

Abstract
Lewy bodies (LB) and Lewy neurites (LN), which are primarily composed of α‐synuclein (α‐syn), are neuropathological hallmarks of Parkinson's disease (PD) and dementia with Lewy bodies (DLB). We recently found that the neuronal phosphoprotein synapsin III (syn III) controls dopamine release via cooperation with α‐syn and modulates α‐syn aggregation. Here, we observed that LB and LN, in the substantia nigra of PD patients and hippocampus of one subject with DLB, displayed a marked immunopositivity for syn III. The in situ proximity ligation assay revealed the accumulation of numerous proteinase K‐resistant neuropathological inclusions that contained both α‐syn and syn III in tight association in the brain of affected subjects. Most strikingly, syn III was identified as a component of α‐syn‐positive fibrils in LB‐enriched protein extracts from PD brains. Finally, a positive correlation between syn III and α‐syn levels was detected in the caudate putamen of PD subjects. Collectively, these findings indicate that syn III is a crucial α‐syn interactant and a key component of LB fibrils in the brain of patients affected by PD.
Keywords: Lewy bodies, Lewy body dementia, Parkinson's disease, α‐synuclein, synapsin III
Introduction
Parkinson's disease (PD) is the most common age‐related neurodegenerative movement disorder. The clinical symptoms include resting tremor, bradykinesia, rigidity and gait disturbances. The primary neuropathological hallmarks are a progressive degeneration of dopaminergic neurons in the pars compacta of the substantia nigra (SN) that projects to the striatum and intraneuronal and intraneuritic insoluble protein inclusions termed Lewy bodies (LB) and Lewy neurites (LN), respectively 1, 25. The main protein constituent of LB and LN is fibrillary aggregated α‐synuclein (α‐syn) 58. This is a 140 amino acid protein enriched at the synaptic terminals of nigral dopaminergic neurons 61. Αlpha‐synuclein is a crucial modulator of synaptic dopamine release 1 and vesicle dynamics 14, 54, 62. In particular, α‐syn interacts with and regulates several key factors that control these functions, such as the dopamine transporter (DAT) 5, 6, 40, 56, Rab small GTP‐binding proteins 11, 18, 22, 28, 66, soluble N‐ethylmaleimide sensitive fusion attachment protein receptor (SNARE) complex, vesicle associated membrane protein‐2 (VAMP‐2) 13, 23, 27 and vesicular monoamine transporter 2 (VMAT2) 42. The pathological deposition of α‐syn is considered a causative factor for PD 19, 20, because the topographical pattern of LB spreading in the brain and the progression of PD symptoms are correlated 10. Experimental evidence supports that α‐syn accumulation and aggregation at the synapse determines the onset of dopaminergic terminal failure, which collapses axonal trafficking and might gradually lead to retrograde cell degeneration 2, 8, 15, 53.
We recently found that a member of the synapsin (syn) family of phosphoprotein 16, 50, synapsin III (syn III), interacts and cooperates with α‐syn in the control of dopamine release from mesencephalic neurons 68.
The present study investigated alterations in syn III levels and its distribution and interaction with α‐syn in the SN and caudate putamen (CP) of patients affected by PD and age‐matched controls. Hippocampal slices of one patient affected by dementia with LB (DLB) were also analyzed. We found that LB and LN in the SN of PD patients, and in the hippocampus of the DLB subject, exhibited a marked immunopositivity for syn III. The immunoreactivity for this protein was increased in the CP of PD patients where we also observed a significant correlation between syn III levels and α‐syn using western blot (WB) analysis. Notably, double immunogold transmitted electron microscopy (TEM), revealed that fibrils extracted from LB exhibited α‐syn and syn III immunopositivity, which indicates that these proteins co‐aggregate in insoluble fibrillary structures and suggests the occurrence of a marked interaction between them. The in situ proximity ligation assay (PLA) revealed a pronounced accumulation of proteinase K‐resistant inclusions that contained tightly associated α‐syn and syn III in PD and DLB brains compared with controls. No changes in the distribution of synapsin Ia/b (syn Ia/b) or synapsin II (syn II) were detected in the brain of affected subjects.
Materials and Methods
Human tissues and CSF
Paraffin embedded sections as well as fresh frozen tissue from ten patients with PD (Age 79 ± 6) and ten age‐matched control subjects (Age 78 ± 9), kindly supplied by the Parkinson's UK Brain Bank, a Charity funded by Parkinson's UK (Imperial College London, UK), were used for immunostainings and biochemical studies, respectively. Sections from the SN and CP of PD patients and age‐matched control subjects were paraffin embedded and supplied at 5‐µm thick. Hippocampal sections (5‐µm thick) from one patient with DLB, from the Pathological Anatomy Division of the Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy), were also employed for the immunohistochemical studies. The study on human brain samples was performed in accordance of the local clinical research regulations and obtained approval from the Ethics Committee of Brescia District.
Animals
C57BL/6J and syn III knockout mice (a kind gift of Prof. Fabio Benfenati, Italian Institute of Technology, Genoa, Italy) 46 were used to prepare brain protein extracts. Animals were bred and housed in the Animal House facility of the Department of Molecular and Translational Medicine of the University of Brescia with food and water and maintained on a 12‐h light/dark cycle at a room temperature 23°C. All experiments and surgical procedures were approved by the Animal Research Committees of the University of Brescia and were conformed and approved to the National Research Guide for the Care and Use of Laboratory Animals (Aut. Min. 719/2015‐PR) according to art.31 D.Lgs.26/2014 of the Italian Ministry of Health. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Immunohistochemistry
For immunohistochemical studies we analyzed CP and SN sections from ten PD patients and ten age‐matched controls as well as hippocampal sections from the DLB patient. Briefly, following deparaffinization and antigen retrieval with 10 mM sodium citrate buffer, sections were incubated for 1 h at room temperature (RT) in blocking solution made up by 2% w/vol bovine serum albumin (BSA, Sigma Aldrich), 3% vol/vol normal goat serum (NGS, Sigma Aldrich), 0.3% Triton X‐100 diluted in phosphate buffer saline (PBS) 0.1 M pH 7.4 and then with the primary antibody diluted at optimal working concentration in the above described blocking solution overnight (o.n.) at 4°C. Sections were then washed twice with PBS 0.1 M pH 7.4 and incubated with the opportune biotin‐conjugated secondary antibody (Vector Laboratories) diluted at optimal working concentration for 45 min at RT. The immunostaining was visualized with an avidin–biotin system (Vector Laboratories) and 3,3‐diaminobenzidine as the chromogen (DAB kit, Vector Laboratories).
Counterstaining was performed using Hematoxylin for 3 min and Eosin Y (0.5%) for 30 s. Sections were then washed and dehydrated before mounting using the Vectamount mounting medium (Vector Laboratories). Specificity of the syn III antibody was confirmed by performing pre‐adsorption of the antibody with recombinant human syn III (NovusBio) according to previously described methods 4.
