

Abstract
Nowadays, amyotrophic lateral sclerosis (ALS) is considered as a multisystem disorder, characterized by a primary degeneration of motor neurons as well as neuropathological changes in non‐motor regions. Neurodegeneration in subcortical areas, such as the thalamus, are believed to contribute to cognitive and behavioral abnormalities in ALS patients. In the present study, we investigated neurodegenerative changes including neuronal loss and glia pathology in the anterodorsal thalamic nucleus (AD) of SOD1(G93A) mice, a widely used animal model for ALS. We detected massive dendrite swelling and neuronal loss in SOD1(G93A) animals, which was accompanied by a mild gliosis. Furthermore, misfolded SOD1 protein and autophagy markers were accumulating in the AD. Since innate immunity and activation inflammasomes seem to play a crucial role in ALS, we examined protein expression of Nod‐like receptor protein 3 (NLRP3), apoptosis‐associated speck‐like protein containing a caspase‐1 recruitment domain (ASC) and the cytokine interleukin 1 beta (IL1β) in AD glial cells and neurons. NLRP3 and ASC were significantly up‐regulated in the AD of SOD1(G93A) mice. Finally, co‐localization studies revealed expression of NLRP3, ASC and IL1β in neurons. Our study yielded two main findings: (i) neurodegenerative changes already occur at an early symptomatic stage in the AD and (ii) increased inflammasome expression may contribute to neuronal cell death. In conclusion, neurodegeneration in the anterior thalamus may critically account for cognitive changes in ALS pathology.
Keywords: amyotrophic lateral sclerosis, anterodorsal nucleus, autophagy, interleukin 1 beta, misfolded SOD1, NLRP3
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common motoneuron disease in adults affecting upper motoneurons in the cerebral cortex and lower motoneurons in the spinal cord. Patients usually die within 3–5 years from the onset of symptoms. Most ALS cases occur sporadically with no obvious related risk factors and only about 10% account for familiar forms including mutations in diverse genes, including chromosome 9 open reading frame 72 (C9ORF72) repeat expansions, Cu2+/Zn2+ superoxide dismutase (SOD1) and TAR DNA binding protein (TDP‐43) 32. Although generally thought to exclusively affect the executive motor system, there is a growing body of evidence that ALS is a multisystem disease with widespread neurodegeneration in extra‐motor areas. Actually, up to 40% of ALS patients show cognitive impairment, ranging from mild cognitive deficits to frontotemporal dementia (FTD) 6. Thus, ALS patients may develop cognitive problems and behavioral changes including apathy and disorientation 80, impaired verbal fluency, decision‐making and memory deficits because of neuropathological alterations within the prefrontal and temporal lobe cortex and subcortical structures, including basal ganglia, substantia nigra, subthalamic nucleus and thalamus 6, 14, 74. An emerging body of evidence highlights the role of thalamus dysfunctions contributing to cognitive impairment in ALS 37, 44. The thalamus is reciprocally connected with numerous cortical and subcortical areas and involved in the integration of sensory as well as motor information. Because of its widespread integration, the thalamus can be thought of as a “relay station” which is sensitive to neuropathological changes in various brain regions 60. Pathological alterations within the thalamus of ALS patients have been confirmed by in vivo studies using positron emission tomography (PET) and neuropsychological examinations 37, 76, diffusion tensor magnetic resonance imaging (DTI) and event‐related functional magnetic resonance imaging (fMRI) 42, 62 as well as 1H MRS techniques 64. However, information about the cellular pathology and molecular mechanisms of ALS‐associated neurodegeneration within the thalamus are sparse. In the present work, we investigated neurodegenerative changes and neuroinflammation in the anterodorsal nucleus (AD) of the anterior thalamus (ANT) in SOD1(G93A) mice, a widely used animal model for ALS. The ANT consists of three different nuclei: the anteroventral (AV), anterodorsal (AD) and anteromedial (AM) nucleus 60. The ANT projects to the subiculum, hippocampus, cingulate and retrosplenial cortex. Main inputs arise from the subiculum, retrosplenial cortex and mammillary bodies (MB) (via the mammillothalamic tract, MTT). The fiber network from the hippocampus (via the fornix), MB, ANT (via the MTT), cingulum, entorhinal cortex and back to the hippocampus is also known as the “Papez circuit” 58. Interruption of this pathway clearly leads to deficits in spatial orientation, memory acquisition and storage emotional behavior 34. There is substantial evidence that the neuronal circuit from the dorsal tegmental nucleus of Gudden (passing the lateral MB to the AD) plays a critical role in the generation and propagation of head‐direction signals 5, 9. These head direction cells are thought to convey information for spatial orientation 15. In murine models of ANT lesions impaired spatial and non‐spatial memory have been demonstrated 3, 51, 66, 81. Clinical and experimental data have demonstrated that damage of the ANT or MB is responsible for memory deficits occurring in the Korsakoff syndrome 38, in thalamic stroke 57 and in prion diseases 50. Furthermore, the ANT seems to be involved in the maintenance and propagation of seizures 48, 49. In patients suffering from MS, a relationship of cognitive impairment and focal atrophy of the ANT was described 8. Finally, ALS patients with diminished regional cerebral blood flow (rCBF) in the ANT showed an impaired verbal fluency 37.
Mechanisms involved in the pathology of ALS are complex and poorly understood but may involve excitotoxicity, oxidative stress, protein misfolding and aggregation, RNA mis‐processing and neuroinflammation, resulting in necrotic or programmed neuronal cell death 4, 10, 17, 23, 75. Pattern recognition receptors (PRRs) play a pivotal role in the recognition of damage‐associated molecular patterns (DAMPs) 70. They are widely expressed within the central nervous system (CNS) by glial cells and neurons 18, 24, 69 and exert an important function in the innate immune response. Much attention has focused on the role of inflammasomes during neurodegeneration 63. Inflammasomes consist of a PRR NOD‐like receptor (NLR) molecule, the apoptosis‐associated speck‐like protein (ASC) protein containing a caspase activating and recruitment domain (CARD) and pro‐caspase‐1. After assembly of the inflammasome complex, caspase 1 becomes activated, processes IL1β and interleukin 18 (IL18) and induces pyroptotic cell death 46. The NLRP3 inflammasome is currently the most clinically relevant inflammasome involved in various neurological diseases, including Multiple sclerosis (MS) 16, Alzheimer's disease 31, 70, stroke 24, 40 and spinal cord injury 83. NLRP3 activation occurs in response to numerous DAMPs, including K+ efflux 36, extracellular ATP 45, amyloid‐β 28, and mitochondrial reactive oxygen species (mROS) 1. Recently, we have shown NLRP3 activation in the spinal cord of the SOD1(G93A) mouse model and in human sporadic ALS patients 35.
In the present work we examined neuronal and glial pathology, as well as expression of autophagy markers within the AD of the ANT in SOD1(G93A) mice. Furthermore, we studied the possible involvement of NLRP3 inflammasome activation in AD neurodegeneration.
Material and Methods
Animals
All animal experiments were performed under the terms of the German animal protection law and according to the regulations of the local animal research council and legislation of the State of North Rhine Westphalia (NRW), Germany. High‐copy number B6/SJL‐Tg(SOD1*G93A)1Gur/J mice 27, carrying a mutant human SOD1 gene, were obtained from Jackson Labs (Stock Number 002726; Bar Harbor, USA). The colony was maintained by crossing B6/SJL males carrying the SOD1 transgene with wild type B6/SJL females. Animals were housed in a pathogen free environment under a 12 h light/12 h dark cycle with free access to food and water. Genotyping was performed from tail biopsies by a standardized PCR protocol using primers against the human SOD1 gene 35. Male (m) and female (f) SOD1(G93A) mice were used to study pre‐symptomatic (6–9 weeks old; SOD1 PS) and early symptomatic (11–14 weeks old; SOD1 ES) phases of the disease 35. Wild type (WT) littermates in the same age served as controls. Each of the four experimental groups comprised 8 animals (4m and 4f). Late stage 18 weeks old SOD1 animals (SOD1 18W) and WT (n = 2) were included for representative H&E, immunofluorescence and electron microscopy. In total, 34 animals (n = 34) were included in the present study.
