

Abstract
Peroxisomes play a crucial role in normal neurodevelopment and in the maintenance of the adult brain. This depends largely on intact peroxisomal β‐oxidation given the similarities in pathologies between peroxisome biogenesis disorders and deficiency of multifunctional protein‐2 (MFP2), the central enzyme of this pathway. Recently, adult patients diagnosed with cerebellar ataxia were shown to have mild mutations in the MFP2 gene, hydroxy‐steroid dehydrogenase (17 beta) type 4 (HSD17B4). Cerebellar atrophy also develops in MFP2 deficient mice but the cellular origin of the degeneration is unexplored. In order to investigate whether peroxisomal β‐oxidation is essential within Purkinje cells, the sole output neurons of the cerebellum, we generated and characterized a mouse model with Purkinje cell selective deletion of the MFP2 gene. We show that selective loss of MFP2 from mature cerebellar Purkinje neurons causes a late‐onset motor phenotype and progressive Purkinje cell degeneration, thereby mimicking ataxia and cerebellar deterioration in patients with mild HSD17B4 mutations. We demonstrate that swellings on Purkinje cell axons coincide with ataxic behavior and precede neurodegeneration. Loss of Purkinje cells occurs in a characteristic banded pattern, proceeds in an anterior to posterior fashion and is accompanied by progressive astro‐ and microgliosis. These data prove that the peroxisomal β‐oxidation pathway is required within Purkinje neurons to maintain their axonal integrity, independent of glial dysfunction.
Keywords: ataxia, axonal swellings, multifunctional protein‐2, β‐oxidation, peroxisomes, Purkinje cell
Abbreviations
- BOS
base of support
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DCN
deep cerebellar nuclei
- HSD17B4
17β‐hydroxysteroid dehydrogenase type 4
- LF
left front
- LH
left hind
- MFP2
multifunctional protein‐2
- ML
molecular layer
- PBS
phosphate buffered saline
- PC
Purkinje cell
- PFA
paraformaldehyde
- PPD
p‐phenylenediamine
- RF
right front
- RH
right hind
- VLCFA
very long‐chain fatty acids
- WT
wild type
Background
Peroxisomal multifunctional protein‐2 (MFP2) deficiency is a rare metabolic disease caused by hydroxy‐steroid dehydrogenase (17 beta) type 4 (HSD17B4) gene mutations and characterized biochemically by the accumulation of very long‐chain fatty acids (VLCFAs), bile acid intermediates and branched‐chain fatty acids in the patients plasma 12. MFP2, alternately known as D‐bifunctional protein, is a central node in the peroxisomal β‐oxidation pathway 38. One of the hallmarks of MFP2 deficiency is cerebellar pathology. Ectopic Purkinje cells (PCs), cerebellar demyelination and atrophy are recurrent features in severely affected patients who usually do not survive past one year of age 37. Milder HSD17B4 mutations are primarily linked to leukodystrophies and allow survival into childhood 36.
More recently, exon sequencing revealed compound heterozygous mutations in the HSD17B4 gene in patients surviving into adulthood 25, 26, 27, 31. In general, these patients develop normally without obvious neurological symptoms during early childhood. However, between late childhood and adulthood, they develop progressive ataxia and show cerebellar atrophy on magnetic resonance imaging (MRI). Intriguingly, the typical peroxisomal metabolites were present at (near) normal levels in plasma questioning the origin of their cerebellar degeneration.
We have previously generated mouse models with global and neural selective (oligodendrocytes, astrocytes and neurons) inactivation of MFP2 19, 20, 40. Ataxia and progressive loss of cerebellar PCs are pathological hallmarks of both the constitutive Mfp2−/− and Nestin‐Mfp2−/− mutants. In the latter model, we recently reported that ataxia and an impairment in motor learning were already apparent at the age of 4 weeks. This was associated with altered electrophysiological properties of PCs, PC axonal degeneration and cerebellar wiring abnormalities with subtle morphological alterations of neuronal spines 8. It remains, however, unresolved whether MFP2 is necessary within PCs or whether this enzyme is required in the surrounding cells to support these cerebellar output neurons. In addition, the contribution of developmental vs. purely degenerative mechanisms to cerebellar demise in MFP2 deficiency remains unclear.
According to previous immunohistological investigations MFP2 is enriched in PCs, suggesting an important role in these cells. In the adult human cerebellum MFP2 was detected in PCs, the deep cerebellar nuclei (DCN) and glial cells. It was also shown that in the developing human brain, the enzyme was observed the earliest in PCs and in neurons of the DCN (at 13–15 gestational weeks) 21. Furthermore, the presence of MFP2 in rat PCs was shown in a study using MFP2 auto‐antibodies obtained from patients with Stiff‐man syndrome 9. Also in the porcine brain, MFP2 protein expression was abundant in PCs 28.
In the present study we investigated the hypothesis that MFP2 is crucial within PCs to maintain cerebellar integrity and motor behavior. Therefore, we developed a PC specific Mfp2 knockout model using L7‐Cre mice 32, which induces efficient gene recombination at a postnatal age. With this innovative L7‐Mfp2−/− mouse model we demonstrate that the peroxisomal β‐oxidation is crucial within PCs to maintain axonal homeostasis, which deviates from the glia‐centered view of peroxisomal importance in the central nervous system 2, 39, 40. Moreover, we prove for the first time that the deletion of MFP2 from PCs can result in a postnatal and solely degenerative cerebellar pathology, different from the developmental problems observed earlier in juvenile Mfp2−/− 23 and Nestin‐Mfp2−/− mice 8. This purely degenerative origin of cerebellar deterioration resembles the cerebellar pathology in patients with mild HSD17B4 mutations.
Materials and Methods
Animals
Floxed MFP2 mice in which exon 8 of the HSD17B4 gene is flanked by loxP sequences 40 were crossed with L7‐Cre mice, in which Cre recombinase is solely expressed in the cerebellar PCs and retinal cells 32. These conditional knockout mice are further denoted as L7‐Mfp2−/−. Tomato reporter mice 30 were a kind gift from the Laboratory of Angiogenesis and Vascular Metabolism (VIB‐KU Leuven, Center for Cancer Biology). Mice were bred in the animal housing facility of the KU Leuven, had ad libitum access to standard rodent chow and water and were kept on a 12 h light and dark cycle.
Motor coordination assessment
Rotarod
An accelerating rotarod (Med Associates, Inc.) was used for assessing neuromotor functions and motor skill learning in L7‐Mfp2−/− mice and their respective controls as described 8. Motor coordination was monitored monthly, starting at 2 months of age up until 20 months when further testing became impossible because of the severe ataxic phenotype. Briefly, mice were tested four times a day on a rotating rod with an acceleration of 4–40 rounds per minute in 300 s. A separate group of 4‐months‐old naïve wild type (WT) and L7‐Mfp2−/− mice was used to establish motor skill learning. Following an adaptation period of 30 s, during which the mice were trained to stay on a steady rod, four test trials per day were performed on five consecutive days. Mice were allowed to rest for 1 h in between each trial. For both tests, the average rotarod performance was expressed for each animal. Falling of the rod or reaching 300 s were considered as the trial ending. A cohort of four to eight mice per genotype was used.
Noldus CatWalk
Gait dynamics of L7‐Mfp2−/− mice and WT littermates were studied in detail using the CatWalk system (Noldus Information Technology) as previously described 8. In short, the gait pattern of mice was recorded with a high speed color camera when crossing a glass plate in an unforced manner. Three uninterrupted tracks with at least four cycles of complete steps were included and averaged for each mouse. The speed variation between both genotypes was less than 25%. The CatWalk software was used for data analysis. Information on the parameters analyzed with the CatWalk software is shown in Supporting Information Table S1. Some results are displayed for the individual paws: right front (RF), left front (LF), right hind (RH) and left hind (LH). A cohort of five mice per genotype was tested.
