

Abstract
The present study investigates global transcriptional changes in frontal cortex area 8 in incidental Lewy Body disease (iLBD), Parkinson disease (PD) and Dementia with Lewy bodies (DLB). We identified different coexpressed gene sets associated with disease stages, and gene ontology categories enriched in gene modules and differentially expressed genes including modules or gene clusters correlated to iLBD comprising upregulated dynein genes and taste receptors, and downregulated innate inflammation. Focusing on DLB, we found modules with genes significantly enriched in functions related to RNA and DNA production, mitochondria and energy metabolism, purine metabolism, chaperone and protein folding system and synapses and neurotransmission (particularly the GABAergic system). The expression of more than fifty selected genes was assessed with real time quantitative polymerase chain reaction. Our findings provide, for the first time, evidence of molecular cortical alterations in iLBD and involvement of several key metabolic pathways and gene hubs in DLB which may underlie cognitive impairment and dementia.
Keywords: axonema, cerebral cortex, chaperones, dementia with Lewy bodies, dynein, GABA, Lewy body diseases, mitochondria, neurotransmission, purine metabolism, synapses, taste receptors, transcriptome
Introduction
Lewy body diseases (LBDs), which include Parkinson's disease (PD) and dementia with Lewy Bodies (DLB), are neurodegenerative disorders, characterized by the presence of intracytoplasmic neuronal inclusions named Lewy bodies (LB) and abnormal neurites containing α‐synuclein species and aggregates 32, 41, 46, 49, 64. Nonmotor symptoms such as sleep disorders, loss of olfaction and autonomic alterations may precede the appearance of motor symptoms; cognitive impairment and dementia can occur at advanced stages of PD, and dementia is compulsory in DLB. Braak stages of α‐synuclein pathology distribution are useful to delineate a framework to interlink LBDs into a spectrum. Stages 1–3 can be associated with pre‐motor symptoms although in most cases the presence of LBs and neurites in selected regions of the medulla oblongata, pons and midbrain is an incidental finding at autopsy. The term incidental PD or Lewy Body disease (LBD) (iPD or incidental LBD [iLBD]) refers to those early stages of LBD pathology with no apparent clinical symptoms, corresponding to the early stages of LBD spectrum 22. PD is usually manifested at stages 4 and 5 once the involvement of the substantia nigra reaches determinate thresholds of neuronal loss and dopaminergic denervation of the striatum is manifested. Cognitive impairment can be detected at stages 5 and 6 in PD, whereas DLB cases are categorized as Braak stages 5 and 6 of LB pathology 11, 84. However, no exact correlation exists between Braak stages and clinical symptoms linked to cognitive impairment and dementia 14, 23, 28, 47, 48, 65, 77, this fact suggesting that factors other than LBs and neurites play a cardinal role in the pathogenesis of LBDs. Concomitant pathologies, particularly those linked to Alzheimer's disease (AD), have also been suggested to explain variations in the degree of cognitive impairment in DLB 8, 17, 47, 50, 70, 71.
Decreased dopaminergic, noradrenergic serotoninergic and cholinergic innervation of the cerebral cortex are contributory factors to the appearance of cognitive impairment and dementia in PD. They are due to the loss of vulnerable neurons in the substantia nigra, locus coeruleus, raphe nuclei and nuclei of the basal forebrain including the basal nucleus of Meynert, respectively 13, 43, 45, 53, 54, 62, 89. However, recent studies demonstrate the primary impairment of several metabolic pathways in the cerebral cortex in PD and other LBDs, such as synaptic transmission, mitochondria and energy metabolism, purine metabolism, protein synthesis, lipid composition of membranes and inflammation, among others 19, 20, 26, 28, 31, 34, 35, 36, 61, 63, 68, 74, 79, 94.
Studies of transcriptomic profiling in LBDs have been mainly focused on the typically affected subcortical regions such as substantia nigra, locus coeruleus and striatum in PD 7, 18, 40. Cortical regions have received much less attention with studies limiting their outcome to lists of differentially expressed genes (DEGs) 59. Still, this approach has allowed the discovery of the brain expression of olfactory and taste receptors and their deregulation in the cerebral cortex and substantia nigra in PD 33, 42.
Here, we set out to investigate global transcriptomic changes occurring in the frontal cortex of cases of iLBD, PD and DLB relative to middle‐aged individuals with no neurological symptoms and with no alterations at the postmortem examination. We focused particularly on identifying gene coexpression modules showing correlation with the spectrum of LB disorders. By applying weighted gene coexpression network analysis (WGCNA) 57 to microarray data, we determined the transcriptome structure in the frontal cortex and identified coexpression modules correlated to iLBD, PD and DLB. Validation of hubs and selected altered pathways was carried out with real time quantitative polymerase chain reaction (RT‐qPCR).
Material and Methods Brain Samples
Brain tissue was obtained from the Institute of Neuropathology Brain Bank (HUB‐ICOIDIBELL Biobank) and the Hospital Clinic‐IDIBAPS Biobank following the guidelines of Spanish legislation on this matter and of the local ethics committee. Processing of brain tissue has been detailed elsewhere 29, 80. The postmortem interval between death and tissue processing was between 3 h and 15 h. One hemisphere was cut in 1‐cm‐thick coronal sections, and selected areas of the encephalon were rapidly dissected, frozen on metal plates over dry ice, placed in individual air‐tight plastic bags, numbered with water‐resistant ink and stored at −80°C until use for biochemical studies. The other hemisphere was fixed by immersion in 4% buffered formalin for 3 weeks for morphologic studies. Neuropathological diagnosis was categorized following current staging classifications for LBD 1, 10, 78. For AD‐related pathology, neurofibrillary tangles (NFTs) 9 and phases of AD‐related β‐amyloid plaques 91 were assigned. Only cases with “typical” staging of LBD pathology were selected for study.
Two series of cases were used. The first one served for microarray studies. RNA samples from frontal cortex (area 8) of middle aged (MA) (n = 8, 4 men, 4 women; age: 67.5 ± 12.8 years), iLBD (n = 4, 1 men, 3 women; age: 71.2 ± 4.5 years), PD (n = 8, 4 men, 4 women, age: 67.8 ± 8.8 years) and DLB (n = 8, 5 men, 3 women; age: 72.3 ± 8.6) cases were analyzed using the Affymetrix microarray platform and the Genechip Human Gene 1.1 ST Array (Affymetrix, Santa Clara, CA, USA). The second series of cases was used for RT‐qPCR validation of altered expression of selected genes. Cases used for gene validation were iLDB (n = 5, 4 men, 1 woman; age: 66.8 ± 8.9 years) at stages 3 and 4, and PD cases (n = 9, 3 men, 6 women; age: 77.1 ± 4.7 years) at stages 5 and 6, DLB cases (n = 9, 8 men, 1 woman; age 76.44 ± 5.77 years). PD cases had suffered from parkinsonism and had received treatment during the duration of the disease but did not have dementia. MA cases had not suffered from neurological disease and the neuropathological examination did not reveal abnormalities (n = 15, 6 men, 9 women, age 64.4 ± 15.5 years). In addition, four cases with rapid clinical course DLB (rapid course DLB [rpDLB]; two years of less of disease duration) (2 men and 2 women, age 73.7 ± 2.2 years) were chosen for a few selective studies 37, 38.
Cases with associated pathologies such as vascular diseases (excepting mild atherosclerosis and arteriolosclerosis), TDP‐43 proteinopathy, infection of the nervous system, brain neoplasms, systemic and central immune diseases, metabolic syndrome and hypoxia were excluded from the present study. Regarding AD‐related pathology, rare NFTs and β‐amyloid deposits were found in iLBD (Braak stages 0‐II; Thal phases 0‐I); Braak stages I–III and Thal phases 0–3 were observed in PD; Braak stages 0–V and Thal phases 0–5 occurred in DLB. MA cases had not suffered from neurologic, psychiatric or metabolic diseases (including metabolic syndrome), and did not have abnormalities in the neuropathological examination excepting NFT pathology stages I–II and phases 0–2 of β‐amyloid plaques.
RNA Extraction
Purification of RNA was carried out with RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany) following the protocol provided by the manufacturer. During purification, samples were treated with RNase‐free DNase Set (Qiagen) to avoid later amplification of genomic DNA. The concentration of each sample was obtained from A260 measurements with Nanodrop 1000. RNA integrity was tested using the Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA). Values of RNA quality (RNA integrity number [RIN] values) were from 7 to 8.8 in the first series and from 6.2 to 8.2 in the second series.
Microarray Analysis
Affymetrix microarray platform and the Genechip Human Gene 1.1 ST Array was used to analyze gene expression patterns on a whole‐genome scale on a single array with probes covering several exons on the target genes. Starting material was 200 ηg of total RNA from each sample. Sense ssDNA was generated from total RNA with the Ambion WT Expression Kit from Ambion (Carlsbad, CA, USA), according to the manufacturer's instructions. Sense ssDNA was fragmented, labeled and hybridized to the arrays with the GeneChip WT Terminal Labeling and Hybridization Kit from Affymetrix. Chips were processed on an Affymetrix GeneTitan platform.
Preprocessing of raw data and statistical analyses were performed using Bioconductor packages in R programming environment. We read CEL files from Affymetrix arrays, corrected the background and summarized and normalized the data with the robust microarray method implemented in the Bioconductor Limma package 81. Then, fold change and SEs were assessed by fitting a linear model (using the lmFit function in Limma package) for each gene. Genes with empirical Bayes t test P‐values at a level of 0.01 were selected. Multiple testing correction was performed by adjusting P‐values for false discovery rate (FDR) using the Benjamini and Hochberg method (BH).
Gene Enrichment Score
The average expression values of different transcripts of the same gene were used for gene enrichment scores and weighted gene coexpression network analysis. Only genes in the upper 25% percentile of standard deviation of expression among samples were assessed. For probes mapping to multiple genes we fused all gene ids into one and that was considered a “gene” in the network analysis but not for functional annotation, gene ontology (GO) enrichment and protein–protein interactions (PPI) analysis, for which all individual ids were used.
An enrichment score per gene 5, which is a measure of specificity of a gene for a particular group, disease state in our study, was calculated relative to the rest of the groups tested. Briefly, the method is based on computing linear model coefficients contrasting all groups pair‐wise. These coefficients represent a measure of difference between two groups in which more distant categories present higher coefficient values usually associated with lower P‐values. The enrichment score of a gene in a group is the sum of its significant coefficients against all other groups.
The enrichment score for each of the 5114 genes under study was calculated after obtaining the linear models for microarray data with the LIMMA package 81 considering a linear coefficient statistically significant at uncorrected P‐value lower than 0.01.
Weighted Gene Coexpression Network Analysis
WGCNA was done in R using the WGCNA library 57. We first constructed a gene coexpression network based on pair‐wise correlation of gene expression using all samples at the same time or independently for each disease condition. Not all network topologies fitted with the scale‐free topology model (i.e., the iLBD network). Therefore, all subsequent analysis based on the coexpression network constructed with all samples used a soft‐power threshold of 5. We identified modules of genes based on their topological overlap dissimilarity with their connection strengths in the weighted network 44, 66. Using the dynamic tree‐pruning algorithm, 23 initial modules were obtained; genes not assigned to any module were labeled in gray. After merging all modules with highly correlated eigengenes (Pearson correlation >/= 0.8), 13 final modules were obtained. Module eigengenes were correlated to LBD diagnostic. The P‐values were obtained from a general multivariate lineal model including additional control variables (i.e., age, sex, RIN, PMI and batch).
Gene enrichment analysis
GO enrichment analysis was performed using GOstats 27. Differentially expressed genes with uncorrected P‐values <0.01 for each contrast were used for GO analysis. All genes belonging to a particular gene coexpression module were also used for independent GO analyses. P‐values for categories were adjusted considering FDR using BH with the p.adjust function in R.
Protein–Protein Interaction Network
Assumed protein–protein interaction was obtained with BioGrid, latest release. Subnetworks of genes were obtained using the function‐induced subgraph from the R library rTRM 73. Only nodes with evidence of physical interaction in humans were considered. Networks were analyzed and visualized using Cytoscape 87.