Electron microscopy (EM)
The SOD1 (n = 1), and the correspondent WT animals (n = 1) were anaesthetized and transcardially perfused with 1% glutaraldehyde and 1% paraformaldehyde in a 0.1 M phosphate buffer (PB, pH 7.4), and the tissue samples were processed for electron microscopy. The brains were removed and brain slices (800 μm) were postfixed in 1% OsO4 in PB, pH 7.4, for 2 h, dehydrated in graded ethanol, and embedded in Epon 820 (Serva, Heidelberg, Germany). The blocks were trimmed to contain the anterior thalamus. Semi‐thin sections (1µm) were taken to find the region of interest and stained with toluidine blue. Thin sections were cut on a Reichert–Jung OmU3 ultramicrotome were stained with uranyl acetate, followed by lead citrate. Thin sections were examined with a Philipps EM 100 electron microscope and photos were taken using a computer assisted digital camera (Olympus, Quemesa).
Immunohistochemistry and immunofluorescence
Brain tissue from SOD1 G93A mice was fixed and processed as described previously 35. Coronal sections of 5 μm were prepared within the region 245–255 according to the mouse brain atlas by Sidmann et al (http://www.hms.harvard.edu/research/brain/atlas.html). After heat‐unmasking and blocking with 5% normal serum sections were incubated overnight with the primary antibody diluted in blocking solution (Table 1). After rinsing in PBS, sections were exposed to the appropriate biotinylated secondary antibody (1:50, Vektor Laboratories, USA). Finally, immunoreaction was visualized using 3,3'‐diaminobenzidine (DAB) as substrate (DAKO, USA). For immunofluorescence studies, secondary antibodies conjugated with the fluorescent dyes AlexaFluor 488 or 594 (1:500; Invitrogen, Germany) were applied. Cell nuclei were counterstained with Hoechst 33342 (Invitrogen, Germany). To reduce autofluorescence, samples were treated with 0.5% Sudan Black solved in 70% ethanol for 5min at room temperature. Sections were viewed using a Leica DMI6000 B inverted microscope.
Table 1.
List of primary antibodies.
| Antibody | Host |
Company Order Number |
Dilution |
|---|---|---|---|
| ASC | Rabbit | Santa Cruz, USA | 1:100 |
| sc‐22514‐R | |||
| GFAP | Goat | Santa Cruz, USA | 1:100 |
| sc‐6170 | |||
| Iba1 | Mouse | Millipore, USA | 1:600 |
| MABN92 | |||
| Iba1 | Rabbit | Wako, USA | 1:5000 |
| 019‐19741 | |||
| IL1β | Mouse | Cell signaling, USA | 1:100 |
| 12242S | |||
| MAP2 | Rabbit | Cell signaling, USA | 1:500 |
| 8707S | |||
| NeuN | Mouse | EMD Millipore, USA | 1:250 |
| MAB 377 | |||
| NLRP3 | Rabbit | Bioss, USA | 1:500 |
| Bs10021R | |||
| NLRP3 | Mouse | Adipogen, USA | 1:300 |
| AG‐20B‐0014‐C100 | |||
| Olig2 | Rabbit | EMD Millipore, USA | 1:1000 |
| AB9610 | |||
| LAMP2 | Rat | BD Pharmingen, USA | 1:600 |
| 550292 | |||
| hSOD1 | Mouse | Santa Cruz, USA sc‐17767 | 1:500 |
| B8H10 | Mouse | Medimabs, USA | 1:250 |
| MM‐0070 | |||
| LC3B | Rabbit | Sigma, USA | 1:300 |
| L7543 | |||
| p62/SQSTM1 | Rabbit | MBL, USA | 1:300 |
| PM045 | |||
| Ubiquitin | Rabbit | Sigma, USA | 1:200 |
| U5379 |
Cell counting and fluorescence intensity measurements
Cell counting was executed to determine the number of positive stained cells within the area of the AD. Numbers of neuronal nuclei‐positive (NeuN+) neurons, glial fibrillary acidic protein‐positive (GFAP+) astrocytes, ionized calcium‐binding adapter molecule‐positive (Iba1+) microglia and Olig2+ oligodendrocytes were counted in the AD. Cell nuclei were counterstained with Hoechst (blue). Two slides (with a distance of ∼100 μm), each containing 2 brain sections, per animal were sampled. In total, all cells within the whole area of 8 AD (4 brain sections: left and right AD) were counted to estimate the number of cells per mm2. Only cells containing a visible nucleus were counted.
Intensity measurements were performed to determine relative changes in protein expression independently of the cell number. Analysis of GFAP, LAMP2, NLRP3, ASC and IL1β (N = 8 per group) and LC3B, p62/SQSTM1 and Ub (N = 4–5 per group) were performed using 20× magnification and Leica Application Suite X software. The whole area of both AD was selected in each brain section and integrated density was measured. Background readings were recorded from each individual nucleus and subtracted from the integrated density values to obtain corrected total fluorescence. For intensity measurements one slide per animal containing 2 brain sections were sampled.
Statistical analysis
Parametric statistics were applied with data that met Shapiro–Wilk criteria for normal distribution and passed Bartlett's test for equal variances and if necessary a Box–Cox transformation was performed to allow parametric tests. Multiple comparisons with regard to genotype and age were analyzed by one‐ or two‐way ANOVA followed by Bonferroni's post‐hoc analysis. Data were evaluated using PASW Statistics 22 (Chicago, IL) and GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). For histological quantification 4–8 animals per group were analyzed. All data represent the means ± standard error of the mean (SEM). Differences were considered significant when P ≤ 0.05 (exact P‐values are given). Furthermore, a correlation analysis was performed and Pearson correlation (r) is given in the results.
Results
Neuropathological changes in the AD of SOD1(G93A) mice
Pathological changes in the AD were first investigated by H&E staining (Figure 1B–I). In stained sections, the AD was easily assignable because of its anatomical characteristics: the triangular shaped AD is localized left beside the stria medularis (sm) and densely packed with large neurons (Figure 1A–I). In comparison with WT, AD morphology of SOD1 14W exhibited obvious histopathological changes, such as a prominent parenchymal vacuolization (spongiform‐like degeneration) (Figure 1F, H). Similar pathological features were found in the AD of 18W old, late disease stage, animals (Figure 1I). In SOD1 9W, tissue morphology of the AD appeared normal without apparent vacuolization (Figure 1G). To better characterize the spongiform pathology, electron microscopy of the AD from 18W old WT and SOD1 mice was performed (Figure 2). The primary neuropathological hallmark was a massive enlargement of dendrites in SOD1 18W compared with WT (Figure 2A,B) (green asterisk mark representative dendrites). In higher magnification, remnants of mitochondrial membranes (Figure 2E, arrow) and vesicular compartments of the autophagic pathway such as multilamellar bodies (MLBs; Figure 2E and G) were detected in dystrophic dendrites. Furthermore, vacuolated mitochondria (Figure 2E,F, black arrowheads) are frequently detected in AD tissue of SOD1 18W. In WT, the dendrite diameter is regular and mitochondria exhibit a normal morphology (Figure 2B,C).
Figure 1.
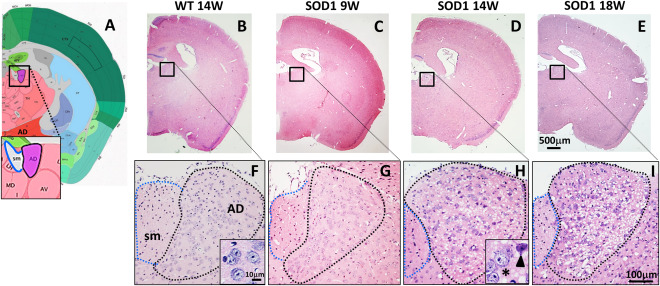
AD morphology of wild type mice (WT), pre‐symptomatic 9 weeks (SOD1 9W), early symptomatic 14 weeks (SOD1 14W) and late symptomatic 18 weeks (SOD1 18W) old SOD1 mice. (A) Location of the AD next to the stria medullaris (sm) (modified from http://www.brain-map.org/). (B–E) Representative H&E staining of the brain from WT, 9W, 14W and 18W SOD1 mice (4× magnification) and (F–I) with higher magnification (20×) of the AD. Morphology of the AD appeared normal in WT and 9W SOD1 animals. Massive vacuolization (spongiform degeneration), abnormal morphology and loss of neurons were noticed in 14W SOD1 animals (H, detail: normal (asterisk) and atypical (arrowhead) shaped neurons).
