

Abstract
Aβ immunization of Alzheimer's disease (AD) patients in the AN1792 (Elan Pharmaceuticals) trial caused Aβ removal and a decreased density of neurons in the cerebral cortex. As preservation of neurons may be a critical determinant of outcome after Aβ immunization, we have assessed the impact of previous Aβ immunization on the expression of a range of apoptotic proteins in post‐mortem human brain tissue. Cortex from 13 AD patients immunized with AN1792 (iAD) and from 27 nonimmunized AD (cAD) cases was immunolabeled for proapoptotic proteins implicated in AD pathophysiology: phosphorylated c‐Jun N‐terminal kinase (pJNK), activated caspase3 (a‐casp3), phosphorylated GSK3β on tyrosine 216 (GSK3βtyr216), p53 and Cdk5/p35. Expression of these proteins was analyzed in relation to immunization status and other clinical data. The antigen load of all of these proapoptotic proteins was significantly lower in iAD than cAD (P < 0.0001). In cAD, significant correlations (P < 0.001) were observed between: Cdk5/p35 and GSK3βtyr216; a‐casp3 and Aβ42; p53 and age at death. In iAD, significant correlations were found between GSK3βtyr216 and a‐casp3; both spongiosis and neuritic curvature ratio and Aβ42; and Cdk5/p35 and Aβ‐antibody level. Although neuronal loss was increased by immunization with AN1792, our present findings suggest downregulation of apoptosis in residual neurons and other cells.
Keywords: Alzheimer, anti‐amyloid immunotherapy, brain, impact, neurons, treatment
Abbreviations
- a‐casp3
activated caspase 3
- Aβ
β‐amyloid
- AD
Alzheimer's disease
- ATG5
autophagy‐related gene 5
- cAD
nonimmunized Alzheimer's disease brains
- CDK5
cyclin dependent Kinase 5
- GSK3βtyr216
glycogen Synthetase Kinase 3 phosphorylated at tyrosine 216
- iAD
immunized Alzheimer's Disease brains
- JNK
c‐Jun N Terminal Kinase
- LC3
microtubule‐associated protein light chain 3
- p53
tumor protein 53
- PKR
double‐stranded RNA dependent protein kinase
- pTau
phosphorylated tau.
Introduction
Alzheimer's disease (AD) is characterized by the accumulation of β‐amyloid (Aβ) peptide and hyperphosphorylated tau protein, and eventually synaptic and neuronal loss. The pathophysiology of the neuronal death remains unclear and controversial. Neuropathological studies have provided evidence of apoptotic neuronal death compatible with the slow progression of neuronal degeneration 15, 27, 32, in addition to possible deregulated autophagic activity 3, 14, 16, 24, 44. Apoptosis is a sequence of programmed events leading to the activation of caspases and cell disintegration 15, 27, 32, whereas autophagy is an intracellular catabolic process leading to the removal of aggregated proteins within cells 21, 28, 38. Both autophagy and apoptosis are highly regulated, play critical roles in tissue homeostasis and tend to be upregulated in response to extracellular or intracellular stress and in neurodegenerative diseases 26. In AD, both processes have been extensively studied but their contribution to neuronal death remains unclear. Apoptotic cell death in AD may result from an imbalance between proapoptotic and antiapoptotic proteins 15. The expression of several proapoptotic kinases such as activated GSK3β phosphorylated at tyrosine 216 (GSK3βtyr216) 1, 6, 37, pPKR 6, 7, 10, 29, 33, 34, 36, pJNK 9, 18, 42, 43, p53 (8) and activated caspase‐3 (a‐casp3) 2, 15, 17, 41 is increased in AD brains. In AD, autophagic activity is increased but may be dysfunctional, with failure of substrate clearance reflected by the presence of vacuoles 3, 14, 16, 24, 44.
Active Aβ42 immunization (AN1792, Elan Pharmaceuticals) in AD patients led to Aβ removal 19, 30, 31 associated with a decrease in phosphorylated tau (pTau) 4, long‐term downregulation of inflammation 46, reduction in the number of neurons and reduced neuritic abnormalities 34, 39. To investigate possible mechanisms underlying the observed neuronal loss after immunotherapy, we have explored the expression of apoptotic and autophagic proteins in the unique cohort of immunized AD patients from the AN1792 trial.
Materials and Methods
Case selection
Immunized AD cases
The brains of clinical AD patients enrolled in the initial Elan Pharmaceuticals Aβ immunization trial AN1792 (19) were obtained following consent to post‐mortem neuropathology. The study received ethical approval from Southampton and South West Hampshire Local Research Ethics Committees (Reference No: LRC 075/03/w). Thirteen post‐mortem brains in which the cause of the dementia was confirmed as AD neuropathologically were included in this study. All patients had received Aβ42 plus adjuvant and had died between 4 and 162 months after the first immunization (mean 72.8 months, median 63 months), with Braak tangle stage V/VI disease, as previously described 34 (Table 1). The post‐mortem delay was between 6 h and 48 h (mean 18.5 h; median 6 h). In addition to dementia, the most common clinical diagnoses recorded in the death certificate were bronchopneumonia, cerebrovascular accident and myocardial infarction. Other diagnoses included ruptured aortic aneurysm, pulmonary embolism, carcinoma of the breast, carcinoma of the bronchus and carcinoma of the pancreas. Neurodegenerative pathology was assessed by standard histological methods including hematoxylin and eosin (H&E), Luxol fast blue/cresyl violet and modified Bielschowsky silver impregnation. Selected sections were immunolabeled for Aβ, tau, α‐synuclein and TDP43 to confirm AD.
