

Abstract
The imbalance between production and clearance of amyloid‐beta (Aβ) is a key step in the onset and development of Alzheimer’s disease (AD). Therefore, reducing Aβ accumulation in the brain is a promising therapeutic strategy for AD. The recently discovered glymphatic system and meningeal lymphatic vasculature have been shown to be critical for the elimination of interstitial waste products, especially Aβ, from the brain. In the present study, ligation of deep cervical lymph nodes was performed to block drainage of this system and explore the consequences on Aβ‐related pathophysiology. Five‐month‐old APP/PS1 mice and their wild‐type littermates received deep cervical lymphatic node ligation. One month later, behavioral testing and pathological analysis were conducted. Results demonstrated that ligation of dcLNs exacerbated AD‐like phenotypes of APP/PS1 mice, showing more severe brain Aβ accumulation, neuroinflammation, synaptic protein loss, impaired polarization of aquaporin‐4 and deficits in cognitive and exploratory behaviors. These results suggest that brain lymphatic clearance malfunction is one of the deteriorating factors in the progression of AD, and restoring its function is a potential therapeutic target against AD.
Keywords: Alzheimer’s disease, amyloid β, dural lymphatic vessels, glymphatic system; aquaporin 4
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder. Unfortunately, there are no effective drugs to reverse or even slow the progress of this disease 11, 28. Therefore, it is crucial to determine valuable therapeutic targets against the progression of AD.
The imbalance between production and clearance of amyloid‐beta (Aβ) is a key step in the onset and development of both early and late forms of AD. To date, restoring Aβ clearance function is regarded as a promising strategy for treating AD 43, 44. Several mechanisms, such as enzymatic degradation and cellular uptake, transport across the brain–blood barrier (BBB) and blood–cerebrospinal fluid barrier are involved in clearance of extracellular Aβ from the brain 9, 45, 48, 56. However, despite considerable efforts devoted to target at these Aβ clearance systems, the scientific community has thus far be ineffective in determining ways to reduce excess Aβ deposits and prevent AD onset.
Recent findings suggest that the brain lymphatic system contributes to clearing the majority of extracellular Aβ from the brain 31, 38, 50, 61, 64, 67. The cerebral lymphatic system consists of two parts: glymphatic system in the brain parenchyma and lymphatic networks located in the dura mater 31, 32, 41. Glymphatic clearance is dependent on perivascular astrocyte aquaporin‐4 (AQP4), which mediates rapid transport of water from the para‐arterial spaces into the brain parenchyma, before finally entering the para‐venous spaces 31. Meningeal lymphatic vasculatures are connected to deep cervical lymph nodes (dcLNs), draining both macromolecules and immune cells from the CSF into the peripheral circulation 1, 42. These two systems seem to play a synergetic role in driving Aβ out of the brain, with each alteration potentially influencing the AD process 50. Suppression of glymphatic fluid transport has been observed in APP/PS1 transgenic AD model mice, with glymphatic malfunction seen prior to presence of substantial Aβ deposits 50, 67.
In the present study, we examined whether meningeal lymphatic drainage is also impaired in APP/PS1 mice, and whether blocking its drainage aggravates glymphatic dysfunction, in turn promoting AD‐like progress. The results suggest that alleviating brain lymphatic clearance deficits could potentially be a therapeutic target to slow the onset and progression of AD.
Materials and Methods
Animals
Male APP695/PS1‐dE9 (APP/PS1) transgenic mice with C57/BL6 background, and their wild type (WT) littermates, were used in this experiment. Mice were housed under standard conditions (room temperature 18 °C ~ 22 °C, humidity 30% ~ 50%, well‐ventilated, a 12‐h light‐dark cycle) until 5 months of age. Animals were randomly divided into four groups (n = 16 per group): WT‐sham, WT‐ligation, APP/PS1‐sham and APP/PS1‐ligation. The experimental design is available in Figure S1. Animal experiments were approved by Nanjing Medical University (NMU), Animal Ethical and Welfare Committee (Approval No. IACUC‐1601106) and conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of NMU.
