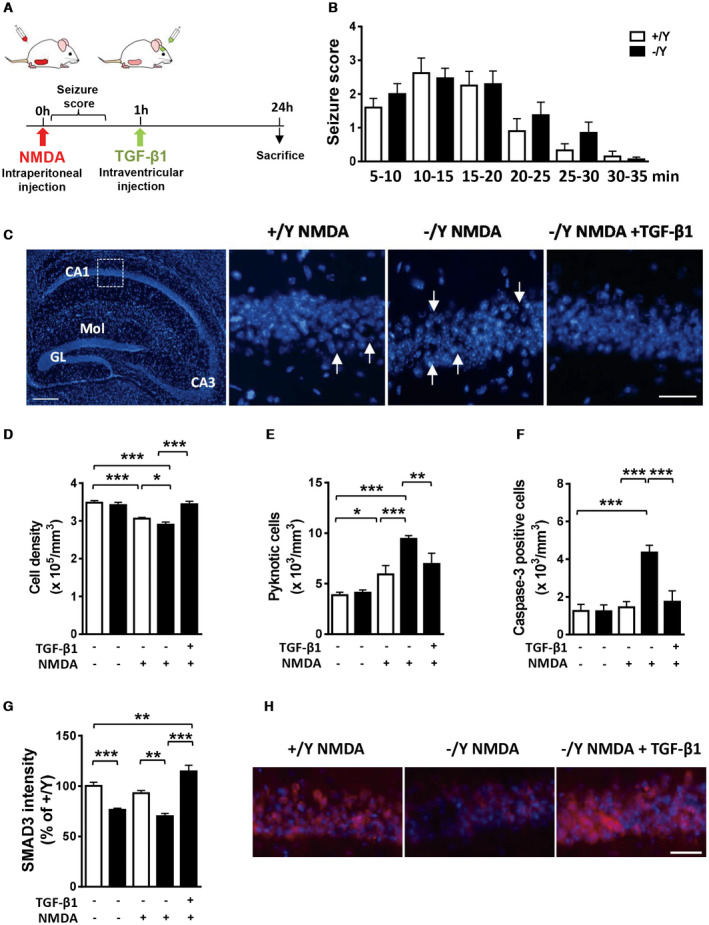
Effect of treatment with TGF‐β1 on NMDA‐induced hippocampal neuron cell death in Cdkl5 KO mice. A. Schematic view of in vivo treatments and analysis schedule. B. Graph represents NMDA‐induced seizure score for wild‐type (+/Y, n = 13) and Cdkl5 −/Y (n = 17) mice at indicated time points after NMDA injection. C: Left panel: representative fluorescence image of a hippocampal section processed for Hoechst staining. Abbreviations: GL, granule cell layer; Mol, molecular layer. Scale bar = 150 μM. The dotted box in the panel indicates the analyzed region (CA1). Magnifications in the right‐hand side panels show examples of the pyramidal neuron layer in CA1 of a Cdkl5 +/Y and a Cdkl5 −/Y mouse treated with NMDA (60 mg/kg), and of a Cdkl5 −/Y mouse treated with NMDA and TGF‐β1 (50 ng). Arrows indicate the neuronal damage sites. Scale bar = 100 μM. D–F. Quantification of Hoechst‐positive cells (D), number of pyknotic nuclei (E), and number of cleaved caspase‐3 positive cells (F) in CA1 of hippocampal sections from untreated (+/Y n = 5, −/Y n = 5), NMDA‐treated (+/Y n = 8, −/Y n = 9), and NMDA + TGF‐β1 treated (−/Y n = 5) mice. G: Quantification of SMAD3 signal intensity in the CA1 pyramidal neuron layer of Cdkl5 +/Y and Cdkl5 −/Y mice treated as in D. H. Representative fluorescent images of the pyramidal neuron layer in CA1 of a Cdkl5 +/Y and a Cdkl5 −/Y mouse treated with NMDA (60 mg/kg), and of a Cdkl5 −/Y mouse treated with NMDA and TGF‐β1 (50 ng) immunostained for SMAD3 and counterstained with Hoechst. Scale bar = 50 μM. Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Fisher’s LSD after ANOVA).
Neuronal damage was assessed 24 h after NMDA injection using Hoechst staining and immunohistochemistry for cleaved caspase‐3. In the CA1 layer of the hippocampus, treated Cdkl5 −/Y mice showed a lower cell density (Figure 6C,D) and a higher number of pyknotic (Figure 6E) and cleaved caspase‐3 positive cells (Figure 6F) in comparison with treated wild‐type mice, indicating that Cdkl5 −/Y mice are more vulnerable to neurotoxicity/neurodegeneration induced by treatment with NMDA. To test whether TGF‐β1 is sufficient to protect Cdkl5 KO neurons against NMDA‐induced cell death, we administered TGF‐β1 to Cdkl5 −/Y mice via intracerebroventricular infusion 1‐h after NMDA treatment (Figure 6A). Treatment with TGF‐β1 prevented neurodegeneration of CA1 pyramidal neurons in Cdkl5 −/Y mice (Figure 6C–F).