Balance beam test
Fine motor behavior and balancing potential were tested using the balance beam test as described 8. Mice had to cross a series of square beams with a flat surface and one meter in length, positioned 50 cm above the table top. Beams had a cross section of 30, 10 or 5 mm. Mice were trained four times a day for three consecutive days on the 30 mm beam until they were able to cross within 30 s without hesitation. Time necessary for traversing the 10 and 5 mm beams and the number of hind paw slips were analyzed in triplicate on a monthly basis. Results for 2‐ and 4‐months‐old mice of the same genotype were not significantly different and were pooled together. The same was done for 6‐ and 8‐months‐old mice and 10‐ to 12‐months‐old mice. A cohort of six animals per genotype was used.
Clinical scoring of cerebellar ataxia
Disease severity and progression were scored using a series of rapid but sensitive tests, previously applied in a set of mouse models with cerebellar ataxia. The procedure involves four different experiments 15. For each of the tests, animals were assessed three consecutive times and the average score per animal was calculated. A cohort of four to eight mice per genotype was used.
Ledge test
The ledge test was applied to assess motor coordination. Briefly, mice were positioned on the ledge of a cage and were scored while walking along the edge and while lowering themselves into the cage. Mice that walked along the edge without losing balance and that got into the cage smoothly, using their paws, received a score of 0. Scores of 1 and 2 were given to occasional slips and non‐coordinated use of the hind paws or falling into the cage, respectively. Mice that (nearly) fell off the ledge or refused to move back into the cage were given a score of 3.
Hind limb clasping
Disease progression was scored by observing hind limb clasping. Mice were lifted by their tails and the position of their hind limbs was observed during ten seconds. Mice received a score of 0 when they spread their hind limbs away from the abdomen. A score of 1 was given to mice that pulled their hind legs partially toward their abdomen for more than 5 s. When this was observed for both hind limbs, the mouse received a score of 2. A score of 3 was assigned to mice that retracted both hind limbs completely for more than 50% of the observation time.
Gait analysis
Gait analysis of mice placed on a flat surface, heading away from the researcher, gives a read‐out for muscle function and coordination. Mice receive a score of 0 when equally using their four paws in a way that the abdomen did not touch the surface. If tremor was observed or if the mouse appeared to limp, a score of 1 was assigned. A score of 2 was given to mice showing one or more of the following symptoms: severe tremor or limp, lowered pelvis or feet point away from the body. Mice that dragged their abdomen along the surface were scored 3.
Kyphosis
Kyphosis refers to an excessive convex curvature of the spine. It can result from different neurodegenerative diseases and is caused by a loss of muscle tone in the spinal muscles. Mouse posture was observed while walking on a flat surface. Scores from 0 to 3 were assigned according to kyphosis severity.
Immunofluorescence
Mice were deeply anesthetized using a mix of Dormitor (1 mg/kg) and Nimatek (75 mg/kg) and perfused transcardially with phosphate buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde (PFA). Brains were isolated, post‐fixed overnight in 4% PFA and cryopreserved in 20% (wt/vol) sucrose until further processing. Cerebella were embedded in TissueTek (Thermo Scientific) in a sagittal or coronal orientation and free floating sections (40 µm) were immunohistochemically processed for calbindin (mouse antibody, Sigma; 1:200) single stainings (overnight incubation at 4ºC) or for calbindin and Aldolase C (rabbit antibody, Santa Cruz; 1:200) double stainings (48 h incubation at 4ºC) as described in Ref. 8. The following secondary antibodies were used: goat anti‐mouse Alexa‐Fluor‐568 (Invitrogen, 1:200) and/or goat ant‐rabbit Alexa‐Fluor‐488 (Invitrogen; 1:200). Immunofluorescent imaging was performed using a Zeiss confocal laser scanning microscope 780 with a 63× objective, the pinhole set at 1 arbitrary unit and a step size of 2 µm in the z‐direction. Detection of Tomato and GFP was performed as described 30. A cohort of four to five mice per genotype was used for all histological analyses unless stated otherwise.
Quantitative measurements
PC density was quantified in lobule IV (representative for the anterior cerebellum) and lobules IX and X (representative for the posterior cerebellum) on 40× maximal intensity projection confocal images stained for calbindin. The number of cells was normalized to the length of the PC layer analyzed. The thickness of the molecular layer (ML) and PC soma size were measured as described 8, 35. All measurements were performed using the FiJi software. A cohort of four to five mice per genotype was used.
Golgi staining
The Golgi staining kit (FD Neurotechnologies) was used according to the manufacturer's protocol. In short, cerebella were dissected quickly from 7‐months‐old WT and L7‐Mfp2−/− mice. Cerebella were cut in the sagittal plane and processed, imaged and analyzed as described previously 8. A cohort of three mice per genotype was used. Analyses of PC spine number and length were performed using the FiJi software on 15 PCs per genotype.
Electron microscopy
Mice were fixed by cardiac perfusion with 2% paraformaldehyde, 2% glutaraldehyde (v/v) in 0.1 M sodium cacodylate pH 7.4. The brain was dissected and the cerebellum isolated and post‐fixed by immersion in a solution of 4% glutaraldehyde in 0.1 M sodium cacodylate pH 7.4. Post‐fixation was at room temperature for 1 day followed by 2 days at 4ºC. A midline sagittal section (∼2 mm thick) was processed for electron microscopy as previously described 6. Semi‐thin sections (1‐μm thick) were stained with p‐phenylenediamine (PPD) to select an area, within the ML and IGL, to perform ultra‐thin sections that were visualized on a JEOL JEM‐1400 transmission electron microscope (TEM). After the analysis at the ML and IGL, the tissue was trimmed to reach the DCN and a new area was selected to cut ultra‐thin sections and perform the TEM analysis. All images were acquired with an Orious Sc1000 digital camera.
Statistical analysis
Data analysis was done using the GraphPad Prism software (version 5.0). Statistical analysis was performed using the unpaired, two‐sided Student's t‐test or with two‐way ANOVA repeated measures followed by the Bonferroni post hoc test. Data are shown as the mean ± SEM and statistical significance was set at P < 0.05.
Results
Generation of L7‐Mfp2−/− mice
To study whether cell autonomous mechanisms contribute to PC degeneration in Mfp2−/− and Nestin‐Mfp2−/− mice 8, 40, we created conditional knockout mice by crossing floxed Mfp2 mice with a PC specific (L7‐)Cre line, further denoted as L7‐Mfp2−/− mice. The L7‐Cre transgenic mouse strain has previously been shown to allow highly selective and efficient recombination in PCs. CRE expression in PCs occurs from postnatal day 6 onward but it is not fully established until 2–3 weeks after birth 3. Therefore, this knockout strategy enables both a cell type restricted and a post‐developmental deletion of MFP2. We confirmed the specificity of the L7‐Cre line used in our study, by crossing them with a homozygous Tomato reporter mouse (mT/mG) 30. Whereas all cell types in the cerebellum of the non‐recombined reporter mice display red fluorescence (Supporting Information Figure S1A), only PCs express green fluorescence in the offspring, following CRE‐mediated recombination, proving a successful cell‐specific action of the L7‐cre line (Supporting Information Figure S1B). L7‐Mfp2−/− mice developed normally and were indistinguishable from their littermates in the home cage during the first months of life. The life span of L7‐Mfp2−/− mice extended past 2 years.