Ligation of dcLNs
Mice were anesthetized and fixed on cardboard with abdomen upwards. Following shaving and disinfection with iodophor, a longitudinal incision was made on the neck from the mandible to the sternum. The muscles and fascia were carefully separated along the trachea using a blunt tip tweezer under the stereomicroscope. dcLNs were located below the sternothyroid muscle and their afferent and efferent vessels were carefully ligated using 8‐0 nylon suture 1. Sham‐operated mice were only surgically exposed dcLNs without ligation. After the animals became conscious, they were returned to their cages and fed for 1 month, followed by behavioral testing.
Open field test
The open‐field test was used to assess autonomous and exploration behaviors in a novel environment. The apparatus consists of the open field experiment box (60 cm × 60 cm × 25 cm) with an outlined center area (30 cm × 30 cm). At the start of the experiment, mice were placed in the middle of the bottom. They were allowed to move freely for 5 minutes within the box and the time spent and distance traveled in, as well as the number of entrances into the center area, were recorded. After each test, the box was cleaned to avoid odors of the last mice which may affect the next animal 29.
Y‐maze test
The Y‐maze test was carried out one day after the open field test to evaluate short‐term working memory 4. The maze is randomly defined as three arms, including the novel arm (NA), the starting arm and other arm. This test contains two 5‐minute stages with an interval of 2 h. During the training period, a baffle with the same color as the maze was used to block the NA, and mice were placed in the starting arm to explore activities freely in the SA and OA for 5 minutes. During the second stage, the NA was opened, allowing the mouse to move freely throughout 3 arms for 5 minutes. The percentage of time traveled in the NA and the number of entries into NA were recorded and analyzed.
Intracisternal injection of fluorescent tracer
Deeply anesthetized mice were fixed in a stereotaxic instrument. 5 μL Texas Red‐dextran‐3 (TR‐d3) (ThermoFisher, Cat. No. D3328) was injected at a concentration of 5 mg/mL into the cisterna magna using a microinjection needle. Injection speed was 1 μL/min, and the needle was retained for 10 minutes after the injection to prevent leakage of the tracer 31. After 30 minutes, the brain and dcLNs were quickly dissected and fixed with 4% paraformaldehyde (PFA) for 12 h. Sections were cut on a vibratome at 100 μm and mounted onto poly‐l‐lysine‐coated slides in sequence.
Brain section preparation
After deep anesthesia, mice were transcardially perfused with 0.9% saline, followed by 4% PFA for 15 minutes. The forebrain was dissected from the skull and post fixed overnight at 4℃. Brain tissues were dehydrated in a series of graded ethanol solutions and embedded in paraffin, which were serially cut at 5 μm using a paraffin slicing machine (Leica, RM2135, Germany).
Cerebral dura mater stripping
The skull was separated into the upper half and lower half utilizing microsurgical scissors. Brain tissue was removed and complete skull was obtained. After placement in 20% sucrose solution for 3 days, the skull was placed into sterilized water for 2 days. Under a stereomicroscope, the dura mater was carefully dissected from the skull using a pair of ophthalmic forceps, then unfolded on glass slide covered with poly‐l‐lysine for immunofluorescence.
Immunohistochemistry
Immunohistochemical staining was performed as previously described 66. Briefly, sections were deparaffinized in xylene and rehydrated in a series of graded ethanol solutions. After inactivating endogenous peroxidase and blockage of antigens, brain sections were incubated overnight at 4°C with the following primary antibodies directed against 6E10 (1:1000; Covance, Cat. No. 803001), glial fibrillary acidic protein (GFAP) (1:1000; Millipore, Cat. No. MAB360), ionized calcium binding adaptor molecule 1 (Iba‐1) (1:1000; Wako, Cat. No. 019‐19741), AQP4 (1:500; Santa, Cat. No. sc9887) and caspase‐3 (1:1000, Cell Science Technology, Cat. No. 9662S). Following incubation with biotinylated IgG and ABC at 37°C for 60 minutes, the reaction was visualized with DAB (Sigma‐Aldrich, Cat. No. D8001). Partial AQP4 stained sections were counterstained with Congo red (Sigma‐Aldrich, Cat. No. C6767).