To determine whether SMAD3 is involved in TGF‐β1‐induced neuroprotection against NMDA‐induced cell death, we quantified SMAD3 expression in the CA1 layer of the hippocampus of Cdkl5 −/Y mice 1 h after TGF‐β1 treatment (Figure 6A). NMDA‐treated Cdkl5 −/Y mice showed lower SMAD3 levels in comparison with NMDA‐treated Cdkl5 +/Y mice, with a difference of the same amplitude as that found between vehicle‐treated Cdkl5 +/Y and Cdkl5 −/Y mice (Figure 6G–H). Treatment with TGF‐β1 strongly increased SMAD3 levels in CA1 pyramidal neurons of NMDA‐treated Cdkl5 −/Y mice (Figure 6G–H), suggesting that SMAD3 plays a functional role in TGF‐β1‐induced neuroprotection.
Discussion
Reduced SMAD3 levels caused by Cdkl5 loss of function impair neuron survival and maturation
SMADs play a central role in neuronal survival, maturation, and specification of cell types, by integrating transforming growth factor (TGF)‐β signaling with other essential pathways 21, 32, 47. In the current study, we provide novel evidence that SMAD3 is a direct phosphorylation target of CDKL5. Despite the extensive sequence similarity between SMAD2 and SMAD3, we found that CDKL5 specifically phosphorylates SMAD3 at the MH1 domain. While the MH2 domain is highly conserved among all SMADs, the different structure of the MH1 domain of SMAD3, which does not contain the 30‐amino‐acid insert that is present in SMAD2 15, could explain the specificity of the CDKL5‐dependent phosphorylation of SMAD3. Though recent observation has raised the possibility that the RPXSA motif might represent a consensus sequence for phosphorylation by CDKL5 3, 41, 50, other studies have identified CDKL5 phosphorylation targets that do not contain this consensus motif 40, 48, 55, 64, suggesting the presence of a different consensus sequence for CDKL5 phosphorylation or of a protein folding that creates a noncontiguous CDKL5 phosphorylation motif 19. SMAD3 seems to belong to this latter group because the MH1 domain does not present a RPXSA motif. Further studies will be needed to identify the CDKL5 phosphorylation site on SMAD3.
We additionally found that CDKL5‐dependent phosphorylation of SMAD3 does not directly affect SMAD3 activity, but that CDKL5 deficiency causes a reduction in SMAD3 protein levels and, consequently, in activity in Cdkl5 KO neurons. Phosphorylation at different sites of SMAD3 contributes to its stability 28, 29. Most SMAD proteins can be polyubiquitinated and degraded in either a ligand‐dependent or a ligand‐independent manner 38. Although it still remains to be established how CDKL5‐dependent phosphorylation affects SMAD3 protein stability, we found that treatment with TGF‐β1, similarly to CDKL5 re‐expression, normalized SMAD3 levels, indicating a reversible SMAD3 stability/activity in Cdkl5 KO neurons. We found that CDKL5 does not phosphorylate SMAD3 at the C‐terminal residues Ser425 in the MH2 domain or at the Ser213 site in the linker region, phosphorylation sites that drive TGF‐β type I receptor‐induced SMAD3 nuclear localization and stability 8. On the other hand, there is no evidence that TGF‐β has a direct action on the SMAD3 MH1 domain 8. The finding that TGF‐β1 can rescue SMAD3 levels and transcriptional activity in the absence of CDKL5 suggests that TGF‐β1 modulates SMAD3 stability through a CDKL5‐independent pathway.
Importantly, we provide novel evidence that loss of Cdkl5 increases cell death of differentiating hippocampal neurons. The restoration of SMAD3 activity through TGF‐β1 treatment fully rescued survival of Cdkl5 KO neurons, suggesting that SMAD3 signaling dysregulation is involved in the reduced survival of Cdkl5 KO neurons. Consistent with our findings, primary neurons lacking TGF‐β1 showed a reduced survival rate compared with wild‐type controls 6. Moreover, studies on SMAD3‐deficient mice have revealed that SMAD3 plays a role in trophic support for nigral dopaminergic neurons 61 in addition to the maintenance of survival of newborn granule cells in the hippocampal dentate gyrus 62.