Real‐Time PCR
RT‐qPCR assays were conducted in duplicate on 1000 ηg of cDNA samples obtained from the retrotranscription reaction, diluted 1:20 in 384‐well optical plates (Kisker Biotech, Steinfurt, GE) utilizing the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Parallel amplification reactions were carried out using 20x TaqMan Gene Expression Assays and 2x TaqMan Universal PCR Master Mix (Applied Biosystems). TaqMan probes used in the study are shown in Supporting Information Table 1. The reactions were performed using the following parameters: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles at 95°C for 15 s and at 60°C for 1 minute. TaqMan PCR data were captured using the Sequence Detection Software (SDS version 2.2, Applied Biosystems). Subsequently, threshold cycle (CT) data for each sample were analyzed with the double delta CT (ΔΔCT) method. First, delta CT (ΔCT) values were calculated as the normalized CT values for each target gene in relation to the endogenous controls β‐glucuronidase (GUS‐β) and X‐prolyl aminopeptidase P1 (XPNPEP1). These housekeeping genes were selected because they show no modifications in several neurodegenerative diseases in human postmortem brain tissue 4, 25. A similar pattern was observed using GUS‐β and XPNPEP1 for normalization (data not shown). The mean of GUS‐β and XPNPEP1 was used for correction and representation. Finally, ΔΔCT values were obtained with the ΔCT of each sample minus the mean ΔCT of the population of control samples (calibrator samples). The fold‐change was determined using the equation 2‐ΔΔCT 33.
Statistical Analysis for RT‐QPCR
The normality of distribution of the mean fold‐change values obtained with RT‐qPCR for every region and stage between controls and PD cases was analyzed with the Kolmogorov–Smirnov test. The nonparametric Mann–Whitney test was performed to compare each group when the samples did not follow a normal distribution and the unpaired student's T‐test was used for normal variables. Statistical analysis was performed with GraphPad Prism version 5.01 (La Jolla, CA, USA) and Statgraphics Statistical Analysis and Data Visualization Software version 5.1 (Warrenton, VA, USA). Differences between groups were considered statistically significant at P‐values: *P < 0.05, **P < 0.01 and ***P < 0.001. Additionally, BH‐FDR adjusted P‐values were obtained using the p.adjust function in R.
Results
Differential gene expression and gene enrichment score in frontal cortex area 8 in iLBD, PD and DLB
We contrasted gene expression values of all possible pair‐wise comparisons among all diagnostic groups. We selected differentially expressed genes or DEGs (see methods). We found most DEGs occurred between DLB and the rest of groups; in particular, most were genes downregulated in DLB compared to controls, followed by genes upregulated in DLB. Considering nominal P‐values lower than 0.01, PD and iLBD showed fewer DEGs than DLB. Considering multiple‐testing adjusted P‐values < 0.05, only DLB cases produced differential expressed genes (DEG) genes comparable to controls, and no gene survived that threshold in iLBD and PD (Supporting Information Table 2).
We performed GO enrichment analysis focused on biological process categories for each group of DEGs independently in upregulated and downregulated genes (Supporting Information Table 3).
To summarize all pair‐wise group comparison in one statistic, we calculated a gene enrichment score for each of the 5114 genes analyzed. The advantage of this approach is the possibility of highlighting genes that are differentially expressed in one group relative to all other groups. The score of a given gene summarizes differences in expression levels between comparisons among all four groups of samples. Positive or negative scores are associated with an increase or a reduction in expression, respectively, of each gene in a given group relative to the rest of the groups. We obtained 1914 genes with an enrichment score other than 0 in at least one of the groups. As revealed by clustering analysis, we found that DLB presented the highest number of genes with higher enrichment scores (Figure 1A). In agreement with the proportion of DEGs described above, gene scores in DLB were mostly negative (Figure 1A). The fifty top upregulated and top downregulated genes in DLB are listed in Supporting Information Table 4.
Figure 1.
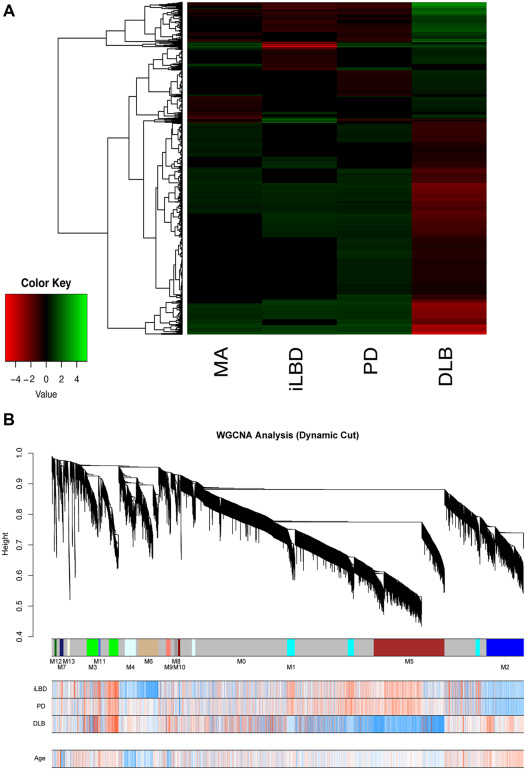
A. Heatmap representation of the enrichment scores of 1914 genes having at least one group of samples with enrichment scores other than 0. MA: middle‐aged, iLBD: incidental Lewy body disease, PD: Parkinson disease, DLB: dementia with Lewy bodies. B. Weighted gene coexpression network analysis of the frontal cortex transcriptome using 5114 gene expression values of 28 assessed samples identifies 13 gene modules. Modules are labeled by color and number (M1 to M13). Genes not assigned to any particular coexpression modules were labeled M0 or gray. Dendrogram obtained by hierarchical clustering of genes based on their topological overlap is shown at the top. Bottom rows indicate the correlation value of each gene expression and the spectrum of LBD pathology. Blue to red indicates negative to positive correlation values. None of our identified modules correlated with age.
The main upregulated genes in frontal cortex in iLBD were associated with axonemal dynein complex assembly and taste receptors, whereas the main downregulated genes were linked to inflammation (Table 1).
Table 1.
Main deregulated gene clusters in frontal cortex in iLBD.
| Cluster | Gene names | Count | Size | Odds Ratio | P‐value | Deregulation |
|---|---|---|---|---|---|---|
| Adaptive immune response | C3AR1 C3 C1QC C1QB INPP5D PTPN6 ADA FCER1G CTSC TGFB1 | 10 | 58 | 12.64 | 0.00 | Down |
| Antigen processing and presentation | SEC23A RAB3B PSMD8 PSMC2 PSMB2 ACTR1B LGMN RAB7A PSMB3 RAB3C DCTN6 AP1S1 PSMD12 DCTN2 DCTN3 PSMA5 AP2M1 PSMB6 PSMC4 PSMA3 PSMA4 DYNC1I1 KIF3A AP1M1 | 24 | 65 | 2.85 | 0.00 | Down |
| Axonemal dynein complex assembly | LRRC6 | 1 | 2 | 353.08 | 0.01 | Up |
| Cell activation involved in immune response | APBB1IP TYROBP VAMP8 ADA LCP1 FCER1G HLA‐DMB TGFB1 | 8 | 52 | 10.72 | 0.00 | Down |
| Detection of chemical stimulus involved in sensory perception of bitter taste | TAS2R4 TAS2R14 | 2 | 5 | 256.67 | 0.00 | Up |
| Granulocyte activation | TYROBP FCER1G | 2 | 7 | 21.94 | 0.01 | Down |
| Innate immune response | CSF1R HLA‐DPB1 AIF1 C3 TYROBP C1QC C1QB HLA‐DPA1 TREM2 TRIM22 ITGB2 PTPN6 CYBB FCER1G RPS6KA1 VSIG4 TGFB1 CXCL16 | 18 | 277 | 4.53 | 0.00 | Down |
| Lymphocyte activation involved in immune response | APBB1IP ADA LCP1 FCER1G HLA‐DMB TGFB1 | 6 | 31 | 13.83 | 0.00 | Down |
| Mononuclear cell proliferation | HLA‐DPB1 AIF1 LST1 HLA‐DPA1 INPP5D ITGB2 PTPN6 ADA HLA‐DMB VSIG4 TGFB1 | 11 | 54 | 15.77 | 0.00 | Down |
| Myeloid dendritic cell activation | TGFBR2 CD37 TGFB1 | 3 | 8 | 33.34 | 0.00 | Down |
| Positive regulation of mast cell activation | VAMP8 FCER1G | 2 | 7 | 21.94 | 0.01 | Down |
| Regulation of B cell mediated immunity | C3 PTPN6 FCER1G TGFB1 | 4 | 8 | 56.34 | 0.00 | Down |
| Regulation of immunoglobulin production | CD37 TGFB1 | 2 | 8 | 18.28 | 0.01 | Down |
| T cell activation involved in immune response | APBB1IP LCP1 FCER1G HLA‐DMB | 4 | 13 | 25.01 | 0.00 | Down |
The main upregulated genes in frontal cortex in DLB compared with controls were categorized into cellular development and DNA/RNA metabolism genes. The main downregulated genes in frontal cortex in DLB compared with controls were grouped into synapsis and neurotransmission, chaperone and protein folding, mitochondria and energy metabolism, purine metabolism and inflammation (Table 2).
Table 2.
Main deregulated gene clusters in frontal cortex in DLB.