Late‐onset cerebellar phenotype in L7‐Mfp2−/− mice
To investigate whether L7‐Mfp2−/− mice develop cerebellar ataxia, mice of both genotypes were monitored at different time points using a previously developed scoring system 15. At the age of 2 and 4 months, mutant mice showed normal motor behavior but at 6 and 9 months L7‐Mfp2−/− mice scored significantly higher on a subset of tests. Individual scores for each test are not shown, but mutant mice especially performed worse on the ledge test and showed pronounced clasping of the front and hind limbs (Figure 1A).
Figure 1.
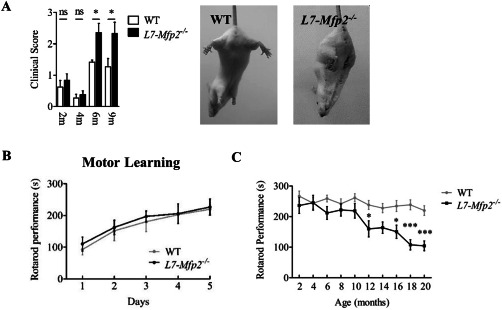
Unaltered motor learning but progressive motor phenotype in L7‐Mfp2 −/− mice. A. The increased clinical score of 6‐ and 9‐months‐old L7‐Mfp2−/− mice is mainly determined by the presence of pronounced hind limb clasping. B. Rotarod testing of naive WT and L7‐Mfp2 knockout mice on five consecutive days reveals a similar pattern of motor learning in both groups. C. The accelerating rotarod shows progressive motor deterioration in L7‐Mfp2−/− mice from the age of 12 months. N = 4–8 mice per group. Results are displayed as mean ± SEM. NS: not significant; *P < 0.05; ***P < 0.001.
To study the effect of MFP2 deletion from PCs on motor learning and behavior, mice of both genotypes were tested on the accelerating rotarod. We found that the ability to learn a new motor skill was not affected in 4‐months‐old L7‐Mfp2−/− mice as rotarod performance improved equally in both genotypes (Figure 1B). Subsequent follow‐up demonstrated impaired rotarod performance from the age of 12 months in L7‐Mfp2−/− mice that progressively worsened thereafter (Figure 1C).
To monitor cerebellar defects in a more sensitive way, the gait pattern of 6‐months‐old L7‐Mfp2−/− and WT mice was assessed using the Noldus CatWalk system. As many gait parameters are strongly speed‐related, we first assured that the run duration was not significantly different between WT and L7‐Mfp2−/− mice. Computer‐assisted analysis of the static and dynamic gait parameters showed that most gait parameters were unaffected, with the exception of diagonal support. Although not significant, a clear tendency for a decreased stride length was also observed at this age (Figure 2A; Supporting Information Figure S2A). Stride length and stance deteriorated markedly in 10‐months‐old L7‐Mfp2−/− mice. Moreover, the base of support (BOS) of the hind and front paws, the diagonal as well as the three point support were significantly altered at this age whereas no differences were observed in the swing phase of gait. Together, this reflects worsening postural alterations in 6–10‐months‐old L7‐Mfp2−/− mice (Figure 2A–B; Supporting Information Figure S2B).
Figure 2.
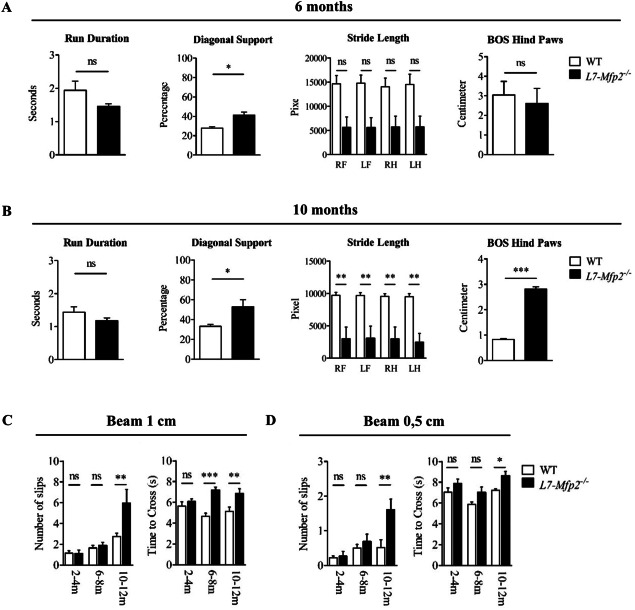
Late‐onset cerebellar ataxia in L7‐Mfp2−/− mice. A,B. CatWalk analysis at the age of 6 and 10 months reveals worsening postural changes and gait abnormalities in L7‐Mfp2−/− mice. C,D. Significant increases in the number of paw slips and the time needed to cross on beams with an increasing degree of difficulty (1 and 0.5 cm) indicates that ataxia initiates in 6‐ to 8‐months‐old L7‐Mfp2−/− mice. N = 4–5 mice per group. Results are displayed as mean ± SEM. NS: not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
The balance beam was implemented as a measure for fine motor coordination and equilibrium. Although the number of slips on the 1 and 0.5 cm beams did not differ between 6‐ and 8‐months‐old WT and L7‐Mfp2−/− mice, the significant increase in time needed to cross the 1 cm beam indicated the presence of subtle motor deficits in the knockout group. From the age of 10 months the hind paws of L7‐Mfp2−/− mice slipped significantly more off the 1 and 0.5 cm beams and for both beams they needed more time to cross (Figure 2C,D).
The combined results of these motor tests demonstrate that specific deletion of MFP2 from PCs causes cerebellar ataxia, starting around 6–8 months of age and progressively aggravating thereafter.
PC loss in L7‐Mfp2−/− cerebella occurs in an anterior to posterior fashion
We questioned whether PC loss might underlie cerebellar ataxia in L7‐Mfp2−/− mice. Based on gross morphology, cerebella of L7‐Mfp2−/− animals were indistinguishable from their control littermates at 4, 6 and 9 months of age as histology showed normal‐sized cerebella with an unaffected foliation pattern (Figure 3A). Calbindin immunoreactivity of PC somata revealed an uninterrupted monolayer in 4‐ and 6‐months‐old WT and L7‐Mfp2−/− mice suggesting the absence of early histopathological changes (Figure 3B). Quantification of calbindin stained sections confirmed this visual perception as PC number and thickness of the ML in lobules IV and IX were equal in both genotypes (Figure 3C,D). From the age of 9 months, calbindin staining became less intense in L7‐Mfp2−/− cerebella (Figure 3A,B) and PC countings revealed partial PC loss restricted to the anterior cerebellar lobules, represented by lobule IV. However, the significant decrease in PC density in lobule IV had no effect on the ML thickness that was possibly preserved by the elaborate dendritic trees of the remaining PCs (Figure 3C,D). Histological examination at 24 months of age demonstrated that the majority of the PCs disappeared in the L7‐Mfp2−/− mice, both in the anterior and the posterior cerebellar lobules (Figure 3A–D). Remarkably, PCs in lobule X did not degenerate (Figure 3E) and the soma size of the PCs did not significantly differ from that of WT PCs. These results indicate a clear‐cut pattern of PC loss following peroxisomal β‐oxidation blockade, initiating around 9 months of age and progressing from the anterior to the posterior lobules with apparent sparing of lobule X.
Figure 3.