For double labeling immunofluorescence staining, after the incubation with the primary antibody, sections were washed with 0.3% Triton X‐100 diluted in PBS 0.1 M pH 7.4 and incubated with the appropriate fluorophore‐conjugated secondary antibody (Jackson Immuno Research) for 45 min at RT. This was followed by three washes in 0.3% Triton X‐100 diluted in PBS 0.1 M, pH 7.4 and then by incubation with a second primary antibody diluted at optimal working concentration in blocking solution for 2 h at RT. Sections were finally incubated with the optimal fluorophore‐conjugated secondary antibody and then washed three times with 0.3% Triton X‐100 diluted in PBS 0.1 M, pH 7.4 for 1 h at RT. Cell nuclei were then counterstained with Hoechst 33258 (Sigma‐Aldrich), and finally sections were mounted using the Vectashield mounting medium (Vector Laboratories).
In situ PLA
The in situ PLA allows the detection of protein‐protein interactions in situ in intact tissues 6, 7, 57, 68. For the in situ PLA studies we analyzed human paraffin embedded brain sections from the SN and CP of PD patients and age‐matched controls as well as hippocampal sections from a patient affected by DLB by using the Duolink assay kit (O‐LINK Bioscience) with a protocol adapted from the manufacturer's instruction. Briefly, following deparaffinization and antigen retrieval with proteinase K (50 µg/mL) digestion for 10 min at RT followed by 5 min incubation in 80% formic acid, sections were incubated in blocking solution (provided by the kit) for 1 h at RT and then with the primary antibodies recognizing syn III and α‐syn at 1:100 dilution o.n. at 4°C. On the following day, samples were washed and then incubated with PLA probe solution for 2 h at 37°C. Sections were then washed and incubated with the ligation solution for 1 h at 37°C, and then with the amplification solution at 37°C for 3 h. Finally, counterstaining was performed using Hematoxylin for 3 min and sections were washed, dehydrated, mounted using Vectamount mounting medium (Vector Laboratories).
Bright field and confocal microscopy
Sections were observed by means of either an inverted light/epifluorescence microscope (Olympus BX41; Olympus) or a Zeiss confocal laser microscope (Carl Zeiss S.p.A.), with the laser set on λ = 405–488–543 nm and the height of the sections scanning = ∼1 μm. Images (512 × 512 pixels) were then reconstructed using Zen lite 2.3 (Carl Zeiss S.p.A.) and Adobe Photoshop CC (Adobe systemCA) software.
Immunoblotting on human brain tissues
For protein extraction, frozen human brain tissue samples or striatal brain tissue from C57BL/6J wild type and C57BL/6J syn III knockout mice, were lysed in radioimmunoprecipitation (RIPA) buffer containing 50 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 1% NP‐40 (Sigma‐Aldrich), 0.5% sodium deoxycholate, 1 mM NaF, 1 mM Na3VO4 and 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM ethylenediaminetetraacetic acid (EDTA) plus complete proteasome inhibitor mixture (Roche Diagnostics), followed by centrifugation at 10 000 × g for 20 min at 4°C. Protein concentration in the samples was measured using the Bio‐Rad DC™ protein assay kit (Bio‐Rad Laboratories). Equal amounts of proteins (30 μg) were separated on 10% SDS‐Page gels and blotted on onto a polyvinylidene difluoride membrane (PVDF, Merck Millipore). Membranes were blocked in blocking buffer made up by Tris buffer saline (TBS) containing 0.1% Tween 20 (Sigma Aldrich) and 5% non‐fat dried milk (Gene Spin) for 1 h at RT and then incubated o.n. at 4°C with the appropriate primary antibody diluted at optimal working concentration in blocking buffer. On the following day membranes were washed in TBS containing 0.1% Tween 20 and incubated for 1 h at RT with the appropriate peroxidase‐conjugated secondary antibody diluted at optimal working concentration in TBS containing 0.1% Tween 20 (Santa Cruz Biotechnology). Blots were developed by using enhanced chemiluminescence (Gene Spin). Densitometry analysis of bands was performed by means of ImageJ Software (NIH Image). All bands were normalized to GAPDH levels as a control of equal loading of the different samples analyzed. For densitometry analysis of bands, each experimental condition was performed in triplicate.
Purification of LB‐enriched fractions from PD brains
LB were extracted from fresh frozen SN sections of four PD patients using a previously described method 51. Briefly, tissues were homogenized in 9 vol (w/vol) ice‐cold membrane signal extraction (MSE) buffer [10 mM MOPS/KOH, pH 7.4, 1 M sucrose, 1 mM ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid (EGTA) and 1 mM EDTA with proteasome inhibitor mixture (Roche Diagnostics)]. For LB purification, a sucrose step gradient was prepared by overlaying 2.2 M with 1.4 M and finally with 1.2 M sucrose in volume ratios of 1:2:2 (vol/vol). The homogenate was layered on the gradient and centrifuged at 160 000 × g for 3 h using a SW40 rotor (Beckman Coulter). Twelve fractions of 1 mL were collected from each gradient from top (fraction 1) to bottom (fraction 12), and analyzed for the presence of protein aggregates by immunoblotting.
Immunogold transmission electron microscopy
To examine LB by electron microscopy, we used negative staining technique. Briefly, 25 µL of the indicated LB‐enriched fraction were absorbed onto carbon coated grid, and negatively stained with 0.5% aqueous uranile acetate for 5 min, air‐dried and examined using a Tecnai G2 12 Twin instrument (FEI).
For immunogold labeling, 25 µL of the indicated LB‐enriched fraction were absorbed onto carbon coated grid, the excess was removed using a filter paper. Subsequently, a blocking step was performed in which grids were incubated with the following primary antibodies. First, α‐syn (1:1000) and syn III (1:1000), for 1 h at RT. After removing the unbound antibodies by washing, anti‐mouse IgG conjugated to 15 nm gold particles and anti‐rabbit IgG conjugated to 5 nm gold particles (1:100 dilution) were used to reveal α‐syn and syn III, respectively. Once the antibody reactions had taken place, grids were washed twice with PBS and three times with water, then double stained for 10 min first with 0.5% aqueous uranile acetate, and then 1% Pb citrate.
Antibodies
A list of the primary antibodies and of their working concentrations for immunohistochemistry and WB studies is summarized in Table 1. The secondary antibodies used for fluorescence immunohistochemistry were a goat anti‐mouse IgG Cy3‐conjugated and a goat anti‐rabbit Alexa488‐conjugated (Jackson ImmunoResearch). For bright‐field microscopy we used biotinylated goat anti‐rabbit IgG or goat anti‐mouse IgG (Vector Laboratories). The secondary antibodies used for WB were goat anti‐rabbit IgG‐HRP or goat anti‐mouse IgG‐HRP (Santa Cruz Biotechnology).
Table 1.