Table 1.
Characteristics of the immunized (iAD) and nonimmunized (cAD) Alzheimer's disease cohorts.
| ID case | Gender | Age | Braak stage | Dementia duration (years) | APOE status | Mean antibody response (ELISA units) | Survival time from 1st injection (months) | Post‐mortem delay (h) |
|---|---|---|---|---|---|---|---|---|
| iAD1 | F | 74 | VI | 6 | 3.4 | 1:119 | 20 | 48 |
| iAD2 | M | 83 | V | 11 | 3.3 | <1:100 | 4 | 6 |
| iAD3 | M | 63 | VI | 6 | 3.3 | <1:100 | 41 | 6 |
| iAD4 | F | 71 | VI | 10 | 3.3 | 1:4072 | 44 | 24 |
| iAD5 | M | 81 | VI | 7 | 3.4 | 1:1707 | 57 | 6 |
| iAD6 | M | 82 | VI | 6 | 3.4 | 1:4374 | 60 | 24 |
| iAD7 | M | 63 | VI | 10 | 3.4 | 1:6470 | 64 | 6 |
| iAD8 | M | 81 | VI | 11 | 4.4 | 1:491 | 63 | ? |
| iAD9 | F | 88 | VI | 11 | 3.3 | 1:137 | 86 | 24 |
| iAD10 | M | 88 | VI | 12 | 3.4 | 1:142 | 94 | 6 |
| iAD11 | F | 89 | VI | 15 | 3.4 | 1:142 | 111 | ? |
| iAD12 | F | 86 | VI | 13 | 4.4 | <1:100 | 141 | 6 |
| iAD13 | F | 75 | VI | 19 | ? | 1:221 | 162 | 48 |
| cAD (n = 28) | 15F:13M | 63–88 | V/VI | 3–17 | 21ɛ4+:6 ɛ4− | n/a | n/a |
mean 39 median 26 |
Abbreviations: n/a = nonapplicable.
?: unknown.
Nonimmunized AD cases
Twenty‐seven AD cases provided by the South West Dementia Brain Bank (SWDBB, Bristol, UK) were identified and used as a control unimmunized AD cohort (Supporting Information Table 1). All nonimmunized AD (cAD) cases had a clinical diagnosis of AD made during life by an experienced clinician, a Mini‐Mental State Examination score of <17 prior to death and satisfied post‐mortem neuropathological Consensus Criteria for Alzheimer's disease 20. The post‐mortem delay was between 9 h and 110 h (mean 39 h, median 26 h). The immunized and control AD cases were matched as closely as possible for age, gender, duration of dementia and APOE genotype (Table 1). The SWDBB tissue was used under the ethical approval from North Somerset and South Bristol Hampshire Local Research Ethics Committees (Reference No: REC 08/H0106/28 + 5).
Immunohistochemistry
Middle temporal gyrus, usually markedly affected by AD pathology, was investigated in this study. Four‐µm sections of formalin‐fixed paraffin‐embedded tissue from immunized AD cases (iAD) and cAD cases were immunolabeled together in batches to ensure comparability of staining.
Primary antibodies and immunohistochemistry
To evaluate the impact of active AN1792 immunization on apoptotic and autophagic pathways, we explored by immunohistochemistry the expression of the following proapoptotic proteins: GSK3βtyr216 (polyclonal rabbit anti‐phosphorylated GSK3βtyr216, #ab75745, Abcam) 6, 37, neuron‐specific activator of cyclin‐dependent kinase 5 with its activator p35 (C‐19 polyclonal rabbit anti‐Cdk5/p35, #sc‐820, Santa Cruz) 12, 42, phosphorylated c‐Jun N‐terminal kinase (monoclonal rabbit anti‐pJNK Thr183/Tyr185, clone 81E11, #4668, Cell Signaling) 18, 45, p53 (monoclonal mouse anti‐p53, clone DO‐1, #sc‐126, Santa Cruz) 8 and a‐casp3 (polyclonal rabbit anti‐activated caspase 3 (Asp175), # 9661, Cell Signaling) 15, 40, 41; and of the autophagic proteins ATG5 (initial step) (polyclonal rabbit anti‐ATG5, #AP1812b, Abgent) and microtubule‐associated protein light chain LC3‐II (a marker of the final stage reflecting efficient autophagic activity) (polyclonal rabbit anti‐LC3‐II, #AP1801a, Abgent) 21, 22, 28. The specificity of the antibodies pJNK 18, GSK3βtyr216 (1) and CDK5/p35 22 was previously demonstrated. To demonstrate the specificity of the antibodies p53, ATG5 and LC3II, we performed western blot on human brain tissue homogenates.
Immunohistochemistry was carried out by a standard method as previously described 1, 4, 5, 19, 31, 34, 46. Biotinylated secondary antibodies, normal serum and avidin‐biotin complex were from Vector Laboratories (Peterborough, UK). Immunodetection was performed using the avidin‐biotin‐peroxidase complex method (Vectastain Elite ABC, UK) with 3,3′‐diaminobenzidine (DAB) as chromogen and 0.05% hydrogen peroxide as substrate. All the sections were dehydrated before mounting in DePeX (BDH Laboratory Supplies, UK). Sections from which the primary antibody was omitted were included in each immunohistochemistry run.