In addition to its role in neuronal survival we found that restoration of SMAD3 signaling recovered the primary axon outgrowth and reduced connectivity caused by Cdkl5 loss of function. Contrariwise, treatment with TGF‐β1 did not improve the dendritic hypotrophy that characterizes Cdkl5 KO neurons. This is in agreement with previous evidence that indicates that TGF‐β1 signaling is required for axon specification in the developing brain 77, and for synaptic growth and function 11, 58, while it does not affect the process of branching and the number of dendrite‐like processes in hippocampal neurons 37.
Increased vulnerability of Cdkl5 KO hippocampal neurons to neurotoxic stress is rescued by treatment with TGF‐β1
Our data provide the first evidence that CDKL5 has a key role in neuronal survival, and indicate that CDKL5 deficiency increases the vulnerability of neural cells to apoptosis induced by different types of neurotoxic stress. It is becoming increasingly clear that various types of cell death cascades can share pathways in death execution 76. Our finding that the increased neuronal vulnerability to oxidative stress, VPA, and excitotoxic injury shown by Cdkl5 KO hippocampal neurons is fully recovered by TGF‐β1 treatment, suggests that SMAD3 signaling deregulation might be the common mechanism responsible for the enhanced vulnerability of Cdkl5 KO neurons. The neuroprotective role of TGF‐β/SMAD3 signaling in the injured CNS is increasingly being recognized 17. TGF‐β1 deficiency in adult TGF‐β1−/+ mice does not result in overt neurodegeneration but does increase neuronal cell loss after excitotoxic injury 6. Moreover, SMAD3 deficiency increases cortical and hippocampal neuronal loss following traumatic brain injury 67. Therefore, the reduced SMAD3 activation in NMDA‐treated Cdkl5 KO mice might underlie the increased neurodegeneration observed in the absence of Cdkl5. Moreover, our observations that the increased NMDA‐induced cell death of hippocampal neurons of Cdkl5 KO mice was fully rescued by TGF‐β1 treatment correlates well with previous studies that show that activation of TGF‐β/SMAD3 dependent signaling protects neurons against NMDA‐induced cell death 18, 54.
A recent study in a Cdkl5 KO mouse model [Cdkl5 exon 2 deletion; 52] showed increased seizure susceptibility in response to NMDA that was correlated with an upregulation of the NMDA receptor subunits GluN2B at the CA1 hippocampal glutamatergic synapses 52. We did not observe an increase in NMDA‐induced tonic‐clonic seizure episodes in the Cdkl5 KO mouse model used in this study [deletion of Cdkl5 exon 4; 1]. Therefore, the increased hippocampal neuron vulnerability that we observed in Cdkl5 KO mice in response to NMDA treatment is unlikely to be caused by a higher NMDA‐induced neuronal hyper‐excitability, and is, rather, ascribable to a generalized increased susceptibility to neurotoxic stress because of the impairment of SMAD3 signaling.
Several neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and motor neuron diseases have been associated with disturbed cellular or subcellular SMAD localization and disruption of SMAD‐controlled transcriptional machinery 17. Our findings suggest that, through the deregulation of SMAD3 signaling, CDKL5 loss of function predisposes neurons to multiple forms of cell death. The endangering action of CDKL5 mutations is likely to sensitize neurons in the brain to neurotoxic conditions known to promote neuronal death. Seizures are prominent in CDD patients, and are usually severe and untreatable. Conceivably, individuals with CDKL5 deficiency may be more susceptible to oxidative stress as a direct consequence of seizures 23. Therefore, preventive and therapeutic strategies that target both excitotoxic and apoptotic pathways might be recommended to forestall neurodegenerative processes in CDD.
Disclosures
All authors reported that they had no biomedical financial interests or potential conflicts of interest.
Supporting information
Figure S1. A. Western blot analysis of SMAD2 levels normalized to GAPDH levels in the somatosensory cortex of wild‐type (+/Y; n = 4) and Cdkl5 −/Y (n = 4) adult mice. Immunoblots are examples from two animals of each experimental group. B. Representative images of hippocampal sections at the CA1 field level processed for fluorescent SMAD3 immunostaining of wild‐type (+/Y) and Cdkl5 −/Y mice. Scale bar = 40 μm. C. Western blot analysis of P‐SMAD3 (Ser213 and Ser425) levels normalized to SMAD3 levels in the somatosensory cortex of wild‐type (+/Y; n = 4) and Cdkl5 −/Y (n = 4) adult mice. Immunoblots are examples from one animal of each experimental group. D. Quantification by RT‐qPCR of SMAD3 expression in the somatosensory cortex of wild‐type (+/Y; n = 5) and Cdkl5 −/Y (n = 6) mice. Data are expressed as a percentage of the values of Cdkl5 +/Y mice. Values are represented as means ± SE.