Figure 2.
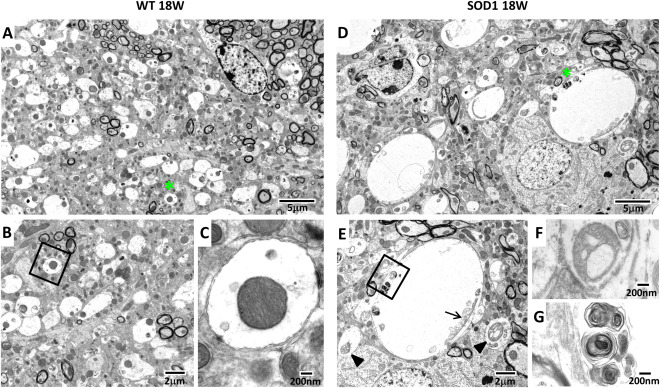
Ultrastructural changes in the AD of SOD1 mice. Representative electron micrographs of the AD from 18 weeks old WT (A–C) and transgenic SOD1 mice (D–G). Tissue from WT exhibits a well preserved tissue structure, including normal sized dendrites (A, asterisks) and mitochondria (B, C). Dendrites from SOD1 mice were expanded (D, asterisk) and mitochondria show various abnormalities, including vacuolation (E, arrowheads) and mitochondrial remnants (E, arrow). Higher magnifications of E show a vacuolated mitochondrion (F) and MLBs (G).
Expression of mutant SOD1(G93A) protein
An antibody specific to the human SOD1 protein (hSOD1) was applied to visualize expression of the mutant protein (Figure 3A–D). Immunofluorescence of hSOD1 revealed a massive accumulation of the protein in the AD of symptomatic animals (Figure 1A–D). Annular structures between neuronal bodies (MAP2 positive), highly immunoreactive for hSOD1 (Figure 3C, detail) represent dystrophic dendrites, as detected by EM (Figure 2). The G93A mutation in the hSOD1 protein leads to conformation changes within the native protein structure, resulting in an undefined toxic gain of function. Several antibodies have been established to specifically target misfolded SOD1 59. To detect misfolded SOD1 protein, we applied the B8H10 antibody (Figure 3 E‐H). Similar to the results obtained by the hSOD1 antibody, B8H10 immunoreactivity was increased in AD neurons and aggregates were detected within enlarged dendrites (Figure 3G, detail). In WT animals, no immunofluorescence of either hSOD1 or B8H10 was detected (Figure 3A, E). Noteworthy, in other nuclei of the ANT, no morphological abnormalities were observed in H&E stained sections (Supporting Information Figure 1A–C). Furthermore, fluorescence signal of hSOD1 and B8H10 was very prominent in the AD nucleus of the ANT (Supporting Information Figure 1D,E).
Figure 3.
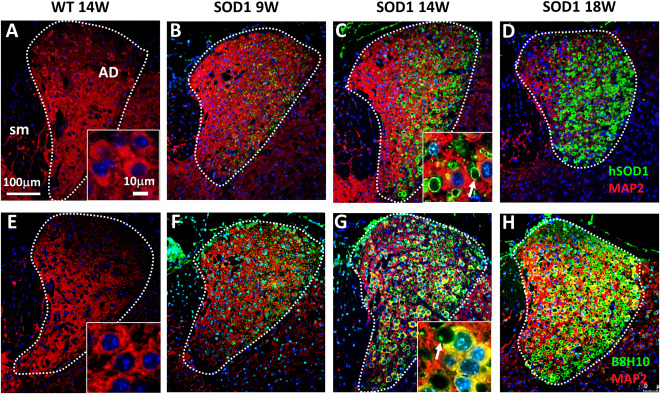
Accumulation of mutant hSOD1 protein. (A–D) Immunofluorescence double staining against hSOD1 (green) and MAP2 (red) revealed strong accumulation of hSOD1 protein especially in the AD of SOD1 animals (B–D). (E–H) Fluorescence staining using B8H10 yielded a similar result with increased accumulation of misfolded SOD1 protein in AD neurons (F–H). Annular structures (white arrows) between the neuronal cell bodies represent dilated dendrites (C and G, detail). SOD1 aggregates were frequently observed within dystrophic dendrites (D). Tissues of WT mice were negative for hSOD1 (A) and B8H10 (E). Cell nuclei were counterstained with Hoechst (blue).
Expression of autophagic markers in AD neurons
Autophagy eliminates misfolded and aggregated proteins associated with neurodegenerative diseases. We have demonstrated strong accumulation of misfolded SOD1 in AD neurons during disease progression, suggesting that protein clearance by autophagy might be impaired. To investigate autophagy, we measured fluorescence intensity of the autophagy markers LC3B, p62/SQSTM1 and Ub (Figure 4A–F). The intensity of all three markers increased during disease progression (Figure 4G–I), although significant differences between WT and SOD1 were only detected for p62/SQSTM1 (**P = 0.004458) and Ub (*P = 0.022119) (Figure 4H–I). Furthermore, p62 positive puncta and Ub aggregates were detected in AD neurons of SOD1 animals (Figure 4D and F, detail) whereas the LC3B staining pattern was more diffuse (Figure 4A–B). Additionally, a positive correlation was detected between p62 intensity and Ub intensity (r = 0.579; *P = 0.030125) (Supporting Information Table 1).
Figure 4.
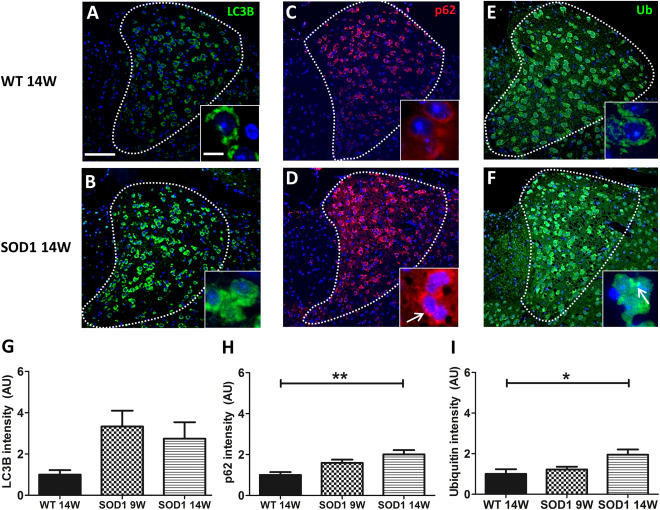
Increased expression of autophagy markers in AD neurons. Representative immunofluorescence labeling of LC3B (A,B), p62 (C,D) and Ubiquitin (Ub) (E,F) are shown. (G–I) Quantitative analysis of fluorescence intensity demonstrated no significant changes in LC3B (G) but a significant difference of p62 (H; **P = 0.004458 vs. WT 14W) and Ub (I; *P = 0.022119 vs. WT 14W) immunoreactivity in 14W SOD1 mice. Accumulation of p62 positive puncta (D, arrow) and Ub aggregates (F, arrow) was observed in AD neurons of 14W SOD1 animals. Cell nuclei were counterstained with Hoechst (blue). Data represent means ± SEM from n = 4–5. One‐way‐ANOVA followed by Tukey's post‐hoc test.
Neuronal loss and glia pathology in the AD
To get a deeper knowledge of early neuropathological changes, we have focused in particular on AD‐related changes in 9 and 14W old animals.