Immunofluorescence
For immunofluorescence, the tissue sections were prepared by adding 0.3% PBST with 5% serum blocking solution at room temperature for 1 h. Sections were incubated in a mixture of a polyclonal rabbit anti‐6E10 antibody (1:1000, Covance, Cat. No. 803001) and a monoclonal mouse anti‐lymphatic vessel endothelial hyaluronan receptor 1 (lyve‐1) antibody (1:1000, Abcam, Cat. No. ab33682) at 4°C overnight. Subsequently, sections were incubated for 2 h at room temperature in a mixture of Texas Red‐conjugated goat anti‐mouse IgG (1:200; Invitrogen, Cat. No. A32732) and FITC‐conjugated goat anti‐rabbit IgG (1:200; Invitrogen, Cat. No. 31635). After PBS washing, the sections were incubated for 5 minutes at room temperature in 1.5 μM 4′,6‐diamidino‐2‐phenylindole (DAPI, Invitrogen, Cat. No. D1306) and sealed with buffered PBS/glycerol.
Thioflavin‐S staining
Deparaffinized sections were stained with 1% Thioflavin‐S (Sigma‐Aldrich) for 5 minutes. After a running water wash, 70% alcohol was used to differentiate for 2 minutes. The sections were rinsed with PBS and cover‐slipped with buffered PBS/glycerol.
Image analysis
All sections were visualized using a digital microscope (Leica Microsystems, Wetzlar, Germany) and captured with constant exposure time, offset and gain for each staining marker. The cerebral cortex and hippocampal areas in each section were manually delineated and the area of positive signal was measured by Image J (NIH) using the interest grayscale threshold analysis with constant settings for minimum and maximum intensities for each staining marker. The positive signal percentage area of TR‐d3, 6E10, Thioflavin‐S, GFAP and Iba‐1 were calculated by dividing the area of positive signal to the total area in the region of interest. For analysis of AQP4 polarization, images at 400× magnification were randomly captured from the superficial layers of the cortex and the lacunosum moleculare layer of the hippocampus on each brain section. The mean integrated optical density of AQP4 immunoreactivity at the domains immediately abutting vessels and adjacent parenchyma was measured, and their ratio was calculated and termed as the polarity of AQP4 66. In addition, caspase‐3 positive apoptotic neurons in the DG region of hippocampus were counted and the percentage of neuronal apoptosis was expressed. Five sections per mouse, and four mice per group, were averaged to provide a mean value for each group. All quantification was done blind to animal genotype and operation.
Western blotting
Forebrain tissues including hippocampus and cortex were homogenized and centrifuged at 4°C and 12 000 rpm for 15 minutes. The extracts were then loaded onto 10%–16% Tris/tricine SDS gels and protein was transferred to PVDF membranes. After blocking in 5% non‐fat milk/TBST for 1 h, membranes were incubated overnight with one of the following primary antibodies directed against Aβ1–42 (1:1000, Abcam, Cat. No. ab201060), Aβ1–40 (1:1000, Abcam, Cat. No. ab20068), total Tau (1:1000, Abcam, Cat. No. ab32057), finger protein 1 (PHF‐1) (1:1000, Abcam, Cat. No. ab80042), glial fibrillary acidic protein (GFAP) (1:1000; Millipore, Cat. No. MAB360), ionized calcium‐binding adaptor molecule 1 (Iba‐1) (1:1000; Wako, Cat. No. 019‐19741), interleukin 1 beta (IL‐1β) (1:1000, Millipore, Cat. No. AB1832P), interleukin 6 (IL‐6) (1:1000, abcam, Cat. No. ab83339), tumor necrosis factor‐α (TNF‐α) (1:1000, Abcam, Cat. No. ab9739), aquaporin‐4 (AQP4) (1:1000; Santa, Cat. No. sc9887), caspase‐3 (1:1000, Cell Signaling Technology, Cat. No. 9662S), a disintegrin and metalloproteinase domain‐containing protein 10 (ADAM10) (1:1000, Millipore, Cat. No. AB19026), β‐site amyloid precursor protein–cleaving enzyme 1 (BACE1) (1:2000, Millipore, Cat. No. MAB5308), presenilin1 (PS1) (1:1000; Sigma‐Aldrich, Cat. No. PRS4203), neprilysin (NEP) (1:1000, Millipore, Cat. No. AB4348), insulin degrading enzyme (IDE) (1:1000, Abcam, Cat. No. ab32216), synaptophysin (SYN) (1:1500, Abcam, Cat. No. ab64581), postsynaptic density protein 95 (PSD‐95) (1:1000; Abcam, Cat. No. ab18258), β‐actin (1:500, Boster, Cat. No. BM0627). Horseradish peroxidase‐conjugated secondary antibodies (Vector Laboratories) were used for incubation for 1 h at room temperature. Bands were visualized using ECL plus detection system. β‐actin was used as an internal reference for protein loading.