| Cluster | Gene names | Count | Size | Odds ratio | P‐value | Deregulation |
|---|---|---|---|---|---|---|
| Antigen processing and presentation | SEC23A RAB3B PSMD8 PSMC2 PSMB2 ACTR1B LGMN RAB7A PSMB3 RAB3C DCTN6 AP1S1 PSMD12 DCTN2 DCTN3 PSMA5 AP2M1 PSMB6 PSMC4 PSMA3 PSMA4 DYNC1I1 KIF3A AP1M1 | 24 | 65 | 2.85 | 0.00 | Down |
| Antigen processing and presentation of exogenous peptide antigen via MHC class II | SEC23A ACTR1B LGMN RAB7A DCTN6 AP1S1 DCTN2 DCTN3 AP2M1 DYNC1I1 KIF3A AP1M1 | 12 | 29 | 3.40 | 0.00 | Down |
| Apoptotic mitochondrial changes | SLC25A4 YWHAZ MLLT11 DNM1L PARK2 FAM162A YWHAB MAPK9 BLOC1S2 YWHAG GSK3B GGCT OPA1 PIM2 | 14 | 40 | 2.60 | 0.01 | Down |
| ATP hydrolysis coupled proton transport | ATP6V1F ATP5B ATP6V1E1 ATP6AP1 ATP6V1B2 ATP5G1 ATP6V0D1 ATP6V1C1 ATP6V1A ATP1A1 ATP6V0A1 ATP1A3 ATP6V1H ATP5A1 | 14 | 17 | 22.66 | 0.00 | Down |
| ATP metabolic process | COX8A NDUFB5 ATP5B OLA1 NDUFA5 ATP6V1B2 ATP5G1 ENTPD5 DLD HTR2A DNM1L PRKAG1 ATP6V1A CYCS ENO2 NDUFA9 UQCRC1 PGK1 UQCRC2 ATP6V0A1 BPGM HK1 NDUFS3 GBAS NDUFS4 NDUFV2 ATP5H PPP2R5D NDUFAB1 ATP5A1 | 30 | 77 | 3.13 | 0.00 | Down |
| Cell proliferation | AKR1C2 PRKX GJA1 NUPR1 P2RX7 FGF17 TYK2 TOB2 HDAC4 MSX1 RXRA IKBKB FLCN PTCH1 YAP1 CD151 NACC2 LRP2 ID4 TNS3 KAT2A CDC14A GPER1 FOXO1 FOXC1 RELA EPS8 | 27 | 536 | 2.04 | 0.00 | Up |
| Coenzyme metabolic process | IDH3G PNPO MDH2 PDHB IDH3B ACLY RFK AASDHPPT ENTPD5 DLD HTR2A DLAT PRKAG1 ENO2 NMNAT2 MDH1 NDUFA9 SLC25A32 PDP1 PGK1 ELOVL6 ELOVL4 BPGM FAR2 HK1 PTS PDK3 ACSL4 SUCLA2 KCNAB2 PPP2R5D MVK MAT2B | 33 | 113 | 2.01 | 0.00 | Down |
| Covalent chromatin modification | EYA2 RBM14 HDAC4 FLCN NACC2 KAT2A DOT1L | 7 | 80 | 3.41 | 0.01 | Up |
| Developmental process | PRELP KLF15 PNPT1 AKR1C2 PRKX LRP4 GJA1 WWC3 SLC5A3 NUPR1 P2RX7 FGF17 EYA2 TYK2 AK4 CPQ TOB2 HDAC4 CAPN2 ADGRV1 MSX1 MRAS PPP2R1B RXRA IKBKB SASH1 ANKRD11 CDC42EP4 ZIC2 FLCN PTCH1 PCID2 MT1G YAP1 S100A1 ID3 CA2 DNAJB6 GNA12 ROCK1 LRP2 RAMP1 PPP1R13L SPR FRYL ARAP1 ID4 HIF3A SRGAP1 SLC39A12 VCAN TNS3 KAT2A CDC14A GPER1 FOXO1 FOXC1 MAN2A1 EHD2 PI16 RELA EPS8 | 62 | 1671 | 1.65 | 0.00 | Up |
| Electron transport chain | COX8A NDUFB5 ATP5B NDUFA5 ATP5G1 ALDH5A1 DLD CYCS NDUFA9 UQCRC1 SLC25A14 UQCRFS1 UQCRC2 SLC25A12 NDUFS3 NDUFS4 NDUFV2 ATP5H NDUFAB1 ATP5A1 | 20 | 43 | 4.23 | 0.00 | Down |
| Exocytosis | VPS33B TUBA4A SNX4 CDK5 NAPA RAB3B RPH3A PLCB1 PFN2 SYT5 ATP6AP1 PAK1 CHP1 SCAMP1 SCFD2 RAB7A RAB3C RASGRP1 SCAMP5 NSF SNAP25 HABP4 RAB3A VPS33A KIT VAMP1 PPIA VSNL1 STXBP1 RAB27B VPS4B SYT13 CRHBP VAMP2 RALB BLOC1S6 SYT16 CDK5R2 SYT1 DOC2A ARHGAP44 PPP3CB | 42 | 160 | 1.74 | 0.00 | Down |
| Gluconeogenesis | GOT1 MDH2 RBP4 SLC35B4 ENO2 MDH1 PRKACA PGK1 G6PC3 BPGM SLC25A12 GOT2 | 12 | 31 | 3.04 | 0.00 | Down |
| Histone modification | EYA2 RBM14 HDAC4 FLCN NACC2 KAT2A DOT1L | 7 | 80 | 3.41 | 0.01 | Up |
| Innate immune response activating cell surface receptor signaling pathway | PAK1 PSMD8 PSMC2 PSMB2 PSMB3 PRKACA PSMD12 PSMA5 PSMB6 UBE2N PSMC4 PSMA3 PSMA4 TAB3 PPP3CB | 15 | 44 | 2.50 | 0.01 | Down |
| miRNA metabolic process | PNPT1 RELA | 2 | 4 | 34.69 | 0.00 | Up |
| Mitochondrial electron transport, NADH to ubiquinone | NDUFB5 NDUFA5 DLD NDUFA9 NDUFS3 NDUFS4 NDUFV2 NDUFAB1 | 8 | 16 | 4.81 | 0.00 | Down |
| Mitochondrial translation | MRPL15 MRPL30 MRPL37 HARS MTFMT LRPPRC CHCHD1 MRPL45 MPV17L2 MRPS21 MRPL42 | 11 | 27 | 3.31 | 0.00 | Down |
| Mitochondrial transport | TOMM20 VPS11 MPC2 ATP5B YWHAZ PSMD8 ATP5G1 TIMM17A HAX1 PARK2 TOMM34 TOMM70A TOMM22 SLC25A32 BAG4 NRG1 SLC25A14 YWHAB MAPK9 UGCG BLOC1S2 YWHAG DNAJC19 SLC25A12 MTX3 GSK3B MICU1 KIF1BP ATP5H ATP5A1 | 30 | 104 | 1.97 | 0.00 | Down |
| mRNA polyadenylation | PNPT1 PAPOLA | 2 | 4 | 34.69 | 0.00 | Up |
| NADH metabolic process | IDH3G MDH2 IDH3B ENO2 MDH1 PGK1 HK1 PPP2R5D | 8 | 18 | 3.85 | 0.01 | Down |
| Neuron‐neuron synaptic transmission | CDK5 NAPA RAB3B PAK1 CHRNB2 CAMK4 CACNB4 GABRA1 HTR2A GRIN2A SLC6A1 PARK2 PRKACA GABRG2 STXBP1 GLRA2 MEF2C GRIA3 CRHBP GRIN3A SYT1 DNM1 PNKD | 23 | 80 | 1.95 | 0.01 | Down |
| Neurotransmitter transport | SLC32A1 CDK5 GAD1 SV2C GAD2 NAPA RAB3B RPH3A PFN2 SYT5 PAK1 ALDH5A1 SV2A NRXN3 PPT1 SLC6A1 SLC6A17 RAB3C PARK2 SNAP25 RAB3A ATP2A2 STXBP1 SLC38A1 SYT13 MEF2C VAMP2 BLOC1S6 SYT16 SYT1 DOC2A SV2B ICA1 | 33 | 112 | 2.04 | 0.00 | Down |
| Nucleobase‐containing compound metabolic process | KLF15 PNPT1 ZNF471 AHNAK GJA1 PAN2 WWC3 NUPR1 P2RX7 EYA2 RBM14 AK4 HDAC4 SAFB2 MSX1 RBMS2 RXRA IKBKB TRIM52 ZIC2 FLCN RAD52 IFI27 PTCH1 PCID2 YAP1 S100A1 ID3 SNRNP48 ZFHX2 DNAJB6 NACC2 CRY1 RAMP1 PPP1R13L FRYL ID4 HIF3A PAPOLA KAT2A RFX2 ZNF347 GPER1 TCP10L FOXO1 POLN TOP3B FOXC1 RELA GABPB2 DOT1L RBM4B MICAL3 | 53 | 1235 | 1.94 | 0.00 | Up |
| Phagosome maturation | ATP6V1F ATP6V1E1 ATP6V1B2 ATP6V0D1 ATP6V1C1 RAB7A ATP6V1A ATP6V0A1 ATP6V1H ATP6V1D | 10 | 18 | 6.03 | 0.00 | Down |
| Proteasome‐mediated ubiquitin‐dependent protein catabolic process | ERLEC1 USP5 KCTD13 PSMD8 PSMC2 PSMB2 USP11 PSMB3 PARK2 USP10 UBXN11 FBXO9 PRICKLE1 FBXL2 PSMD12 PLK2 RNF185 PSMA5 PSMB6 PSMC4 UCHL1 UBE2W PSMA3 GSK3B PSMA4 GLMN BBS7 RNF14 RNF4 NEDD4L | 30 | 99 | 2.12 | 0.00 | Down |
| Protein K11‐linked ubiquitination | UBE2E2 PARK2 UBE2W UBE2T RNF4 | 5 | 7 | 11.99 | 0.00 | Down |
| Protein K6‐linked ubiquitination | PARK2 UBE2T RNF4 | 3 | 3 | Inf | 0.01 | Down |
| Protein monoubiquitination | PARK2 FBXL2 UBE2W TRIM37 RNF2 UBE2T | 6 | 11 | 5.76 | 0.01 | Down |
| Protein ubiquitination involved in ubiquitin‐dependent protein catabolic process | PSMD8 PSMC2 PSMB2 PSMB3 PARK2 PSMD12 PSMA5 PSMB6 PSMC4 PSMA3 PSMA4 RNF14 NEDD4L | 13 | 37 | 2.61 | 0.01 | Down |
| Proton transport | COX8A ATP6V1F ATP5B ATP6V1E1 SLC9A7 ATP6AP1 ATP6V1B2 ATP5G1 ATP6V0D1 CHP1 ATP6V1C1 ATP6V1A SLC9A6 COX7A2 UQCRC1 SLC25A14 ATP1A1 UQCRFS1 SLC2A13 ATP6V0A1 ATP1A3 ATP6V1H ATP5H SLC9B2 ATP6V1D ATP5A1 | 26 | 59 | 3.86 | 0.00 | Down |
| Purine nucleoside metabolic process | DGUOK COX8A NDUFB5 ATP5B PRPS1 OLA1 MPP1 NDUFA5 ATP6V1B2 HPRT1 ATP5G1 ACLY ENTPD5 DLD HTR2A DNM1L PRKAG1 ATP6V1A CYCS ENO2 NDUFA9 UQCRC1 DLG3 PGK1 UQCRC2 ATP6V0A1 BPGM HK1 NDUFS3 GBAS NDUFS4 NDUFV2 ATP5H PPP2R5D NDUFAB1 MAT2B ATP5A1 | 37 | 111 | 2.46 | 0.00 | Down |
| Regulation of cell cycle | PNPT1 NUPR1 RBM14 KLHL21 MSX1 FLCN PTCH1 PCID2 ID3 NACC2 CRY1 CENPJ CDC14A GPER1 FOXC1 CTDSP2 CAB39L DOT1L | 18 | 259 | 2.82 | 0.00 | Up |
| Regulation of gene expression | KLF15 ZNF471 AHNAK GJA1 WWC3 NUPR1 P2RX7 EYA2 RBM14 TOB2 HDAC4 SAFB2 MSX1 RXRA IKBKB TRIM52 ZIC2 FLCN IFI27 PTCH1 PCID2 YAP1 S100A1 ID3 ZFHX2 DNAJB6 NACC2 CRY1 PPP1R13L FRYL ID4 HIF3A PAPOLA KAT2A RFX2 ZNF347 GPER1 TCP10L FOXO1 FOXC1 RELA GABPB2 DOT1L RBM4B MICAL3 | 45 | 908 | 2.24 | 0.00 | Up |
| Regulation of macromolecule biosynthetic process | KLF15 ZNF471 GJA1 WWC3 NUPR1 MARCH8 EYA2 RBM14 HDAC4 SAFB2 MSX1 RXRA IKBKB TRIM52 ZIC2 FLCN IFI27 PTCH1 PCID2 YAP1 S100A1 ID3 ZFHX2 DNAJB6 NACC2 CRY1 RAMP1 PPP1R13L FRYL ID4 HIF3A KAT2A RFX2 ZNF347 GPER1 TCP10L FOXO1 FOXC1 RELA GABPB2 DOT1L RBM4B MICAL3 | 43 | 863 | 2.23 | 0.00 | Up |
| Regulation of mitochondrion organization | VPS11 BECN1 YWHAZ PSMD8 MLLT11 DDHD2 DNM1L HAX1 PARK2 FAM162A BAG4 NRG1 YWHAB UGCG LRPPRC YWHAG HK1 GSK3B OPA1 MPV17L2 | 20 | 66 | 2.10 | 0.01 | Down |
| Regulation of ubiquitin‐protein transferase activity | PSMD8 PSMC2 PSMB2 PSMB3 PIN1 PSMD12 PSMA5 PSMB6 UBE2N PSMC4 PSMA3 DYNC1LI1 LCMT1 PSMA4 | 14 | 40 | 2.60 | 0.01 | Down |
| Synaptic transmission | SLC32A1 CDK5 GAD1 NCALD CALB1 GAD2 NAPA RAB3B GNG3 RPH3A PLCB1 PFN2 SYT5 PAK1 KCND2 ASIC2 ALDH5A1 GABRA3 HCN1 NRXN3 LRRTM1 CHRNB2 CAMK4 CACNB4 PPT1 GABRA1 ARHGEF9 HTR2A GRIN2A SLC6A1 KCNAB1 RAB3C PARK2 CACNG3 SEZ6 PRKACA SST NSF SNAP25 GABRA4 GABRG2 KCNC2 RAB3A LRRTM2 KIT PLK2 DLG3 GRIN2B RASGRF1 HTR4 ATP2A2 GABRA5 STXBP1 SNCB SLC38A1 AP2M1 SYT13 GLRA2 MEF2C GRIA3 CRHBP VAMP2 YWHAG GRIN3A FGF12 KCNQ5 NETO1 NCDN BLOC1S6 SYT16 CACNB1 SYT1 DOC2A CHRM4 GABRB3 KCNH7 GNAL KCNAB2 GABBR2 CACNG2 DNM1 PCDH8 PANX1 AKAP5 PNKD PPP3CB SYP FGF14 | 88 | 390 | 1.44 | 0.00 | Down |
| Synaptic transmission, dopaminergic | CDK5 RAB3B CHRNB2 PARK2 CRHBP PNKD | 6 | 12 | 4.80 | 0.01 | Down |
| Tissue development | KLF15 AKR1C2 PRKX LRP4 GJA1 NUPR1 P2RX7 EYA2 CPQ HDAC4 MSX1 RXRA IKBKB ZIC2 PTCH1 YAP1 ID3 CA2 ROCK1 PPP1R13L KAT2A FOXC1 MAN2A1 PI16 RELA | 25 | 493 | 2.04 | 0.00 | Up |
| Transcription from RNA polymerase II promoter | KLF15 WWC3 RBM14 HDAC4 MSX1 RXRA IKBKB FLCN IFI27 PTCH1 YAP1 S100A1 ID3 NACC2 CRY1 PPP1R13L ID4 HIF3A PAPOLA KAT2A RFX2 GPER1 TCP10L FOXO1 FOXC1 RELA GABPB2 MICAL3 | 28 | 461 | 2.57 | 0.00 | Up |
| Transcription, DNA‐templated | KLF15 ZNF471 WWC3 NUPR1 EYA2 RBM14 HDAC4 SAFB2 MSX1 RXRA IKBKB TRIM52 ZIC2 FLCN IFI27 PTCH1 PCID2 YAP1 S100A1 ID3 ZFHX2 DNAJB6 NACC2 CRY1 PPP1R13L FRYL ID4 HIF3A PAPOLA KAT2A RFX2 ZNF347 GPER1 TCP10L FOXO1 FOXC1 RELA GABPB2 DOT1L MICAL3 | 40 | 785 | 2.24 | 0.00 | Up |
| Ubiquitin‐dependent protein catabolic process | ERLEC1 USP5 KCTD13 PSMD8 PSMC2 PSMB2 USP11 PSMB3 PARK2 NDFIP1 USP10 UBXN11 FBXO9 PRICKLE1 FBXL2 PSMD12 PLK2 RLIM RNF185 VPS25 PSMA5 VPS4B PSMB6 UBE2N PSMC4 COPS3 UCHL1 UBE2W PSMA3 GSK3B PSMA4 GLMN BBS7 RNF14 RNF4 UCHL5 NEDD4L | 37 | 123 | 2.11 | 0.00 | Down |
| Vesicle docking | VPS33B RAB3B SCFD2 RAB3C NSF SNAP25 RAB3A VPS33A STXBP1 RAB27B RALB STX12 BLOC1S6 | 13 | 28 | 4.19 | 0.00 | Down |
| Vesicle organization | VPS11 VPS33B SNX4 SEC23A COPG1 NSG1 RAB7A SNAP25 COPZ1 RAB3A VAMP1 PRKCI PTPRN STXBP1 VPS4B SNX10 BLOC1S2 VAMP2 STX12 VTA1 BLOC1S6 HMP19 AP1M1 SYP | 24 | 79 | 2.12 | 0.00 | Down |
Weighted gene coexpression network analysis
Weighted gene coexpression network was constructed using the expression values of 5114 genes with variable expression among the 28 samples. Thirteen uncorrelated (r < 0.8 gene modules were identified and labeled by colors and numbers (M1 to M13). The genes not assigned to any particular coexpression module were assigned to M0 or “gray” (Figure 1B).