To further address the nature of the observed morphological changes, immunofluorescence staining using neuronal and glial markers was performed. To identify neurons, sections were stained against NeuN (Figure 5A–C). Evaluation of NeuN‐positive cells revealed a significant genotype × age interaction F(1, 28)=14.50 P = 0.0007. A significant reduction (∼20%) of neurons was detected in SOD1 14W mice compared with WT 14W (***P = 0.000018) and SOD1 9W (### P = 0.000368). No difference was found between WT 9W and SOD1 9W (Figure 5A–D). Because of the fact that neurodegeneration is usually accompanied by activation and proliferation of microglia, staining against the microglia/macrophage marker Iba1 was conducted. Statistical analysis by two‐way ANOVA showed no significant interaction F(1, 28) = 0.22, P = 0.6457. Post‐hoc analysis revealed a significant difference between genotypes (WT vs. SOD1) with F(1, 28) = 11.80 and **P = 0.0030 but not between age. Thus, the number of Iba1 positive cells was in general higher in the AD of SOD1 compared with WT animals. Activation of microglia in response to neurodegeneration is often accompanied by an increased expression of phagocytosis marker. The phagocytic state of AD microglia was analyzed by fluorescence staining of the lysosome marker LAMP2 (Figure 5I–K). No significant difference of LAMP2 immunoreactivity was detected in the AD of SOD1 compared with WT animals (Figure 5L). In a next step the glial marker GFAP and Olig2 were applied to determine the number of astrocytes and oligodendroglia, respectively (Figure 6). No statistically significant differences were found in GFAP‐positive cell numbers with respect to genotype and/or age (Figure 6A–D). These results are in accordance with data obtained from fluorescence intensity measurements for GFAP (Figure 6E). Quantification of Olig2‐positive cells yielded similar numbers of oligodendrocytes in the AD of all investigated groups with no statistical differences (Figure 6F–I).
Figure 5.
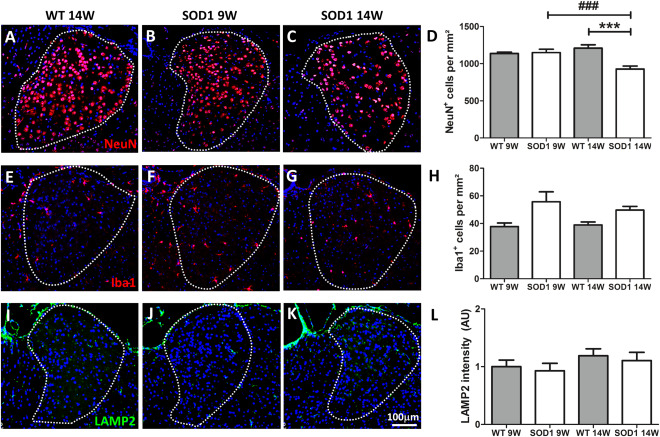
Neuronal loss and microgliosis in the AD. (A–C) Representative immunofluorescence of NeuN+ cells in the AD of WT and 9W and 14W SOD1 mice (20× magnification) are shown. Cell nuclei were counterstained with Hoechst (blue). (D) A significant loss of neurons was detected in 14W SOD1 but not 9W SOD1 mice compared with the age‐matched control group (***P = 0.000018 vs. WT 14W and ###P = 0.000368 vs. SOD1 9W). (E–G) Representative images of immunofluorescence staining of Iba1 (20× magnification) are shown. The number of Iba1‐positive cells was elevated in SOD1 compared with WT mice. A significant genotype difference (**P = 0.0030 WT vs. SOD1) but no significance between age was found for Iba1 (H). Expression of LAMP2 (I–K) was not significantly altered (J). Cell nuclei were counterstained with Hoechst (blue). Data represent means ± SEM from n = 8. Two‐way‐ANOVA followed by Bonferroni's post‐hoc test.
Figure 6.
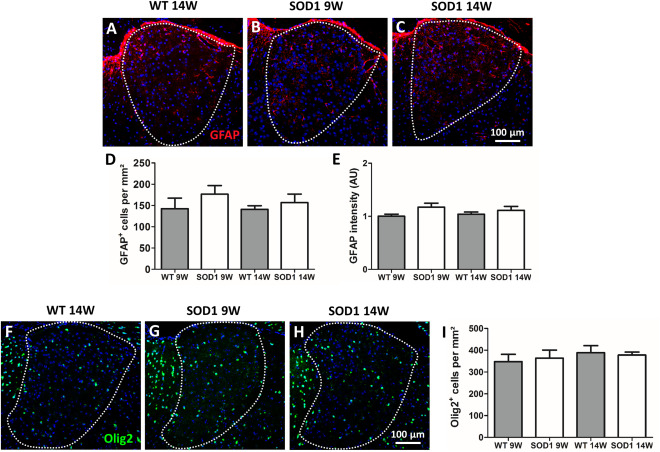
Astrocytes and oligodendrocytes cell numbers and GFAP fluorescence intensity. (A–C) Representative images of fluorescence staining of GFAP (20× magnification) are presented. Quantification of GFAP+ astrocytes in the AD revealed no change in the cell number in all investigated groups (D). Similar results were obtained from fluorescence density measurements (E). (F–H) Representative images of immunofluorescence staining of Olig2 (20× magnification) are shown. The number of Olig2‐positive cells remained unchanged in all groups (I). Cell nuclei were counterstained with Hoechst (blue). Data represent means ± SEM from n = 8. Two‐way‐ANOVA followed by Bonferroni's post‐hoc test.
Expression of NLRP3 inflammasome components in the AD
Recently, we reported NLRP3 inflammasome activation in the spinal cord of SOD1(G93A) mice (Johann et al 2015). Therefore, we investigated the presence of inflammasome proteins in the AD. Immunofluorescence staining revealed that NLRP3 and ASC, two major components of the inflammasome complex, are expressed in SOD1 animals (Figure 7A–H). Fluorescence intensity of NLRP3 and ASC was significantly increased in 14W‐old SOD1 mice compared with WT and SOD1 9W‐old animals (Figure 7A–H). Statistical analysis of NLRP3 fluorescence intensity revealed a significant interaction of genotype × age (F(1,28) = 5.53, P = 0.0260). A significant increase of NLRP3 fluorescence signals was found in 14W SOD1 mice in comparison with WT 14W (**P = 0.001771) and 9W SOD1 animals (### P = 0.000264) (Figure 7D). Evaluation of ASC total fluorescence in the AD revealed increased ASC staining in 9W SOD1 mice that became significant in SOD1 animals at 14 weeks compared with WT (***P = 0.000658) and 9W SOD1 (## P = 0.003891) (Figure 7H). No interaction of genotype × age was found for ASC F(1,28) = 3.34 P = 0.0784). To identify cells expressing NLRP3 (Figure 7I–L) and/or ASC (Figure 7M–P), double immunofluorescence staining was performed in 14W SOD1 mice by using specific markers for astrocytes (GFAP), microglia (Iba1), neurons (NeuN) and oligodendrocytes (Olig2). Co‐staining of NLRP 3 was detected in astrocytes and neurons (Figure 7I and K; white arrowheads), whereas microglia and oligodendrocytes were NLRP3 negative (Figure 7J and L). Double staining using ASC revealed positive co‐staining with astrocytes, microglia and neurons (Figure 7M–O, white arrowheads) but not oligodendrocytes (Figure 7P). Additionally, ASC speck formation (ASC aggregation) was frequently detected in neurons (Figure 7O, white arrow). Table Q in Figure 7 summarizes the results of the co‐localization study were a positive co‐localization is indicated with a plus (+) and a negative co‐localization with a minus (−). Finally, correlation studies revealed negative correlations between neuronal cell number (NeuN) and fluorescence intensity of NLRP3 (r = −0.490; **P = 0.004417) and ASC (r = −0.483; **P = 0.005073) (Supporting Information Table 1). The number of Iba1 positive cells was positively correlated with ASC intensity (r = 0.458; **P = 0.008382) (Supporting Information Table 1). At last, ASC and NLRP3 were also positively correlated (r = 0.537; **P = 0.001523). With respect to autophagy markers we found a positive correlation between NLRP3 intensity and Ub intensity (r = 0.557; *P = 0.038356).
Figure 7.