Quantification of immunolabeling
Quantification was performed blind to the identity of the cases. Thirty fields of cortical gray matter at objective magnification x20 were acquired for each case from the same anatomical regions in a zigzag sequence along the cortical ribbon to ensure that all cortical layers were represented. Slides were marked by the same neuropathologist to ensure consistency in the location of acquisition of the images. Protein “load” defined as the percentage of the field immunopositive for the marker of interest was determined using ImageJ (developed by W.S. Rasband National Institutes of Health, Bethesda MD, USA, version 1.47 g), as in our previous studies 1, 4, 5, 19, 34, 46.
Statistical analysis
The normality of distribution of each marker across the cohort was assessed by examination of quantile‐quantile plots (not shown). Levels of each marker were compared between cAD and iAD cases in two‐sample two‐sided t‐tests or nonparametric Mann–Whitney U‐tests (depending on the normality of the data). In both groups, correlations were analyzed by Pearson's or Spearman's test, depending on the normality of distribution of the markers. We analyzed the correlation between the apoptosis and autophagy‐associated markers and (i) indicators of disease severity and neuronal integrity as reported in our previous published studies as follows: Aβ42 load, pTau load, tangles density by image, dystrophic neurites, spongiosis, number neuronal NeuN+ density by image, neuritic curvature ratio assessed by neurofilament immunohistochemistry, phosphorylated (p)PKR (a marker of early neurodegeneration) 5, 19, 34, 46; and (ii) available clinical indicators of disease course and antibody response—duration of dementia, survival time after immunization, age at death, mean and peak antibody level. The threshold for statistical significance was set at 5% for intergroup comparisons and 1% for correlations, as determined by use of SPSS 21.0.
Results
The immunolabeling of all of the antigens was neuronal, with additional labeling of glial cells for some proteins as described in Table 2. Of note, the immunolabeling of activated‐caspase 3 was cytoplasmic with the nuclei of the stained neurons morphologically normal, without the karyorrhexis classically associated with apoptosis.
Table 2.
Topographical distribution of the apoptotic and autophagic proteins.
| cAD | Neurons | Glial cells | ||
|---|---|---|---|---|
| Cytoplasm | Nuclear | Cytoplasm | Nuclear | |
| a‐casp3 | + | – | + | – |
| Cdk5/p35 | + | – | + | – |
| pJNK | + | – | + | – |
| GSK3βtyr216 | + | + | – | – |
| P53 | + | – | – | – |
| LC3 | + | – | + | – |
| ATG5 | + | – | – | + |
| Iad | Neurons | Glial cells | ||
|---|---|---|---|---|
| Cytoplasm | Nuclear | Cytoplasm | Nuclear | |
| a‐casp3 | + | – | – | – |
| Cdk5/p35 | + | – | + | – |
| pJNK | + | – | – | – |
| GSK3βtyr216 | + | + | – | – |
| P53 | + | – | – | – |
| LC3 | + | – | – | – |
| ATG5 | + | – | – | + |
The expression of all apoptotic kinases was significantly lower in iAD than cAD cases: a‐casp3 load, P < 0.001; Cdk5/p35 load, P = 0.013; p53 load, P < 0.001; GSK3βtyr216 load, P < 0.01; and pJNK, P < 0.001 (Figure 1). Of the two autophagic markers examined, LC3‐II load was significantly lower in iAD than cAD (P < 0.001) while ATG5 load did not differ between the cohorts (P = 0.130, Figure 1).
Figure 1.
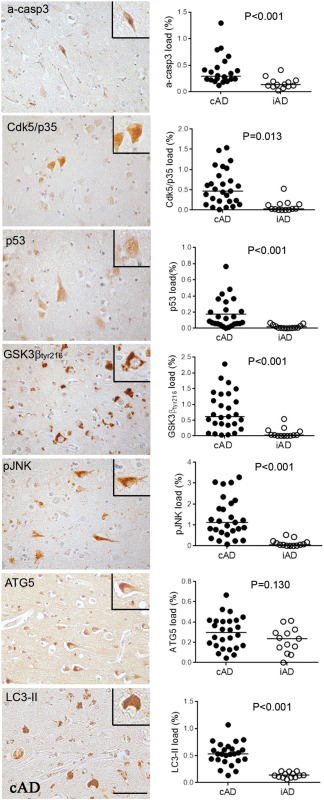
On the left, illustration of the immunolabeling of proapoptotic and autophagic proteins as observed in Alzheimer's disease. On the right, quantification of the proteins in the nonimmunized AD (cAD) compared to immunized AD (iAD) cases showing a significant decrease in all apoptotic proteins and of LC3II after immunization. Scale bar = 50 μm.
The expression of apoptotic and autophagic markers was analyzed for correlation with other aspects of AD pathology (Aβ42 load, pTau load, dystrophic neurite counts, spongiosis, NeuN+ neurons and curvature ratio) in the same anatomical region and also with a range of clinical parameters (age, gender, age at death, dementia duration, peak antibody, survival time). We did not observe any modification in the distribution of the proteins between both cohorts except for the GSK3βtyr216, which was detected mainly in granulo‐vacular degeneration (GVD) in the iAD group but not in the cAD group. To take account of possible variations in neuronal density, we also assessed the percentage of all neurons that was immunopositive for a‐casp3. This confirmed the striking decrease in neuronal expression of a‐casp3 in iAD compared with cAD (P < 0.0001, data not shown).