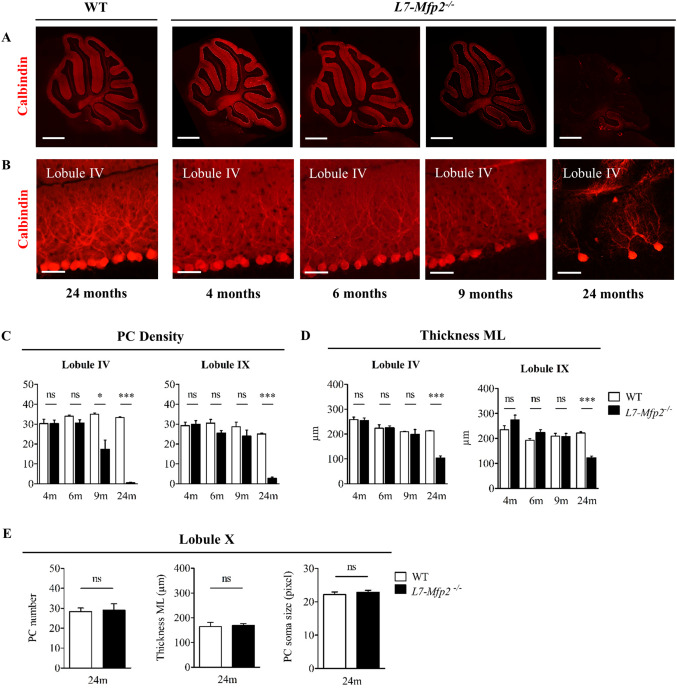
Anterior to posterior PC degeneration in the absence of MFP2. A. Overview pictures of calbindin stained sagittal sections and B. magnified pictures of anterior lobule IV show progressive PC loss in cerebella of L7‐Mfp2−/− mice. C,D. Quantifications of PC density and ML thickness in lobules IV and IX demonstrate the selective vulnerability of PCs in the anterior cerebellum. E. PCs localized in lobule X are spared from degeneration as their number, soma size and ML thickness are unaltered in both genotypes. N = 4–5 mice per group. Results are displayed as mean ± SEM. NS: not significant, *P < 0.05; ***P < 0.001. Scale bars A: 750 µm, B: 50 µm.
Patterned PC degeneration in the absence of MFP2
Amongst other markers, the expression patterns of zebrin II/aldolase C subdivides the transverse zones of the cerebellar cortex into well‐defined parasagittal bands. By processing a series of coronal cerebellar sections for anti‐calbindin immunohistochemistry, we investigated if PC loss in 11‐months‐old L7‐Mfp2−/− mice occurred in a patterned manner. In the WT cerebellum, an uninterrupted monolayer of PCs was present in all lobules. In contrast, the PC layer in L7‐Mfp2−/− mice was characterized by narrow stripes of calbindin negative gaps (Figure 4A). Parasagittal bands of surviving PCs were seen in the anterior region of L7‐Mfp2−/− cerebella (Figure 4A,B), while little PC loss was observed in the posterior lobules (data not shown). In the anterior lobe, the residual PCs stained positive for Aldolase C (Figure 4B). These observations indicate the selective vulnerability of a subset of PCs to MFP2 deletion.
Figure 4.
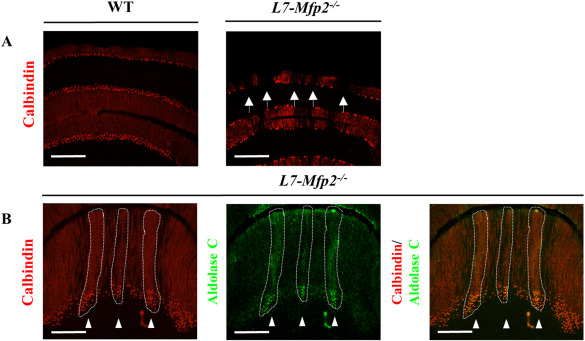
Patterned PC death in L7‐Mfp2 −/− cerebella. A,B. Calbindin stainings on coronal sections of 11‐months‐old L7‐Mfp2−/− mice show striped PC degeneration in the anterior cerebellar lobules. B. Aldolase C positive PCs of L7‐Mfp2−/− mice are resistant to degeneration. N = 2 mice per group. Scale bars: 500 µm.
Pronounced astro‐ and microgliosis accompanies PC loss in L7‐Mfp2−/− mice
Gliosis is a sensitive readout for pathological changes in the brain that might disclose neuronal abnormalities in an early stage. We therefore analyzed astrocyte and microglia reactivity in L7‐Mfp2−/− mice by glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule 1 (Iba1) immunofluorescent stainings, respectively. At the age of 4 months, the GFAP immunoreactivity of Bergmann glia (BG), present in the cerebellar PC layer, and of astrocytes in the DCN of L7‐Mfp2−/− cerebella was similar to that observed in WT mice (Figure 5B,C). At the age of 6 months, a small subset of GFAP positive cells localized in the DCN of L7‐Mfp2 mutants showed an increase in size (Figure 5B). In 9‐months‐old L7‐Mfp2−/− cerebella, evident alterations in astroglial morphology and a dramatic increase in GFAP staining intensity were observed, which were more pronounced in the DCN than in the ML (Figure 5A–C). At the age of 15 months, reactive astrocytes were present in the cerebellar white matter tracts and in the DCN (Figure 5A,B), and an unorganized pattern of highly reactive BG fibers was visible in the ML (Figure 5C).
Figure 5.
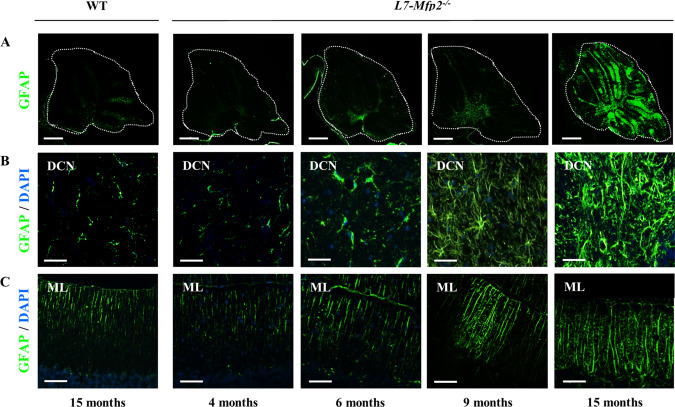
Progressive astrogliosis accompanies PC degeneration in L7‐Mfp2 −/− mice. A,B. At early stages of degeneration (6–9 months), astrogliosis is mainly present at the level of the DCN. C. Isolated regions with reactive BG are also occasionally detected in the ML of 9‐months‐old L7‐Mfp2−/− cerebella. A–C. In L7‐Mfp2−/− mice aged 15 months, reactive astrocytes are increased in number and are spread throughout the cerebellum. DCN: deep cerebellar nuclei; ML: molecular layer. N = 4–5 mice per group. Scale bars A: 750 µm; B, C 50 µm.
Similar to astrocytes, obvious changes in the distribution pattern of microglia were observed in 9‐ and 15‐months‐old L7‐Mfp2−/− mice, in particular accumulating at the level of the DCN (Supporting Information Figure S3A). At the same ages, reactive microglia with hypertrophic cell bodies and thickened processes were also localized within the PC layer, in the ML (Supporting Information Figure S3B) and in the cerebellar white matter tracts (data not shown) suggesting their interaction with the soma and axons of these degenerating neurons. In contrast to the heavily activated BG expanding in the ML of 9‐ and 15‐months‐old L7‐Mfp2−/− mice, reactive microglia in the ML were rather scarce (Supporting Information Figure S3B). Together, the data obtained from L7‐Mfp2−/− mice showed progressive cerebellar gliosis, starting in the DCN and progressing toward the cerebellar white matter, the PC layer and the ML.