Figure S2. A. Western blot analysis of SMAD3 levels normalized to GAPDH levels in SKNBE cells co‐transfected with CDKL5ΔN (n = 4) or wild‐type CDKL5 (n = 4) and SMAD3. Immunoblots (upper panel) are three examples from each experimental group. B. Western blot analysis of P‐SMAD3 (Ser213) levels normalized to SMAD3 levels in untreated hippocampal cultures (+/Y = 9; −/Y = 14) or treated for 1h with TGF‐β1 (−/Y = 8). C. Representative fluorescent images of 10‐day (DIV10) differentiated hippocampal neurons from wild‐type (+/Y) mice immunopositive for P‐SMAD3 (Ser213) and counterstained with Hoechst. Cdkl5 +/Y hippocampal cultures were treated with TGF‐β1 (1 ng/ml) for 1h. Scale bar = 2.5 µm. Quantification of P‐SMAD3 (Ser213) signal intensity in untreated (+Y n = 2) or TGF‐β1 treated (+Y n = 2) hippocampal neurons. Values are represented as means ± SE. *P < 0.05; ***P < 0.001 (Unpaired t‐test in A; Fisher’s LSD after ANOVA in B).
Figure S3. Primary hippocampal cultures were infected with adenoviral particle for GFP (Ad‐GFP) or CDKL5 (Ad‐CDKL5) on the third day (DIV3) in culture, and immunostained for cleaved caspase‐3 on day 4 or for TAU1, MAP2 or MAP2 + SYN on day 10. A. Percentage of cleaved caspase‐3 positive neurons in 4‐day differentiated hippocampal neurons from wild‐type (+/Y n = 5) and Cdkl5 −/Y (n = 5) mice. B–E. Quantification of the length of the primary axon (B, TAU1‐positive; +/Y = 4, −/Y = 4), the total length of MAP2‐positive neurites (C, +/Y = 6, −/Y = 6), the number of SYN‐immunoreactive puncta per 10 μm in proximal dendrites (D, +/Y = 6, −/Y = 6), and the number of MAP2‐positive spines (E, +/Y = 6, −/Y = 6) from differentiated hippocampal cultures from Cdkl5 +/Y and Cdkl5 −/Y mice infected with GFP or CDKL5 adenoviral particles. F. Representative fluorescence images of proximal dendrite segments of hippocampal neurons that were immunopositive for MAP2 showing spine protrusions. Scale bar = 2.5 μm. Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Fisher’s LSD after ANOVA).
Acknowledgments
This work was supported by the Telethon foundation [grant number GGP15098 to E. C.] and by the Italian parent association “CDKL5 insieme verso la cura.”
Data Availability Statement
Data are stored and available on University servers.
Data Availability Statement
Data are stored and available on University servers.
References
- 1. Amendola E, Zhan Y, Mattucci C, Castroflorio E, Calcagno E, Fuchs C et al (2014) Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PloS one 9:e91613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bahi‐Buisson N, Bienvenu T (2012) CDKL5‐related disorders: from clinical description to molecular genetics. Mol Syndromol 2:137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baltussen LL, Negraes PD, Silvestre M, Claxton S, Moeskops M, Christodoulou E et al (2018) Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J 37:e99763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbiero I, Peroni D, Tramarin M, Chandola C, Rusconi L, Landsberger N, Kilstrup‐Nielsen C (2017) The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5‐IQGAP1 complex. Hum Mol Gen 26:3520–3530. [DOI] [PubMed] [Google Scholar]
- 5. Bertani I, Rusconi L, Bolognese F, Forlani G, Conca B, De Monte L et al (2006) Functional consequences of mutations in CDKL5, an X‐linked gene involved in infantile spasms and mental retardation. J Biol Chem 281:32048–32056. [DOI] [PubMed] [Google Scholar]
- 6. Brionne TC, Tesseur I, Masliah E, Wyss‐Coray T (2003) Loss of TGF‐beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 40:1133–1145. [DOI] [PubMed] [Google Scholar]
- 7. Brown KA, Pietenpol JA, Moses HL (2007) A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF‐beta signaling. J Cell Biochem 101:9–33. [DOI] [PubMed] [Google Scholar]
- 8. Bruce DL, Sapkota GP (2012) Phosphatases in SMAD regulation. FEBS Lett 586:1897–905. [DOI] [PubMed] [Google Scholar]
- 9. Calvo M, Sanz‐Blasco S, Caballero E, Villalobos C, Nunez L (2015) Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non‐steroidal anti‐inflammatory drugs: role of mitochondrial calcium. J Neurochem 132:403–417. [DOI] [PubMed] [Google Scholar]
- 10. Chen X, Zhang Q, Cheng Q, Ding F (2009) Protective effect of salidroside against H2O2‐induced cell apoptosis in primary culture of rat hippocampal neurons. Mol Cell Biochem 332:85–93. [DOI] [PubMed] [Google Scholar]