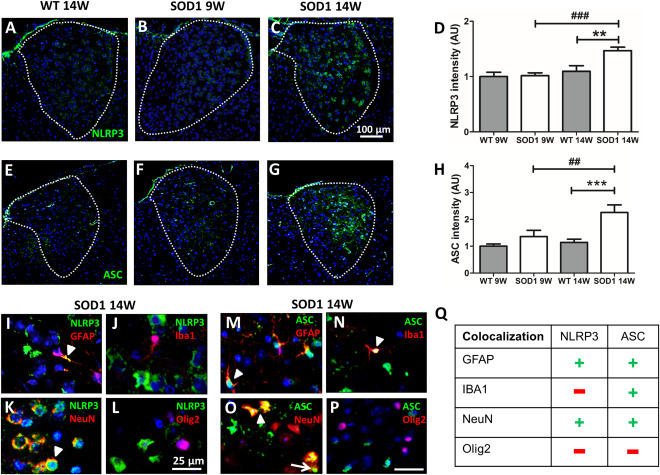
NLRP3 and ASC expression and cellular co‐localization in the AD. (A–C) Representative images of immunofluorescence staining of NLRP3 (20× magnification) are depicted. Fluorescence intensity of NLRP3 in the AD was significantly higher in 14W SOD1 mice (**P = 0.001771 WT 14W vs. SOD1 14W; ###P = 0.000264 SOD1 14W vs. SOD1 9W) (D). (E–G) Representative images of immunofluorescence staining of ASC (20× magnification) are shown. Intensity of ASC in the AD was slightly increased in 9W SOD1 and significantly elevated in 14W SOD1 animals (***P = 0.000658 WT 14W vs. SOD1 14W; ##P = 0.003891 SOD1 14W vs. SOD1 9W) (H). Data represent means ± SEM from n = 8. Two‐way‐ANOVA followed by Bonferroni's post‐hoc test. Co‐localization of NLRP3 (I–L) and ASC (M–P) with neuronal cell marker revealed expression of NLRP3 and ASC in astrocytes and neurons (I, K; M, O), whereas microglia only turned out to be ASC‐positive (N). NLRP3 and ASC were absent in oligodendrocytes (L, P). (Q) Summary of the co‐localization of NLRP3 and ASC with cell type specific markers (+ co‐localization; − no co‐localization). Note that only the merge of respective immunofluorescence of SOD1 14W is shown (63× magnification). Cell nuclei were counterstained with Hoechst (blue).
Expression of IL1β in the AD
NLRP3 activation is characterized by increased expression of inflammasome components and processing of pro‐IL1β to the mature peptide. Because NLRP3 and ASC were significantly up‐regulated in symptomatic SOD1 mice IL1β fluorescence staining was performed in 14W old animals. IL1β immunoreactivity was noticed in the AD of WT and 14W SOD1 mice (Figure 8A,B) but mean fluorescence was not significantly different in SOD1 compared with WT (Figure 8C). However, MAP2‐positive neurons turned out to be the major source of IL1β (Figure 8D). Individual astrocytes (Figure 8E) but not microglia (Figure 8F) were slightly positive for IL1β.
Figure 8.
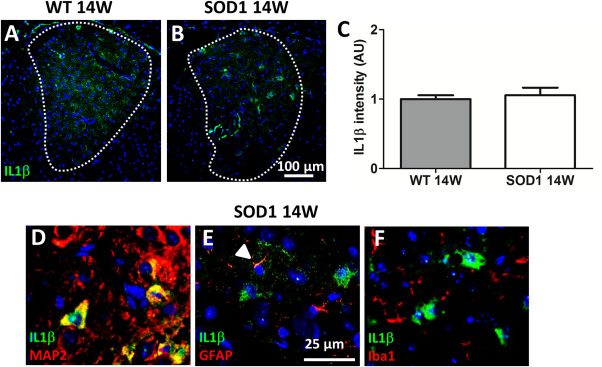
Co‐localization of IL1β with and MAP2‐, GFAP‐ and Iba1‐ positive cells. Representative pictures of IL1β fluorescence staining of the AD (A,B), (20× magnification). Immunoreaction of IL1β was detected in WT and SOD1 animals but mean intensity was not significantly different. Data represent means ± SEM from n = 8. Student's t‐test (C). Higher magnification (63×) from 14W SOD1 shows IL1β co‐localization with MAP2‐positive neurons (D). Individual astrocytes (E), but not microglia (F) were positive for IL1β. Only the merge of respective immunofluorescence is shown. Cell nuclei were counterstained with Hoechst (blue).
Discussion
In the present study, we describe neuropathological changes in the ANT of the SOD1(G93A) ALS mouse model. Neurodegenerative changes were most prominent in the AD of the ANT and occurred early in disease progression. We observed massive dendrite swelling, accumulation of multilaminar structures of autophagic origin, misfolded hSOD1 and neuronal loss in the AD of SOD1 animals. This pathology was accompanied by a mild gliosis. To elucidate the molecular nature of neuronal loss in the AD, NLRP3 inflammasome expression and autophagy markers were analyzed in pre‐ and early symptomatic mice. Accumulation of p62 and Ub suggest an impaired autophagic elimination of misfolded hSOD1 protein, thereby promoting neurodegeneration. Furthermore, ASC and NLRP3 immunoreactivity was significantly increased during disease progression. Inflammasome proteins and IL1β were primarily co‐localized with neuronal markers. Together, our results clearly demonstrate ongoing neurodegeneration of a sensory thalamic nucleus and a possible involvement of the NLRP3 inflammasome in neuronal cell death in the SOD1(G93A) mouse model for ALS.
Neurodegeneration of the ANT
Although less severe than in SOD1 mice, morphological changes in the ANT have been detected in ALS patients. However, most studies applied functional imaging methods, such as PET studies 2, 37, 79, whereas histopathological information is sparse. Kew et al (1993) have further demonstrated that abnormalities in the ANT significantly correlated with verbal fluency and picture recall scores. Our study presents evidence for ANT pathology in ALS using the well‐established SOD1(G93A) mouse model. Interestingly, neuronal loss and spongiform changes in SOD1 animals were only detected in the AD, whereas the AV and AM did not show signs of neurodegeneration. So far, we do not have a satisfying explanation for the specific vulnerability of AD neurons. Recently, regional atrophic changes of the thalamus have been described in ALS patients, which might be at least in part because of AD degeneration 79. Similar observations of selective AD degeneration have been shown in patients suffering from Alzheimer's disease where a bilateral atrophy of the AD region was found in patients 82. These findings are in part in accordance with increased Aβ deposits and neurofibrillary tangles (NFT) in limbic nuclei of the ANT in Alzheimer's disease patients showing the most severe NFT pathology 11. Besides neuronal loss, the most striking neuropathological feature was the profound spongiform neurodegeneration in the AD of SOD1 animals. Whereas the neuropil in pre‐symptomatic mice appeared unremarkable, a massive vacuolization was observed in symptomatic mice. Electron microscopy revealed that the vacuole‐like structures are dilated and degenerating dendrites of AD neurons. Dendrite swelling has been described previously in the spinal cord of SOD1(G93A) mice 53. Furthermore, neuropil vacuolization is also a typical hallmark of prion diseases, such as sporadic Creutzfeldt–Jakob disease (sCJD), but similar spongiform changes have also been described for other neurodegenerative diseases including Alzheimer's disease 67, dementia with Lewy bodies 33 and FTD 54, 68. Though, artifacts produced by poor fixation and tissue processing 78 can be ruled out, since no vacuolization was observed in WT mice. Ultrastructurally, accumulation of MLBs and mitochondrial remnants in dilated dendritic processes are indicative of intense autophagy in the AD nucleus 29, 39. Generally, MLBs have been suggested to be involved in the storage, transport and digestion of autophagic and endosomal cargo/waste 21. Therefore, high level of misfolded hSOD1 and accumulation of MLBs, LC3B p62 and Ub suggest that autophagic flux is affected in AD neurons. Various studies have described impaired protein clearance in disease pathology of SOD1 mice and human ALS patients 72, 84. Additionally, mutations in autophagy related genes including p62 can cause fALS 41, 71.