Axonal spheroids are the first histopathological sign of PC degeneration in L7‐Mfp2 mutants
Because motor impairment of L7‐Mfp2−/− mice already occurred at 6 months, clearly preceding the loss of PCs, we sought to determine whether more subtle PC morphological alterations manifested at a younger age. The Golgi impregnation method was used to evaluate the PC dendritic tree of 7‐months‐old WT and L7‐Mfp2−/− mice (Figure 6A,B). Detailed analysis of dendrite thickness, spine length and number revealed no significant differences between both genotypes (Figure 6C–E).
Figure 6.
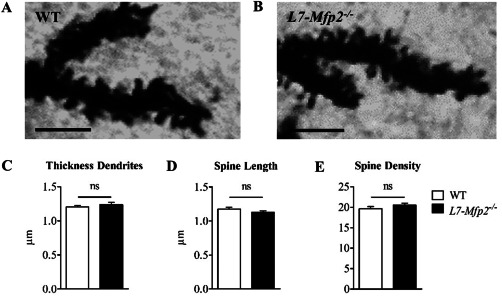
Deletion of MFP2 from PCs does not affect the dendritic morphology. A–E. PC dendrite thickness, spine length and spine density are unaffected in L7‐Mfp2−/− mice at the age of 7 months. N = 15 cells per group, obtained from three mice per genotype. Results are displayed as mean ± SEM. NS: not significant. Scale bars: 5 µm.
Given the early gliosis in the DCN, we subsequently examined PC axons relying on calbindin staining but did not find abnormalities in the granular layer or the DCN of 4‐months‐old L7‐Mfp2−/− mice (Figure 7A). However, at 6 months of age calbindin staining revealed PC axonal spheroids in L7‐Mfp2−/− cerebella. Remarkably, these axonal spheroids were more abundant in the DCN (indicated by the white arrows) suggesting that swellings preferably occurred at the PC axon terminals. More pronounced spheroids on PC axons in the DCN and closer to the cell soma were observed in 9‐months‐old L7‐Mfp2−/− mice (Figure 7A,B). These observations indicate that PC axon terminal swellings represent an early pathological event in L7‐Mfp2−/− cerebella which coincides with the development of their ataxic features.
Figure 7.
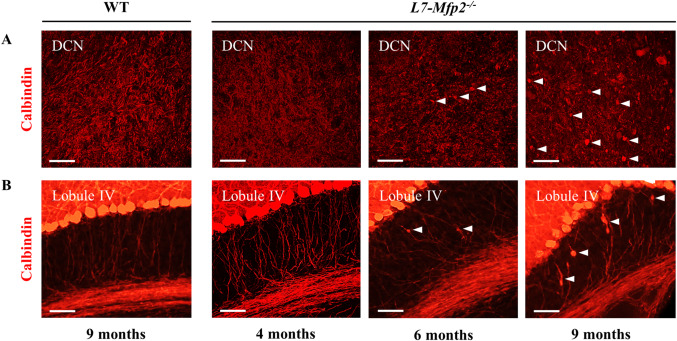
PC axonal swellings as a first histopathological sign in L7‐Mfp2 −/− cerebella. A,B. Calbindin staining in the DCN and cerebellar lobule IV in 4‐month‐old WT and L7‐Mfp2−/− cerebella are indistinguishable. Calbindin positive axonal spheroids are present in both regions of 6‐months‐old L7‐Mfp2−/− mice, but numbers are higher in the DCN (white arrowheads). At 9 months of age PC swellings are very pronounced in the DCN and also occur closer to the cell soma (white arrowheads). N = 4–5 mice per group. Scale bars: 50 µm.
Axonal swellings and distal axon loss of PC from L7‐Mfp2 mutants
To understand PC degeneration in L7‐Mfp2−/− mice we performed an ultrastructural characterization of cerebella in 11‐month‐old WT and L7‐Mfp2−/− mice (Figure 8). No overt structural changes were observed within the ML. Nevertheless, axonal swellings with accumulation of organelles were detected in PC axons within the cerebellar IGL of L7‐Mfp2−/− mice (Figure 8A). The localization of these axonal enlargements was in accordance with the immunological detection of axonal spheroids using calbindin antibodies (Figure 7B). The cerebellar DCN of L7‐Mfp2−/− mice were more severely affected when compared with other cerebellar areas (Figure 8B,C). In the DCN, axonal swellings were widespread and appeared to affect both un‐myelinated regions of axons (eg, giant axonal swelling at the node of Ranvier; left panel of Figure 8B) and myelinated portions of axons (right panels of Figure 8B). The axonal swellings were characterized by the accumulation of mitochondria, vesicles and membrane stacks. The increased pathology at the DCN of L7‐Mfp2−/− mice was also evident by the generalized occurrence of myelin ovoids (Figure 8C). These abnormal myelin figures are caused by the complete degeneration of the underlying axon, and highlight the distal loss of PC axons in L7‐Mfp2−/− mice. As a consequence of myelin fragmentation following axonal degeneration in L7‐Mfp2−/− mice, we could observe myelin‐laden cells (presumably microglia) in attempts to clear the myelin debris caused by axon degeneration (Supporting Information Figure S4). The results clearly demonstrate the distal loss of PC axons at the DCN of L7‐Mfp2−/− mice, which combined with detection the axonal swellings at the DCN and IGL suggests a dying‐back process that contributes to PC death.
Figure 8.
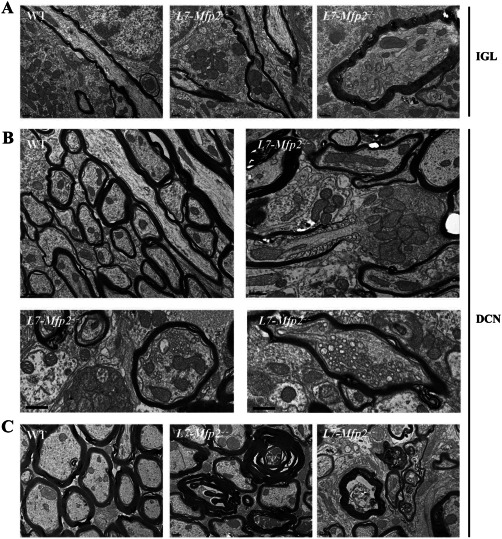
Distal degeneration of PC axons at the DCN disrupts cerebellar circuitry. A–C. Ultrastructural analysis of cerebella from 11‐month‐old WT and L7‐Mfp2−/− mice revealed axonal degeneration of PC from L7‐Mfp2−/− mice. Axon swellings with accumulation of organelles were observed in the internal granular layer (IGL) of L7‐Mfp2−/− mice (A). At the deep cerebellar nuclei (DCN), the pathology was more prominent (B and C). Axonal swellings containing mitochondria, membrane stacks and vesicles were observed at the node of Ranvier (left panel) or within myelinated segments of axons (right panels). The DCN region showed abundant myelin ovoids (C), the hallmark of axon degeneration that triggers myelin fragmentation. Scale bars in A–C: 0.5 μm.
