

Abstract
Amyloidomas are rare amyloid‐containing lesions, which may also occur in the central nervous system. Etiology, pathogenesis and clinical course are poorly understood. To gain more insight into the biology of cerebral amyloidoma, they aimed to characterize its histopathological, molecular and clinical features in a retrospective series of seven patients. FFPE tissue specimens were examined using immunohistochemistry, chromogenic in situ hybridization (CISH) for light chains kappa and lambda as well as an IgH gene clonality analysis. Follow‐up information was gathered by reviewing patient records and imaging results. Median age of the three males and four females was 50 years (range: 35–53 years). All cerebral amyloidomas were located supratentorially and were classified as lambda light chain amyloidosis (AL‐λ; n = 6) and kappa light chain amyloidosis (AL‐κ; n = 1) on immunohistochemistry and CISH. B‐cell clonality was confirmed by IgH gene clonality assay in all cases examined. After a median follow‐up of 21 months, all patients were alive and showed stable disease. No progression to systemic disease was observed. In conclusion, their data suggest that cerebral amyloidoma is a local disease characterized by B‐cell clonality and associated with a stable clinical course.
Keywords: amyloidoma, B‐cell, central nervous system, light chain amyloidosis, neoplasm
Introduction
Amyloids are insoluble fibrillar proteinaceous aggregates accumulating in intra‐ and extracellular compartments under pathological conditions 8, 22. The term “amyloid” has been coined by Virchow based on positive iodine staining of tissue abnormalities 19. A large number of proteins including the immunoglobulin light chains kappa and lambda may form amyloid locally or systemically 12, 22. Amyloidomas are rare tumor‐forming deposits of amyloid, which may occur in various locations, including respiratory and gastrointestinal tract 4, 20, bone 14, soft tissues 7, 15 and the central nervous system 9.
At least 30 cases of cerebral amyloidoma have been reported 1, 2, 9, 18. However, most of them represent case reports focusing on neuroradiological aspects 3, 21 and relatively little is known on the etiology and pathogenesis 17. As summarized by Fisher et al 1, the majority of cerebral amyloidomas appears to originate from immunoglobulin‐derived kappa and lambda light chains, probably produced by clonal plasma cells 1, 9, 18. Immunohistochemical studies on light chain restriction, however, may yield inconsistent results 2 and clonality of the immunoglobulin heavy chain (IgH) has been demonstrated only in few cases 9. Furthermore, the clinical course of cerebral amyloidomas has not been systematically addressed: the largest series of cerebral amyloidomas reported to date provided only short follow‐up information of two patients 9. To gain more insight into the biology of cerebral amyloidoma, molecular and clinical features were analyzed in a series of seven patients. Here we show that cerebral amyloidoma is characterized by B‐cell clonality and a stable clinical course.
Materials and Methods
Patients
Formalin‐fixed and paraffin‐embedded (FFPE) tissue samples from seven patients treated at the authors' institutions over a period of 15 years (2001–2016) were evaluated retrospectively. The histopathology of one case (patient 2) had been previously reported 1. Clinical information was retrieved from medical records and included sex, age at time of surgery, date of biopsy and postoperative treatment. Tumor localization and size were determined by magnetic resonance imaging (MRI) and/or computed tomography (CT). Follow‐up information was gathered by reviewing patient records and imaging results. Progression free survival (PFS) was defined as the time from first diagnosis until amyloidoma clinical and/or radiological progression and/or death from any cause. The study was done in accordance with the ethical principles of the involved hospitals; the tumor bank of the Institute of Neuropathology Münster received approval from the local ethical committee.
Histopathology
Representative paraffin sections were stained with hematoxylin and eosin (H&E), Elastica‐van Gieson (EvG) and Congo red. The presence of amyloid was examined by polarization microscopy on Congo red‐stained sections. Immunohistochemistry was carried out using the avidin‐biotin peroxidase technique. Antibodies against transthyretin (dilution 1:4,000, Acris GmbH, Herford, Germany), light chains kappa (1:30,000 Dako, Glostrup, Denmark) and lambda (1:60,000 Dako) were applied to characterize the subtype of amyloid. Further, lymphocytic cells were characterized using antibodies against CD3 (1:25, Dako), CD5 (1:100, Dako), CD10 (1:20, NovoCastra, Wetzlar, Germany), CD20 (1:700, Dako), CD138 (1:500, Dako), cyclin D1 (1:10, Thermo Scientific, Darmstadt, Germany) and IgG4 (1:1,000, Abcam, Cambridge, United Kingdom). Proliferation was assessed by labeling for Ki67/MiB1 (1:100, Dako). Secondary antibodies were a mixture of anti‐rabbit and anti‐goat mouse sera. Diaminobenzidine (Leica Biosystems Nussloch, Germany) served as chromogen.