Nonetheless, even if we can clearly demonstrate neurodegenerative changes in the AD, the fact that significant gliosis is absent remains elusive. Gliosis is a cardinal feature of neurodegeneration in neurological disorders, including ALS 13. In vivo live imaging studies have demonstrated that microglia become rapidly activated and migrate to injured site 56. Astrocytes, in contrast, do not migrate to the injury site but they can osmotically swell and, depending on the severity of the insult, can become reactive and hypertrophic 7. To our surprise the number of Iba1 positive microglia was not significantly different in age‐matched WT and SOD1 mice. However, with regard only to the genotype (irrespective of the age), the numbers of Iba1 positive cells were significantly higher in SOD1 compared with WT mice. This result suggests an early microglia reaction in response to hSOD1 induced pathology. In the spinal cord, microglia react in response to motoneuron loss with proliferation and a morphological change from a ramified resting cell to an activated amoeboid macrophage‐like cell type 35. Other phenotypic alterations include the up‐regulation of lysosomal markers, such as LAMP2, characterizing the cell as phagocytic active. However, we could not detect morphological changes or an up‐regulation of LAMP2 in the AD of SOD1 mice. Thus, the very mild microgliosis and absent LAMP2 up‐regulation suggests a dysfunctional microglial reaction to neuropathological changes in the AD. In ALS rats, it has been reported that in contrast to spinal cord microglia, cortical microglia seemed to be unaffected by the disease 55, implicating differences in microglia depending on their specific CNS area. Microglia dysfunction is suggested to play an important role in neurodegenerative diseases, including ALS 12, 52. However, possible reasons leading to impaired microglia function are diverse and further studies are needed to investigate microglia phenotypes and genotypes in ALS with respect to different CNS regions.
Because of the fact that we only observed a slight increase in GFAP+ cells and GFAP− immunoreactivity, we suggest that a more pronounced gliosis may be present in later stages of the disease when tissue remodelling takes place 13. Dendrite degeneration may cause damage of neuropil, shrinkage and loss of neurons, but significant changes may virtually be absent until excessive neuronal damage occurs. It is further possible that AD astrocytes exhibit a dysfunctional phenotype. In the AD cell number and GFAP expression is unaffected in contrast to spinal cord astroglia where increased GFAP intensity is observed with no differences in the cell number between WT and SOD1 animals 25, 35. But it is still unclear how the supportive functions of astrocytes depend on their reactive state. Recent studies have described the role of aberrant astrocytes (AbA) in SOD1(G93A) rats 22, 73. These astrocyte‐like cells express high level of S100β and Connexin‐43 (Cx‐43) and low levels of GFAP. Furthermore these cells lack the glutamate transporter GLT1 and release toxic factors in cell culture.
In conclusion, the low grade of gliosis in the AD of SOD1 animals reflects somehow an atypical glial reaction. A potential malfunction of microglia and astrocytes might be an important factor and other functional markers behind Iba1, LAMP2 and GFAP need to be investigated. Another reason could be that cells may not play an active role during neurodegeneration in the AD but be rather a secondary reaction because of disease processing in neurons.
Expression of NLRP3 inflammasome proteins in the AD
NLRs are involved in forming multiprotein complexes, so called “Inflammasomes.” In the CNS, NLRs are highly expressed by glial cells such as microglia 77 and astrocytes 35, 47. However, several studies have shown the presence of NLRs in neurons 20, 24, 83. In an earlier report, we have investigated NLRP3 inflammasome activation in the spinal cord of SOD1 mice and human sALS patients. Therein, we described increased levels of inflammasome proteins together with elevated levels of IL1β and IL18, suggesting a critical role of inflammasome activation in the pathogenesis of ALS 35. The present data suggest that neurodegeneration in the AD is accompanied by NLRP3 activation. We detected NLRP3 expression at low levels in most of the AD neurons in WT mice. In symptomatic SOD1 animals, NLRP3 protein was markedly up‐regulated in neurons and only few astrocytes, but neither microglia nor oligodendrocytes were NLRP3 positive. These results reflect in part observations from our recent study, where NLRP3 was significantly up‐regulated in the spinal cord of 14W SOD1 animals. However, in the spinal cord NLRP3 was mainly detected in astrocytes 35. In a set of studies, microglia and astrocytes have been shown to respond to CNS damage with the activation of inflammasomes. More recently, NLR expression was also reported for neurons, e.g. NLRP1 in cerebellar granule neurons 43 and cortical neurons 19, 69 and NLRP3 in neurons of the cerebral cortex and in the spinal cord 40, 83 in various models of neurodegeneration. Together, these observations suggest that neurons have the ability to respond to DAMPs with the formation of inflammasome complexes. Regarding the adaptor molecule ASC, constitutive expression was detected in microglia and endothelial cells in the AD of WT mice. During disease progression, ASC immunoreactivity increased and significant differences were observed in symptomatic animals. By using co‐staining, we confirmed ASC expression in astrocytes, microglia and neurons, but not oligodendrocytes. Increased ASC expression might be in part because of increased number of micro‐ and astroglial cells in the AD of SOD1 mice compared with WT mice. Though, the significant increase of ASC observed in symptomatic animals is rather because of increased expression in damaged neurons. This assumption is further supported by the observation of a very mild gliosis and that ASC‐like speck formation was only seen in AD neurons. To analyze inflammasome activation, we investigated IL1β immunoreactivity in the AD of 14W old WT and SOD1 mice. We detected IL1β immunoreactivity in the AD of symptomatic mice, mainly co‐localized with the neuronal marker MAP2. Some astrocytes, but not microglia, were immunoreactive for IL1β. However, quantification revealed no statistical difference in IL1β fluorescence between WT and SOD1. This result is explained by the fact that all commercial available antibodies detect both, the pro‐ and mature IL1β peptide. To distinguish between the two peptides Western Blot analysis might be necessary. A limiting factor of the present study is the small size and the very local pathology of the AD. Thus, a specific isolation of the AD from the ANT proved to be difficult and tissue amount was insufficient for Western Blot analysis. Nevertheless, several studies have demonstrated a mutual relationship between NLRP3 activation and autophagy. By removal of potential activators, such as misfolded protein and damaged mitochondria 26, 85, autophagy contributes to the inhibition of NLRP3 inflammasome activation. Furthermore, autophagy plays a crucial role in the sequestration of pro‐IL1β into autophagosomes 30 and degradation of inflammasome proteins via p62 61, 65. Therefore, increased expression of inflammasome proteins along with the presence of damaged mitochondria and accumulation of p62 and Ub in SOD1 animals support the hypothesis of impaired autophagy in AD neurons.
Conclusion
To our knowledge, this is the first study describing early neurodegeneration with the focus on the histological/morphological pathogenesis in the AD of the ANT in SOD1(G93A) mice. Our findings indicate a selective vulnerability of sensory thalamic neurons, which is accompanied by massive dendrite swelling, accumulation of misfolded hSOD1, impaired autophagy and neuronal loss. Furthermore, ineffective autophagy and increased NLRP3 inflammasome expression in damaged neurons may be responsible for cell death and further spreading of the disease along the “Papez circuit.” According to that, a couple of studies have already reported neurodegenerative changes associated with cognitive deficits in the hippocampus, subiculum, retrosplenial cortex and mammillary bodies in SOD1 transgenic animals and human ALS patients 31, 36, 51, 58, 59. Neuropathological changes in any of these structures may also be involved in cognitive failure in a subset of ALS patients. Finally, the SOD1(G93A) mouse may provide a suitable model to study the involvement of limbic structures, especially the ANT and related cognitive function in ALS pathology.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Morphological examination of the anterior, ventral and medial group of the thalamus. (A) Representative brain maps (anterior thalamic group is colored in purple; modified from http://www.brain-map.org/) and H&E staining of the brain from 14W WT (B) and SOD1 (C) mice at 4× magnification. (D–E) Representative double immunofluorescence against (D) hSOD1 or (E) B8H10 (green) and MAP2 (red). Cell nuclei were counterstained with Hoechst (blue). In the anterior group (pale blue dotted line) of 14W SOD1 animals, only the AD showed histopathological abnormalities (white dotted line). No abnormalities were observed in other nuclei of the anterior thalamic group.
Table S1. Pearson's correlation analysis of investigated markers in the AD. Significant negative correlation was observed between the number of NeuN positive neurons and ASC intensity (r = −0.483; **P = 0.005073) or NLRP3 intensity (r = −0.490; **P = 0.004417) respectively. Also the number of Iba1 positive cells was negatively correlated with LC3B intensity (r = −0.597; *P = 0.018683). Positive correlations were observed between the number of Iba1 positive cells and ASC intensity (r = 0.458; **P = 0.008382), ASC intensity and NLRP3 intensity, (r = 0.537; **P = 0.001523) NLRP3 intensity and Ub intensity (r = 0.557; *P = 0.038356) and p62 intensity and Ub intensity (r = 0.579; *P = 0.030125). Significant correlations are marked in bold characters.