Discussion
While we recently demonstrated the primordial role of the pivotal peroxisomal β‐oxidation enzyme MFP2 for the functioning, morphology and innervation of cerebellar PCs 8, the question remained what cell types are at the origin of cerebellar degeneration in MFP2 deficiency. In this study, we explored the cell autonomous role of peroxisomal β‐oxidation in PCs by generating a PC specific knockout mouse model of MFP2, L7‐Mfp2−/−. With this novel model we not only prove the importance of the peroxisomal β‐oxidation in neurons, and more specifically in PCs, we also demonstrate that peroxisomal β‐oxidation deficiency causes neurodegeneration independent of developmental anomalies. This mimics the degenerative cerebellar histopathology and the progressive ataxic phenotype of mild MFP2 deficient patients who are diagnosed with cerebellar ataxia at a later age 25, 26, 27, 31. This neuronal centered cause of brain pathology deviates from the established insights that peroxisomal β‐oxidation dysfunction primarily causes white matter disease as for example shown in patients with X‐linked adrenoleukodystrophy (X‐ALD) 11.
PC axonal spheroids are the first histopathological lesion in the cerebellum of L7‐Mfp2 mutants, initiating in the DCN and from there on progressing to the cerebellar white matter tracts. The fact that PC axonal abnormalities preceded neurodegeneration in L7‐Mfp2−/− mice is compatible with a dying‐back neuropathy whereby the cells start degenerating at the synapse and provoke retrograde PC death 5 (Figure 9). It is striking that axonal spheroids were also the earliest histological anomalies in cerebella of Nestin‐Mfp2−/− mice 8. While previously we mainly put our attention on myelin and oligodendrocyte abnormalities as factors likely causing swellings on PC axons in MFP2 deficiency 8, 40, we here show that MFP2 in PCs itself is crucial to prevent their axons from degenerating. In contrast to the axonal compartment of the PCs, we did not find differences in dendritic spine morphology and density in L7‐Mfp2−/− mice at the same age.
Figure 9.
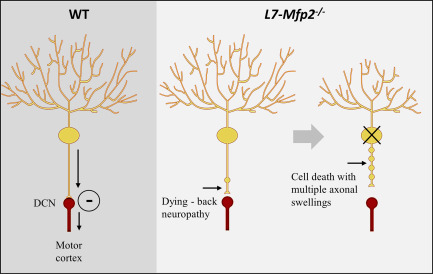
Pathological events in Mfp2−/− mice. Cerebellar ataxia initiates in 6‐months‐old L7‐Mfp2−/− mice at the same time when the first PC axonal torpedoes are detected. This progresses with increased numbers of axonal swellings, distal loss of PC axons and death of PC.
It is compelling that the late onset motor problems in L7‐Mfp2−/− mice occurred simultaneously with the first PC axonal swellings, preceding PC loss with several months. The distal axonal loss interrupts the neuronal circuit and thereby disrupts the inhibitory action of PC on DCN neurons. This causes neuronal hyperexcitability, which may mediate the postural and gait abnormalities of L7‐Mfp2−/− mice. In agreement, it has been shown that, in some models, only massive PC loss provokes ataxia. In PC degeneration and Lurcher mutant mice, for example, virtually all PCs degenerated while they only displayed mild motor signs 10, 24, 29. The fact that the early‐onset motor phenotype of Mfp2−/− and Nestin‐Mfp2−/− mice 8, 40 is not recapitulated in the L7‐Mfp2−/− model strengthens the necessity of an intact peroxisomal β‐oxidation in structures within the neural circuit innervating the PCs and/or in the neighboring glial cells. This once again proves the importance of an intact peroxisomal metabolism during brain development. We here show that on top of that, MFP2 is required within PCs to maintain axonal integrity.
Interestingly, PC degeneration in elder L7‐Mfp2 mutants progresses in a clear‐cut anterior to posterior gradient with apparent sparing of lobule X. This characteristic pattern of PC loss is well known and occurs in several disease models. In Niemann‐Pick C (NPC) disease for example, a lipid trafficking disorder, PCs located in lobule X are typically resistant to the toxic effects of Npc1 deficiency 18, 22. Similarly, in PC degeneration and Leaner mutants, surviving neurons reside respectively within the nodulus and the posterior cerebellum 17, 29, 41. The specific expression pattern of the small heat shock protein 25 has been put forward as an explanation for this cell‐selective resistance to death 10, 33. Through its chaperone function it may protect neurons in the posterior cerebellum from degeneration 1.
In addition, the striped pattern of PC loss observed in 11‐months‐old L7‐Mfp2−/− mice shows that not all PCs are equally vulnerable to degeneration when MFP2 is absent. This characteristic “banded pattern” of PC degeneration is related to the expression of aldolase C, also known as zebrin II, a key enzyme involved in glycolysis 14. In several other mouse models, it has been described that either zebrin II+ or zebrin II‐ PCs degenerate first, depending on the gene defect. Nervous and PC degeneration mutations, for example, primarily affect the zebrin II+ neurons, while zebrin II− PCs seem more vulnerable in mouse models of Leaner, NPC disease and Slc9a6−/− (Angelman syndrome) 16, 33, 34. In some cases, this patterned loss of PCs is eventually followed by widespread degeneration of all neurons, independent of their zebrin II expression 33. Of importance, each set of PCs co‐express other molecular markers that also may influence their vulnerability to cell death 4. Therefore, the restricted pattern of PC loss in L7‐Mfp2−/− cerebella raises the question on whether MFP2 might be expressed in a banded pattern too. This is however difficult to investigate since, in our hands, all available anti‐MFP2 antibodies do not stain specifically in the murine brain as they also produce signals in knockout mice (unpublished observations). Given that zebrin II+ PCs are spared in L7‐Mfp2−/− mice, these results may refer to the interaction of the peroxisomal β‐oxidation with other metabolic processes, such as glycolysis, or with the molecular markers specifically expressed by zebrin II+ and zebrin II− PC populations 4.
The present data in fact highlight and further obscure our limited understanding of the role of peroxisomal β‐oxidation in the CNS and particularly in neurons. Cerebellar degeneration in peroxisomal disorders was mostly linked to aberrant morphogenesis and leukodystrophy 7. With regard to peroxisomal metabolites, primarily the branched‐chain fatty acid, phytanic acid was related to ataxic features in men and mice 7. For example, in Phyh−/− mice, a model for Refsum disease, phytol supplementation induced loss of PCs and ataxia 13. In the L7‐Mfp2 mutant it is highly unlikely that phytanic acid plays a role because the mice were fed with regular chow, which is almost devoid of phytanic acid precursors, and any uptake should readily be degraded in the liver. It remains therefore enigmatic which metabolic alterations take place within MFP2 deficient PCs that cannot be compensated by intact peroxisomes in neighboring cells. This setting seems similar to MFP2 deficient patients presenting with cerebellar ataxia as primary symptom, in which peroxisomal metabolites are (near) normal in plasma. We therefore assume that similar mechanisms govern the cerebellar atrophy in these patients and in the L7‐Mfp2−/− mice.
Conclusions
We provide unprecedented insight of a cell autonomous role of peroxisomal β‐oxidation in PCs for the homeostasis of the axon and cell body and for cerebellar integrity. In addition, we prove that MFP2 deficiency can initiate cerebellar deterioration through a purely degenerative process. Together with our previous findings it seems that peroxisomal β‐oxidation is required in several cell types both for the formation and for the maintenance of an intact cerebellum. These data shed light on the cerebellar pathology in an increasing number of patients diagnosed with mild MFP2 deficiency who have normal levels of peroxisomal metabolites in plasma.
Declarations
Ethics approval: All animal experiments were performed in accordance with the “Guidelines for Care and Use of Experimental Animals” and fully approved by the Research Advisory Committee (Research Ethical committee) of the KU Leuven (#122/2015).
Consent for publication: not applicable
Availability of data and material: All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Conflicts of interests
The authors declare that they have no competing interests
Funding
This work was funded by grants from Fonds Wetenschappelijk Onderzoek Vlaanderen (G.OA15.13) and Bijzonder Onderzoeksfonds KU Leuven (OT12/078).