Chromogenic in situ hybridization (CISH)
mRNA expression of Ig‐kappa and Ig‐lambda was performed using the ZytoFasthuman Ig‐kappa/Ig‐lambda CISH Kit (ZytoVision, Bremerhaven, Germany). The probe contains digoxigenin‐labeled oligonucleotides targeting Ig‐kappa mRNA and biotin‐labeled oligonucleotides targeting Ig‐lambda mRNA. FFPE sections were incubated at 70°C and pretreated with pepsin. Slides were denaturized with ZytoFast human Ig‐kappa/Ig‐lambda DNA probes at 75°C for 5 minutes and at 55°C for 2 hours. After washing with Tris‐buffered saline (TBS 20×, Zytomed System GmbH, Berlin, Germany), 3‐amino‐9‐ethylcarbazol (AEC, ZytoChem Plus Double Stain Polymer Kit) and alkaline phosphatase (AP), conjugated with streptavidin (ZytoFast AP‐Streptavidin Detection Kit, ZytoVision GmbH, Bremerhaven, Germany) were applied and incubated subsequently for 20 minutes at 37°C in a humidity chamber. Diaminobenzidine (Leica Biosystems) served as chromogen.
IgH gene clonality assay
DNA was extracted from FFPE tissue (Maxwell® 16 FFPE Plus LEV DNA Purification Kit, Mannheim, Germany) and subjected to IgH gene clonality analysis using the IndentiClone IgH Gene Clonality Assay and protocols provided by the manufacturer (Invivoscribe Technologies Inc. San Diego, CA). Clonality was analyzed by running samples on capillary electrophoresis (Applied Biosystems, Waltham, MA). Evaluation was performed using the Peak Scanner™ Software v1.0 (ThermoFisher Scientific, Waltham, MA).
Results
Median age of the three male and four female patients was 50 years (range: 35–53 years; see Table 1). The most common presenting symptom was seizures (n = 4), but headache, visual deficits, vertigo, hemiparesis and hemiparesthesia were also reported. On neuroimaging, all patients showed supratentorial cerebral lesions. Maximum diameter accounted for 46 ± 23 mm (mean± standard deviation, Figure 1). In one patient (patient #4) additional smaller lesions were encountered. The majority of patients underwent biopsy (n = 3) or partial resection (n = 3) while gross total resection of the tumor was only achieved in one patient.
Table 1.
Patient characteristics.Characteristics of seven patients harboring cerebral amyloidoma. Progression free survival (PFS) was defined as the time from first diagnosis until clinical and/or radiological amyloidoma progression and/or death from any cause (IHC, immunohistochemistry; CISH, chromogenic in situ‐hybridization; IgH, IgH Gene Clonality Assay; AL‐λ, lambda light chain amyloidosis; AL κ, kappa light chain amyloidosis; CT, computer tomography; MRI, magnetic resonance imaging; *pseudo‐clonality could not be excluded, probably because of low cellularity of the sample).