We obtained the most representative pattern of gene expression across all samples for each one of these modules by calculating the eigengene (i.e., the first principal component). The eigengene of each module was then correlated with the LBD spectrum (iLBD, PD and DLB) obtaining an eigengene significance for each module (Figure 2A). Three modules, M4‐lightcyan (157 genes), M6‐tan (215 genes) and M10‐darkred (23 genes) were negatively correlated with iLBD. M3‐green (226 genes), M11‐royalblue (23 genes) were positively and M10‐darkred negatively, correlated to PD were negatively correlated with iLBD. M3‐green (226 genes) and M11‐royalblue (23 genes) were positively and M10‐darkred negatively, correlated to PD. Three other modules, M3‐green, M5‐brown and M9‐salmon, correlated significantly with DLB. M5‐brown correlated negatively with DLB and contained 766 genes. M3‐green and M9‐salmon correlated positively with DLB and contained 226 and 51 genes, respectively (Figure 2B). None of our identified disease‐correlated modules were also correlated with age, indicating that our disease associated modules are not significantly confounded by transcriptomic changes produced by normal ageing. Of note, one module, M7‐midnightblue, was negatively correlated with age.
Figure 2.
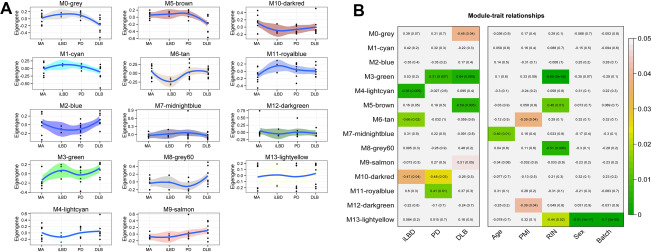
A. Module eigengenes for each module by LB spectrum. B. Correlation values (univariate) and P‐values for the relationship between each module eigengene and each LBD stage compared separately with MA or various control variables. P‐values for LBD stages correspond to partial coefficients in the multivariate analysis. Color code indicates the significance of the correlation. Seven modules significantly correlate with different stages of LBD.
Module characterization
Four strategies were used: 1 Identification of top highly connected genes (or hubs) in each module, 2 Assessment of overlaps with available published brain modules to correlate present LBD‐associated modules to brain‐related biological categories, 3 Identification of enriched GO among genes in each module and 4 Subdivision of each module into subnetworks based on available knowledge concerning physical PPI networks of human proteins.
Top hub genes
MSN, UGT2B11, lysosomal protein transmembrane 5 (LAPTM5) gene was the top hub genes in the iLBD‐associated modules M4‐lightcyan, M10‐darkred and M6‐tan, respectively. MSN encodes for Moesin, a molecule that links cytoskeleton to membrane and which is a suggested phosphorylation target of LRRK2. UGT2B11 encodes a UDP‐glucuronosyltransferase and LAPTM5 is a lysosomal gene recently identified as a hub node in the protein‐interaction network obtained from DEGs in locus ceruleus in PD patients vs. healthy donors 18. Hub genes in M3‐green and M11‐royalblue, PD‐associated modules, are genes with unknown function. ATP6V1B2 and ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A (ATP6V1A) and stress‐induced‐phosphoprotein 1 (STIP1) were top hub genes for M5‐brown and M9‐ salmon, respectively, linked to DLB modules. ATP6V1A encodes a component of vacuolar ATPase which mediates the acidification of intracellular organelles including endosomes, lysosomes, the trans‐Golgi network and synaptic vesicles, thus enabling a plethora of functions such as zymogen activation, endocytosis, synaptic transmission and protein transport 12. STIP1 (HOP) is implicated in assisting the function of chaperone protein interaction with HSP70 and HSP90 72 (Supporting Information Table 5).
Overlap of modules with reported brain‐related categories: specific cell populations and particular diseases
Module M6‐tan and M4‐lightcyan were highly enriched in markers of microglia 15. M5‐ brown and M9‐salmon DLB‐associated modules were found enriched in neuronal and microglial markers 24, 58 (Figure 3, Supporting Information Table 6). Other modules identified in our study, but not correlated with LBD, showed enrichment in particular cell types. For instance, M2‐blue module was enriched in oligodendrocyte markers whereas M1‐cyan and M7‐midnightblue were enriched in neuronal and glial cell markers. These observations support the biological consistency of our network and the robustness of the method to build similar transcriptomic modules using different sources of data.
Figure 3.
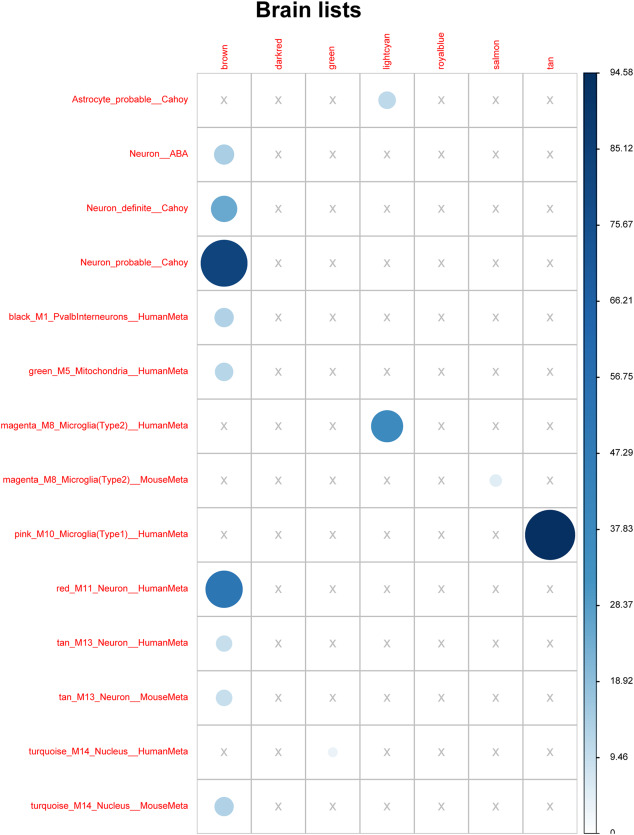
Enrichment score (‐log10 P‐value) of genes in selected modules in previously published brain gene sets associated with cell types or brain regions. Module‐brown (M6) is enriched in neuronal genes; module‐tan (M5) is enriched in microglial genes; module‐salmon (M6) has nonspecific enrichment.
M6‐tan modules presented enrichment in genes that was also deregulated at early stages of AD 75. Moreover, module M5‐brown showed overlap with deregulated gene sets and modules reported in AD 6, 60. These confluences suggest commonalities involving multiple processes in these neurodegenerative diseases. In fact, DLB is largely associated with AD‐related pathology, and DLB cases in the present series had associated AD pathology.
Identification of enriched GO among genes in each module
We also attempted to relate genes in modules to particular GO. Consistent with the microglial nature of this module, genes in M6‐tan were highly enriched in GO categories related to immune system and inflammation. Similarly, M4‐lightcyan was also enriched in categories related to inflammatory defence response. Module M5‐brown showed highly significant enrichment in GO categories related to synaptic transmission and energy metabolism, among others. Genes in M9‐salmon module presented a highly significant enrichment in GO categories related to protein folding and heat‐shock chaperone activity. Interestingly, the age associated M7‐midnightblue module was enriched in GO categories such as hormone response, oxidative stress and learning, among others. Supporting Information Table 7 shows the full list of categories in modules 3, 5, 6, 9 and 11. Only few genes composed M11‐royalblueand M10‐darkred for proper GO analysis, but those modules contained taste 2 receptor members (TAS2R10, TAS2R4, TAS2R50) and nine olfactory receptors, respectively.
Putative subnetworks of protein–protein interactions
We intersected genes from each coexpression module with PPI databases to identify putative subnetworks with biological relevance within modules. Genes in M6‐tan, M4‐lightcyan, M5‐brown and M9‐salmon each produced notable PPI subnetworks composed of 44, 51, 302 and 21 genes, respectively (representing 25.7%, 35.4%, 47.2% and 45.7% of the genes in the module listed in the PPI database). Regarding M6‐tan module PPI subnetwork, spleen tyrosine kinase (encoded by SYK) appeared with 16 interacting partners as the nucleating gene with highest degree (Figure 4A). M4‐lightcyan included a subnetwork with CDK2, MSN and TP53 as top degree nodes (Figure 4B). Heat‐shock related proteins encoded by genes STIP1, heat shock 70 kDa protein 5 (glucose‐regulated protein, 78 kDa) (HSPA5), AHSA1 and heat shock 70 kDa protein 1A (HSPA1A) were the highest connected nodes in the M9‐salmon PPI subnetwork (Figure 4C). M5‐brown module contained a large PPI subnetwork with several nodes including proteins encoded by tyrosine 3‐monooxygenase/tryptophan 5‐monooxygenase activation protein, beta polypeptide (YWHAB), TP1A, glycogen synthase kinase 3 beta (GSK3B), proteasome (prosome, macropain) subunit, alpha type, 3 (PSMA3) and SNCA, among others (Figure 4D).
Figure 4.