- 11. Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH (2002) Transforming growth factor beta1 alters synapsin distribution and modulates synaptic depression in Aplysia. J Neurosci 22:RC220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daly AC, Vizan P, Hill CS (2010) Smad3 protein levels are modulated by Ras activity and during the cell cycle to dictate transforming growth factor‐beta responses. J Biol Chem 285:6489–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das DK, Mehta B, Menon SR, Raha S, Udani V (2013) Novel mutations in cyclin‐dependent kinase‐like 5 (CDKL5) gene in Indian cases of Rett syndrome. NeuroMol Med 15:218–225. [DOI] [PubMed] [Google Scholar]
- 14. Della Sala G, Putignano E, Chelini G, Melani R, Calcagno E, Michele Ratto G et al (2016) Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin‐like growth factor 1. Biol Psychiatry 80:302–311. [DOI] [PubMed] [Google Scholar]
- 15. Dennler S, Huet S, Gauthier JM (1999) A short amino‐acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 18:1643–1648. [DOI] [PubMed] [Google Scholar]
- 16. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGF beta‐inducible elements in the promoter of human plasminogen activator inhibitor‐type 1 gene. EMBO J 17:3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dobolyi A, Vincze C, Pal G, Lovas G (2012) The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci 13:8219–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Docagne F, Nicole O, Gabriel C, Fernandez‐Monreal M, Lesne S, Ali C et al (2002) Smad3‐dependent induction of plasminogen activator inhibitor‐1 in astrocytes mediates neuroprotective activity of transforming growth factor‐beta 1 against NMDA‐induced necrosis. Mol Cell Neurosci 21:634–644. [DOI] [PubMed] [Google Scholar]
- 19. Duarte ML, Pena DA, Nunes Ferraz FA, Berti DA, Paschoal Sobreira TJ, Costa‐Junior HM et al (2014) Protein folding creates structure‐based, noncontiguous consensus phosphorylation motifs recognized by kinases. Sci Signal 7:ra105. [DOI] [PubMed] [Google Scholar]
- 20. Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S et al (2013) The CDKL5 disorder is an independent clinical entity associated with early‐onset encephalopathy. Eur J Hum Genet 21:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feng XH, Derynck R (2005) Specificity and versatility in tgf‐beta signaling through Smads. Annu Rev Cell Dev Biol 21:659–693. [DOI] [PubMed] [Google Scholar]
- 22. Flanders KC, Ren RF, Lippa CF (1998) Transforming growth factor‐betas in neurodegenerative disease. Prog Neurogibol 54:71–85. [DOI] [PubMed] [Google Scholar]
- 23. Frantseva MV, Perez Velazquez JL, Tsoraklidis G, Mendonca AJ, Adamchik Y, Mills LR et al (2000) Oxidative stress is involved in seizure‐induced neurodegeneration in the kindling model of epilepsy. Neuroscience 97:431–435. [DOI] [PubMed] [Google Scholar]
- 24. Fuchs C, Fustini N, Trazzi S, Gennaccaro L, Rimondini R, Ciani E (2018) Treatment with the GSK3‐beta inhibitor Tideglusib improves hippocampal development and memory performance in juvenile, but not adult, Cdkl5 knockout mice. Eur J Neuorsci 47:1054–1066. [DOI] [PubMed] [Google Scholar]
- 25. Fuchs C, Rimondini R, Viggiano R, Trazzi S, De Franceschi M, Bartesaghi R, Ciani E (2015) Inhibition of GSK3beta rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol Dis 82:298–310. [DOI] [PubMed] [Google Scholar]
- 26. Fuchs C, Trazzi S, Roberta T, Viggiano R, De Franceschi M, Amendola E et al (2014) Loss of Cdkl5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK‐3beta signaling. Neurobiol Dis 70:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuchs C, Trazzi S, Torricella R, Viggiano R, De Franceschi M, Amendola E et al (2014) Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK‐3beta signaling. Neurobiol Dis 70:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao H, Yan P, Zhang S, Huang H, Huang F, Sun T et al (2016) Long‐term dietary alpha‐linolenic acid supplement alleviates cognitive impairment correlate with activating hippocampal CREB signaling in natural aging rats. Mol Neurobiol 53:4772–4786. [DOI] [PubMed] [Google Scholar]