Enzyme‐linked immunosorbent assay (ELISA)
Forebrain tissues including hippocampus and cortex were homogenized and sonicated in ice‐cold TBS buffer containing 0.5 mM PMSF, 0.5 mM benzamidine, 1.0 mM DTT, 1.0 mM EDTA, 1% Triton × 100 and 0.6% SDS, followed by centrifugation at 350 000 × g for 20 minutes. Supernatants were used for measurements utilizing total Aβ1–40 and Aβ1–42. The above indexes were quantified with ELISA kits according to the manufacturer’s instructions (Biolegend Corporation, USA).
Flow cytometry
Mice were perfused from the left cardiac ventricle with cold PBS for 10 minutes. Forebrains were quickly removed, dissociated in DMEM containing 10% fetal calf serum. Tissue was homogenized in RPMI, and then a cell suspension was gently made. After filtering with 70‐µm filters and adding 70% and 30% SIP into the suspension, brain homogenate was centrifuged for 30 minutes at ×500 g at 18°C. Suspension containing cells was removed from the centrifuge tube and washed twice to be pelleted 3. Meninges were dissected with fine forceps and digested in RPMI + 1.4 U/mL Collagenase VIII (Worthington) + 1 mg/mL DNase1 (Sigma‐Aldrich) for 15 minutes at 37°C. DcLNs were removed and smashed in DMEM containing 10% fetal calf serum. Digested meninges and smashed dcLNs were dissociated using pipetting, and were passed through 70‐µm filters. Cells were washed twice before the next step 14, 27. For surface staining, the anti‐CD3 antibody (1:400, Bioscience, Cat. No. 145‐2C11) was used according to the manufacturer’s instructions. Data were acquired on an LSRII cytometer (BD Biosciences) and analyzed using the Flowjo Pro software (FlowJo, LLC). The number of cells used for analysis each time was 10 000 for dura mater and 50 000 for brain and dcLNs. The absolute number together with the percentage of CD3+ T cells in the lymphocyte population were also recorded.
Statistical analysis
All data were expressed as means ± SEM. Using GraphPad Prism, version 5.02 software (GraphPad software, San Diego, CA, USA), data were analyzed by two‐way ANOVA, followed by Tukey’s post hoc test or Student’s t‐test. P < 0.05 was considered to have statistical significance.
Results
Ligation of dcLNs blocks brain lymphatic drainage
We first verified whether dcLNs ligation would block brain lymphatic drainage, then investigated its effect on ISF bulk flow by intracisternal injection of TR‐d3. As shown in Figure 1A, TR‐d3 fluorescent signal was clearly observed in the dcLNs of WT‐sham mice, while virtually no signal was present in the ligated dcLNs of WT mice and APP/PS1 mice. Notably, the percentage of TR‐d3 positive area in the dcLNs of APP/PS1‐sham mice was significantly lower than that of WT‐sham controls (P = 0.0401; Figure 1C), indicating impaired extracranial lymphatic drainage in this AD model. Consistent with previous studies 40, 50, APP/PS1‐sham mice showed reduced glymphatic influx of intracisternal TR‐d3 to the brain parenchyma (P = 0.0428, APP/PS1‐sham vs. WT‐sham). Impairment of extracranial lymphatic drainage was further exacerbated after ligation of dcLNs in APP/PS1 mice (P = 0.0292, APP/PS1‐ligation vs. APP/PS1‐sham; Figure 1B,D). Furthermore, 6E10 immunoreactive products consisting of APP and its hydrolysis products, including Aβ, were significantly increased within the lyve‐l positive meningeal lymphatic vessels in APP/PS1 mice after ligation of dcLNs (Figure 2A). Quantitative data revealed a synergistic effect of the genotype and ligation on enhancing the percentage of 6E10 positive area within meningeal lymphatic vessels (P = 0.0034, APP/PS1‐sham vs. WT‐sham; P = 0.0039, APP/PS1‐ligation vs. WT‐ligation; Figure 2B).