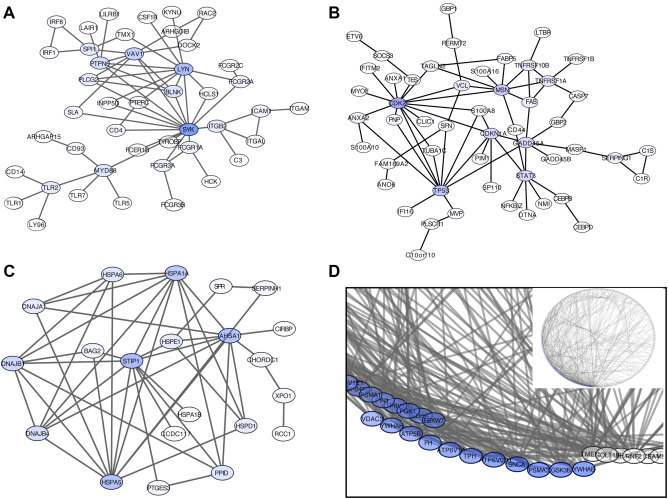
Putative subnetworks of protein–protein interactions. A. M6‐tan module PPI subnetwork showing that spleen tyrosine kinase (encoded by SYK) interacting with 16 partners is the gene with the highest degree of nucleation. B. M4‐lightcyan module subnetwork showing high degree proteins such as CDK2, MSN and TP53. C. M9‐salmon PPI subnetwork shows a large connected node related to heat shock proteins encoded by STIP1, DNAJA1, DNAJB1, DNAJB4, HSPA5, HSAPA6, AHSA1 and HSPA1A, among others. D. M5‐brown module contains a large PPI subnetwork with several nodes including proteins encoded by YWHAB, GSK3B, PSMA3, ATP6V0D1, TPI1, ATP6V1A and SNCA, among others (circular network representation is shown in the small panel proteins ordered by degree, a zoom in the top degree proteins is shown in the larger panel).
These results allow for better characterization of selected LBD‐associated modules. M5‐brown globally downregulated in DLB and enriched in neuronal markers and in genes mostly related to synaptic transmission and energy metabolism presented ATP6V1B2 and YWHAB as top hub genes in the transcriptomic and PPI networks, respectively. Other highly connected molecules were GSK3B, PSMA3, ATP6V0D1, triosephosphate isomerase 1 (TPI1), ATP6V1A and SNCA. Finally, an M9‐salmon module, globally upregulated in DLB, was characterized by significant enrichment in genes involved in heat‐shock protein folding and microglial markers, and was nuclearized around the hub gene STIP1 in both transcriptomic and PPI networks. Other highly interacting proteins were encoded by DnaJ (Hsp40) homolog, subfamily A, member 1 (DNAJA1), DnaJ (Hsp40) homolog, subfamily B, member 1 (DNAJB1), DnaJ (Hsp40) homolog, subfamily B, member 4 (DNAJB4), HSPA5, HSAPA6, AHSA1 and HSPA1A.
RT‐qPCR validation
LAPTM5, a top gene in M6, was found not to be deregulated in iLBD, PD and DLB. ATP6V1A, one of the top genes in M5, was downregulated in DLB (1.03 ± 0.25 vs. 0.57 ± 0.52, P = 0.007). The expression of STIP1, one top gene in M9, was not significantly altered in DLB but only in a subset of rpDLB (see later) and in PD (1.03 ± 0.28 vs. 1.75 0.70, P = 0.006); however, mRNA expression of DNAJA1 and DNAJB1, other top genes in the same module, was significantly increased in DLB (see later). SYK, the principal PPI in M6 was not deregulated in iLBD, PD and DLB (MA: 1.06 ± 0.51, iLBD: 0.81 ± 0.58, PD: 1.02 ± 0.38, DLB: 1.47 ± 0.93).
TPI1, a PPI member of module 5, was upregulated in PD (1.02 ± 0.20 vs. 1.29 ± 0.14, P = 0.004). No significant modifications in the expression mRNA levels were observed for selected PPI members of M5 and M9 modules in DLB. However, several members were deregulated in a subpopulation of DLB cases characterized by their rapid course and classified as rpDLB. Regarding M5, upregulated genes in rpDLB were GSK3B (1.03 ± 0.22 vs. 1.62 ± 0.88, P =0.046), whereas PSMA3 (1.01 ± 0.15 vs. 0.57 ± 0.05, P =0.000) and YWHAB (1.01 ± 0.15 vs. 0.69 ± 0.22, P = 0.007) were downregulated. Genes representative of PPI in M9 were upregulated in rpDLB: HSPA1A (1.01 ± 0.40 vs. 3.10 ± 2.00, P = 0.007), HSPA5 (1.06 ± 0.38 vs. 2.14 ± 0.83, P = 0.003) and STIP1 (1.03 ± 0.28 vs. 2.07 ± 1.10, P = 0.009).
Abnormal regulation of the dynein cluster was assessed by RT‐qPCR in LBDs with particular attention on iLBD. TASRs were assessed in iLBD, PD and DLB. Three biological functions, RNA/DNA metabolism, chaperone and protein folding, and synaptic neurotransmission, were selected for validation in DLB. We extracted those pathways and clusters from each of those categories that appeared in the corresponding module and disease in which the category appeared as significantly enriched.
Regarding dyneins, dynein axonemal assembly factor 1 (DNAAF1), dynein axonemal heavy chain 11 (DNAH11), dynein axonemal heavy chain 2 (DNAH2), dynein axonemal heavy chain 7 (DNAH7) and dynein axonemal intermediate chain 1 (DNAI1), but not dynein axonemal heavy chain 5 (DNAH5) and dynein axonemal heavy chain 9 (DNAH9), were significantly upregulated in iLBD when compared with MA. Interestingly, upregulation also occurred in PD and DLB. Taste receptor TAS2R5 and TAS2R13 were upregulated and downregulated, respectively, in iLBD; TAS2R10 upregulated in PD, and TAS2R4, TAS2R5, TAS2R14, TAS2R10 and TAS2R13 significantly upregulated in DLB (Table 3).
Table 3.
Quantification of the expression of selected genes corresponding to axonema and taste receptors clusters in MA, iLBD, PD and DLB.
| P‐value | P‐value | P‐value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Probes | MA | iLBD | PD | DLB | MA vs iLBD | Adjusted | MA VS PD | Adjusted | MA vs DLB | Adjusted | |
| Axonema | DNAAF1 | 1.04 ± 0.31 | 2.18 ± 0.72 | 2.06 ± 1.03 | 1.04 ± 0.36 | 0.000481 | 0.013524 | 0.005583 | 0.078162 | 0.987973 | 0.987973 |
| DNAH11 | 1.16 ± 0.57 | 3.17 ± 1.94 | 1.96 ± 0.51 | 3.27 ± 2.43 | 0.005601 | 0.039207 | 0.003889 | 0.078162 | 0.011763 | 0.043544 | |
| DNAH2 | 1.07 ± 0.41 | 2.03 ± 0.97 | 2.40 ± 1.50 | 1.05 ± 0.38 | 0.012770 | 0.061973 | 0.010576 | 0.085253 | 0.899477 | 0.944451 | |
| DNAH5 | 1.08 ± 0.44 | 1.76 ± 0.89 | 1.69 ± 0.83 | 1.05 ± 0.51 | 0.064296 | 0.245494 | 0.060820 | 0.196495 | 0.886887 | 0.944451 | |
| DNAH7 | 1.08 ± 0.42 | 1.84 ± 0.69 | 1.35 ± 0.51 | 1.67 ± 0.45 | 0.016352 | 0.068678 | 0.227373 | 0.471895 | 0.007938 | 0.039284 | |
| DNAH9 | 1.16 ± 0.63 | 2.39 ± 2.25 | 2.36 ± 1.25 | 1.67 ± 0.56 | 0.105147 | 0.339706 | 0.012179 | 0.085253 | 0.084009 | 0.147016 | |
| DNAI1 | 1.07 ± 0.43 | 2.13 ± 0.32 | 3.09 ± 2.22 | 2.96 ± 1.44 | 0.000644 | 0.013524 | 0.008326 | 0.085253 | 0.000592 | 0.021154 | |
| Taste receptors | TAS2R4 | 1.03 ± 0.27 | 1.48 ± 1.12 | 1.33 ± 0.80 | 1.82 ± 0.78 | 0.203630 | 0.503086 | 0.247183 | 0.471895 | 0.004187 | 0.035171 |
| TAS2R5 | 1.03 ± 0.25 | 0.61 ± 0.33 | 1.14 ± 0.67 | 2.25 ± 1.03 | 0.013280 | 0.061973 | 0.639661 | 0.814114 | 0.001261 | 0.021154 | |
| TAS2R14 | 1.15 ± 0.57 | 0.89 ± 0.54 | 2.44 ± 2.31 | 2.51 ± 1.59 | 0.427225 | 0.690133 | 0.077225 | 0.231675 | 0.012441 | 0.043544 | |
| TAS2R50 | 1.08 ± 0.40 | 0.68 ± 0.42 | 0.65 ± 0.65 | 1.54 ± 1.15 | 0.091428 | 0.319998 | 0.089294 | 0.250023 | 0.229848 | 0.321787 | |
| TAS2R10 | 1.13 ± 0.50 | 1.49 ± 1.09 | 4.50 ± 3.33 | 1.68 ± 0.64 | 0.371381 | 0.628649 | 0.002611 | 0.078162 | 0.038324 | 0.080480 | |
| TAS2R13 | 1.11 ± 0.49 | 7.27 ± 5.99 | 0.99 ± 0.50 | 1.63 ± 0.62 | 0.002092 | 0.021966 | 0.625557 | 0.814114 | 0.042350 | 0.080850 | |
Green, indicates downregulation; Red, indicates upregulation.
Data are expressed as mean values ± SD; significant comparisons between groups are expressed by P‐values. Upregulated and downregulated genes are represented by different colors. P‐values have been adjusted using the BH method in each contrast and are indicated next to nominal P‐values.
Moving on to iLBD, expression of allograft inflammatory factor 1 (AIF1) (which encodes IBA‐1) was downregulated, as predicted, in frontal cortex (control: 1.07 ± 0.41, iLBD: 0.57 ± 0.20, P = 0.018). Focusing on DLB, polyribonucleotide nucleotidyltransferase 1 (PNPT1), poly(A) polymerase alpha (PAPOLA), RELA (proto‐oncogene, NF‐kB subunit) and DOT1 like histone lysine methyltransferase (DOT1L) mRNAs, all of them involved in RNA/DNA metabolism, but not lysine acetyltransferase 2A (KAT2A), were significantly upregulated in DLB (Table 4). Importantly, PNPT1 and RELA were upregulated in iLBD, and KAT2A was added in PD (Table 4).
Table 4.
Quantification of the expression of selected genes corresponding to RNA/DNA processing, chaperones and neurotransmitters and synapses in MA, iLBD, PD and DLB.