- 29. Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF (2008) Axin and GSK3‐ control Smad3 protein stability and modulate TGF‐ signaling. Genes Dev 22:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He HJ, Zong Y, Bernier M, Wang L (2009) Sensing the insulin signaling pathway with an antibody array. Proteomics Clin Appl 3:1440–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hector RD, Dando O, Landsberger N, Kilstrup‐Nielsen C, Kind PC, Bailey ME, Cobb SR (2016) Characterisation of CDKL5 transcript isoforms in human and mouse. PLoS One 11:e0157758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hill CS (2009) Nucleocytoplasmic shuttling of Smad proteins. Cell Res 19:36–46. [DOI] [PubMed] [Google Scholar]
- 33. Hwang JJ, Lee SJ, Kim TY, Cho JH, Koh JY (2008) Zinc and 4‐hydroxy‐2‐nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J Neurosci 28:3114–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inman GJ (2005) Linking Smads and transcriptional activation. Biochem J 386(Pt 1):e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inoue Y, Kitagawa M, Onozaki K, Hayashi H (2004) Contribution of the constitutive and inducible degradation of Smad3 by the ubiquitin‐proteasome pathway to transforming growth factor‐beta signaling. J Interferon Cytokine Res 24:43–54. [DOI] [PubMed] [Google Scholar]
- 36. Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S et al (2011) USP15 is a deubiquitylating enzyme for receptor‐activated SMADs. Nat Cell Biol 13:1368–1375. [DOI] [PubMed] [Google Scholar]
- 37. Ishihara A, Saito H, Abe K (1994) Transforming growth factor‐beta 1 and ‐beta 2 promote neurite sprouting and elongation of cultured rat hippocampal neurons. Brain Res 639:21–25. [DOI] [PubMed] [Google Scholar]
- 38. Izzi L, Attisano L (2006) Ubiquitin‐dependent regulation of TGFbeta signaling in cancer. Neoplasia 8:677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalscheuer VM, Tao J, Donnelly A, Hollway G, Schwinger E, Kubart S (2003) Disruption of the serine/threonine kinase 9 gene causes severe X‐linked infantile spasms and mental retardation. Am J Hum Genet 72:1401‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kameshita I, Sekiguchi M, Hamasaki D, Sugiyama Y, Hatano N, Suetake I et al (2008) Cyclin‐dependent kinase‐like 5 binds and phosphorylates DNA methyltransferase 1. Biochem Biophys Res Commun 377:1162–1167. [DOI] [PubMed] [Google Scholar]
- 41. Katayama S, Sueyoshi N, Kameshita I (2015) Critical determinants of substrate recognition by cyclin‐dependent kinase‐like 5 (CDKL5). Biochemistry 54:2975–2987. [DOI] [PubMed] [Google Scholar]
- 42. Kilstrup‐Nielsen C, Rusconi L, La Montanara P, Ciceri D, Bergo A, Bedogni F, Landsberger N (2012) What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast 2012:728267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim SG, Kim HA, Jong HS, Park JH, Kim NK, Hong SH et al (2005) The endogenous ratio of Smad2 and Smad3 influences the cytostatic function of Smad3. Mol Biol Cell 16:4672–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Levy L, Howell M, Das D, Harkin S, Episkopou V, Hill CS (2007) Arkadia activates Smad3/Smad4‐dependent transcription by triggering signal‐induced SnoN degradation. Mol Cell Biol 27:6068–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin C, Franco B, Rosner MR (2005) CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet 14:3775–3786. [DOI] [PubMed] [Google Scholar]
- 46. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. [PubMed] [Google Scholar]
- 47. Macias MJ, Martin‐Malpartida P, Massague J (2015) Structural determinants of Smad function in TGF‐beta signaling. Trends Biochem Sci 40:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R et al (2005) CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early‐onset seizure variant of Rett syndrome. Hum Mol Genet 14:1935–1946. [DOI] [PubMed] [Google Scholar]
- 49. Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M et al (1998) Identification and characterization of a novel serine‐threonine kinase gene from the Xp22 region. Genomics 51:427–433. [DOI] [PubMed] [Google Scholar]
- 50. Munoz IM, Morgan ME, Peltier J, Weiland F, Gregorczyk M, Brown FC et al (2018) Phosphoproteomic screening identifies physiological substrates of the CDKL5 kinase. EMBO J 37:e99559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nawaz MS, Giarda E, Bedogni F, La Montanara P, Ricciardi S, Ciceri D et al (2016) CDKL5 and shootin1 interact and concur in regulating neuronal polarization. PloS One 11:e0148634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Okuda K, Kobayashi S, Fukaya M, Watanabe A, Murakami T, Hagiwara M et al (2017) CDKL5 controls postsynaptic localization of GluN2B‐containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol Dis 106:158–170. [DOI] [PubMed] [Google Scholar]