| Case | Sex | Age | Presenting symptoms | Location | Neuroimaging | Surgery | Postoperative Treatment | PFS (months) | Medical condition at last follow‐up | IHC | CISH | IgH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 50 years | Confusion, headache, vomiting and visual deficits of the lower right quadrant, dyslexia | Left occipital | MRI: 75 x 50 mm lesion with minor contrast enhancement | Biopsy | Steroids | 17 | Stable. Minor residual visual impairment. | Inconclusive | AL‐κ | Clonality |
| 2 | Male | 35 years | Intermittent headache, right leg paresis | Left pre‐central | CT and MRI: subcortical 20 x 15 mm lesion with contrast enhancement and minor perifocal edema | Biopsy | Anticonvulsants | 180 | Stable. Epileptiform activity on EEG. | Inconclusive | AL‐λ | Not evaluable |
| 3 | Female | 49 years | Seizures | Right frontal | MRI: 58 x 49 x 35 mm lesion with inhomogeneous contrast enhancement | Gross total resection | Anticonvulsants | 11 | Stable. Epileptiform activity on EEG. | Inconclusive | AL‐λ | Clonality |
| 4 | Female | 53 years | Seizures, left homonymous hemianopsia, hemiparesis and numbness of left hand and foot | Right parieto‐occipital | PET‐MRI: 66 x 38 x 31 mm right parieto‐occipital lesion with inhomogeneous contrast enhancement. Multiple additional smaller lesions. | Partial resection | Steroids | 24 | Stable. Minor residual left‐sided hemiparesis, dysphagia and visual impairment. | Inconclusive | AL‐λ | Clonality |
| 5 | Female | 52 years | Seizures | Right temporal | CT and MRI: 35 x 22 x 25 mm lesion with contrast enhancement. Focal calcifications and minor perifocal edema | Partial resection | Steroids, anticonvulsants | 71 | Stable. No further seizures. | AL‐λ | AL‐λ | Clonality* |
| 6 | Female | 49 years | Depression, frontal lobe disorder, seizures, numbness of the right half of the body | Left frontal | CT and MRI: 50 x 50 x 35 mm lesion with inhomogeneous contrast enhancement, midline shift, perifocal edema | Biopsy | Steroids, radiation (20Gy) | 21 | Stable. Minor residual numbness and spasticity. | AL‐λ | AL‐λ | Clonality |
| 7 | Male | 52 years | Vertigo | Right thalamus | MRI: 15 x 13 x 12 mm thalamic lesion with midline shift | Partial resection | None | 4 | Stable. No symptoms. | Inconclusive | AL‐λ | Clonality |
Figure 1.
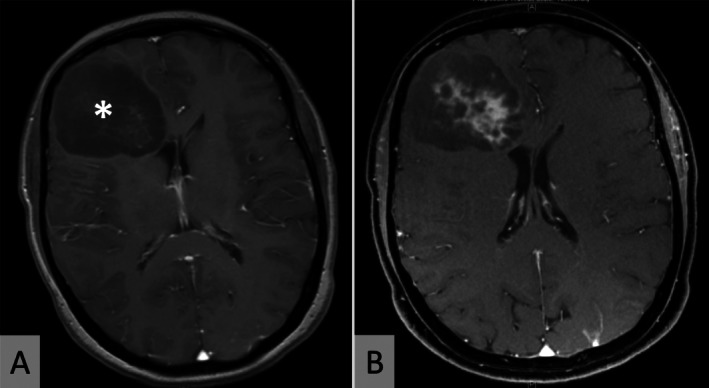
Neuroimaging. Representative axial MRI scans (patient 3) showing a hypointense right frontal expansive mass (*) on T1‐weighted sequences (A). The lesion shows inhomogeneous contrast enhancement (B).
Histological examination of all specimens showed accumulation of homogeneous eosinophilic masses upon H&E staining (Figure 2A) with yellow‐green birefringence under polarized light in Congo red‐stained sections (Figure 2B). Amyloid deposits were surrounded by infiltrates comprised of lymphocytes, macrophages and plasma cells. Most of the infiltrates contained CD138‐positive plasma cells, CD20‐positive B‐cells and CD3‐positive T‐cells. There were only few scattered CD5 or CD10 positive cells and staining for cyclinD1, IgG4 and transthyretin was negative. The Ki67/MIB1 proliferation index was relatively low and accounted for 4.0% ± 3.3% (mean ± standard deviation; Figure 2C). On immunohistochemistry, light chain restriction could only be demonstrated in two cases (AL‐λ), thus ancillary molecular methods were applied. On CISH examination (Figure 3A, B), light chain clonality at the mRNA level could be demonstrated in all cases, six amyloidomas showing lambda and one amyloidoma showing kappa mRNA, respectively. Furthermore, using an IgH gene clonality assay, monoclonal rearrangement of the IgH gene was confirmed in five amyloidomas, from which DNA of sufficient quality could be extracted (Figure 3C). In one additional case, pseudo‐clonality could not be excluded, probably because of the low cellularity of the sample. Of note, this patient had received steroids preoperatively.
Figure 2.