| P‐value | P‐value | P‐value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Probes | MA | iLBD | PD | DLB | MA vs iLBD | Adjusted | MA vs PD | Adjusted | MA vs DLB | Adjusted | |
| RNA/DNA | KAT2A | 1.08 ± 0.39 | 1.38 ± 0.80 | 1.68 ± 0.75 | 1.65 ± 1.22 | 0.318894 | 0.608798 | 0.032546 | 0.113911 | 0.162525 | 0.252817 |
| PNPT1 | 1.03 ± 0.26 | 1.73 ± 0.45 | 1.63 ± 0.74 | 2.18 ± 0.99 | 0.001300 | 0.018200 | 0.020982 | 0.106943 | 0.001511 | 0.021154 | |
| PAPOLA | 1.01 ± 0.18 | 1.02 ± 0.55 | 1.01 ± 0.33 | 1.52 ± 0.68 | 0.966720 | 0.990299 | 0.987272 | 0.997676 | 0.029792 | 0.069238 | |
| RELA | 1.01 ± 0.14 | 1.42 ± 0.39 | 1.62 ± 0.79 | 2.39 ± 1.84 | 0.009412 | 0.056472 | 0.027832 | 0.106943 | 0.029333 | 0.069238 | |
| DOT1L | 1.01 ± 0.16 | 1.28 ± 0.62 | 1.24 ± 0.60 | 1.34 ± 0.48 | 0.179901 | 0.472240 | 0.241060 | 0.471895 | 0.046556 | 0.085015 | |
| Chaperones | DNAJA1 | 1.02 ± 0.19 | 1.24 ± 0.58 | 1.32 ± 0.36 | 1.51 ± 0.75 | 0.249723 | 0.525382 | 0.027383 | 0.106943 | 0.041181 | 0.080850 |
| DNAJA4 | 1.28 ± 0.76 | 1.62 ± 1.15 | 1.75 ± 0.83 | 2.53 ± 1.19 | 0.479652 | 0.719478 | 0.199588 | 0.471895 | 0.008306 | 0.039284 | |
| DNAJB1 | 1.21 ± 0.65 | 1.13 ± 0.58 | 1.14 ± 0.54 | 2.47 ± 0.54 | 0.827989 | 0.921038 | 0.822303 | 0.933425 | 0.020325 | 0.056910 | |
| DNAJB4 | 1.21 ± 0.73 | 1.32 ± 0.70 | 1.13 ± 0.28 | 1.64 ± 0.82 | 0.788308 | 0.921038 | 0.776413 | 0.905815 | 0.223143 | 0.321787 | |
| HSPA1A | 1.06 ± 0.40 | 1.16 ± 0.55 | 1.28 ± 0.88 | 1.63 ± 1.55 | 0.727662 | 0.901719 | 0.490696 | 0.775885 | 0.283092 | 0.349702 | |
| HSPA4 | 1.01 ± 0.12 | 1.05 ± 0.37 | 1.34 ± 0.42 | 1.63 ± 0.72 | 0.727987 | 0.901719 | 0.016296 | 0.097776 | 0.007965 | 0.039284 | |
| HSPA5 | 1.06 ± 0.38 | 0.84 ± 0.29 | 1.70 ± 0.78 | 1.48 ± 0.82 | 0.270249 | 0.540498 | 0.028009 | 0.106943 | 0.143620 | 0.232002 | |
| HSPA6 | 1.06 ± 0.39 | 0.86 ± 0.29 | 1.24 ± 0.91 | 1.63 ± 0.47 | 0.374196 | 0.628649 | 0.557809 | 0.780933 | 0.017922 | 0.056910 | |
| HSPD1 | 1.02 ± 0.22 | 1.01 ± 0.66 | 1.17 ± 0.46 | 1.98 ± 1.28 | 0.950284 | 0.990299 | 0.324922 | 0.568614 | 0.018989 | 0.056910 | |
| HSPE1 | 1.01 ± 0.13 | 1.04 ± 0.47 | 1.25 ± 0.50 | 1.04 ± 0.29 | 0.827808 | 0.921038 | 0.114919 | 0.301662 | 0.721566 | 0.819075 | |
| Neurotransmitters and synapses | GABBR2 | 1.05 ± 0.32 | 1.15 ± 0.48 | 1.05 ± 0.46 | 0.64 ± 0.32 | 0.616222 | 0.862711 | 0.281410 | 0.513879 | 0.012003 | 0.043544 |
| GABRA1 | 1.07 ± 0.40 | 0.88 ± 0.56 | 1.05 ± 0.52 | 0.60 ± 0.50 | 0.451273 | 0.701980 | 0.913177 | 0.958836 | 0.031322 | 0.069238 | |
| GAD1 | 1.08 ± 0.43 | 0.73 ± 0.45 | 1.08 ± 0.45 | 0.50 ± 0.34 | 0.161648 | 0.452614 | 0.997676 | 0.997676 | 0.004059 | 0.035171 | |
| GRIN2A | 1.04 ± 0.28 | 1.04 ± 0.43 | 1.18 ± 0.45 | 1.14 ± 0.51 | 0.999447 | 0.999447 | 0.393711 | 0.661434 | 0.560841 | 0.654315 | |
| GRIN2B | 1.03 ± 0.27 | 0.99 ± 0.81 | 0.93 ± 0.41 | 1.19 ± 0.56 | 0.869824 | 0.936734 | 0.534280 | 0.775885 | 0.409171 | 0.491005 | |
| NETO1 | 1.05 ± 0.32 | 0.47 ± 0.14 | 1.12 ± 0.75 | 0.64 ± 0.44 | 0.005157 | 0.039207 | 0.771798 | 0.905815 | 0.027200 | 0.069238 | |
| RAB3A | 1.03 ± 0.25 | 1.09 ± 0.04 | 1.14 ± 0.48 | 0.79 ± 0.53 | 0.636911 | 0.862912 | 0.535730 | 0.775885 | 0.218554 | 0.321787 | |
| RPH3A | 1.03 ± 0.25 | 1.10 ± 0.55 | 1.13 ± 0.53 | 0.78 ± 0.41 | 0.729963 | 0.901719 | 0.594324 | 0.805213 | 0.117159 | 0.196827 | |
| SNAP25 | 1.02 ± 0.22 | 1.06 ± 0.42 | 1.23 ± 0.49 | 0.85 ± 0.43 | 0.833320 | 0.921038 | 0.220065 | 0.471895 | 0.263212 | 0.334997 | |
| STXBP1 | 1.02 ± 0.20 | 1.12 ± 0.46 | 1.22 ± 0.49 | 0.86 ± 0.39 | 0.530886 | 0.768869 | 0.230966 | 0.471895 | 0.262784 | 0.334997 | |
| SYP | 1.02 ± 0.20 | 0.82 ± 0.31 | 1.09 ± 0.48 | 0.69 ± 0.29 | 0.131839 | 0.395517 | 0.674822 | 0.833604 | 0.008418 | 0.039284 | |
| SYT1 | 1.01 ± 0.18 | 0.88 ± 0.42 | 1.15 ± 0.63 | 0.83 ± 0.48 | 0.357715 | 0.628649 | 0.509828 | 0.775885 | 0.238214 | 0.322742 | |
| SYT13 | 1.04 ± 0.30 | 0.83 ± 0.38 | 1.07 ± 0.45 | 1.05 ± 0.54 | 0.246964 | 0.525382 | 0.868258 | 0.958836 | 0.981224 | 0.987973 | |
| SYT16 | 1.03 ± 0.25 | 0.85 ± 0.36 | 1.05 ± 0.49 | 1.00 ± 0.59 | 0.250182 | 0.525382 | 0.895977 | 0.958836 | 0.868671 | 0.944451 | |
Green, indicates downregulation; Red, indicates upregulation.
Data are expressed as mean values ± SD; significant comparisons between groups are expressed by P‐values. Upregulated and downregulated genes are represented by different colors. P‐values have been adjusted using the BH method in each contrast and are indicated next to nominal P‐values.
Chaperone members DNAJA1, DnaJ (Hsp40) homolog, subfamily A, member 4 (DNAJA4), DNAJB1, heat shock 70 kDa protein 4 (HSPA4), heat shock 70 kDa protein 6 (HSP70B) (HSPA6) and heat shock 60 kDa protein 1 (chaperonin) (HSPD1), but not DNAJB4, HSPA1A, HSPA5 and MOB family member 4, phocein (HSPE1), were significantly upregulated in frontal cortex area 8 in DLB when compared with MA (Table 4). No modification in the expression of these genes was found in iLBD, but DNAJA1, HSPA4 and HSPA5 mRNA expression was significantly increased in PD (Tables 3 and 4).
Finally, neurotransmission‐related components gamma‐aminobutyric acid B receptor, 2 (GABBR2), gamma‐aminobutyric acid A receptor, alpha 1 (GABRA1), glutamate decarboxylase 1 (brain, 67 kDa) (GAD1) and synaptic proteins neuropilin and tolloid‐like 1 (NETO1), as well as synaptophysin (SYP) mRNAs, were found to be significantly downregulated in DLB when compared with MA. The expression of glutamate receptor, ionotropic‐methyl d‐aspartate 2A (GRIN2A) and glutamate receptor, ionotropic‐methyl d‐aspartate 2B (GRIN2B), and synaptic proteins member RAS oncogene family (RAB3A), Rabphilin 3A homolog (mouse) (RPH3A), synaptosomal‐associated protein, 25 kDa (SNAP25), syntaxin binding protein 1 (STXBP1), synaptotagmin I (SYT1), synaptotagmin XIII (SYT13) and Synaptotagmin XVI (SYT16), was not altered in DLB (Table 4).
Discussion
This study presents the first transcriptome analysis of the frontal cortex integrating the whole spectrum of LBDs. In addition to performing a traditional gene‐based differential expression analysis we considered a system biology approach based on coexpression networks. In particular, we applied WGNCA which allows the identification of modules of genes that are coexpressed across samples. These modules have to be interpreted with the understanding that such coexpression reflects a common biological function (e.g., from biochemical pathways to cell type defining signatures). We have identified some modules that correlate with LBDs. A proportion of genes included in those modules are also differentially expressed in a traditional gene‐based comparative approach using the same microarray data and using independent case series for RTqPCR quantification and validation. The present study has also identified several hubs and PPI which raise the alert about putative biomarkers and targets for therapeutic intervention.
Lack of comorbidities in the present series implies the selection of a limited number of cases which minimizes the risk of bias. PD and DLB cases have concomitant AD‐related pathology which was variable from one case to another. Thal phases were used to evaluate β‐amyloid plaques; however, no distinction was made between diffuse and neuritic plaques.
Discussion is centred on selected genes, among those identified by differential expression analysis and weighted correlation networks, whose mRNA expression was assessed with RT‐qPCR. The transcriptome is relatively conserved in iLBD and PD as no genes surpass the threshold of significance when P‐values are adjusted for multiple testing. This is in contrast with DLB where hundreds of genes show multiple testing‐corrected significant expression differences when compared with MA individuals.
Dyneins are one deregulated cluster in LBDs. Five of seven assessed members are upregulated in iLBD and PD, and three of seven in DLB. Because of their ATP hydrolysis‐mediated involvement in cytoplasmic transport 82, early alteration of the cargo transport along neurites may be suggested in the frontal cortex within the LBD spectrum.
Expression of taste and olfactory receptors and down‐stream obliged functional signaling pathways in the CNS and their deregulation in neurodegenerative diseases is intriguing. It is possible that these receptors in brain are not involved in the perception of odors and taste but rather correspond to new central chemoreceptors looking for putative ligands or interacting complementary receptors 30. TASRs are significantly upregulated in frontal cortex in PD and DLB 33, 38, and our findings further indicate that deregulation of TASRs in frontal cortex occurs in iLBD as well. Previous studies have shown altered protein synthesis machinery in the frontal cortex in PD and DLB from the nucleolus to the ribosome 36, 38. Nuclear alterations include increased nucleolar stress and altered synthesis of ribosomal RNAs, in addition to altered production of mRNAs. Our findings show altered mRNA expression of different molecules involved in RNA and DNA metabolism such as KAT2A which is upregulated in PD, and PNPT1, PAPOLA, RELA and DOT1Ls which are upregulated in DLB. PNPT1 and RELA are also upregulated in iLBD. Therefore, genes which encode proteins involved in the acetylation and methylation of histones, RNA processing and the modulation of NF‐κB‐mediated gene transcription are abnormally regulated in DLB and, to a lesser degree, in other disorders within the LBD spectrum. A primary effect of α‐synuclein can be postulated, as α‐synuclein is abnormally localized in the nucleus in neurons, and probably also in glial cells, in the frontal cortex in PD 36.
Another deregulated cluster in LBD is related to innate inflammatory responses. This has been the subject of two detailed studies in PD and DLB 34, 38. An interesting aspect was the downregulation of cytokines and several mediators of the inflammatory response in the substantia nigra at early stages of PD 34 and the relatively low inflammatory response in the frontal cortex in DLB 38, which is in line with previous observations pointing out the relatively low inflammatory response and enhanced dystrophic microglia in DLB 2, 88, 90. This is further supported in the present study by downregulation of AIF1, which encodes the microglial marker IBA‐1 in the frontal cortex in iLBD, thus suggesting early microglial alteration in LBDs.
Another DLB‐associated module is related to heat‐shock/chaperone proteins and is globally upregulated in DLB. Three out of ten assessed genes are upregulated in the frontal cortex in PD and six out of ten in DLB. These include DNAJA1, DNAJ4 and DNAJB1, the products of which act as heat shock protein 70 cochaperones, and chaperones HSPA4, HSPA6 and HSPD1.
Moreover, STIP1, a copartner in the HSP70/HSP90 activity in protein folding, is the top hub gene in this module and PPI together with HSPA1A and HSPA5. DNAJA1 and DNAJB1 are also among the top hub genes in this module. Importantly, the expression of this gene is increased in PD and DLB but significantly upregulated only in PD. Refolding and clearance of α‐synuclein aggregates requires chaperones and the proteasome system 55. Our findings are consistent with the presence of abnormally aggregated α‐synuclein in the cortex in DLB and also with previous observations of increased folding proteins and unfolded protein response in DLB 3, 16.