Figure 1.
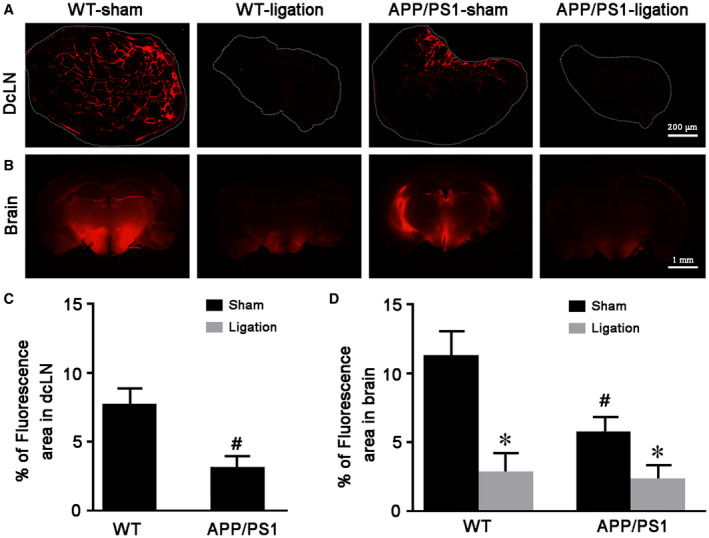
Ligation of dcLNs blocks peripheral drainage of intracisternally injected TR‐d3 and decreased its influx into the rain parenchyma. A, B. Representative images showing TR‐d3 fluorescence within dcLNs and forebrain 30 minutes after injection into cisterna magna. C, D. The percentage of TR‐d3 fluorescent area of dcLNs and forebrain, respectively. Data represent mean ± SEM from four mice per group. Data in Figure 1C were analyzed by paired‐student t test, #P < 0.05, WT vs. APP/PS1. Data in Figure 1D were analyzed by the two‐way ANOVA with Tukey’s post hoc test. Ligation: F1,12 = 15.792, P = 0.002; Genotype: F1,12 = 5.396, P = 0.039; Interaction: F1,12 = 3.052, P = 0.106; #P < 0.05, WT vs. APP/PS1; *P < 0.05, sham vs. dcLNs.
Figure 2.
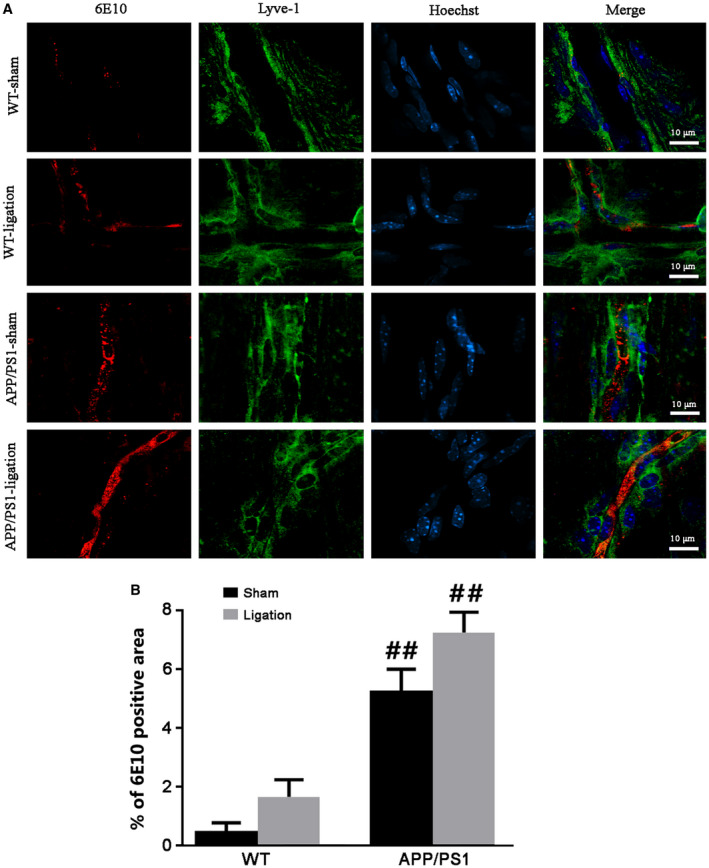
Analysis of APP and its hydrolysis products within the meningeal lymphatic vessels. A. Representative images showing immunoreactive products of 6E10 antibody that can detect APP and its hydrolysis products within the meningeal lymphatic vessels of mice with four groups. B. The percentage of 6E10 positive area within the meningeal lymphatic vessels. Data represent mean ± SEM from four mice per group and analyzed by the two‐way ANOVA with Tukey’s post hoc test. Ligation: F1,12 = 6.084, P = 0.030; Genotype: F1,12 = 41.405, P = 0.000; Interaction: F1,12 = 0.896, P = 0.362; #P < 0.05, ##P < 0.01, WT vs. APP/PS1.