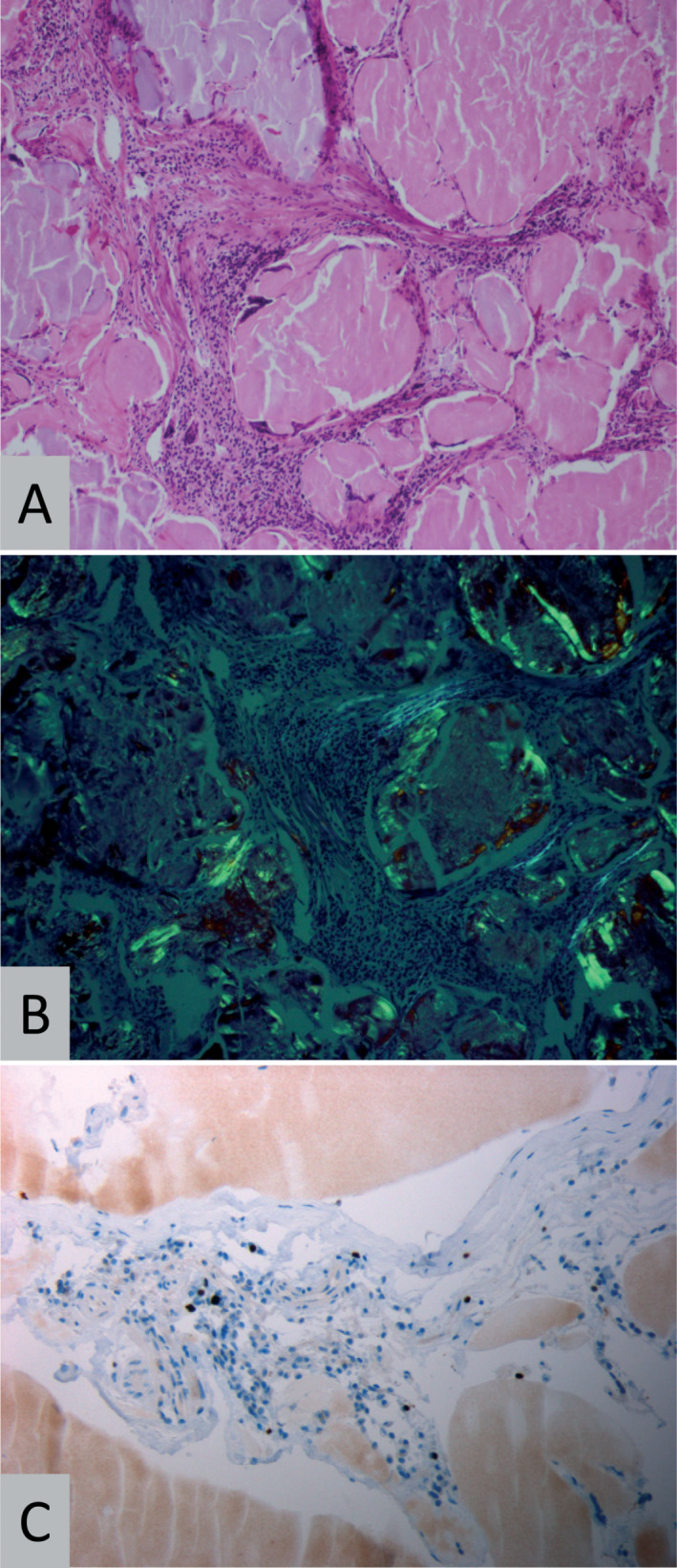
Histopathology. Representative histopathology (patient 2) showing amyloid deposits surrounded by lymphocytes on H&E staining (A, original magnification 200×) and birefringence under polarized light (B) when stained with Congo red (original magnification 200×). On immunohistochemistry for MiB1/Ki67 (C), few nuclei of lymphoid cells stain positive (original magnification 400×).
Figure 3.