We found a large gene coexpression module downregulated in DLB that is enriched in neuronal markers and in genes mainly involved in synaptic transmission. Synapses are altered in the cortex in PD and DLB 19, 20, 74, 85, 86. α‐synuclein is abundant in the pre‐synaptic terminals, where it plays a role in synaptic function. Abnormal folding and oligomerization and formation of certain types of aggregates may directly produce synaptic damage, and may do so indirectly by exerting toxicity on mitochondria, lysosomes and cytoskeleton 56, 74, 85. Notably, a top degree gene of the PPI subnetwork from this module is α‐synuclein. In spite of being a PPI hub, SNCA is not a DEG between DLB and CTL as assessed by Limma, which highlights the importance of the present higher‐complexity level study in revealing genes otherwise veiled in conventional differential expression methods.
Gene downregulation related to neurotransmission in the frontal cortex in DLB particularly involves GAD1, the product of which catalyzes the synthesis of γ‐ aminobutyric acid from l‐glutamic acid, and GABA receptors GABBR2 and GABRA1, showing that the GABAergic system in frontal cortex is vulnerable to DLB. Other downregulated genes are NETO1, which encodes a protein involved in synaptic N‐methyl‐d‐aspartic acid receptor complexes, and SYP, which encodes SYP, a major synaptic protein. In line with the present findings, loss of post‐synaptic GABAreceptor markers has been reported in the occipital cortex in DLB 52. SYP expression was previously reported as decreased in the occipital cortex in DLB as well 67. The identification of deregulated M5 top hub gene, ATP6V1A, the product of which mediates the acidification of several intracellular organelles including synaptic vesicles, thereby facilitating synaptic transmission 12, further points to altered synaptic function.
Other altered pathways identified in the M5‐brown module, namely energy metabolism and mitochondrial function, and purine metabolism, have been the subject of previous analysis. Mitochondria and energy metabolism is altered in the frontal cortex in PD and DLB 37, 38, 39, 51, 68, 69, 76, 92. Moreover, decreased enzymatic activity of complexes I–IV has been demonstrated in a subset of PD cases with dementia 38, 69 and in cases with DLB 37, 68. Abnormal α‐synuclein is localized in mitochondria in the frontal cortex in PD 38 and in the nucleus 36; thus, a possible link between altered nuclear and mitochondrial DNA/RNA processing and aberrant α‐synuclein may be considered. The present study also shows two additional PPIs in module 5. Cytochrome c, somatic (CYCS) encodes a small heme protein localized in the inner membrane of the mitochondria which transports electrons from cytochrome b to the cytochrome oxidase complex 21. Second, TPI1 encodes a protein which participates in glycolysis and glucogenesis 93.
Regarding purine metabolism, our previous RT‐qPCR studies revealed abnormal expression of several enzymes involved in purine metabolism in PD and DLB 35, 38. No attempt was made to validate other important altered pathways such as those inked to the ubiquitin proteasome system (UPS) in LBDs revealed by microarrays, and interpretation of significantly altered regulation of certain top hub genes in M5 such as GSK3B and YWHAB, and M6 as LAPTM5 remains speculative.
Concluding Comments
The study of human cases is irreplaceable in the effort to gain understanding of the neurodegenerative diseases that are exclusive to human beings. It can be criticized the limited number of available cases suitable for molecular studies in the present series. This limitation may account for certain discrepancies between the observations obtained with arrays and the subsequent validations with RT‐qPCR, and also by the observation of isolated deregulated genes which are apparently disconnected from highly represented deregulated clusters. In spite of these constraints, the present study, using selected samples with no comorbidity and adequate RNA preservation, has documented an extraordinary amount of information. Validation of the present findings should be expected in other independent series.
We used a “network medicine” approach to the study of LBDs aiming at unveiling changes in the transcriptome in frontal cortex area 8 which may affect particular metabolic pathways and biological functions. This approach is being increasingly used to discover relevant pathogenic effectors in neurodegenerative diseases 83. Here, we show early alteration in the regulation of the axonema and particularly of dyneins, and of taste receptors in iLBD, maintained and augmented along with the progression of the LBD spectrum. Interestingly, innate inflammation is downregulated in iLBD as manifested by decreased AIF1 expression. It is worth mentioning that the expression of several cytokines and mediators of the inflammatory response is also reduced in iLBD (unpublished observations); the small number of cases assessed in this series precludes further evaluations, but our findings provide insights that will allow deepening to into the study of reduced innate inflammatory regulation at early stages of LBDs. We have also identified and validated deregulation of several pathways in the frontal cortex in DLB; this includes upregulation of RNA/DNA processing and chaperones, and downregulation of neurotransmission thereby revealing the vulnerability of the GABAergic system. Altered mitochondria and energy metabolism, protein synthesis, and altered purine metabolism regulation were the subject of previous studies. Our approach has also identified robust deregulated modules represented by several genes, hub genes and PPI subnetworks within these modules. These observations reveal potential candidates for further analysis as they may involve key pathogenic molecular events taking place in the frontal cortex in LBDs.
Author's Contribution
GS performed WGCNA, PG‐E carried out RT‐qPCR, PA‐B the analysis of arrays, BL‐G performed the gene enrichment score analysis, AN refined the design, IF designed the study, selected the cases, supervised the work and wrote the manuscript which was circulated among all the authors to add comments and suggestions. All the authors agreed with the contents of the manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Supplementary Table 1. TaqMan probes used in the study, gene abbreviation, full name of the genes and references.
Supplementary Table 2. Number of DEGs for different thresholds for each comparison of iLBD, PD, DLB and middle aged individuals.
Supplementary Table 3. Gene ontology analysis comparing upregulated and downregulated genes in the different categories.
Supplementary Table 4. Fifty top upregulated and top downregulated genes, frontal cortex area 8 in DLB.
Supplementary Table 5. Top 20 hub genes per modules.
Supplementary Table 6. Enrichment of modules in neuronal and glial cell markers, and commonalities with neurological diseases such as Alzheimer's disease and autism.
Supplementary Table 7. Identification of enriched GO among genes in each module.
Acknowledgments
This study was funded by the Ministerio de Econom.a y Competitividad, Instituto de Salud Carlos III – Fondos FEDER, a Way to Build Europe FIS (grant PI14/00757 to IF and grants BFU‐2012‐38236 and BFU‐2015‐68649‐P to AN), by the Spanish National Institute of Bioinformatics of the Instituto de Salud (PT13/0001/0026) and by FEDER (Fondo Europeo de Desarrollo Regional)/FSE (Fondo Social Europeo), and coordinated Intraciber 2014: Rapid Dementias. We wish to thank Gerard Muntané. for technical assistance and valuable discussion and T. Yohannan for editorial help.
Compliance with ethical standards No relevant data; no conflicts of interest.
References
- 1. Alafuzoff I, Ince PG, Arzberger T, Al‐Sarraj S, Bell J, Bodi I et al (2009) Staging/typing of Lewy body related alphasynuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol 117:635–652. [DOI] [PubMed] [Google Scholar]
- 2. Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ et al (2015) Disease‐related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baek JH, Whitfield D, Howlett D, Francis P, Bereczki E, Ballard C et al (2016) Unfolded protein response is activated in Lewy body dementias. Neuropathol Appl Neurobiol 42:352–365. [DOI] [PubMed] [Google Scholar]
- 4. Barrachina M, Castaño E, Ferrer I (2006) TaqMan PCR assay in the control of RNA normalization in human post‐mortem brain tissue. Neurochem Int 49:276–284. [DOI] [PubMed] [Google Scholar]
- 5. Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ (2010) Gene enrichment profiles reveal T‐cell development, differentiation, and lineage‐specific transcription factors including ZBTB25 as a novel NF‐AT repressor. Blood 115:5376–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW (2004) Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA 101:2173–21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Botta‐Orfila T, Sànchez‐Pla A, Fernández M, Carmona F, Ezquerra M, Tolosa E (2012) Brain transcriptomic profiling in idiopathic and LRRK2‐associated Parkinson'sdisease. Brain Res 1466:152–157. [DOI] [PubMed] [Google Scholar]
- 8. Boyle PA, Yu L, Wilson RS, Gamble K, Buchman AS, Bennett DA (2012) Poor decision making is a consequence of cognitive decline among older persons without Alzheimer's disease or mild cognitive impairment. PLoS One 7:e43647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braak H, Braak E (1991) Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 10. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 11. Braak H, Rüb U, Del Tredici K (2006) Cognitive decline correlates with neuropathological stage in Parkinson's disease. J Neurol Sci 248:255–258. [DOI] [PubMed] [Google Scholar]
- 12. Breton S, Brown D (2013) Regulation of luminal acidification by the V‐ATPase. Physiology 28:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buddhala C, Loftin SK, Kuley BM, Cairns NJ, Campbell MC, Perlmutter JS, Kotzbauer PT (2015) Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann Clin Transl Neurol 2:949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burke RE, Dauer WT, Vonsattel JP (2008) A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 64:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cantuti‐Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP et al (2005) Alpha‐synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol 64:1058–1066. [DOI] [PubMed] [Google Scholar]
- 17. Colom‐Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Martí MJ et al (2013) Confluence of α‐synuclein, tau, and β‐amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 72:1203–1212. [DOI] [PubMed] [Google Scholar]
- 18. Cui S, Sun H, Gu X, Lv E, Zhang Y, Dong P, Fu C et al (2015) Gene expression profiling analysis of locus coeruleus in idiopathic Parkinson's disease by bioinformatics. Neurol Sci 36:97–102. [DOI] [PubMed] [Google Scholar]
- 19. Dalfó E, Albasanz JL, Martin M, Ferrer I (2004) Abnormal metabotropic glutamate receptor expression and signaling in the cerebral cortex in diffuse Lewy body disease is associated with irregular alpha‐synuclein/phospholipase C (PLCbeta1) interactions. Brain Pathol 14:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dalfó E, Barrachina M, Rosa JL, Ambrosio S, Ferrer I (2004) Abnormal alphasynuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol Dis 16:92–97. [DOI] [PubMed] [Google Scholar]
- 21. Delgado JY, Owens GC (2012) The cytochrome c gene proximal enhancer drives activity‐dependent reporter gene expression in hippocampal neurons. Front Mol Neurosci 5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dickson DW, Fujishiro H, DelleDonne A, Menke J, Ahmed Z, Klos KJ et al (2008) Evidence that incidental Lewy body disease is pre‐symptomatic Parkinson's disease. Acta Neuropathol 115:437–444. [DOI] [PubMed] [Google Scholar]
- 23. Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y (2010) Evidence in favor of Braak staging of Parkinson's disease. Mov Disord 25: S78–S82. [DOI] [PubMed] [Google Scholar]
- 24. Diez D, Hutchins AP, Miranda‐Saavedra D (2014) Systematic identification of transcriptional regulatory modules from protein‐protein interaction networks. Nucleic Acids Res 42:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durrenberger PF, Fernando FS, Magliozzi R, Kashefi SN, Bonnert TP, Ferrer I et al (2012) Selection of novel reference genes for use in the human central nervous system: a BrainNet Europe Study. Acta Neuropathol 124:893–903. [DOI] [PubMed] [Google Scholar]
- 26. Fabelo N, Martín V, Santpere G, Marín R, Torrent L, Ferrer I, Diaz M (2011) Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson's disease and incidental Parkinson's disease. Mol Med 17:1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Falcon S, Gentleman R (2007) Using GOstats to test gene lists for GO term association. Bioinformatics 23:257–258. [DOI] [PubMed] [Google Scholar]
- 28. Ferrer I (2009) Early involvement of the cerebral cortex in Parkinson's disease: convergence of multiple metabolic defects. Prog Neurobiol 88:89–103. [DOI] [PubMed] [Google Scholar]
- 29. Ferrer I, Martinez A, Boluda S, Parchi P, Barrachina M (2008) Brain banks: benefits, limitations and cautions concerning the use of post‐mortem brain tissue for molecular studies. Cell Tissue Bank 9:181–194. [DOI] [PubMed] [Google Scholar]