List of the primary antibody used for the study and of their working dilutions.
| Primary antibody | Manufacturer | Working concentrations | |
|---|---|---|---|
| WB | IHC | ||
| DAT | Millipore | 1:2000 | / |
| GAPDH | Sigma‐Aldrich | 1:5000 | / |
| Syn Ia/b | SYSY | 1:3000 | 1:600 |
| Syn II | AbCam | / | 1:600 |
| Syn III | SYSY | 1:3000 | 1:600 |
|
α‐syn (Syn211) aa 121–125 |
Santa Cruz | / | 1:500 |
|
α‐syn (Syn211) aa 121–125 |
Thermo Fisher | 1:1000 | / |
| TH | Millipore | 1:2000 | 1:600 |
Statistical analysis
Differences between PD and control SN and CP samples in the levels of TH, DAT, syn III, α‐syn and syn Ia/b, when normalized against GAPDH, were assayed by using unpaired Student t‐test. Correlations between syn III and α‐syn levels were expressed as Spearman rank correlation coefficient. All calculations were performed by using GraphPad Prism version 6 for Windows (GraphPad Prism Software). All data were presented as mean + s.d. Statistical significance was established at P < 0.05.
Results
Synapsin III immunoreactivity in the brain of patients affected by PD and DLB or age‐matched controls
The distribution of syn III and α‐syn was evaluated in the post‐mortem brains of patients with PD and age‐matched controls. Specificity of the staining was confirmed by pre‐adsorption of the antibody against syn III with human recombinant syn III protein (Supporting Information Figure 1C) and resulted in line with our previously published observations 68. Intracellular (Figure 1E,F, black arrowheads) and parenchymal (Figure 1G,H, black arrowheads) LB and LN with a varicose (Figure 1I, black arrowhead) or filiform shape (Figure 1J, black arrowhead) exhibited α‐syn (Figure 1C,E,G,I) and syn III (Figure 1D,F,H,J) immunopositivity in the SN. Control brains did not show LB or LN (Figure 1A,B). Remarkably, we observed that some of the syn III‐immunopositive LB exhibited a round‐shaped, dark‐stained core and a clearer halo, which is a morphology that is typical of the LB of the brainstem 26, 32 (Figure 1H).
Figure 1.
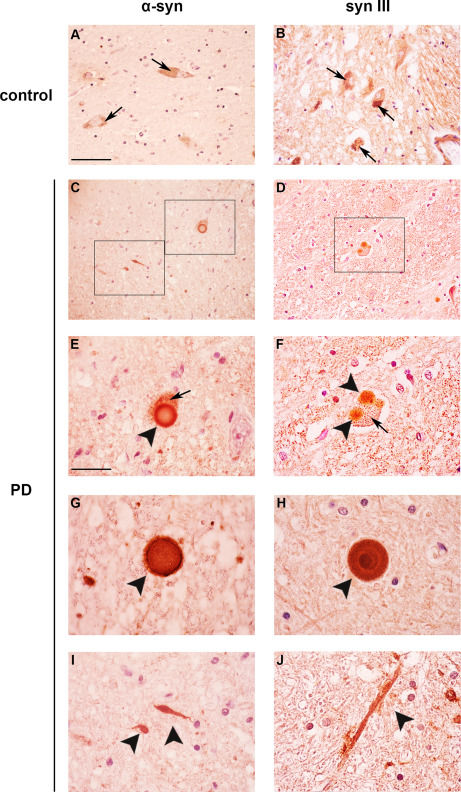
Synapsin III staining in the SN of PD patients and age‐matched controls. Representative images showing α‐syn (A,C,E,G,I) and syn III (B,D,F,H,J) immunolabeling in the SN of patients affected by sporadic PD and age‐matched control subjects (CTR). Please note the presence of neuromelanin positive neurons in both control and PD subjects (A,B,E,F, arrows). In the PD samples α‐syn was detected in LB (E–G, black arrowheads) and LN of different shapes (I, black arrowheads). Please note that in the SN of PD patients both LB (D,F,H) and swollen LN (J) showed a marked immunopostivity for syn III. Scale bars: (A–D) 50 µm, (E–J) 20 µm.
We then examined α‐syn distribution in the CP. Control subjects only exhibited a diffused α‐syn staining in the gray matter (Figure 2A, arrowheads). However, this protein showed a dot‐like, sparse distribution in the white matter of the PD brain (Figure 2C, black arrowhead) and a more intense and widespread accumulation in the gray matter. Several serpentine‐shaped LN were also visible (Figure 2E, black arrowhead). Synapsin III exhibited a diffused staining in the gray matter and granular‐like syn III‐positive dots with a tight distribution were detected in the white matter (Figure 2D,F). Conversely, syn III in the CP of control subjects was primarily localized within immunopositive untidy areas that were reminiscent of the scattered distribution described in the mouse brain 47 (Figure 2B). The differences observed between syn III and α‐syn distribution in the CP of control and PD subjects are in agreement with our previous observations 68 showing that the proteins do not completely co‐localize in this brain area.
Figure 2.
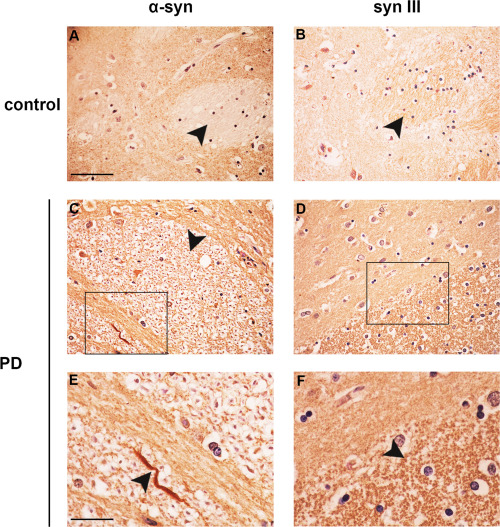
Synapsin III immunolabeling in the CP of PD patients and age‐matched controls. Panels are showing α‐syn (A,C,E) and syn III (B,D,F) immunolabeling in the CP of PD patients (C–F) when compared with that of age‐matched control subjects (CTR) (A,B). Please note that the distribution of α‐syn and syn III in the PD brains was very different from that of controls. In particular, in the brains of PD patients α‐syn resulted to be present both in the parenchyma, in sparsely distributed dots within white matter (E, arrowhead) and several LN (E, arrowhead), while syn III staining was accumulated both in the parenchyma and in densely packed dots within white matter areas (F, arrowhead). Conversely, in the CP of healthy controls α‐syn showed a widely diffused staining in the gray matter with white matter areas that were found to be almost devoid of signal (A), while the immunopositivity for syn III was very scarce and poorly organized (B). Scale bars: (A–D) 50 µm, (E,F) 20 µm.
Further analysis of the hippocampus of one patient affected by DLB (Supporting Information Figure 1C–H) confirmed the presence of syn III‐ and α‐syn‐immunopositive LB. Alpha‐synuclein‐immunopositive structures reminiscent of coiled bodies were also visible in the brain of this patient (Supporting Information Figure 1G).
Synapsins can form heterodimers 31. Therefore, we evaluated whether syn Ia/b or syn II were detectable in LB or showed a distribution similar to syn III. Only a slight decrease in immunopositivity for syn Ia/b and syn II was observed in the SN (Figure 3C–F) and CP (Figure 3I–J) of PD brains compared with controls (Figure 3A,B). No syn Ia/b‐ (Figure 3C,E) or syn II‐positivity (Figure 3D,F) was observed in LB in the SN of PD patients. To date, in the PD tissues that were immunolabeled by using syn I a/b and II antibodies, very few LB showed a very faint brownish unspecific staining (Figure 3C–F) that is in line with that of unstained LB 3 and was comparable to that observed in the pre‐adsorbed negative controls (Supporting Information Figure 1C).