In the cAD group, a‐casp3 load correlated positively with Aβ42 (r = 0.561, P = 0.005), and Cdk5/p35 correlated positively with pGSK3βtyr216 (r = 0.642, P < 0.001) (Table 3). Comparison of present findings with the clinical data revealed positive correlations between p53 and age at death (r = 0.564, P = 0.003), and between LC3‐II and dementia duration (r = 0.691, P = 0.001) (Table 3).
Table 3.
Results of correlation analyses within the nonimmunized AD control group.
| pJNK | Cdk5/p35 | p53 | a‐casp3 | GSK3βtyr216 | ATG5 | LC3‐II | |
|---|---|---|---|---|---|---|---|
| Aβ42 | r = 0.141 | r = −0.238 | r = 0.142 | r = 0.561 ** | r = −0.079 | r = −0.173 | r = −0.346 |
| P = 0.483 | P = 0.232 | P = 0.497 | P = 0.005 | P = 0.696 | P = 0.399 | P = 0.090 | |
| ptau | r = −0.228 | r = 0.178 | r = 0.052 | r = −0.224 | r = 0.365 | r = −0.214 | r = 0.060 |
| P = 0.252 | P = 0.374 | P = 0.804 | P = 0.303 | P = 0.061 | P = 0.295 | P = 0.777 | |
| tangles | r = −0.088 | r = 0.092 | r = −0.254 | r = −0.070 | r = 0.008 | r = −0.387 | r = −0.046 |
| P = 0.662 | P = 0.648 | P = 0.221 | P = 0.750 | P = 0.970 | P = 0.050 | P = 0.828 | |
| dystrophic neurites | r = 0.157 | r = 0.001 | r = 0.094 | r = 0.068 | r = −0.010 | r = −0.235 | r = 0.027 |
| P = 0.433 | P = 0.998 | P = 0.655 | P = 0.758 | P = 0.959 | P = 0.248 | P = 0.898 | |
| spongiosis | r = −0.181 | r = 0.404 | r = 0.048 | r = −0.327 | r = 0.166 | r = 0.231 | r = 0.084 |
| P = 0.365 | P = 0.037 | P = 0.818 | P = 0.128 | P = 0.409 | P = 0.256 | P = 0.690 | |
| NeuN | r = 0.008 | r = −0.039 | r = 0.413 | r = −0.118 | r = 0.361 | r = 0.232 | r = 0.160 |
| P = 0.971 | P = 0.860 | P = 0.063 | P = 0.610 | P = 0.090 | P = 0.298 | P = 0.489 | |
| NFP curvature ratio | r = −0.042 | r = 0.180 | r = 0.182 | r = −0.059 | r = 0.174 | r = −0.055 | r = 0.134 |
| P = 0.837 | P = 0.369 | P = 0.383 | PP = 0.790 | P = 0.384 | P = 0.788 | P = 0.524 | |
| pPKR | r = −0.267 | r = 0.085 | r = −0.081 | r = 0.094 | r = 0.337 | r = 0.110 | r = −0.075 |
| P = 0.178 | P = 0.673 | P = 0.701 | P = 0.670 | P = 0.085 | P = 0.593 | P = 0.723 | |
| pJNK | r = 0.426 | r = 0.055 | r = 0.177 | r = 0.311 | r = −0.226 | r = 0.202 | |
| P = 0.027 | P = 0.792 | P = 0.419 | P = 0.115 | P = 0.266 | P = 0.334 | ||
| Cdk5/p35 | r = 0.277 | r = −0.146 | r = 0.648 ** | r = −0.196 | r = 0.300 | ||
| P = 0.18 | P = 0.505 | P < 0.001 | P = 0.338 | P = 0.144 | |||
| p53 | r = 0.172 | r = 0.280 | r = −0.055 | r = 0.319 | |||
| P = 0.457 | P = 0.175 | P = 0.795 | P = 0.120 | ||||
| a–casp3 | r = −0.136 | r = −0.492 | r = −0.157 | ||||
| P = 0.536 | P = 0.020 | P = 0.496 | |||||
| GSK3βtyr216 | r = −0.01 | r = 0.128 | |||||
| P = 0.927 | P = 0.542 | ||||||
| ATG5 | r = −0.062 | ||||||
| P = 0.770 | |||||||
| Age at death | r = 0.210 | r = 0.289 | r = 0.564 ** | r = 0.389 | r = 0.438 | r = −0.287 | r = 0.220 |
| P = 0.294 | P = 0.144 | P = 0.003 | P = 0.0670 | P = 0.022 | P = 0.156 | P = 0.291 | |
| Dementia duration | r = 0.057 | r = 0.372 | r = 0.388 | r = −0.062 | r = −0.008 | r = 0.049 | r = 0.691 |
| P = 0.796 | P = 0.080 | P = 0.082 | P = 0.795 | P = 0.970 | P = 0.830 | P = 0.001 | |
| Peak antibody | r = 0.033 | r = 0.840 ** | r = −0.175 | r = −0.431 | r = −0.284 | r = 0.459 | r = −0.386 |
| P = 0.914 | P < 0.001 | P = 0.569 | P = 0.142 | P = 0.348 | P = 0.115 | P = 0.193 | |
| Survival time | r = 0.455 | r = 0.162 | r = −0.077 | r = 0.252 | r = 0.446 | r = 0.280 | r = 0.568 |
| P = 0.119 | P = 0.590 | P = 0.802 | P = 0.406 | P = 0.126 | P = 0.354 | P = 0.043 |
Bold: ** correlation significant at the 0.01 level (2‐tailed).