- 30. Ferrer I, Garcia‐Esparcia P, Carmona M, Carro E, Aronica E, Kovacs G et al (2016) Olfactory receptors in non‐chemosensory organs: the nervous system in health and disease. Front Aging Neurosci 8:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferrer I, López‐Gonzalez I, Carmona M, Dalfó E, Pujol A, Martínez A (2012) Neurochemistry and the non‐motor aspects of PD. Neurobiol Dis 46:508–526. [DOI] [PubMed] [Google Scholar]
- 32. Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff‐Radford NR, Uitti RJ et al (2008) Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol 67:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garcia‐Esparcia P, Schlüter A, Carmona M, Moreno J, Ansoleaga B, Torrejón‐Escribano B et al (2013) Functional genomics reveals dysregulation of cortical olfactory receptors in Parkinson disease: novel putative chemoreceptors in the human brain. J Neuropathol Exp Neurol 72:524–539. [DOI] [PubMed] [Google Scholar]
- 34. Garcia‐Esparcia P, Llorens F, Carmona M, Ferrer I (2014) Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson's disease brain is region dependent. Brain Pathol 24:584–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garcia‐Esparcia P, Hernández‐Ortega K, Ansoleaga B, Carmona M, Ferrer I (2015) Purine metabolism gene deregulation in Parkinson's disease. Neuropathol Appl Neurobiol 41:926–940. [DOI] [PubMed] [Google Scholar]
- 36. Garcia‐Esparcia P, Hernández‐Ortega K, Koneti A, Gil L, Delgado‐Morales R, Castaño E et al (2015) Altered machinery of protein synthesis is region‐ and stage‐dependent and is associated with α‐synuclein oligomers in Parkinson's disease. Acta Neuropathol Commun 3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garcia‐Esparcia P, Koneti A, Rodríguez‐Oroz MC, Lago B, del Rio JA, Ferrer I (2017) Mitochondrial activity in the frontal cortex area 8 and angular gyrus in Parkinson's disease and Parkinson's disease with dementia. Brain Pathol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garcia‐Esparcia P, López Gonzalez I, Grau‐Rivera O, García‐Garrido MF, Konetti A, Llorens F, et al (2017) Dementia with Lewy bodies: age‐related molecular pathology in the frontal cortex in typical and rapidly progressive forms. Front Neurol (in press). [DOI] [PMC free article] [PubMed]
- 39. Gatt AP, Duncan OF, Attems J, Francis PT, Ballard CG, Bateman JM (2016) Dementia in Parkinson's disease is associated with enhanced mitochondrial complex I deficiency. Mov Disord 31:352–359. [DOI] [PubMed] [Google Scholar]
- 40. Glaab E, Schneider R (2015) Comparative pathway and network analysis of brain transcriptome changes during adult aging and in Parkinson's disease. Neurobiol Dis 74:1–13. [DOI] [PubMed] [Google Scholar]
- 41. Goedert M (2001) Alpha‐synuclein and neurodegenerative diseases. Nat RevNeurosci 2:492–501. [DOI] [PubMed] [Google Scholar]
- 42. Grison A, Zucchelli S, Urzì A, Zamparo I, Lazarevic D, Pascarella G et al (2014) Mesencephalic dopaminergic neurons express a repertoire of olfactory receptors and respond to odorant‐like molecules. BMC Genomics 15:729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grothe MJ, Schuster C, Bauer F, Heinsen H, Prudlo J, Teipel SJ (2014) Atrophy of the cholinergic basal forebrain in dementia with Lewy bodies and Alzheimer's disease dementia. J Neurol 261:1939–1948. [DOI] [PubMed] [Google Scholar]
- 44. Hawrylycz MJ, Lein ES, Guillozet‐Bongaarts AL, Shen EH, Ng L, Miller JA et al (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang SH, Chang CC, Lui CC, Chen NC, Lee CC, Wang PW, Jiang CF (2015) Cortical metabolic and nigrostriatal abnormalities associated with clinical stage‐specific dementia with Lewy bodies. Clin Nucl Med 40:26–31. [DOI] [PubMed] [Google Scholar]
- 46. Ince PG (2011) Dementia with Lewy bodies and Parkinson's disease with de6entia. In: Neurodegeneration, the Molecular Pathology of Dementia and Movement Disorders. Dickson DW, Weller RO (eds), pp. 224–237. Wiley‐Blackwell: Oxford: [Google Scholar]
- 47. Jellinger KA (2007) Morphological substrates of parkinsonism with and without dementia: a retrospective clinico‐pathological study. J Neural Transm 72:91–104. [DOI] [PubMed] [Google Scholar]
- 48. Jellinger KA (2009) Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci 24:114–125. [DOI] [PubMed] [Google Scholar]
- 49. Jellinger KA (2011) Parkinson's disease. In: Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Dickson DW, Weller RO (eds), pp. 194–233. Wiley‐Blackwell: Oxford. [Google Scholar]
- 50. Jellinger KA, Attems J (2008) Prevalence and impact of vascular and Alzheimer athologies in Lewy body disease. Acta Neuropathol 115:427–436. [DOI] [PubMed] [Google Scholar]
- 51. Keeney PM, Xie J, Capaldi RA, Bennett JP (2006) Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26:5256–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khundakar AA, Hanson PS, Erskine D, Lax NZ, Roscamp J, Karyka E et al (2016) Analysis of primary visual cortex in dementia with Lewy bodies indicates GABAergic involvement associated with recurrent complex visual hallucinations. Acta Neuropathol Commun 4:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim HJ, Lee JE, Shin SJ, Sohn YH, Lee PH (2011) Analysis of the substantia innominata volume in patients with Parkinson's disease with dementia, dementia with Lewy bodies, and Alzheimer's disease. J Mov Disord 4:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klein JC, Eggers C, Kalbe E, Weisenbach S, Hohmann C, Vollmar S et al (2010) Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease in vivo. Neurology 74:885–892. [DOI] [PubMed] [Google Scholar]
- 55. Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ (2004) Hsp70 Reduces α‐synuclein aggregation and toxicity. J Biol Chem 279:25497–25502. [DOI] [PubMed] [Google Scholar]
- 56. Kramer ML, Schulz‐Schaeffer WJ (2007) Presynaptic alpha‐synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A et al (2006) Genome‐wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. [DOI] [PubMed] [Google Scholar]
- 59. Lewis PA, Cookson MR (2012) Gene expression in the Parkinson's disease brain. Brain Res Bull 88:302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K et al (2008) Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics 33:240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lim S, Chun Y, Lee JS, Lee SJ (2016) Neuroinflammation in synucleinopathies. Brain Pathol 26:404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lim SM, Katsifis A, Villemagne VL, Best R, Jones G, Saling M et al (2009) The 18F‐FDG PET cingulate island sign and comparison to 123I‐beta‐CIT SPECT for diagnosis of dementia with Lewy bodies. J Nucl Med 50:1638–1645. [DOI] [PubMed] [Google Scholar]
- 63. Marin R, Fabelo N, Martín V, Garcia‐Esparcia P, Ferrer I, Quinto‐Alemany D, Diaz M (2016) Anomalies occurring in lipid profiles and protein distribution in frontal cortex lipid rafts in dementia with Lewy bodies disclose neurochemical traits partially shared by Alzheimer's and Parkinson's diseases. Neurobiol Aging 49:52–59. [DOI] [PubMed] [Google Scholar]
- 64. McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen‐Mansfield J et al (2004) Dementia with Lewy bodies. Lancet Neurol 3:19–28. [DOI] [PubMed] [Google Scholar]
- 65. Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, Duda JE (2012) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79:2307–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Miller J, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH (2013) Genes and pathways underlying regional and cell type changes in Alzheimer's disease. Genome Med 5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mukaetova‐Ladinska EB, Andras A, Milne J, Abdel‐All Z, Borr I, Jaros E et al (2013) Synaptic proteins and choline acetyltransferase loss in visual cortex in dementia with Lewy bodies. J Neuropathol Exp Neurol 72:53–60. [DOI] [PubMed] [Google Scholar]
- 68. Navarro A, Boveris A (2009) Brain mitochondrial dysfunction and oxidative damage in Parkinson's disease. J Bioenerg Biomembr 41:517–521. [DOI] [PubMed] [Google Scholar]
- 69. Navarro A, Boveris A, Bández MJ, Sánchez‐Pino MJ, Gómez C, Muntané G, Ferrer I (2009) Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic Biol Med 46:1574–1580. [DOI] [PubMed] [Google Scholar]
- 70. Nelson PT, Kryscio RJ, Jicha GA, Abner EL, Schmitt FA, Xu LO et al (2009) Relative preservation of MMSE scores in autopsy‐proven dementia with Lewy bodies. Neurology 73:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD et al (2010) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Odunuga OO, Longshaw VM, Blatch GL (2004) Hop: more than an Hsp70/Hsp90 adaptor protein. BioEssays 26:1058–1068. [DOI] [PubMed] [Google Scholar]
- 73. Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH (2009) Functional organization of the transcriptome in human brain. Nat Neurosci 11:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Overk CR, Masliah E (2014) Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem Pharmacol 88:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Parachikova A, Agadjanyan MG, Cribbs DH, Blurton‐Jones M, Perreau V, Rogers J et al (2007) Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging 28:1821–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Parker WD, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson's disease frontal cortex. Brain Res 1189:215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Parkkinen L, Kauppinen T, Pirttilä T, Autere JM, Alafuzoff I (2005) αsynuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 57:82–91. [DOI] [PubMed] [Google Scholar]
- 78. Parkkinen L, Pirttilä T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha‐synuclein pathology and their clinical relevance. Acta Neuropathol 115:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Price DL, Rockenstein E, Ubhi K, Phung V, MacLean‐Lewis N, Askay D et al (2010) Alterations in mGluR5 expression and signaling in Lewy body disease and in transgenic models of alpha‐synucleinopathy–implications for excitotoxicity. PLoS One 5:e14020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80. Ravid R, Ferrer I (2012) Brain banks as key part of biochemical and molecular studies on cerebral cortex involvement in Parkinson's disease. FEBS J 279:1167–1176. [DOI] [PubMed] [Google Scholar]
- 81. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA (2013) Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol 14:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Santiago JA, Potashkin JA (2014) A network approach to clinical intervention in neurodegenerative diseases. Trends Mol Med 20:694–703. [DOI] [PubMed] [Google Scholar]
- 84. Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, Bennette DA (2012) Cognitive impairment, decline and fluctuations in older community‐dwelling subjects with Lewy bodies. Brain 135:3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schulz‐Schaeffer WJ (2010) The synaptic pathology of α‐synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol 120:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S (2010) A pathologic cascade leading to synaptic dysfunction in alpha‐synuclein‐induced neurodegeneration. J Neurosci 30:8083–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2005) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Am Assoc Cancer Res Educ B 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shepherd CE, Thiel E, McCann H, Harding AJ, Halliday GM (2000) Cortical inflammation in Alzheimer disease but not dementia with Lewy bodies. Arch Neurol 57:817–822. [DOI] [PubMed] [Google Scholar]
- 89. Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N et al (2009) Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73:273–278. [DOI] [PubMed] [Google Scholar]
- 90. Streit WJ, Xue QS (2016) Microglia in dementia with Lewy bodies. Brain Behav Immun 55:191–201. [DOI] [PubMed] [Google Scholar]
- 91. Thal DR, Rü b. U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 92. Thomas RR, Keeney PM, Bennett JP (2012) Impaired complex‐I mitochondrial biogenesis in Parkinson disease frontal cortex. J Parkinsons Dis 2:67–76. [DOI] [PubMed] [Google Scholar]
- 93. Trentham DR, McMurray CH, Pogson CI (1969) The active chemical state of D‐glyceraldehyde 3‐phosphate in its reactions with D‐glyceraldehyde 3‐phosphate dehydrogenase, aldolase and triose phosphate isomerase. Biochem J 114:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Walker DG, Lue LF, Serrano G, Adler CH, Caviness JN, Sue LI, Beach TG (2016) Altered expression patterns of inflammation‐associated and trophic molecules in substantia nigra and striatum brain samples from Parkinson's disease, incidental Lewy body disease and normal control cases. Front Neurosci 9:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Supplementary Table 1. TaqMan probes used in the study, gene abbreviation, full name of the genes and references.
Supplementary Table 2. Number of DEGs for different thresholds for each comparison of iLBD, PD, DLB and middle aged individuals.
Supplementary Table 3. Gene ontology analysis comparing upregulated and downregulated genes in the different categories.
Supplementary Table 4. Fifty top upregulated and top downregulated genes, frontal cortex area 8 in DLB.
Supplementary Table 5. Top 20 hub genes per modules.
Supplementary Table 6. Enrichment of modules in neuronal and glial cell markers, and commonalities with neurological diseases such as Alzheimer's disease and autism.
Supplementary Table 7. Identification of enriched GO among genes in each module.