Figure 3.
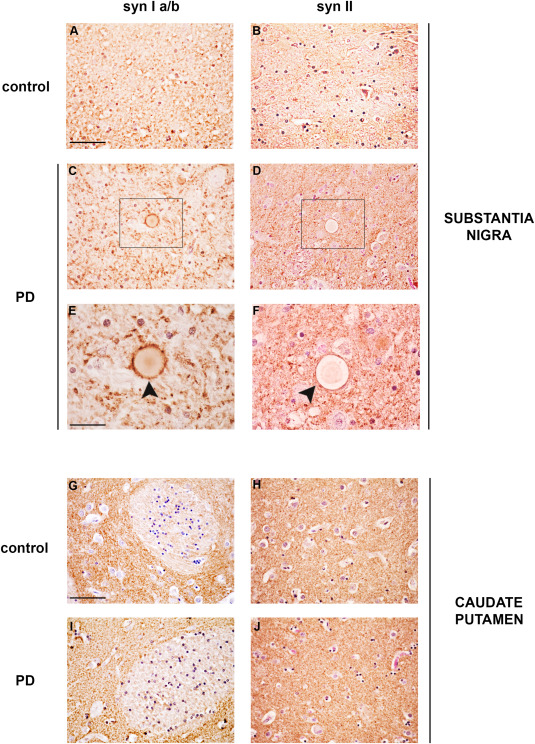
Synapsin Ia/b and syn II immunolabeling in the CP and SN of PD patients and age‐matched controls. Representative images showing syn Ia/b (A,C,E,G,I) and syn II (B,D,F,H,J) immunolabeling in the SN (A–F) and CP (G–J) of PD patients and age‐matched control subjects (CTR). Please note that LB in the SN of PD subjects did neither show positivity for syn Ia/b (C,E, black arrowhead) nor for syn II (D,F, black arrowhead). Scale bars: (A–D, G–J) 50 µm, (E,F) 20 µm.
Synapsin III and α‐syn co‐localization in the brain of PD, DLB and control subjects
We previously demonstrated a marked increase in the co‐localization of syn III and α‐syn in the CP of PD patients 68 which is consistent with the above observations. We used double fluorescence immunolabeling and found that many LB in the SN of PD brains exhibited a positive signal for syn III and α‐syn (Figure 4H,L white arrowheads). The distribution of α‐syn and syn III immunolabeling varied within LB in the SN. Several LB displayed a more marked signal for syn III at the borders with widespread α‐syn immunopositivity (Figure 4E,F,H), but α‐syn/syn III co‐localization was detectable at the edges and appeared to envelop a syn III‐positive core in other LB (Figure 4I,J,L). Synapsin III also co‐localized with α‐syn in hippocampal LB of the patient affected by DLB (Supporting Information Figure 2A). Numerous LN in PD brains exhibited α‐syn and syn III immunolabeling (Figure 4M,N,P, white arrowheads) while control individuals did not show inclusions or aggregates (Figure 4A–D).
Figure 4.
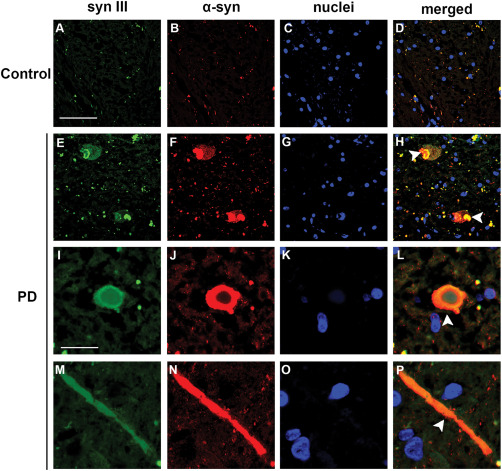
Co‐localization of α‐syn and syn III in LB and LN in the SN of PD subjects. Representative confocal images showing syn III and α‐syn fluorescence immunolabeling in the SN of patients affected by PD (E‐P) and age‐matched controls (CTR) (A–D). Please note LB (H–L, arrowheads) and a filiform LN (P, arrowhead) that were positive for both syn III and α‐syn. Scale bars: (A–H) 50 µm, (I–P) 20 µm.
Alpha‐synuclein‐positive fibrils from LB‐enriched protein extracts exhibited syn III positivity
We investigated the presence of syn III within the LB‐composing fibrils. We produced LB‐enriched protein extracts from the SN of PD brains using a sucrose gradient separation protocol 51. Given the elevated homology between human and mouse syn III, the antibody that we used for this study recognizes both of them. For this reason, we could assay the specificity of the syn III WB immunolabeling by analyzing mouse brain extracts from wild type and syn III knockout mice (Supporting Information Figure 1L). WB analysis showed that LB‐enriched fractions 10–12 and 7 exhibited a strong immunopositive signal for α‐syn (Figure 5A), which is consistent with previous findings 51. We found that the highest molecular weight LB‐enriched fractions (11 and 12) also exhibited a mild and marked immunopositivity for syn III, respectively. These fractions did not contain syn Ia/b and syn II (Figure 5A), which further confirms the above described specificity of syn III immunohistochemical positivity within LB. The LB‐enriched protein fractions were then analyzed using double immunogold TEM. Synapsin III was labeled using small (5 nm) gold particles and α‐syn was labeled using large (15 nm) gold particles. These studies demonstrated that α‐syn fibrils were also syn III‐immunopositive (Figure 5B–E). Synapsin III was detected in close proximity (Figure 5D,E) or further away from α‐syn (Figure 5B,C). However, we cannot exclude that the experimental protocol that we used for the TEM double labeling could have led to an underestimation of the effective rate of the α‐syn/syn III interaction. A previous study described that antibodies directed against different portions of α‐syn resulted in a diverse estimation of the protein content within LB fibrils 58. Here, we used Syn211 antibody which binds to aa 121–125 of the α‐syn protein and it is likely to lower TEM immunopositive signals compared with antibodies that recognize the full‐length protein 58. The steric footprint of 15‐nm gold particles may also have contributed to a lower detection of α‐syn using TEM.
Figure 5.
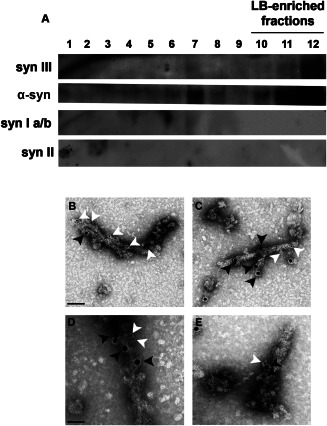
WB and TEM analysis of LB‐enriched protein extracts from the SN of PD patients. Representative immunoblotting of syn III, α‐syn, syn Ia/b and syn II in the different fractions obtained by sucrose gradient fractionation of freshly frozen postmortem nigral brain tissue from one of the PD patients analyzed (A). Please note the presence of both α‐syn and syn III in the LB‐enriched fractions no. 11–12. B–E. representative images showing TEM analysis of the LB‐enriched protein fractions no. 11–12 from one of the PD patients analyzed where syn III was labeled by using small (5 nm) gold particles (white arrowheads) and α‐syn by large (15 nm) gold particles (black arrowheads). Please note that fibrils resulted to be positive both for α‐syn and syn III. Scale bars: (B,C) 100 nm, (D,E) 50 nm.