- 53. Poncelet AC, Schnaper HW, Tan R, Liu Y, Runyan CE (2007) Cell phenotype‐specific down‐regulation of Smad3 involves decreased gene activation as well as protein degradation. J Biol Chem 282:15534–15540. [DOI] [PubMed] [Google Scholar]
- 54. Prehn JH, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ (1994) Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide‐ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA 91:12599–12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D et al (2012) CDKL5 ensures excitatory synapse stability by reinforcing NGL‐1‐PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC‐derived neurons. Nat Cell Biol 14:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rusconi L, Salvatoni L, Giudici L, Bertani I, Kilstrup‐Nielsen C, Broccoli V, Landsberger N (2008) CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C‐terminal tail. J Biol Chem 283:30101–30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shi Y, Massague J (2003) Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 113:685–700. [DOI] [PubMed] [Google Scholar]
- 58. Sweeney ST, Davis GW (2002) Unrestricted synaptic growth in spinster‐a late endosomal protein implicated in TGF‐beta‐mediated synaptic growth regulation. Neuron 36:403–416. [DOI] [PubMed] [Google Scholar]
- 59. Tang S, Wang IJ, Yue C, Takano H, Terzic B, Pance K et al (2017) Loss of CDKL5 in glutamatergic neurons disrupts hippocampal microcircuitry and leads to memory impairment in mice. J Neurosci 37:7420–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tao J, Van Esch H, Hagedorn‐Greiwe M, Hoffmann K, Moser B, Raynaud M et al (2004) Mutations in the X‐linked cyclin‐dependent kinase‐like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 75:1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tapia‐Gonzalez S, Giraldez‐Perez RM, Cuartero MI, Casarejos MJ, Mena MA, Wang XF, Sanchez‐Capelo A (2011) Dopamine and alpha‐synuclein dysfunction in Smad3 null mice. Mol Neurodegener 6:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tapia‐Gonzalez S, Munoz MD, Cuartero MI, Sanchez‐Capelo A (2013) Smad3 is required for the survival of proliferative intermediate progenitor cells in the dentate gyrus of adult mice. Cell Commun Signal 11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tarasewicz E, Jeruss JS (2012) Phospho‐specific Smad3 signaling: impact on breast oncogenesis. Cell Cycle 11:2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Trazzi S, Fuchs C, Viggiano R, De Franceschi M, Valli E, Jedynak P et al (2016) HDAC4: a key factor underlying brain developmental alterations in CDKL5 disorder. Hum Mol Genet 25:3887–3907. [DOI] [PubMed] [Google Scholar]
- 65. Unsicker K, Krieglstein K (2000) Co‐activation of TGF‐ss and cytokine signaling pathways are required for neurotrophic functions. Cytokine Growth Factor Rev 11:97–102. [DOI] [PubMed] [Google Scholar]
- 66. Valli E, Trazzi S, Fuchs C, Erriquez D, Bartesaghi R, Perini G, Ciani E (2012) CDKL5, a novel MYCN‐repressed gene, blocks cell cycle and promotes differentiation of neuronal cells. Biochem Biophys Acta 1819:1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Villapol S, Wang Y, Adams M, Symes AJ (2013) Smad3 deficiency increases cortical and hippocampal neuronal loss following traumatic brain injury. Exp Neurol 250:353–365. [DOI] [PubMed] [Google Scholar]
- 68. Waddell DS, Liberati NT, Guo X, Frederick JP, Wang XF (2004) Casein kinase Iepsilon plays a functional role in the transforming growth factor‐beta signaling pathway. J Biol Chem 279:29236–29246. [DOI] [PubMed] [Google Scholar]
- 69. Wang C, Luan Z, Yang Y, Wang Z, Cui Y, Gu G (2011) Valproic acid induces apoptosis in differentiating hippocampal neurons by the release of tumor necrosis factor‐alpha from activated astrocytes. Neurosci Lett 497:122–127. [DOI] [PubMed] [Google Scholar]
- 70. Wang IT, Allen M, Goffin D, Zhu X, Fairless AH, Brodkin ES et al (2012) Loss of CDKL5 disrupts kinome profile and event‐related potentials leading to autistic‐like phenotypes in mice. Proc Natl Acad Sci USA 109:21516–21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang Y, Wang W, Li D, Li M, Wang P, Wen J et al (2014) IGF‐1 alleviates NMDA‐induced excitotoxicity in cultured hippocampal neurons against autophagy via the NR2B/PI3K‐AKT‐mTOR pathway. J Cell Physiol 229:1618–1629. [DOI] [PubMed] [Google Scholar]