Authors' contributions
SDM and MB designed experiments and wrote the major part of the manuscript, SDM, RKB and DB performed behavioral and histological studies, ARM and PB analyzed and interpreted EM data. All authors read and approved the final manuscript.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Figure S1. Specificity of the L7‐Cre line. (A) Red fluorescence in all layers of the cerebellar cortex of 4‐ to 5‐weeks‐old Cre reporter mice (mT/mG) prior to Cre recombinase exposure. (B) Green fluorescence following Cre‐mediated recombination in 4‐ to 5‐weeks‐old mice is limited to the PCs and proves the specificity of the L7‐Cre line used. The PC selective deletion of the mT cassette allows the expression of the mG cassette in the PCL (PC somata) and the ML (PC dendritic trees) of the reporter (mT/mG) mice. In the IGL, PC axons show a moderate green fluorescence (arrow). ML: molecular layer, PCL: Purkinje cell layer, IGL: internal granule cell layer. N = 2 mice per group. Scale bars: 50 µm.
Figure S2. Worsening ataxia in 6‐ and 10‐months‐old L7‐Mfp2 −/− mice. Additional gait parameters of the CatWalk analysis not shown in Figure 2. N = 4–5 mice per group. Results are displayed as mean ± SEM. NS: not significant, * P < 0.05.
Figure S3. PC degeneration induces microgliosis in the L7‐Mfp2 −/− cerebellum. (A–C) Microglial cells with small cell bodies and numerous, thin extensions are dispersed in the DCN and the cerebellar cortex of 15‐months‐old WT and 4‐months‐old L7‐Mfp2−/− mice. (B) Iba1 positive cells are increased in number and size in the DCN of 6‐, 9‐ and 15‐months‐old L7‐Mfp2−/− mice. DCN: deep cerebellar nuclei; ML: molecular layer. N = 4–5 mice per group. Scale bars A and D: 750 µm; B, C, E, F: 50 µm.
Figure S4. Myelin‐laden cells at the DCN of L7‐Mfp2 −/− mice. (A–C) Three examples of cells (presumably microglia) containing myelin debris. Following complete axonal degeneration, the leftover myelin undergoes fragmentation. In the central nervous system removal of myelin debris is very inefficient, but in a few cases microglia and/or macrophages will attempt to clear myelin debris through endocytosis. Scale bars are 1 μm in A, B and 0.5 μm in C.
Table S1. Gait parameters analyzed with the Noldus CatWalk
Acknowledgments
The authors wish to thank Benno Das for his excellent technical assistance and Prof. J. Parys for providing the IP3R1 antibody.
References
- 1. Armstrong CL, Duffin CA, McFarland R, Vogel MW (2011) Mechanisms of compartmental purkinje cell death and survival in the lurcher mutant mouse. Cerebellum 10:504–514. [DOI] [PubMed] [Google Scholar]
- 2. Aubourg P (2007) Axons need glial peroxisomes. Nat Genet 39:936–938. [DOI] [PubMed] [Google Scholar]
- 3. Barski JJ, Dethleffsen K, Meyer M (2000) Cre recombinase expression in cerebellar Purkinje cells. Genesis 28:93–98. [PubMed] [Google Scholar]
- 4. Bottelbergs A, Verheijden S, Hulshagen L, Gutmann DH, Goebbels S, Nave KA et al (2010) Axonal integrity in the absence of functional peroxisomes from projection neurons and astrocytes. Glia 58:1532–1543. [DOI] [PubMed] [Google Scholar]
- 5. Cerminara NL, Lang EJ, Sillitoe RV, Apps R (2015) Redefining the cerebellar cortex as an assembly of non‐uniform Purkinje cell microcircuits. Nat Rev Neurosci 16:79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coleman M (2005) Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 6:889–898. [DOI] [PubMed] [Google Scholar]
- 7. da Silva TF, Eira J, Lopes AT, Malheiro AR, Sousa V, Luoma A et al (2014) Peripheral nervous system plasmalogens regulate Schwann cell differentiation and myelination. J Clin Invest 124:2560–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Munter S, Verheijden S, Regal L, Baes M (2015) Peroxisomal disorders: a review on cerebellar pathologies. Brain Pathol 25:663–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Munter S, Verheijden S, Vanderstuyft E, Malheiro AR, Brites P, Gall D et al (2016) Early‐onset Purkinje cell dysfunction underlies cerebellar ataxia in peroxisomal multifunctional protein‐2 deficiency. Neurobiol Dis 94:157–168. [DOI] [PubMed] [Google Scholar]
- 10. Dinkel K, Rickert M, Moller G, Adamski J, Meinck HM, Richter W (2002) Stiff‐man syndrome: identification of 17 beta‐hydroxysteroid dehydrogenase type 4 as a novel 80‐kDa antineuronal antigen. J Neuroimmunol 130:184–193. [DOI] [PubMed] [Google Scholar]
- 11. Duffin CA, McFarland R, Sarna JR, Vogel MW, Armstrong CL (2010) Heat shock protein 25 expression and preferential Purkinje cell survival in the lurcher mutant mouse cerebellum. J Comp Neurol 518:1892–1907. [DOI] [PubMed] [Google Scholar]
- 12. Dusart I, Guenet JL, Sotelo C (2006) Purkinje cell death: differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum 5:163–173. [DOI] [PubMed] [Google Scholar]
- 13. Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P et al (2012) X‐linked adrenoleukodystrophy (X‐ALD): clinical presentation and guidelines for diagnosis, follow‐up and management. Orphanet J Rare Dis 7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferdinandusse S, Denis S, Mooyer PA, Dekker C, Duran M, Soorani‐Lunsing RJ et al (2006) Clinical and biochemical spectrum of D‐bifunctional protein deficiency. Ann Neurol 59:92–104. [DOI] [PubMed] [Google Scholar]
- 15. Ferdinandusse S, Zomer AW, Komen JC, van den Brink CE, Thanos M, Hamers FP et al (2008) Ataxia with loss of Purkinje cells in a mouse model for Refsum disease. Proc Natl Acad Sci U S A 105:17712–17717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujita H, Aoki H, Ajioka I, Yamazaki M, Abe M, Oh‐Nishi A et al (2014) Detailed expression pattern of aldolase C (Aldoc) in the cerebellum, retina and other areas of the CNS studied in Aldoc‐Venus knock‐in mice. PloS One 9:e86679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA (2010) A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 39:1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heckroth JA, Abbott LC (1994) Purkinje cell loss from alternating sagittal zones in the cerebellum of leaner mutant mice. Brain Res 658:93–104. [DOI] [PubMed] [Google Scholar]
- 19. Herrup K, Wilczynski SL (1982) Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience 7:2185–2196. [DOI] [PubMed] [Google Scholar]
- 20. Higashi Y, Murayama S, Pentchev PG, Suzuki K (1993) Cerebellar degeneration in the Niemann‐Pick type C mouse. Acta Neuropathol 85:175–184. [DOI] [PubMed] [Google Scholar]
- 21. Huyghe S, Mannaerts GP, Baes M, Van Veldhoven PP (2006) Peroxisomal multifunctional protein‐2: the enzyme, the patients and the knockout mouse model. Biochim Biophys Acta 1761:973–994. [DOI] [PubMed] [Google Scholar]
- 22. Huyghe S, Schmalbruch H, Hulshagen L, Veldhoven PV, Baes M, Hartmann D (2006) Peroxisomal multifunctional protein‐2 deficiency causes motor deficits and glial lesions in the adult central nervous system. Am J Pathol 168:1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Itoh M, Suzuki Y, Akaboshi S, Zhang Z, Miyabara S, Takashima S (2000) Developmental and pathological expression of peroxisomal enzymes: their relationship of D‐bifunctional protein deficiency and Zellweger syndrome. Brain Res 858:40–47. [DOI] [PubMed] [Google Scholar]