Semiquantitative WB analysis of α‐syn, syn III and syn Ia/b in the SN and CP of PD patients and controls
We used WB to probe the levels of monomeric α‐syn, syn III and syn Ia/b in the SN (Figure 6A) and CP (Figure 6B) of PD patients and control subjects to validate immunohistochemical data. The amounts of TH in the SN and DAT in the CP were also evaluated as an index of dopaminergic neuron degeneration in the samples analyzed (Figure 6A,B).
Figure 6.
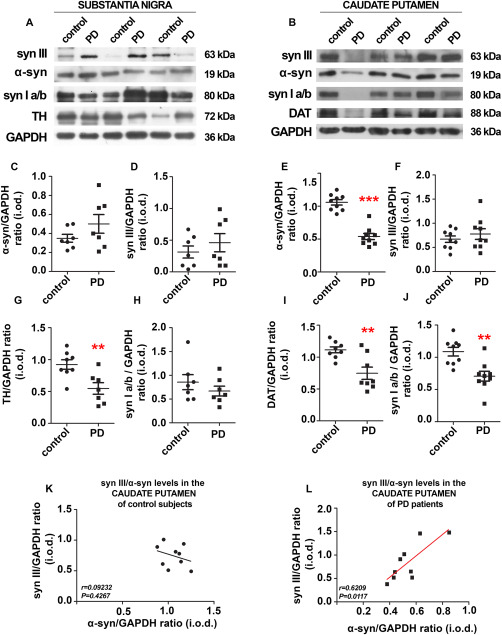
Analysis of protein levels in the SN and CP of PD patients and age‐matched controls. Representative immunoblotting showing syn III, syn Ia/b, α‐syn, TH ad DAT‐positive bands in the SN (A) (n = 7) and CP (B) (n = 8) of PD patients and age‐matched controls (CTR). GAPDH‐immunopositive bands are reported as a control for equal loading. C–J. Histograms showing the densitometric analysis of monomeric α‐syn, syn III, TH and syn Ia/b in the SN (C,D,G,H) as well as in the CP (E,F,I,J). Protein levels were normalized against those of GAPDH. ** P < 0.01, *** P < 0.001 Student's t‐test. K,L. Graphs are showing the correlation between syn III and monomeric α‐syn levels in the CP (n = 9) of control subjects (K) and PD patients (L) expressed as Spearman rank correlation coefficient.
We observed that the levels of monomeric α‐syn (Figure 6C) were not altered in the SN, despite a significant decrease in TH in PD patients compared with controls (Figure 6G), which is consistent with previous findings 60, 65. Similarly, the levels of syn III (Figure 6D) and syn Ia/b (Figure 6H) were not significantly altered in the brains of PD patients compared with controls.
The levels of monomeric α‐syn (Figure 6E), syn Ia/b (Figure 6J) and DAT (Figure 6I) were significantly diminished in the CP of PD patients compared with age‐matched controls. The decrease in monomeric α‐syn is consistent with previous studies 24, 60, and it may reflect the rate of dopamine terminal degeneration. In contrast, the amount of syn III (Figure 6F) in PD and control subjects was comparable. The lack of syn III reduction observed in the PD brain may be indicative of the accumulation of this protein within the remaining dopamine terminals. Synapsin I and syn II mislocalize with VMAT2 in the striatum 9, which suggests that syn III is the only isoform regulating striatal dopamine release. This is supported by studies on syn III knockout mice 38 and is consistent with our previous study on primary dopaminergic mesencephalic neurons 68. By using in vitro and in vivo experimental models we also found that absence or aggregation of α‐syn induces a significant increase and redistribution of syn III in dopaminergic neurons 68, which supports a link between the dysregulation of α‐syn and syn III. Consistently, we found a significant positive correlation between α‐syn and syn III levels in the CP of PD patients (Figure 6L), but not in the CP of control subjects (Figure 6K).
To further probe the interplay between syn III and α‐syn, that was supported by the correlation between the levels of the proteins observed in the CP of PD patients, we probed the presence of syn III in Urea/SDS detergent‐insoluble protein fractions (Supporting Information Figure 1M). The results showed that syn III could be detected only in the Urea/SDS fraction of PD patients that has been previously described to contain an elevated content of α‐syn aggregates 59. Conversely, from the analysis of protein extracts from the CP of control subjects we found that syn III resulted to localize within the TBS+ detergent‐soluble fraction. These observations further support the occurrence of syn III aggregation in the PD brains and are consistent with data deriving from the analysis of LB‐enriched protein fractions described above.
In situ PLA detects syn III/α‐syn neuropathology in the brain of patients affected by PD and DLB
We assessed the α‐syn/syn III interaction using bright‐field in situ PLA to visualize α‐syn/syn III complexes in the brains of PD and DLB patients and controls. This technique allows the visualization of a single protein‐protein interaction in situ in intact tissues 6, 7, 57, 68 and it is useful for the detection of α‐syn protein complexes in the brains of α‐synucleinopathy patients 52. The results revealed a PLA‐positive signal, which indicates a direct interaction between α‐syn and syn III in proteinase K and formic acid pretreated sections of the SN and CP of PD patients and age‐matched controls (Figure 7). The brownish dot‐like PLA signal appeared increased in the SN of PD subjects (Figure 7C–E) compared with controls (Figure 7A,B). Numerous neuromelanin‐containing cells exhibited α‐syn/syn III PLA‐positive inclusions in the PD brains (Figure 7C), but a marked PLA‐positivity was also present within neurons devoid of neuromelanin (Figure 7E, black arrowheads), neurite varicosities and parenchymal LB (Figure 7D, red arrowheads). These observations support an increase in α‐syn/syn III pathological aggregates in PD. A similar pattern of PLA distribution was detected in the hippocampus of the DLB patient, in whom the PLA positivity was localized within pyramidal neurons and the parenchyma (Supporting Information Figure 2B–D). Moreover, we observed an accumulation of PLA positivity in numerous dots localized within neuronal cells (Figure 7I) and the parenchyma (Figure 7J) in PD brains.
Figure 7.