Within the iAD cohort, a‐casp3 and GSK3βtyr216 correlated positively with severity of spongiosis, a marker of neuropil degeneration (r = 0.789, P = 0.004 and r = 0.761, P = 0.007, respectively) (Table 2). ATG5 correlated negatively with Aβ42 load (r = –0.845, P = 0.001) and positively with the curvature ratio (abnormal tortuosity of neuritic processes) (r = 0.841, P = 0.001) (Table 4). Cdk5/p35 correlated positively with peak antibody titer (r = 0.840, P < 0.001) as well as with mean antibody titer (data not shown) (Table 4).
Table 4.
Results of correlation analyses within the immunized AD control group.
| pJNK | Cdk5/p35 | p53 | a‐casp3 | GSK3βtyr216 | ATG5 | LC3‐II | |
|---|---|---|---|---|---|---|---|
| Aβ42 | r = −0.237 | r = −0.491 | r = −0.361 | r = 0.413 | r = 0.324 | r = –0.845 ** | r = 0.484 |
| P = 0.482 | P = 0.125 | P = 0.276 | P = 0.207 | P = 0.331 | P = 0.001 | P = 0.131 | |
| ptau | r = 0.397 | r = 0.082 | r = 0.164 | r = 0.089 | r = −0.231 | r = 0.036 | r = −0.174 |
| P = 0.226 | P = 0.811 | P = 0.629 | P = 0.794 | P = 0.494 | P = 0.915 | P = 0.610 | |
| tangles | r = 0.301 | r = 0.464 | r = −0.050 | r = −0.089 | r = −0.207 | r = 0.155 | r = −0.507 |
| P = 0.368 | P = 0.151 | P = 0.883 | P = 0.796 | P = 0.541 | P = 0.650 | P = 0.112 | |
| dystrophic neurites | r = 0.037 | r = −0.246 | r = −0.165 | r = 0.667 | r = 0.654 | r = −0.269 | r = 0.547 |
| P = 0.915 | P = 0.466 | P = 0.628 | P = 0.025 | P = 0.029 | P = 0.424 | P = 0.082 | |
| spongiosis | r = 0.479 | r = 0.009 | r = −0.087 | r = 0.789 ** | r = 0.761 ** | r = −0.055 | r = 0.128 |
| P = 0.136 | P = 0.979 | P = 0.800 | P = 0.004 | P = 0.007 | P = 0.873 | P = 0.708 | |
| NeuN | r = 0.662 | r = −0.353 | r = 0.107 | r = 0.691 | r = 0.337 | r = 0.170 | r = 0.055 |
| P = 0.037 | P = 0.318 | P = 0.769 | P = 0.027 | P = 0.340 | P = 0.638 | P = 0.880 | |
| NFP curvature ratio | r = 0.448 | r = 0.377 | r = 0.194 | r = −0.152 | r = 0.137 | r = 0.841 ** | r = −0.418 |
| P = 0.167 | P = 0.253 | P = 0.568 | P = 0.656 | P = 0.687 | P = 0.001 | P = 0.201 | |
| pPKR | r = 0.201 | r = −0.564 | r = 0.213 | r = 0.297 | r = 0.258 | r = −0.176 | r = 0.701 |
| P = 0.577 | P = 0.090 | P = 0.555 | P = 0.405 | P = 0.471 | P = 0.627 | P = 0.024 | |
| pJNK | r = 0.11 | r = 0.083 | r = 0.534 | r = 0.078 | r = 0.529 | r = −0.300 | |
| P = 0.720 | P = 0.788 | P = 0.060 | P = 0.801 | 0 P =.063 | P = 0.319 | ||
| Cdk5/p35 | r = −0.223 | r = −0.049 | r = 0.102 | r = 0.363 | r = −0.342 | ||
| P = 0.464 | P = 0.873 | P = 0.739 | P = 0.223 | P = 0.253 | |||
| p53 | r = 0.052 | r = −0.233 | r = 0.165 | r = 0.268 | |||
| P = 0.865 | P = 0.444 | P = 0.589 | P = 0.375 | ||||
| a–casp3 | r = 0.546 | r = −0.165 | r = −0.069 | ||||
| P = 0.054 | p = 0.590 | P = 0.823 | |||||
| GSK3βtyr216 | r = −0.108 | r = −0.218 | |||||
| P = 0.726 | P = 0.474 | ||||||
| ATG5 | r = −0.303 | ||||||
| P = 0.314 | |||||||
| Age at death | r = 0.512 | r = −0.502 | r = −0.029 | r = −0.080 | r = 0.082 | r = 0.337 | r = −0.262 |
| P = 0.074 | P = 0.08 | P = 0.925 | P = 0.795 | P = 0.791 | P = 0.261 | P = 0.388 | |
| Dementia duration | r = 0.119 | r = −0.125 | r = −0.297 | r = 0.134 | r = 0.178 | r = 0.008 | r = −0.292 |
| P = 0.700 | P = 0.684 | P = 0.324 | P = 0.661 | P = 0.560 | P = 0.978 | P = 0.333 | |
| Peak antibody | r = 0.033 | r = 0.840 ** | r = −0.175 | r = −0.431 | r = −0.284 | r = 0.459 | r = −0.386 |
| P = 0.914 | P < 0.001 | P = 0.569 | P = 0.142 | P = 0.348 | P = 0.115 | P = 0.193 | |
| Survival time | r = 0.455 | r = 0.162 | r = −0.077 | r = 0.252 | r = 0.446 | r = 0.280 | r = 0.568 |
| P = 0.119 | P = 0.590 | P = 0.802 | P = 0.406 | P = 0.126 | P = 0.354 | P = 0.043 |
Bold: ** correlation significant at the 0.01 level (2‐tailed).