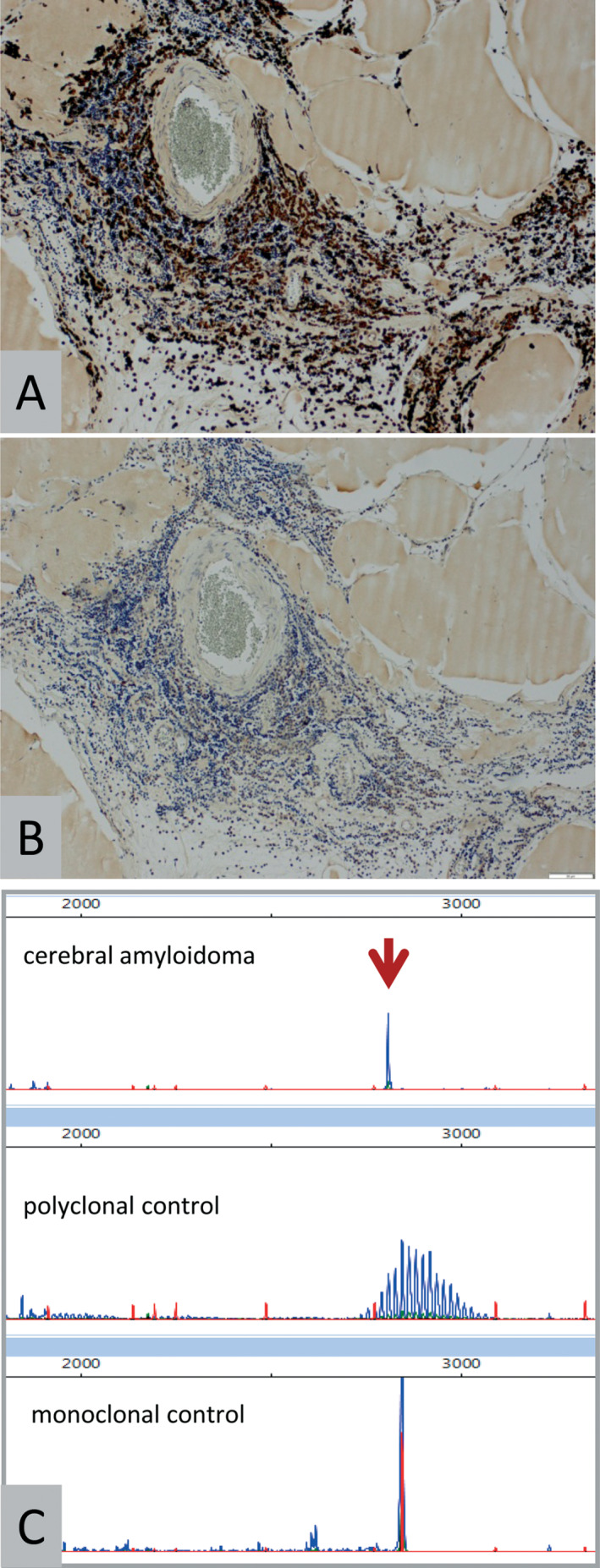
Molecular pathology. Representative chromogenic in situ hybridization (CISH) results for lambda light chain mRNA (A) and kappa light chain mRNA (B) in patient 3. Note positivity for lambda mRNA in lymphoid cells while kappa mRNA is absent (original magnification 200×). C: Representative IgH gene clonality assay results showing a monoclonal peak in the amyloidoma of patient 4 (arrow). Controls included DNA from tonsils (polyclonal control) as well as B‐cell lymphoma (monoclonal control).
Detailed follow‐up information was obtained for all patients. In the absence of consensus guidelines, treatment of the seven patients of this retrospective series was variable. Four patients were treated with steroids for a limited time period, three with anticonvulsants and one patient also received adjuvant radiation therapy. After a median follow‐up of 21 months (range 4–180 months) all patients were alive and showed stable disease (for details see Table 1). Importantly, none of the patients experienced progression to systemic disease, which was excluded by means of neuroimaging, ultrasound examination, electrocardiography, blood and urine screening for free light chains.
Discussion
The results from this study, which represents the largest series of cerebral amyloidomas reported to date, clearly show that cerebral amyloidoma represents a light chain amyloidosis (AL) characterized by B‐cell clonality. Our findings thus extend previous observations in single case reports 1, 18 and a smaller case series 9 and suggest similarities with extracranial amyloidomas, which are also mainly composed of light chains 11. In order to demonstrate B‐cell clonality in cerebral amyloidomas, CISH for light chain mRNA and IgH gene clonality analysis was clearly superior to immunohistochemistry, emphasizing the value of these molecular methods also in the diagnostic setting.
The most common form of AL amyloidosis is a systemic disease caused by a monoclonal plasma cell or rarely B cell dyscrasia. Circulating light chains are deposited as amyloid fibrils in all organs except the brain. In contrast to local amyloidoma, patients with systemic AL amyloidosis often show a progressive disease leading to organ failure or death within months (to years) depending of the involved organ (particularly the heart).