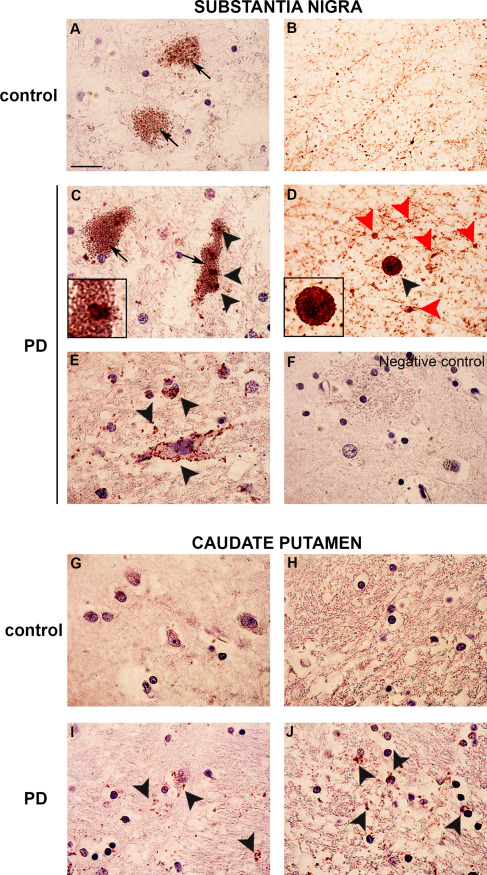
Synapsin III/α‐syn in situ PLA in the human brain. Representative images showing α‐syn/syn III in situ PLA in the SN and CP of PD patients and age‐matched‐controls (CTR). The presence of a positive PLA signal, as brownish spots (black and red arrowheads), indicates the interaction between α‐syn and syn III. Representative images from the SN (A–F). Please note the marked increase in PLA‐positivity in the PD samples, where a marked interaction between α‐syn and syn III was detected within several inclusions localized in neuromelanin‐containing neurons (arrows) as well as in LN and neuropils (C–E, black arrowheads). Numerous neuritic swellings of in the SN of PD patients also resulted to be PLA‐positive (D, red arrowheads). In the SN of control subjects, in situ PLA detected a marked proximity between α‐syn and syn III within neurites (B). However, control neuromelanin‐containing neurons did not show positivity (A). Representative image of a negative control (performed by omitting the syn III antibody) of the in situ PLA assayed on the CP of a PD subject (F). Please note that the absence of PLA‐positive signal is indicative of the specificity of the assay. Representative images of the CP (G–J). Please note the marked increase of the PLA signal in neuropil staining in the PD samples (I,J, black arrowheads) when compared with controls (G,H). Scale bar: 50 μm.
The PLA signal was slight in the CP of control brains, with few positive dots with a sparse distribution in the proximity of neuronal cells (Figure 7G) and a non‐reacting parenchyma (Figure 7H). Negative controls were performed via omission of the syn III antibody. These samples did not exhibit PLA positivity (Figure 7F), which confirms the specificity of the signal. The specificity of the interaction between α‐syn and syn III was further corroborated by double immunogold α‐syn/syn Ia/b, α‐syn/syn II and α‐syn/syn III TEM analysis performed on protein extracts from the brain of PD patients, where only syn III was identified in close association with α‐syn (Supporting Information Figure 2E–G).
Discussion
This study demonstrates that LB fibrils are made up by syn III in complex with α‐syn in PD. Neuropathological analysis in the SN and CP of PD brains showed that LB and LN exhibited a marked immunopositivity for syn III. In addition, a marked accumulation of proteinase K‐resistant α‐syn/syn III PLA‐positive aggregates was observed in the brain of PD patients. Lewy bodies from hippocampal sections from the patient with DLB were also positive for syn III and α‐syn, which again were found to be tightly associated in proteinase K‐resistant neuronal and parenchymal inclusions by in situ PLA. Most strikingly, syn III was found to be present within α‐syn‐positive fibrils extracted from the brain of patients affected by PD. Although syn I, II and III form heterodimers that are relevant for their function 31, LB and LN were not immunopositive for syn Ia/b or syn II.
Although syn III and α‐syn immunolabeling in the SN and CP were found to differ, our in situ PLA and double immunogold TEM studies support a direct interaction between these proteins in the PD and age‐matched control brains. This feature may be ascribed to the fact that the interaction between syn III and α‐syn occurs only in specific neuronal districts beside their distribution is wider. Similarly, although syn III can form functional heterodimers with syn I a/b and syn II 16, the distribution of these proteins is very different in the mouse central nervous system 47.
Synapsin III is the last identified member of the syn phosphoprotein family 31, 33 and few studies characterized its function in the CNS 31, 33, 34, 38, 44, 46, 47, 48, 49, 50, 68. These investigations demonstrated that syn III regulates neurotransmitter release, similar to syn Ia/b and syn II, but the syn III isoform is also strongly involved in brain development and axon elongation 35, 45, 50. Interestingly, syn III‐dependent neuronal survival, neuritic outgrowth and polarization are finely regulated via its phosphorylation by cyclin‐dependent kinase 5 (CDK5) 44, 46, 55, which is localized within LB 12 and may be involved in cell loss in PD 17, 63. The induction of CDK5 by α‐syn mediates apoptotic cell death 21, 36, 63. Synapsin III alterations or changes in the rate of its CDK5‐mediated phosphorylation may thus be involved in the α‐syn‐related apoptosis, a hypothesis that deserves further investigation.
Alterations of syn III are related to the onset of neuropsychiatric disorders, such as attention deficit hyperactivity disorder (ADHD) 37 and schizophrenia 29, 50 as well as to multiple sclerosis 41, 43. No association between syn III genetic variations and PD were detected in two Chinese patient cohorts 67 although this protein was found to be involved in the regulation of dopaminergic neuron synaptic function 38, 68. The present study is the first demonstration that syn III accumulation may represent a pathological hallmark of PD and DLB.
The in situ PLA studies revealed a proteinase K‐resistant α‐syn/syn III‐positive deposits in the brains of PD and DLB patients. These inclusions were composed of both proteins and localized within neuronal cells, neuronal processes and LB‐like structures. This observation is consistent with the results of TEM studies which demonstrated that LB‐fibrils from brains of PD patients were positive for syn III and α‐syn. This is a very first evidence that the commonly described as α‐syn fibrils are indeed α‐syn/syn III fibrils. Numerous studies have previously reported the presence of other proteins within LB and LN in the PD brains, but none of them was found to constitute an α‐syn‐interacting component in fibrils 12, 18, 30, 64.
The positive correlation between α‐syn and syn III levels that was detected in the CP suggests that these proteins are co‐dysregulated at striatal terminals of PD patients although their levels seem to move in different directions. The results of sequential Urea/SDS protein extraction indicate the presence of syn III in detergent‐insoluble Urea/SDS fraction of the PD samples that is highly enriched in aggregated α‐syn 59. This finding is in line with data from LB extraction and in situ PLA and suggests the occurrence of a pathological interplay within these proteins that are both aggregated in the brain of affected patients. The contribution of syn III/α‐syn interaction in the onset and progression of LB pathology is under investigation. Ongoing studies support that experimental syn III knockout hampers α‐syn aggregation and toxicity. These observations entail relevant implications within the synaptic hypothesis of synucleinopathies 39, 53 and hint that microaggregation of α‐syn and syn III, both enriched at synaptic sites, may strictly contribute to the pathogenesis of PD and DLB.
Conflicts of Interest
The authors declare that they have no conflicts of interest to disclose.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Negative controls of the IHC experiments and alpha‐synuclein and syn III immunolabeling in the hippocampus of one patient affected by sporadic DLB.