No other correlation was observed in either group.
Discussion
Our results suggest that active Aβ immunization of AD patients modulates apoptosis and some autophagic cellular signals, causing downregulation of apoptotic proteins and reduction in the final stage of autophagy activity. The decrease of apoptotic protein expression after immunization could have several explanations: 1 downregulation of apoptosis was a consequence of removal of Aβ, consistent with several studies implicating Aβ‐induced apoptosis in neuronal death in AD 6, 8. 2 The reduction in apoptotic proteins may simply reflect the accelerated loss of damaged neurons after immunotherapy, as previously reported by us 34, potentially leaving “healthier” neurons less affected by AD pathophysiology. However, the small magnitude of neuronal loss after immunotherapy (about 10%) could not be the sole explanation for the substantial decrease in apoptotic protein load (between 65% and 85%), and analysis of the percentage of all neurons that was immunopositive for a‐casp3 confirmed the marked reduction in neuronal expression of this antigen in iAD. 3 Immunotherapy may itself downregulate apoptotic proteins. Further studies are needed to clarify the cellular and molecular processes that underlie these findings.
The effects of autophagic proteins are less clear‐cut. The reduction in LC3II suggests downregulation of the later steps of autophagy, potentially explained by reduced metabolic requirement for autophagy or perhaps an aborted or dysfunctional autophagic process. Restrictions on tissue availability did not allow us to explore this mechanistically. Analysis in animal models may help to clarify the influence of immunotherapy on autophagy.
The correlation between a‐casp3 and Aβ42 in the cAD group, is in accordance with previous reports implicating Aβ42 in neuronal apoptosis 6, 15. The link between Cdk5/p35 with GSK3βtyr216 is also consistent with previous studies implicating these proteins in the pathophysiology of AD, particularly in the phosphorylation of Tau protein 13, 23.
Strikingly different associations were observed in the immunized cohort. The relationship between a‐casp3, GSK3βtyr216 loads and the severity of spongiosis, a marker of neuropil degeneration, strengthen the association between these proapoptotic proteins and the neuronal loss detected after immunization 34. This may explain the absence of clinical amelioration in these patients 19. Due to the nature of the post‐mortem study, investigating late‐stage of the disease and treatment, we cannot exclude the possibility that immunotherapy may have induced an early acute apoptotic phase followed by a more quiescent phase several years after the treatment.
The relationship between p53 expression and age at death in the control Alzheimer's cohort is consistent with the documented association between apoptosis and increasing age 11. The increase in LC3‐II with dementia duration may be part of a prosurvival adaptive response by neurons and glia to minimize neurodegeneration 14. After immunization, the anti‐Aβ immune response (mean and peak Aβ antibody titer) was strongly associated with Cdk5/p35 expression. Cdk5/p35 signaling is known to promote microglial phagocytosis of fibrillar Aβ 25, and the present data are in keeping with the enhanced Aβ clearance by phagocytic microglia in the immunized patients who developed an immune response 19, 35, 46. However, it should be noted that the highest Cdk5/p35 level in the immunized cohort was much lower than that in the control group, consistent with the downregulation of microglial activation that occurs when Aβ has been completely removed 46.
This study has some limitations, inherent in the use of post‐mortem tissue. As previously reported 1, 4, 5, 19, 31, 34, 46, the number of placebo immunization cases from which brains could be obtained (n = 1) was far too low to provide useful data for statistical analysis and thus our study used AD brains from patients who were not included in a protocol of immunotherapy, although they were matched as closely as possible to the immunized cohort. Furthermore, this was a retrospective observational study rather than a prospective experimental study, which limited the range of methodological approaches and the comparability of clinical findings. Because this was an end‐stage study, it was not possible to explore the temporal relationship between markers of apoptosis or autophagy and neuronal loss and analysis was limited to assessment of the late‐stage consequences of immunization.
In summary, in this unique human brain series from the first anti‐Aβ42 trial, our results suggest that anti‐Aβ42 immunization downregulates the expression of several proapoptotic proteins in the brain. While these changes might be expected to be beneficial, the absence of cognitive benefit suggests that they occur too late in the disease process or that other mechanisms are responsible for the neuronal death.
Ethical Approval and Consent to Participate
The study received ethical approval from the Southampton and South West Hampshire Local Research Ethics Committees, Reference No. LRC 075/03/w for the use of the iAD cohort. The cAD cases were provided under the SWDBB Ethics (Research Ethics Committee Reference No. 08/H0106/28 + 5).