In contrast, our data confirm that cerebral amyloidoma represents a local disease. In amyloid‐beta‐ and transthyretin‐derived amyloidosis, soluble oligomeric intermediates of fibril assembly have been shown to be cytotoxic both in vitro and in vivo 12. Similar mechanisms may also be operative in AL amyloidoma, causing plasma cell damage, impaired proliferation and eventually self‐limiting disease 23. Indeed, some authors have suggested that amyloidoma might represent burned‐out extramedullary plasmacytoma, overcome by the amyloid deposits and no longer recognizable 5. It seems unlikely, however, that such damage could be related to the type of light chain as suggested by others 23, since the majority of cerebral amyloidomas (as systemic AL amyloidosis) is characterized by lambda light chain restriction. Interestingly, a recently published study suggests different light‐chain variable region (IGVL) gene usage between systemic and localized AL amyloidosis 6.
There is some consensus on the diagnosis and therapy of extracerebral amyloidosis 16, but guidelines for the treatment of cerebral amyloidoma are lacking. Taken into account the general limitations of a retrospective study, the data from the present series suggest that in cerebral amyloidoma extent of tumor resection does not have a major effect on progression‐free survival. Therefore, neurosurgical therapy should be carefully individualized. Because the majority of patients were stable without adjuvant radiotherapy, our data also do not support a role of routine irradiation in the treatment of cerebral amyloidoma.
Even though the clinical course of cerebral amyloidoma seems to be stable and no malignant transformation or progression to systemic disease was observed, the demonstration of B‐cell clonality suggests the presence of an underlying (low‐grade) lymphoid neoplasm. In a large series of patients with extracranial light‐chain amyloidosis, at least a small proportion (1%) experienced progression to systemic amyloidosis 11. Furthermore, in amyloidomas of pulmonary and gastric location, an association with mucosa‐associated lymphoid tissue (MALT) lymphoma is not infrequent 10, 20 and even a case of cerebral amyloidoma associated with lymphoplasmacytic lymphoma is on record 13. Taken together, these observations clearly justify regular and comprehensive follow‐up examinations in patients with cerebral amyloidoma.
In conclusion, cerebral amyloidoma is a local disease characterized by B‐cell clonality. Our data suggest that cerebral amyloidoma is associated with a stable clinical course. Nevertheless, close follow‐up examinations are warranted.
Funding
Jennifer Kollmer received a research grant from the Amyloidosis Foundation (Clarkston, MI).
Conflicts of Interest
G. Reifenberger received grants from Roche and Merck as well as honoraria for advisory boards or lectures from Amgen and Celldex. The other authors have no potential conflict of interest to declare.
Acknowledgment
Susanne Peetz‐Dienhart provided expert technical assistance.
References
- 1. Fischer B, Palkovic S, Rickert C, Weckesser M, Wassmann H (2007) Cerebral AL lambda‐amyloidoma: clinical and pathomorphological characteristics. Review of the literature and of a patient. Amyloid 14:11–19. [DOI] [PubMed] [Google Scholar]
- 2. Foreid H, Barroso C, Evangelista T, Campos A, Pimentel J (2010) Intracerebral amyloidoma: case report and review of the literature. Clin Neuropathol 29:217–222. [DOI] [PubMed] [Google Scholar]
- 3. Gandhi D, Wee R, Goyal M (2003) CT and MR imaging of intracerebral amyloidoma: case report and review of the literature. AJNR Am J Neuroradiol 24:519–522. [PMC free article] [PubMed] [Google Scholar]
- 4. Grogg KL, Aubry MC, Vrana JA, Theis JD, Dogan A (2013) Nodular pulmonary amyloidosis is characterized by localized immunoglobulin deposition and is frequently associated with an indolent B‐cell lymphoproliferative disorder. Am J Surg Pathol 37:406–412. [DOI] [PubMed] [Google Scholar]