Figure S2. Double labeling and in situ PLA in the post‐mortem brain of one patient affected by DLB.
Acknowledgments
We are grateful to Professor Maurizio Memo for insightful discussion. We thank Prof. Fabio Benfenati, for kindly providing syn III knockout mice. This work was supported by Fondazione Cariplo (2014‐0769), by University of Brescia (BIOMANE), by the Michael J. Fox Foundation for Parkinson's Research, NY (Target Advancement Program, grant ID #10742).
Tissue samples and associated clinical and neuropathological data were supplied by the Parkinson's UK Brain Bank, funded by Parkinson's UK, a charity registered in England and Wales (258197) and in Scotland (SC037554). F.L. PhD program was supported by the Fondazione Camillo Golgi, Brescia.
References
- 1. Abeliovich A, Schmitz Y, Farinas I, Choi‐Lundberg D, Ho WH, Castillo PE et al (2000) Mice lacking alpha‐synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25:239–252. [DOI] [PubMed] [Google Scholar]
- 2. Anichtchik O, Calo L, Spillantini MG (2013) Synaptic dysfunction in synucleinopathies. CNS Neurol Disord Drug Targets 999:1094–1100. [PubMed] [Google Scholar]
- 3. Baba M, Nakajo S, Tu P, Tomita H, Nakaya TK, Lee VM et al (1998) Aggregation of alpha‐synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 4. Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG (2004) Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol 165:1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P (2008) Alpha‐synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem 106:560–577. [DOI] [PubMed] [Google Scholar]
- 6. Bellucci A, Navarria Falarti L, Zaltieri E, Bono M, Collo FG, Spillantini M et al (2011) Redistribution of DAT/alpha‐synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson's disease. PLoS One 6:e27959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bellucci A, Fiorentini Zaltieri C, Missale MC, Spano P (2014) The “in situ” proximity ligation assay to probe protein‐protein interactions in intact tissues. Methods Mol Biol 1174:397–405. [DOI] [PubMed] [Google Scholar]
- 8. Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F et al (2016) Review: Parkinson's disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol 42:77–94. [DOI] [PubMed] [Google Scholar]
- 9. Bogen IL, Boulland JL, Mariussen E, Wright MS, Fonnum F, Kao HT, Walaas SI (2006) Absence of synapsin I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J Neurochem 96:1458–1466. [DOI] [PubMed] [Google Scholar]
- 10. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 11. Breda C, Nugent ML, Estranero JG, Kyriacou CP, Outeiro TF, Steinert JR, Giorgini F (2015) Rab11 modulates alpha‐synuclein‐mediated defects in synaptic transmission and behaviour. Hum Mol Genet 24:1077–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brion JP, Couck AM (1995) Cortical and brainstem‐type Lewy bodies are immunoreactive for the cyclin‐dependent kinase 5. Am J Pathol 147:1465–1476. [PMC free article] [PubMed] [Google Scholar]
- 13. Burre J, Sharma Tsetsenis M, Buchman TV, Etherton MR, Sudhof TC (2010) Alpha‐synuclein promotes SNARE‐complex assembly in vivo and in vitro. Science 329:1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burre J (2015) The synaptic function of alpha‐synuclein. J Parkinsons Dis 5:699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Calo L, Wegrzynowicz M, Santivanez‐Perez J, Grazia Spillantini M (2016) Synaptic failure and alpha‐synuclein. Mov Disord 31:169–177. [DOI] [PubMed] [Google Scholar]
- 16. Cesca F, Baldelli P, Valtorta F, Benfenati F (2010) The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91:313–348. [DOI] [PubMed] [Google Scholar]
- 17. Cheung ZH, Ip NY (2012) Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol 22:169–175. [DOI] [PubMed] [Google Scholar]
- 18. Chutna O, Goncalves S, Villar‐Pique A, Guerreiro P, Marijanovic Z, Mendes T et al (2014) The small GTPase Rab11 co‐localizes with alpha‐synuclein in intracellular inclusions and modulates its aggregation, secretion and toxicity. Hum Mol Genet 23:6732–6745. [DOI] [PubMed] [Google Scholar]
- 19. Cookson MR, Xiromerisiou G, Singleton A (2005) How genetics research in Parkinson's disease is enhancing understanding of the common idiopathic forms of the disease. Curr Opin Neurol 18:706–711. [DOI] [PubMed] [Google Scholar]
- 20. Cookson MR, Hardy J, Lewis PA (2008) Genetic neuropathology of Parkinson's disease. Int J Clin Exp Pathol 1:217–231. [PMC free article] [PubMed] [Google Scholar]
- 21. Czapski GA, Gąssowska M, Wilkaniec A, Cieślik M, Adamczyk A (2013) Extracellular alpha‐synuclein induces calpain‐dependent overactivation of cyclin‐dependent kinase 5 in vitro. FEBS Lett 587:3135–3141. [DOI] [PubMed] [Google Scholar]
- 22. Dalfo E, Barrachina M, Rosa JL, Ambrosio S, Ferrer I (2004) Abnormal alpha‐synuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol Dis 16:92–97. [DOI] [PubMed] [Google Scholar]
- 23. Diao J, Burre Vivona JS, Cipriano D, Sharma J, Kyoung MM, Sudhof TC, Brunger AT (2013) Native alpha‐synuclein induces clustering of synaptic‐vesicle mimics via binding to phospholipids and synaptobrevin‐2/VAMP2. Elife 2:e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dunn AR, Stout KA, Ozawa M, Lohr KM, Hoffman CA, Bernstein AI et al (2017) Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc Natl Acad Sci U S A 114:E2253–E2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forno LS (1992) Neuropathologic features of Parkinson's, Huntington's, and Alzheimer's diseases. Ann N Y Acad Sci 648:6–16. [DOI] [PubMed] [Google Scholar]
- 26. Forno LS (1996) Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 55:259–272. [DOI] [PubMed] [Google Scholar]
- 27. Garcia‐Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, et al (2010) SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain 133:2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, et al (2008) The Parkinson's disease protein alpha‐synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A 105:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greco B, Manago F, Tucci V, Kao HT, Valtorta F, Benfenati F (2013) Autism‐related behavioral abnormalities in synapsin knockout mice. Behav Brain Res 251:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guerreiro PS, Huang Y, Gysbers A, Cheng D, Gai WP, Outeiro TF, Halliday GM (2013) LRRK2 interactions with alpha‐synuclein in Parkinson's disease brains and in cell models. J Mol Med (Berl) 91:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hosaka M, Sudhof TC (1999) Homo‐ and heterodimerization of synapsins. J Biol Chem 274:16747–16753. [DOI] [PubMed] [Google Scholar]
- 32. Jellinger KA (2009) A critical evaluation of current staging of alpha‐synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730–740. [DOI] [PubMed] [Google Scholar]
- 33. Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M et al (1998) A third member of the synapsin gene family. Proc Natl Acad Sci U S A 95:4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kao HT, Li P, Chao HM, Janoschka S, Pham K, Feng J et al (2008) Early involvement of synapsin III in neural progenitor cell development in the adult hippocampus. J Comp Neurol 507:1860–1870. [DOI] [PubMed] [Google Scholar]