Acknowledgment
We would like to thank Uta Zahn and Petra Ibold (Institute of Neuroanatomy, RWTH Aachen University) as well as Barbara Holstermann und Brigitte Asmus (Institute of Neuroanatomy, University Medical Centre Hamburg‐Eppendorf) for their technical support. This work was supported by a grant from the Medical Faculty of the RWTH Aachen University (START, SJ).
The authors state no conflict of interest.
References
- 1. Abais JM, Xia M, Zhang Y, Boini KM, Li PL (2015) Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector?. Antioxid Redox Signal 22:1111–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abrahams S, Goldstein LH, Kew JJ, Brooks DJ, Lloyd CM, Frith CD, Leigh PN (1996) Frontal lobe dysfunction in amyotrophic lateral sclerosis. A PET study. Brain: J Neurol 119 (Pt 6):2105–2120. [DOI] [PubMed] [Google Scholar]
- 3. Aggleton JP, Poirier GL, Aggleton HS, Vann SD, Pearce JM (2009) Lesions of the fornix and anterior thalamic nuclei dissociate different aspects of hippocampal‐dependent spatial learning: implications for the neural basis of scene learning. Behav Neurosci 123:504–519. [DOI] [PubMed] [Google Scholar]
- 4. Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS et al (2014) Axonal transport of TDP‐43 mRNA granules is impaired by ALS‐causing mutations. Neuron 81:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bassett JP, Tullman ML, Taube JS (2007) Lesions of the tegmentomammillary circuit in the head direction system disrupt the head direction signal in the anterior thalamus. J Neurosci 27:7564–7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ (2015) The cognitive profile of ALS: a systematic review and meta‐analysis update. J Neurol Neurosurg Psychiatry 87:611–619. [DOI] [PubMed] [Google Scholar]
- 7. Ben Haim L, Carrillo‐de Sauvage MA, Ceyzeriat K, Escartin C (2015) Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci 9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bisecco A, Rocca MA, Pagani E, Mancini L, Enzinger C, Gallo A et al (2015) Connectivity‐based parcellation of the thalamus in multiple sclerosis and its implications for cognitive impairment: a multicenter study. Hum Brain Mapp 36:2809–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blair HT, Cho J, Sharp PE (1999) The anterior thalamic head‐direction signal is abolished by bilateral but not unilateral lesions of the lateral mammillary nucleus. J Neurosci 19:6673–6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boillee S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59. [DOI] [PubMed] [Google Scholar]
- 11. Braak H, Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1:213–216. [DOI] [PubMed] [Google Scholar]
- 12. Brites D, Vaz AR (2014) Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci 8:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81:229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chio A, Pagani M, Agosta F, Calvo A, Cistaro A, Filippi M (2014) Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet Neurol 13:1228–1240. [DOI] [PubMed] [Google Scholar]
- 15. Clark BJ, Taube JS (2011) Intact landmark control and angular path integration by head direction cells in the anterodorsal thalamus after lesions of the medial entorhinal cortex. Hippocampus 21:767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz‐Planillo R, Inserra MC et al (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cozzolino M, Carri MT (2012) Mitochondrial dysfunction in ALS. Prog Neurobiol 97:54–66. [DOI] [PubMed] [Google Scholar]
- 18. de Rivero Vaccari JP, Dietrich WD, Keane RW (2014) Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab 34:369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29:1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW (2008) A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 28:3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y et al (2005) Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J Cell Biol 170:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diaz‐Amarilla P, Olivera‐Bravo S, Trias E, Cragnolini A, Martinez‐Palma L, Cassina P et al (2011) Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 108:18126–18131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans MC, Couch Y, Sibson N, Turner MR (2013) Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol Cell Neurosci 53:34–41. [DOI] [PubMed] [Google Scholar]
- 24. Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M, Cheng YL, Thundyil J, Widiapradja A, Lok KZ, Foo SL, Wang YC, Li YI, Drummond GR, Basta M, Magnus T, Jo DG, Mattson MP, Sobey CG, Arumugam TV (2013) Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome‐mediated neuronal death in ischemic stroke. Cell Death Dis 4:e790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gerber YN, Sabourin JC, Rabano M, Vivanco M, Perrin FE (2012) Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PloS One 7:e36000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gurney ME (1994) Transgenic‐mouse model of amyotrophic lateral sclerosis. N Engl J Med 331:1721–1722. [DOI] [PubMed] [Google Scholar]
- 28. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol 9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hariri M, Millane G, Guimond MP, Guay G, Dennis JW, Nabi IR (2000) Biogenesis of multilamellar bodies via autophagy. Mol Biol Cell 11:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA et al (2011) Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. J Biol Chem 286:9587–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira‐Saecker A et al (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ (2011) Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 122:657–671. [DOI] [PubMed] [Google Scholar]
- 33. Iseki E, Li F, Kosaka K (1997) Close relationship between spongiform change and ubiquitin‐positive granular structures in diffuse Lewy body disease. J Neurol Sci 146:53–57. [DOI] [PubMed] [Google Scholar]
- 34. Jankowski MM, Ronnqvist KC, Tsanov M, Vann SD, Wright NF, Erichsen JT et al (2013) The anterior thalamus provides a subcortical circuit supporting memory and spatial navigation. Front Syst Neurosci 7:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J et al (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63:2260–2273. [DOI] [PubMed] [Google Scholar]
- 36. Kanneganti TD, Lamkanfi M (2013) K(+) drops tilt the NLRP3 inflammasome. Immunity 38:1085–1088. [DOI] [PubMed] [Google Scholar]
- 37. Kew JJ, Goldstein LH, Leigh PN, Abrahams S, Cosgrave N, Passingham RE et al (1993) The relationship between abnormalities of cognitive function and cerebral activation in amyotrophic lateral sclerosis. A neuropsychological and positron emission tomography study. Brain: J Neurol 116 (Pt 6):1399–1423. [DOI] [PubMed] [Google Scholar]
- 38. Kopelman MD, Thomson AD, Guerrini I, Marshall EJ (2009) The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol Alcohol (Oxford, Oxfordshire) 44:148–154. [DOI] [PubMed] [Google Scholar]
- 39. Lajoie P, Guay G, Dennis JW, Nabi IR (2005) The lipid composition of autophagic vacuoles regulates expression of multilamellar bodies. J Cell Sci 118:1991–2003. [DOI] [PubMed] [Google Scholar]
- 40. Lammerding L, Slowik A, Johann S, Beyer C, Zendedel A (2015) Post‐stroke inflammasome expression and regulation in the peri‐infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103:460–475. [DOI] [PubMed] [Google Scholar]
- 41. Lee JK, Shin JH, Lee JE, Choi EJ (2015) Role of autophagy in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta 1852:2517–2524. [DOI] [PubMed] [Google Scholar]
- 42. Li S, Chen Q, Yu B, Xue K, Luo C, Xu Y et al (2009) Structural and functional changes mapped in the brains of amyotrophic lateral sclerosis patients with/without dysphagia: a pilot study. Amyotroph Lateral Scler 10:280–287. [DOI] [PubMed] [Google Scholar]
- 43. Liu F, Lo CF, Ning X, Kajkowski EM, Jin M, Chiriac C et al (2004) Expression of NALP1 in cerebellar granule neurons stimulates apoptosis. Cell Signal 16:1013–1021. [DOI] [PubMed] [Google Scholar]
- 44. Machts J, Loewe K, Kaufmann J, Jakubiczka S, Abdulla S, Petri S et al (2015) Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology 85:1301–1309. [DOI] [PubMed] [Google Scholar]
- 45. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M et al (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232. [DOI] [PubMed] [Google Scholar]
- 46. Miao EA, Rajan JV, Aderem A (2011) Caspase‐1‐induced pyroptotic cell death. Immunol Rev 243:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Minkiewicz J, de Rivero Vaccari JP, Keane RW (2013) Human astrocytes express a novel NLRP2 inflammasome. Glia 61:1113–1121. [DOI] [PubMed] [Google Scholar]