- 5. Kanoh T, Suzuki K, Kawaguchi S (1998) Multifocal nodular AL amyloidosis in primary Sjogren's syndrome. Rinsho Ketsueki 39:1157–1162. [PubMed] [Google Scholar]
- 6. Kourelis TV, Dasari S, Theis JD, Ramirez‐Alvarado M, Kurtin PJ, Gertz MA et al (2017) Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light‐chain amyloidosis by mass spectrometry. Blood 129:299–306. [DOI] [PubMed] [Google Scholar]
- 7. Krishnan J, Chu WS, Elrod JP, Frizzera G (1993) Tumoral presentation of amyloidosis (amyloidomas) in soft tissues. A report of 14 cases. Am J Clin Pathol 100:135–144. [DOI] [PubMed] [Google Scholar]
- 8. Lachmann HJ, Hawkins PN (2006) Systemic amyloidosis. Curr Opin Pharmacol 6:214–220. [DOI] [PubMed] [Google Scholar]
- 9. Laeng RH, Altermatt HJ, Scheithauer BW, Zimmermann DR (1998) Amyloidomas of the nervous system: a monoclonal B‐cell disorder with monotypic amyloid light chain lambda amyloid production. Cancer 82:362–374. [DOI] [PubMed] [Google Scholar]
- 10. Lim JK, Lacy MQ, Kurtin PJ, Kyle RA, Gertz MA (2001) Pulmonary marginal zone lymphoma of MALT type as a cause of localised pulmonary amyloidosis. J Clin Pathol 54:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mahmood S, Bridoux F, Venner CP, Sachchithanantham S, Gilbertson JA, Rowczenio D et al (2015) Natural history and outcomes in localised immunoglobulin light‐chain amyloidosis: a long‐term observational study. Lancet Haematol 2:e241–e250. [DOI] [PubMed] [Google Scholar]
- 12. Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349:583–596. [DOI] [PubMed] [Google Scholar]
- 13. Pace AA, Lownes SE, Shivane A, Hilton DA, Weatherby SJ (2015) A tale of the unexpected: amyloidoma associated with intracerebral lymphoplasmacytic lymphoma. J Neurol Sci 359:404–408. [DOI] [PubMed] [Google Scholar]
- 14. Pambuccian SE, Horyd ID, Cawte T, Huvos AG (1997) Amyloidoma of bone, a plasma cell/plasmacytoid neoplasm. Report of three cases and review of the literature. Am J Surg Pathol 21:179–186. [DOI] [PubMed] [Google Scholar]
- 15. Pasternak S, Wright BA, Walsh N (2007) Soft tissue amyloidoma of the extremities: report of a case and review of the literature. Am J Dermatopathol 29:152–155. [DOI] [PubMed] [Google Scholar]
- 16. Röcken C, Ernst J, Hund E, Michels H, Perz J, Saeger W et al (2006) Interdisciplinary guidelines for diagnosis and therapy of extracerebral amyloidosis. Med Klin (Munich) 101:825–829. [DOI] [PubMed] [Google Scholar]
- 17. Rodriguez FJ, Gamez JD, Vrana JA, Theis JD, Giannini C, Scheithauer BW et al (2008) Immunoglobulin derived depositions in the nervous system: novel mass spectrometry application for protein characterization in formalin‐fixed tissues. Lab Invest 88:1024–1037. [DOI] [PubMed] [Google Scholar]
- 18. Schröder R, Linke RP, Voges J, Heindel W, Sturm V (1995) Intracerebral A lambda amyloidoma diagnosed by stereotactic biopsy. Clin Neuropathol 14:347–350. [PubMed] [Google Scholar]
- 19. Sipe JD, Cohen AS (2000) Review: history of the amyloid fibril. J Struct Biol 130:88–98. [DOI] [PubMed] [Google Scholar]
- 20. Sparks D, Bhalla A, Dodge J, Saldinger P (2014) Isolated gastric amyloidoma in the setting of marginal zone MALT lymphoma: case report and review of the literature. Conn Med 78:277–280. [PubMed] [Google Scholar]
- 21. Symko SC, Hattab EM, Steinberg GK, Lane B (2001) Imaging of cerebral and brain stem amyloidomas. AJNR Am J Neuroradiol 22:1353–1356. [PMC free article] [PubMed] [Google Scholar]
- 22. Wechalekar AD, Gillmore JD, Hawkins PN (2016) Systemic amyloidosis. Lancet 387:2641–2654. [DOI] [PubMed] [Google Scholar]
- 23. Westermark P (2012) Localized AL amyloidosis: a suicidal neoplasm?. Ups J Med Sci 117:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]