- 35. Kao HT, Ryoo Lin KA, Janoschka SR, Augustine GJ, Porton B (2017) Synapsins regulate brain‐derived neurotrophic factor‐mediated synaptic potentiation and axon elongation by acting on membrane rafts. Eur J Neurosci 45:1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kazmierczak A, Czapski G, Adamczyk A, Gajkowska AB, Strosznajder JB (2011) A novel mechanism of non‐Abeta component of Alzheimer's disease amyloid (NAC) neurotoxicity. Interplay between p53 protein and cyclin‐dependent kinase 5 (Cdk5). Neurochem Int 58:206–214. [DOI] [PubMed] [Google Scholar]
- 37. Kenar AN, Edgunlu T, Herken H, Erdal ME (2013) Association of synapsin III gene with adult attention deficit hyperactivity disorder. DNA Cell Biol 32:430–434. [DOI] [PubMed] [Google Scholar]
- 38. Kile BM, Guillot TS, Venton BJ, Wetsel WC, Augustine GJ, Wightman RM (2010) Synapsins differentially control dopamine and serotonin release. J Neurosci 30:9762–9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kramer ML, Schulz‐Schaeffer WJ (2007) Presynaptic alpha‐synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee FJ, Liu F, Pristupa ZB, Niznik HB (2001) Direct binding and functional coupling of alpha‐synuclein to the dopamine transporters accelerate dopamine‐induced apoptosis. faseb J 15:916–926. [DOI] [PubMed] [Google Scholar]
- 41. Liguori M, Cittadella R, Manna I, Valentino P, La Russa A, Serra P et al (2004) Association between Synapsin III gene promoter polymorphisms and multiple sclerosis. J Neurol 251:165–170. [DOI] [PubMed] [Google Scholar]
- 42. Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha‐synuclein. Nat Rev Neurosci 3:932–942. [DOI] [PubMed] [Google Scholar]
- 43. Otaegui D, Zuriarrain O, Castillo‐Trivino T, Aransay A, Ruiz‐Martinez J, Olaskoaga J et al (2009) Association between synapsin III gene promoter SNPs and multiple sclerosis in Basque patients. Mult Scler 15:124–128. [DOI] [PubMed] [Google Scholar]
- 44. Perlini LE, Szczurkowska J, Ballif BA, Piccini A, Sacchetti S, Giovedi S et al (2015) Synapsin III acts downstream of semaphorin 3A/CDK5 signaling to regulate radial migration and orientation of pyramidal neurons in vivo. Cell Rep 11:234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perlini LE, Benfenati F, Cancedda L (2016) Synapsin III in brain development. Oncotarget 7:15288–15289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Piccini A, Perlini LE, Cancedda L, Benfenati F, Giovedi S (2015) Phosphorylation by PKA and Cdk5 mediates the early effects of synapsin III in neuronal morphological maturation. J Neurosci 35:13148–13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pieribone VA, Porton B, Rendon B, Feng J, Greengard P, Kao HT (2002) Expression of synapsin III in nerve terminals and neurogenic regions of the adult brain. J Comp Neurol 454:105–114. [DOI] [PubMed] [Google Scholar]
- 48. Porton B, Kao HT, Greengard P (2002) Characterization of transcripts from the synapsin III gene locus. J Neurochem 73:2266–2271. [DOI] [PubMed] [Google Scholar]
- 49. Porton B, Rodriguiz RM, Phillips LE, Gilbert JWt, Feng J, Greengard P et al (2010) Mice lacking synapsin III show abnormalities in explicit memory and conditioned fear. Genes Brain Behav 9:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Porton B, Wetsel WC, Kao HT (2011) Synapsin III: role in neuronal plasticity and disease. Semin Cell Dev Biol 22:416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Recasens A, Dehay B, Bove J, Carballo‐Carbajal I, Dovero S, Perez‐Villalba A et al (2014) Lewy body extracts from Parkinson disease brains trigger alpha‐synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. [DOI] [PubMed] [Google Scholar]
- 52. Roberts RF, Wade‐Martins R, Alegre‐Abarrategui J (2015) Direct visualization of alpha‐synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain. Brain 138:1642–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schulz‐Schaeffer WJ (2010) The synaptic pathology of alpha‐synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol 120:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scott D, Roy S (2012) alpha‐Synuclein inhibits intersynaptic vesicle mobility and maintains recycling‐pool homeostasis. J Neurosci 32:10129–10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shah K, Rossie S (2017) Tale of the good and the bad Cdk5: remodeling of the actin cytoskeleton in the brain. Mol Neurobiol [Epub ahead of print; doi: 10.1007/s12035-017-0525-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sidhu A, Wersinger C, Vernier P (2004) alpha‐Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson's disease. FEBS Lett 565:1–5. [DOI] [PubMed] [Google Scholar]
- 57. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J et al (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3:995–1000. [DOI] [PubMed] [Google Scholar]
- 58. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG (2003) Ubiquitination of alpha‐synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem 278:44405–44411. [DOI] [PubMed] [Google Scholar]
- 60. Tong J, Wong H, Guttman M, Ang LC, Forno LS, Shimadzu M et al (2010) Brain alpha‐synuclein accumulation in multiple system atrophy, Parkinson's disease and progressive supranuclear palsy: a comparative investigation. Brain 133:172–188. [DOI] [PubMed] [Google Scholar]
- 61. Totterdell S, Hanger D, Meredith GE (2004) The ultrastructural distribution of alpha‐synuclein‐like protein in normal mouse brain. Brain Res 1004:61–72. [DOI] [PubMed] [Google Scholar]
- 62. Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS (2014) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34:9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilkaniec A, Czapski GA, Adamczyk A (2016) Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: facts and hypotheses. J Neurochem 136:222–233. [DOI] [PubMed] [Google Scholar]
- 64. Xia Q, Liao L, Cheng D, Duong DM, Gearing M, Lah JJ et al (2008) Proteomic identification of novel proteins associated with Lewy bodies. Front Biosci 13:3850–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA (2002) Dopamine‐dependent neurotoxicity of alpha‐synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8:600–606. [DOI] [PubMed] [Google Scholar]
- 66. Yin G, Lopes da Fonseca T, Eisbach SE, Anduaga AM, Breda C, Orcellet ML et al (2014) alpha‐Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation‐dependent manner. Neurobiol Dis 70:149–161. [DOI] [PubMed] [Google Scholar]
- 67. Yu WJ, Li NN, Tan EK, Cheng L, Zhang JH, Mao XY et al (2014) No association of four candidate genetic variants in MnSOD and SYNIII with Parkinson's disease in two Chinese populations. PLoS One 9:e88050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zaltieri M, Grigoletto J, Longhena F, Navarria L, Favero G, Castrezzati S et al (2015) alpha‐synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci 128:2231–2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Negative controls of the IHC experiments and alpha‐synuclein and syn III immunolabeling in the hippocampus of one patient affected by sporadic DLB.
Figure S2. Double labeling and in situ PLA in the post‐mortem brain of one patient affected by DLB.