Competing Interest
Prof. PAQUET is member of the International Advisory Boards of Lilly and is involved as investigator in several clinical trials for Roche, Esai, Lilly, Biogen, Astra‐Zeneca, Lundbeck.
Prof. NICOLL is or has been a consultant/advisor relating to Alzheimer immunization programmes for Elan Pharmaceuticals, GlaxoSmithKline, Novartis, Roche, Janssen, Pfizer, Biogen.
Prof. HUGON is investigator in several passive anti‐amyloid immunotherapies and other clinical trials for Roche, Eisai, Lilly, Biogen, Astra‐Zeneca, Lundbeck.
Prof LOVE, Prof HOLMES, Dr BOCHE and Dr MOUTON‐LIGER declare that they have no conflict of interest.
Author's Contributions
Claire PAQUET designed the study, performed the immunohistochemistry experiments, collected and analyzed the data and prepared the manuscript.
Delphine Boche analyzed and interpreted the data and prepared the manuscript.
Seth Love provided the cAD cases from SWDBB and was involved in the preparation of the manuscript.
Clive Holmes provided the clinical data.
François Mouton‐Liger performed Western blot to control for the specificity of the antibodies and prepared the manuscript.
Jacques Hugon advised on the relationship between different apoptotic kinases in Alzheimer's disease.
James Nicoll provided immunized AD brains and was involved in the preparation of the manuscript.
All co‐authors provided input and critically revised the paper.
“All authors read and approved the manuscript.”
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Table 1. Individual characteristics of the non‐immunised (cAD) Alzheimer's disease cohort.
Acknowledgments
All authors had full access to all data and CP and DB have final responsibility for the decision to submit the report for publication. This study was supported jointly by the Fondation Philippe Chatrier (Paris, France), Alzheimer Research UK (ART/PG2006/4 and ART‐EXT2010‐1) and Medical Research Council UK (G0501033). We thank the patients who were involved in this study and their careers. We thank all donors, the president and scientific committee of Fondation Philippe Chatrier. Vivienne Hopkins, David Wilkinson, Anthony Bayer, Roy Jones and Roger Bullock enrolled patients in the original trial. Jim Neal provided 2 immunized cases from Cardiff. We would like to thank the South West Brain Dementia Brain Bank (SWDBB) for providing tissue for this study. The SWDBB is supported by BRACE (Bristol Research into Alzheimer's and Care of the Elderly), Brains for Dementia Research and the Medical Research Council. The Neuropathology Section, Department of Cellular Pathology, University Hospital Southampton NHS Foundation Trust, the Histochemistry Research Unit and the Biomedical Imaging Unit of the Faculty of Medicine, University of Southampton facilitated tissue processing, staining and analysis. Staff at Elan Pharmaceuticals made available original clinical trial data.
References
- 1. Amin J, Paquet C, Baker A, Asuni AA, Love S, Holmes C et al (2014) Effect of Abeta immunisation on hyperphosphorylated tau: a potential role for GSK‐3beta. Neuropathol Appl Neurobiol 41:445–457. [DOI] [PubMed] [Google Scholar]
- 2. Ayala‐Grosso C, Tam J, Roy S, Xanthoudakis S, Da Costa D, Nicholson DW, Robertson GS (2006) Caspase‐3 cleaved spectrin colocalizes with neurofilament‐immunoreactive neurons in Alzheimer's disease. Neuroscience 141:863–874. [DOI] [PubMed] [Google Scholar]
- 3. Barnett A, Brewer GJ (2011) Autophagy in aging and Alzheimer's disease: pathologic or protective? J Alzheimers Dis 25:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM et al (2008) Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain 131:3299–3310. [DOI] [PubMed] [Google Scholar]
- 5. Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, Nicoll JA (2010) Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropathol 120:13–20. [DOI] [PubMed] [Google Scholar]
- 6. Bose A, Mouton‐Liger F, Paquet C, Mazot P, Vigny M, Gray F, Hugon J (2011) Modulation of tau phosphorylation by the kinase PKR: implications in Alzheimer's disease. Brain Pathol 21:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang RC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J (2002) Involvement of double‐stranded RNA‐dependent protein kinase and phosphorylation of eukaryotic initiation factor‐2alpha in neuronal degeneration. J Neurochem 83:1215–1225. [DOI] [PubMed] [Google Scholar]
- 8. Checler F, Alves da Costa C (2014) p53 in neurodegenerative diseases and brain cancers. Pharmacol Ther 142:99–113. [DOI] [PubMed] [Google Scholar]
- 9. Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27:6245–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dumurgier J, Mouton‐Liger F, Lapalus P, Prevot M, Laplanche J‐L, Hugon J et al (2013) Cerebrospinal fluid PKR level predicts cognitive decline in Alzheimer's disease. PLoS One 8:e53587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Engmann O, Giese KP (2009) Crosstalk between Cdk5 and GSK3beta: implications for Alzheimer's Disease. Front Mol Neurosci 2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flaherty DB, Soria JP, Tomasiewicz HG, Wood JG (2000) Phosphorylation of human tau protein by microtubule‐associated kinases: GSK3beta and cdk5 are key participants. J Neurosci Res 62:463–472. [DOI] [PubMed] [Google Scholar]
- 14. Frake RA, Ricketts T, Menzies FM, Rubinsztein DC (2015) Autophagy and neurodegeneration. J Clin Invest 125:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Friedlander RM (2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348:1365–1375. [DOI] [PubMed] [Google Scholar]