- 72. Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H et al (2004) Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 75:1079‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wrighton KH, Lin X, Feng XH (2009) Phospho‐control of TGF‐beta superfamily signaling. Cell Res 19:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu G, Lu ZH, Wang J, Wang Y, Xie X, Meyenhofer MF, Ledeen RW (2005) Enhanced susceptibility to kainate‐induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane‐permeant analog of GM1. J Neurosci 25:11014–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu DJ, Zhao YZ, Wang J, He JW, Weng YG, Luo JY (2012) Smads, p38 and ERK1/2 are involved in BMP9‐induced osteogenic differentiation of C3H10T1/2 mesenchymal stem cells. BMB Rep 45:247–252. [DOI] [PubMed] [Google Scholar]
- 76. Yakovlev AG, Faden AI (2004) Mechanisms of neural cell death: implications for development of neuroprotective treatment strategies. NeuroRx 1:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD (2010) TGF‐beta signaling specifies axons during brain development. Cell 142:144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhu YC, Li D, Wang L, Lu B, Zheng J, Zhao SL et al (2013) Palmitoylation‐dependent CDKL5‐PSD‐95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc Natl Acad Sci USA 110:9118–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A. Western blot analysis of SMAD2 levels normalized to GAPDH levels in the somatosensory cortex of wild‐type (+/Y; n = 4) and Cdkl5 −/Y (n = 4) adult mice. Immunoblots are examples from two animals of each experimental group. B. Representative images of hippocampal sections at the CA1 field level processed for fluorescent SMAD3 immunostaining of wild‐type (+/Y) and Cdkl5 −/Y mice. Scale bar = 40 μm. C. Western blot analysis of P‐SMAD3 (Ser213 and Ser425) levels normalized to SMAD3 levels in the somatosensory cortex of wild‐type (+/Y; n = 4) and Cdkl5 −/Y (n = 4) adult mice. Immunoblots are examples from one animal of each experimental group. D. Quantification by RT‐qPCR of SMAD3 expression in the somatosensory cortex of wild‐type (+/Y; n = 5) and Cdkl5 −/Y (n = 6) mice. Data are expressed as a percentage of the values of Cdkl5 +/Y mice. Values are represented as means ± SE.
Figure S2. A. Western blot analysis of SMAD3 levels normalized to GAPDH levels in SKNBE cells co‐transfected with CDKL5ΔN (n = 4) or wild‐type CDKL5 (n = 4) and SMAD3. Immunoblots (upper panel) are three examples from each experimental group. B. Western blot analysis of P‐SMAD3 (Ser213) levels normalized to SMAD3 levels in untreated hippocampal cultures (+/Y = 9; −/Y = 14) or treated for 1h with TGF‐β1 (−/Y = 8). C. Representative fluorescent images of 10‐day (DIV10) differentiated hippocampal neurons from wild‐type (+/Y) mice immunopositive for P‐SMAD3 (Ser213) and counterstained with Hoechst. Cdkl5 +/Y hippocampal cultures were treated with TGF‐β1 (1 ng/ml) for 1h. Scale bar = 2.5 µm. Quantification of P‐SMAD3 (Ser213) signal intensity in untreated (+Y n = 2) or TGF‐β1 treated (+Y n = 2) hippocampal neurons. Values are represented as means ± SE. *P < 0.05; ***P < 0.001 (Unpaired t‐test in A; Fisher’s LSD after ANOVA in B).
Figure S3. Primary hippocampal cultures were infected with adenoviral particle for GFP (Ad‐GFP) or CDKL5 (Ad‐CDKL5) on the third day (DIV3) in culture, and immunostained for cleaved caspase‐3 on day 4 or for TAU1, MAP2 or MAP2 + SYN on day 10. A. Percentage of cleaved caspase‐3 positive neurons in 4‐day differentiated hippocampal neurons from wild‐type (+/Y n = 5) and Cdkl5 −/Y (n = 5) mice. B–E. Quantification of the length of the primary axon (B, TAU1‐positive; +/Y = 4, −/Y = 4), the total length of MAP2‐positive neurites (C, +/Y = 6, −/Y = 6), the number of SYN‐immunoreactive puncta per 10 μm in proximal dendrites (D, +/Y = 6, −/Y = 6), and the number of MAP2‐positive spines (E, +/Y = 6, −/Y = 6) from differentiated hippocampal cultures from Cdkl5 +/Y and Cdkl5 −/Y mice infected with GFP or CDKL5 adenoviral particles. F. Representative fluorescence images of proximal dendrite segments of hippocampal neurons that were immunopositive for MAP2 showing spine protrusions. Scale bar = 2.5 μm. Values are represented as means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001 (Fisher’s LSD after ANOVA).
Data Availability Statement
Data are stored and available on University servers.