- 24. Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP (2005) Cell‐autonomous death of cerebellar purkinje neurons with autophagy in Niemann‐Pick type C disease. PLoS Genet 1:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr. , Iwata J, Kominami E et al (2007) Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 104:14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krysko O, Bottelbergs A, Van Veldhoven P, Baes M (2010) Combined deficiency of peroxisomal beta‐oxidation and ether lipid synthesis in mice causes only minor cortical neuronal migration defects but severe hypotonia. Mol Genet Metab 100:71–76. [DOI] [PubMed] [Google Scholar]
- 27. Krysko O, Hulshagen L, Janssen A, Schutz G, Klein R, De Bruycker M et al (2007) Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J Neurosci Res 85:58–72. [DOI] [PubMed] [Google Scholar]
- 28. Lalonde R, Strazielle C (2007) Spontaneous and induced mouse mutations with cerebellar dysfunctions: behavior and neurochemistry. Brain Res 1140:51–74. [DOI] [PubMed] [Google Scholar]
- 29. Launay N, Aguado C, Fourcade S, Ruiz M, Grau L, Riera J et al (2015) Autophagy induction halts axonal degeneration in a mouse model of X‐adrenoleukodystrophy. Acta Neuropathol 129:399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lieber DS, Hershman SG, Slate NG, Calvo SE, Sims KB, Schmahmann JD et al (2014) Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4‐deficiency. BMC Med Genet 15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lines MA, Jobling R, Brady L, Marshall CR, Scherer SW, Rodriguez AR et al (2014) Peroxisomal D‐bifunctional protein deficiency: three adults diagnosed by whole‐exome sequencing. Neurology 82:963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McMillan HJ, Worthylake T, Schwartzentruber J, Gottlieb CC, Lawrence SE, Mackenzie A et al (2012) Specific combination of compound heterozygous mutations in 17beta‐hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D‐bifunctional protein deficiency. Orphanet J Rare Dis 7:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moller G, Leenders F, van Grunsven EG, Dolez V, Qualmann B, Kessels MM et al (1999) Characterization of the HSD17B4 gene: D‐specific multifunctional protein 2/17beta‐hydroxysteroid dehydrogenase IV. J Steroid Biochem Mol Biol 69:441–446. [DOI] [PubMed] [Google Scholar]
- 34. Mullen RJ, Eicher EM, Sidman RL (1976) Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci U S A 73:208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double‐fluorescent Cre reporter mouse. Genesis 45:593–605. [DOI] [PubMed] [Google Scholar]
- 36. Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M (2007) Aberrant membranes and double‐membrane structures accumulate in the axons of Atg5‐null Purkinje cells before neuronal death. Autophagy 3:591–596. [DOI] [PubMed] [Google Scholar]
- 37. Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A et al (2010) Mutations in the DBP‐deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet 87:282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saito H, Tsumura H, Otake S, Nishida A, Furukawa T, Suzuki N (2005) L7/Pcp‐2‐specific expression of Cre recombinase using knock‐in approach. Biochem Biophys Res Commun 331:1216–1221. [DOI] [PubMed] [Google Scholar]
- 39. Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R (2003) Patterned Purkinje cell degeneration in mouse models of Niemann‐Pick type C disease. J Comp Neurol 456:279–291. [DOI] [PubMed] [Google Scholar]
- 40. Stromme P, Dobrenis K, Sillitoe RV, Gulinello M, Ali NF, Davidson C et al (2011) X‐linked Angelman‐like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal‐lysosomal dysfunction. Brain 134:3369–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. [DOI] [PubMed] [Google Scholar]
- 42. van der Knaap MS, Wassmer E, Wolf NI, Ferreira P, Topcu M, Wanders RJ et al (2012) MRI as diagnostic tool in early‐onset peroxisomal disorders. Neurology 78:1304–1308. [DOI] [PubMed] [Google Scholar]
- 43. Van Maldergem L, Espeel M, Wanders RJ, Roels F, Gerard P, Scalais E et al (1992) Neonatal seizures and severe hypotonia in a male infant suffering from a defect in peroxisomal beta‐oxidation. Neuromuscul Disord 2:217–224. [DOI] [PubMed] [Google Scholar]
- 44. Van Veldhoven PP (2010) Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res 51:2863–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verheijden S, Beckers L, Casazza A, Butovsky O, Mazzone M, Baes M (2015) Identification of a chronic non‐neurodegenerative microglia activation state in a mouse model of peroxisomal beta‐oxidation deficiency. GLIA 63:1606–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verheijden S, Bottelbergs A, Krysko O, Krysko DV, Beckers L, De Munter S et al (2013) Peroxisomal multifunctional protein‐2 deficiency causes neuroinflammation and degeneration of Purkinje cells independent of very long chain fatty acid accumulation. Neurobiol Dis 58:258–269. [DOI] [PubMed] [Google Scholar]
- 47. Wassef M, Sotelo C, Cholley B, Brehier A, Thomasset M (1987) Cerebellar mutations affecting the postnatal survival of Purkinje cells in the mouse disclose a longitudinal pattern of differentially sensitive cells. Dev Biol 124:379–389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Figure S1. Specificity of the L7‐Cre line. (A) Red fluorescence in all layers of the cerebellar cortex of 4‐ to 5‐weeks‐old Cre reporter mice (mT/mG) prior to Cre recombinase exposure. (B) Green fluorescence following Cre‐mediated recombination in 4‐ to 5‐weeks‐old mice is limited to the PCs and proves the specificity of the L7‐Cre line used. The PC selective deletion of the mT cassette allows the expression of the mG cassette in the PCL (PC somata) and the ML (PC dendritic trees) of the reporter (mT/mG) mice. In the IGL, PC axons show a moderate green fluorescence (arrow). ML: molecular layer, PCL: Purkinje cell layer, IGL: internal granule cell layer. N = 2 mice per group. Scale bars: 50 µm.
Figure S2. Worsening ataxia in 6‐ and 10‐months‐old L7‐Mfp2 −/− mice. Additional gait parameters of the CatWalk analysis not shown in Figure 2. N = 4–5 mice per group. Results are displayed as mean ± SEM. NS: not significant, * P < 0.05.
Figure S3. PC degeneration induces microgliosis in the L7‐Mfp2 −/− cerebellum. (A–C) Microglial cells with small cell bodies and numerous, thin extensions are dispersed in the DCN and the cerebellar cortex of 15‐months‐old WT and 4‐months‐old L7‐Mfp2−/− mice. (B) Iba1 positive cells are increased in number and size in the DCN of 6‐, 9‐ and 15‐months‐old L7‐Mfp2−/− mice. DCN: deep cerebellar nuclei; ML: molecular layer. N = 4–5 mice per group. Scale bars A and D: 750 µm; B, C, E, F: 50 µm.
Figure S4. Myelin‐laden cells at the DCN of L7‐Mfp2 −/− mice. (A–C) Three examples of cells (presumably microglia) containing myelin debris. Following complete axonal degeneration, the leftover myelin undergoes fragmentation. In the central nervous system removal of myelin debris is very inefficient, but in a few cases microglia and/or macrophages will attempt to clear myelin debris through endocytosis. Scale bars are 1 μm in A, B and 0.5 μm in C.
Table S1. Gait parameters analyzed with the Noldus CatWalk