- 48. Mirski MA, McKeon AC, Ferrendelli JA (1986) Anterior thalamus and substantia nigra: two distinct structures mediating experimental generalized seizures. Brain Res 397:377–380. [DOI] [PubMed] [Google Scholar]
- 49. Mirski MA, Rossell LA, Terry JB, Fisher RS (1997) Anticonvulsant effect of anterior thalamic high frequency electrical stimulation in the rat. Epilepsy Res 28:89–100. [DOI] [PubMed] [Google Scholar]
- 50. Montagna P, Gambetti P, Cortelli P, Lugaresi E (2003) Familial and sporadic fatal insomnia. Lancet Neurol 2:167–176. [DOI] [PubMed] [Google Scholar]
- 51. Moreau PH, Tsenkina Y, Lecourtier L, Lopez J, Cosquer B, Wolff M et al, (2013) Lesions of the anterior thalamic nuclei and intralaminar thalamic nuclei: place and visual discrimination learning in the water maze. Brain Struct Funct 218:657–667. [DOI] [PubMed] [Google Scholar]
- 52. Mosher KI, Wyss‐Coray T (2014) Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol 88:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Natale G, Lenzi P, Lazzeri G, Falleni A, Biagioni F, Ryskalin L, Fornai F (2015) Compartment‐dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front Cell Neurosci 9:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neary D, Snowden JS, Mann DM, Northen B, Goulding PJ, Macdermott N (1990) Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatry 53:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nikodemova M, Small AL, Smith SM, Mitchell GS, Watters JJ (2014) Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin‐3 and osteopontin, and blunted inflammatory responses in ALS rats. Neurobiol Dis 69:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 57. Nishio Y, Hashimoto M, Ishii K, Ito D, Mugikura S, Takahashi S, Mori E (2014) Multiple thalamo‐cortical disconnections in anterior thalamic infarction: implications for thalamic mechanisms of memory and language. Neuropsychologia 53:264–273. [DOI] [PubMed] [Google Scholar]
- 58. Papez JW (1995) A proposed mechanism of emotion. 1937. J Neuropsychiatry Clin Neurosci 7:103–112. [DOI] [PubMed] [Google Scholar]
- 59. Pickles S, Vande Velde C (2012) Misfolded SOD1 and ALS: zeroing in on mitochondria. Amyotroph Lateral Scler 13:333–340. [DOI] [PubMed] [Google Scholar]
- 60. Power BD, Looi JC (2015) The thalamus as a putative biomarker in neurodegenerative disorders. Aust New Zeal J Psychiatry 49:502–518. [DOI] [PubMed] [Google Scholar]
- 61. Rodgers MA, Bowman JW, Liang Q, Jung JU (2014) Regulation where autophagy intersects the inflammasome. Antioxid Redox Signal 20:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sach M, Winkler G, Glauche V, Liepert J, Heimbach B, Koch MA et al (2004) Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain: J Neurol 127(Pt 2):340–350. [DOI] [PubMed] [Google Scholar]
- 63. Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832. [DOI] [PubMed] [Google Scholar]
- 64. Sharma KR, Saigal G, Maudsley AA, Govind V (2011) 1H MRS of basal ganglia and thalamus in amyotrophic lateral sclerosis. NMR Biomed 24:1270–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi CS, Shenderov K, Huang NN, Kabat J, Abu‐Asab M, Fitzgerald KA et al (2012) Activation of autophagy by inflammatory signals limits IL‐1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith DM, Mizumori SJ (2006) Learning‐related development of context‐specific neuronal responses to places and events: the hippocampal role in context processing. J Neurosci 26:3154–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smith TW, Anwer U, DeGirolami U, Drachman DA (1987) Vacuolar change in Alzheimer's disease. Arch Neurol 44:1225–1228. [DOI] [PubMed] [Google Scholar]
- 68. Snowden JS, Neary D, Mann DM (2002) Frontotemporal dementia. Br J Psychiatry 180:140–143. [DOI] [PubMed] [Google Scholar]
- 69. Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, Yu JT (2014) Amyloid‐beta induces NLRP1‐dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis 5:e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT (2012) PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 249:158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Teyssou E, Takeda T, Lebon V, Boillee S, Doukoure B, Bataillon G et al (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125:511–522. [DOI] [PubMed] [Google Scholar]
- 72. Tokuda E, Brannstrom T, Andersen PM, Marklund SL (2016) Low autophagy capacity implicated in motor system vulnerability to mutant superoxide dismutase. Acta Neuropathol Commun 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trias E, Ibarburu S, Barreto‐Nunez R, Barbeito L (2016) Significance of aberrant glial cell phenotypes in pathophysiology of amyotrophic lateral sclerosis. Neurosci Lett 636:27–31. [DOI] [PubMed] [Google Scholar]
- 74. Trojsi F, Monsurro MR, Esposito F, Tedeschi G (2012) Widespread structural and functional connectivity changes in amyotrophic lateral sclerosis: insights from advanced neuroimaging research. Neural Plastic 2012:473538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Turner MR, Bowser R, Bruijn L, Dupuis L, Ludolph A, McGrath M et al (2013) Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 14Suppl 1:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ et al (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)‐PK11195 positron emission tomography study. Neurobiol Dis 15:601–609. [DOI] [PubMed] [Google Scholar]
- 77. Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG et al (2014) Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 11:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wells GA, Wells M (1989) Neuropil vacuolation in brain: a reproducible histological processing artefact. J Comp Pathol 101:355–362. [DOI] [PubMed] [Google Scholar]
- 79. Westeneng HJ, Walhout R, Straathof M, Schmidt R, Hendrikse J, Veldink JH, van den Heuvel MP, van den Berg LH (2016) Widespread structural brain involvement in ALS is not limited to the C9orf72 repeat expansion. J Neurol Neurosurg Psychiatry 87:1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Witgert M, Salamone AR, Strutt AM, Jawaid A, Massman PJ, Bradshaw M et al (2010) Frontal‐lobe mediated behavioral dysfunction in amyotrophic lateral sclerosis. Eur J Neurol 17:103–110. [DOI] [PubMed] [Google Scholar]
- 81. Wolff M, Gibb SJ, Dalrymple‐Alford JC (2006) Beyond spatial memory: the anterior thalamus and memory for the temporal order of a sequence of odor cues. J Neurosci 26:2907–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zarei M, Patenaude B, Damoiseaux J, Morgese C, Smith S, Matthews PM et al (2010) Combining shape and connectivity analysis: an MRI study of thalamic degeneration in Alzheimer's disease. NeuroImage 49:1–8. [DOI] [PubMed] [Google Scholar]
- 83. Zendedel A, Johann S, Mehrabi S, Joghataei MT, Hassanzadeh G, Kipp M, Beyer C (2015) Activation and regulation of nlrp3 inflammasome by intrathecal application of sdf‐1a in a spinal cord injury model. Mol Neurobiol 53:3063–3075. [DOI] [PubMed] [Google Scholar]
- 84. Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, Le W (2014) MTOR‐independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure S1. Morphological examination of the anterior, ventral and medial group of the thalamus. (A) Representative brain maps (anterior thalamic group is colored in purple; modified from http://www.brain-map.org/) and H&E staining of the brain from 14W WT (B) and SOD1 (C) mice at 4× magnification. (D–E) Representative double immunofluorescence against (D) hSOD1 or (E) B8H10 (green) and MAP2 (red). Cell nuclei were counterstained with Hoechst (blue). In the anterior group (pale blue dotted line) of 14W SOD1 animals, only the AD showed histopathological abnormalities (white dotted line). No abnormalities were observed in other nuclei of the anterior thalamic group.
Table S1. Pearson's correlation analysis of investigated markers in the AD. Significant negative correlation was observed between the number of NeuN positive neurons and ASC intensity (r = −0.483; **P = 0.005073) or NLRP3 intensity (r = −0.490; **P = 0.004417) respectively. Also the number of Iba1 positive cells was negatively correlated with LC3B intensity (r = −0.597; *P = 0.018683). Positive correlations were observed between the number of Iba1 positive cells and ASC intensity (r = 0.458; **P = 0.008382), ASC intensity and NLRP3 intensity, (r = 0.537; **P = 0.001523) NLRP3 intensity and Ub intensity (r = 0.557; *P = 0.038356) and p62 intensity and Ub intensity (r = 0.579; *P = 0.030125). Significant correlations are marked in bold characters.