- 16. Funderburk SF, Marcellino BK, Yue Z (2010) Cell “self‐eating” (autophagy) mechanism in Alzheimer's disease. Mt Sinai J Med 77:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gastard MC, Troncoso JC, Koliatsos VE (2003) Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann Neurol 54:393–398. [DOI] [PubMed] [Google Scholar]
- 18. Gourmaud S, Paquet C, Dumurgier J, Pace C, Bouras C, Gray F et al (2014) Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J Psychiatry Neurosci 39:140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A et al (2008) Long‐term effects of Abeta42 immunisation in Alzheimer's disease: follow‐up of a randomised, placebo‐controlled phase I trial. Lancet 372:216–223. [DOI] [PubMed] [Google Scholar]
- 20. Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097. [DOI] [PubMed] [Google Scholar]
- 21. Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo‐Arozena A, Adeli K et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee MS, Tsai LH (2003) Cdk5: one of the links between senile plaques and neurofibrillary tangles? J Alzheimers Dis 5:127–137. [DOI] [PubMed] [Google Scholar]
- 24. Li Q, Liu Y, Sun M (2016) Autophagy and Alzheimer's disease. Cell Mol Neurobiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma Y, Bao J, Zhao X, Shen H, Lv J, Ma S et al (2013) Activated cyclin‐dependent kinase 5 promotes microglial phagocytosis of fibrillar beta‐amyloid by up‐regulating lipoprotein lipase expression. Mol Cell Proteomics 12:2833–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marino G, Niso‐Santano M, Baehrecke EH, Kroemer G (2014) Self‐consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1:120–129. [DOI] [PubMed] [Google Scholar]
- 28. Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mouton‐Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F, Hugon J (2012) Oxidative stress increases BACE1 protein levels through activation of the PKR‐eIF2alpha pathway. Biochim Biophys Acta 1822:885–896. [DOI] [PubMed] [Google Scholar]
- 30. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid‐beta peptide: a case report. Nat Med 9:448–452. [DOI] [PubMed] [Google Scholar]
- 31. Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P et al (2006) Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol 65:1040–1048. [DOI] [PubMed] [Google Scholar]
- 32. Obulesu M, Lakshmi MJ (2014) Apoptosis in Alzheimer's disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res 39:2301–2312. [DOI] [PubMed] [Google Scholar]
- 33. Ogolla PS, Portillo JA, White CL, Patel K, Lamb B, Sen GC, Subauste CS (2013) The protein kinase double‐stranded RNA‐dependent (PKR) enhances protection against disease cause by a non‐viral pathogen. PLoS Pathog 9:e1003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paquet C, Amin J, Mouton‐Liger F, Nasser M, Love S, Gray F, et al (2014) Effect of active Aβ immunotherapy on neurons in human Alzheimer's disease. J Pathol 235:721–730. [DOI] [PubMed] [Google Scholar]
- 35. Pareek TK, Lam E, Zheng X, Askew D, Kulkarni AB, Chance MR et al (2010) Cyclin‐dependent kinase 5 activity is required for T cell activation and induction of experimental autoimmune encephalomyelitis. J Exp Med 207:2507–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peel AL (2004) PKR activation in neurodegenerative disease. J Neuropathol Exp Neurol 63:97–105. [DOI] [PubMed] [Google Scholar]
- 37. Pei JJ, Braak E, Braak H, Grundke‐Iqbal I, Iqbal K, Winblad B, Cowburn RF (1999) Distribution of active glycogen synthase kinase 3beta (GSK‐3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol 58:1010–1019. [DOI] [PubMed] [Google Scholar]
- 38. Rubinsztein DC, Shpilka T, Elazar Z (2012) Mechanisms of autophagosome biogenesis. Curr Biol 22:R29–R34. [DOI] [PubMed] [Google Scholar]
- 39. Serrano‐Pozo A, William CM, Ferrer I, Uro‐Coste E, Delisle MB, Maurage CA et al (2010) Beneficial effect of human anti‐amyloid‐beta active immunization on neurite morphology and tau pathology. Brain 133:1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H (1999) Activation of caspase‐3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol 155:1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW (2001) Activated caspase‐3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res 898:350–357. [DOI] [PubMed] [Google Scholar]
- 42. Sun KH, Lee HG, Smith MA, Shah K (2009) Direct and indirect roles of cyclin‐dependent kinase 5 as an upstream regulator in the c‐Jun NH2‐terminal kinase cascade: relevance to neurotoxic insults in Alzheimer's disease. Mol Biol Cell 20:4611–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yarza R, Vela S, Solas M, Ramirez MJ (2015) c‐Jun N‐terminal Kinase (JNK) Signaling as a therapeutic target for Alzheimer's Disease. Front Pharmacol 6:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zare‐Shahabadi A, Masliah E, Johnson GV, Rezaei N (2015) Autophagy in Alzheimer's disease. Rev Neurosci 26:385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA (2001) Activation and redistribution of c‐jun N‐terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem 76:435–441. [DOI] [PubMed] [Google Scholar]
- 46. Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S et al (2013) Inflammatory components in human Alzheimer's disease and after active amyloid‐beta42 immunization. Brain 136:2677–2696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Table 1. Individual characteristics of the non‐immunised (cAD) Alzheimer's disease cohort.