Ligation of dcLNs exacerbates brain Aβ load and phosphorylated tau accumulation
We further observed the consequences of blocking the brain lymphatic pathway on brain Aβ, Tau and phosphorylated tau aggregation 62. Both 6E10‐immunopositive diffuse plaques and thioflavin‐S positive fibrillar plaques noticeably increased in the cerebral cortex and hippocampus of APP/PS1‐ligation mice, compared with APP/PS1‐sham mice (6E10: cortex, P = 0.0489; hippocampus, P = 0.0488; thioflavin‐S: cortex, P = 0.0443; hippocampus, P = 0.0189; Figure 3A–C). No plaque formation was observed in the brain of WT mice regardless of whether or not dcLNs were ligated. ELISA analysis revealed higher levels of Aβ1‐40 and Aβ1‐42 in the forebrain samples of APP/PS1‐sham mice than WT‐sham controls (P = 0.0182, P = 0.0222, respectively; Figure 3D). When APP/PS1 mice were subjected to ligation of dcLNs, brain Aβ1‐40 and Aβ1‐42 levels were dramatically elevated (P = 0.0103, P = 0.0087, respectively, APP/PS1‐ligation vs. WT‐ligation; Figure 3D). Consistently, Western blot results demonstrated that APP/PS1‐ligation mice had high expression levels of Aβ1‐40 and Aβ1‐42, compared with WT‐ligation mice (P = 0.0113, P = 0.0154, respectively; Figure 3E,F). Similarly, dcLNs also increased expression of total Tau and PHF‐1 in both WT mice (P = 0.0241, P = 0.0456, respectively) and APP/PS1 mice (P = 0.0337, P = 0.0348, respectively). APP/PS1‐ligation mice revealed high levels of total Tau and PHF‐1, relative to WT‐ligation mice (P = 0.0146, P = 0.0100, respectively; Figure 3E,F).
Figure 3.
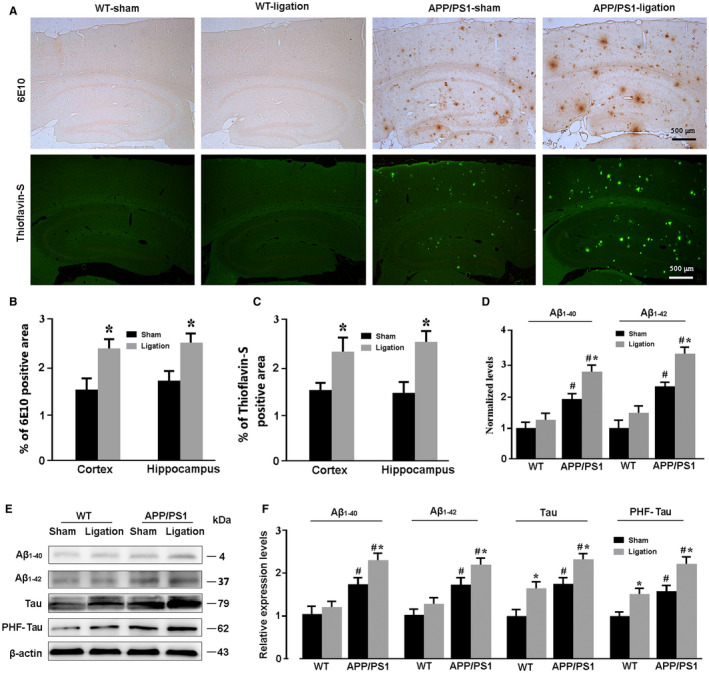
Analysis of brain Aβ and Tau accumulation. A. Representative images showing deposit of Aβ plaques in the cortex and hippocampus of APP/PS1 mice after ligation of dcLNs. B, C. The percentage of positive area of 6E10 and Thioflavin‐S in the cerebral cortex and hippocampus of APP/PS1 mice, respectively. D. Levels of souble Aβ1‐40 and Aβ1‐42 in the forebrain were quantified by ELISA. E, F. Representative Western blot bands and densitometry analysis of Aβ1‐40, Aβ1‐42, Tau and PHF‐1 from the forebrain samples. Data represent mean ± SEM from four mice per group. Data in Figure 3B and C were analyzed by the paired‐student t test. *P < 0.05, sham vs. dcLNs. Data in Figure 3D and F were analyzed by two‐way ANOVA with Tukey’s post hoc test. Figure 3D: Ligation: Aβ1‐40, F1,12 = 5.520, P = 0.037; Aβ1‐42, F1,12 = 6.267, P = 0.028; Genotype: Aβ1‐40, F1,12 = 23.743, P = 0.000; Aβ1‐42, F1,12 = 23.941, P = 0.000; Interaction: Aβ1‐40, F1,12 = 1.073, P = 0.321; Aβ1‐42, F1,12 = 0.815, P = 0.384. Figure 3F: Ligation: Aβ1‐40, F1,12 = 4.778, P = 0.049; Aβ1‐42, F1,12 = 5.025, P = 0.045; Tau, F1,12 = 16.323, P = 0.002); PHF‐1, F1,12 = 13.371, P = 0.03; Genotype: Aβ1‐40, F1,12 = 18.875, P = 0.001; Aβ1‐42, F1,12 = 19.158, P = 0.001; Tau, F1,12 = 22.578, P = 0.000; PHF‐1, F1,12 = 21.833, P = 0.001; Interaction: Aβ1‐40, F1,12 = 1.273, P = 0.281; Aβ1‐42, F1,12 = 0.692, P = 0.422; Tau, F1,12 = 0.002, P = 0.961; PHF‐1, F1,12 = 0.735, P = 0.408. #P < 0.05, WT vs. APP/PS1; *P < 0.05, sham vs. dcLNs.
In addition, we found that ligation of dcLNs did not influence expression of APP secretases, including ADAM10, BACE1 and PS1 53, or Aβ degradation enzymes including, NEP, IDE and LRP‐1, that mainly mediates efflux of Aβ across the BBB in the brain of WT and APP/PS1 mice 20, 54 (Figure S2).
Ligation of dcLNs aggravates glial activation and neuroinflammation in APP/PS1 mice
Reactive gliosis is one of the hallmarks of AD pathology. Activated astrocytes and microglia improve uptake and degradation of Aβ 5, 19. Nevertheless, sustained activation of glial cells is a primary source of neuroinflammation, which exacerbates neurodegenerative progression 25, 33, 63. Both GFAP positive astrocytes and Iba‐1 positive micoglia were in a resting state while activated in the cerebral cortex and hippocampus in the other three groups (Figure 4A). Particularly, compared to WT‐ligation controls, APP/PS1‐ligation mice showed high percentages of positive area for Iba‐1 in the cortex (P = 0.0445) and hippocampus (P = 0.0483) and for GFAP in the hippocampus (P = 0.0404; Figure 4B,C). Western blot also revealed an up‐regulation of GFAP and Ibal‐1 expression in the forebrain of APP/PS1‐ligation mice, compared with APP‐PS1‐sham mice (P = 0.0249, P = 0.0479, respectively) and WT‐ligation controls (P = 0.0477, P = 0.0123, respectively; Figure 4D,E). Consistent with more severe reactive gliosis, forebrain samples of APP/PS1‐ligation mice had higher levels of inflammatory factors including IL‐1β and IL‐6 than APP/PS1‐sham mice (P = 0.0432, P = 0.0464, respectively) and WT‐ligation mice (P = 0.0238, P = 0.0231, respectively; Figure 4D,F).
Figure 4.
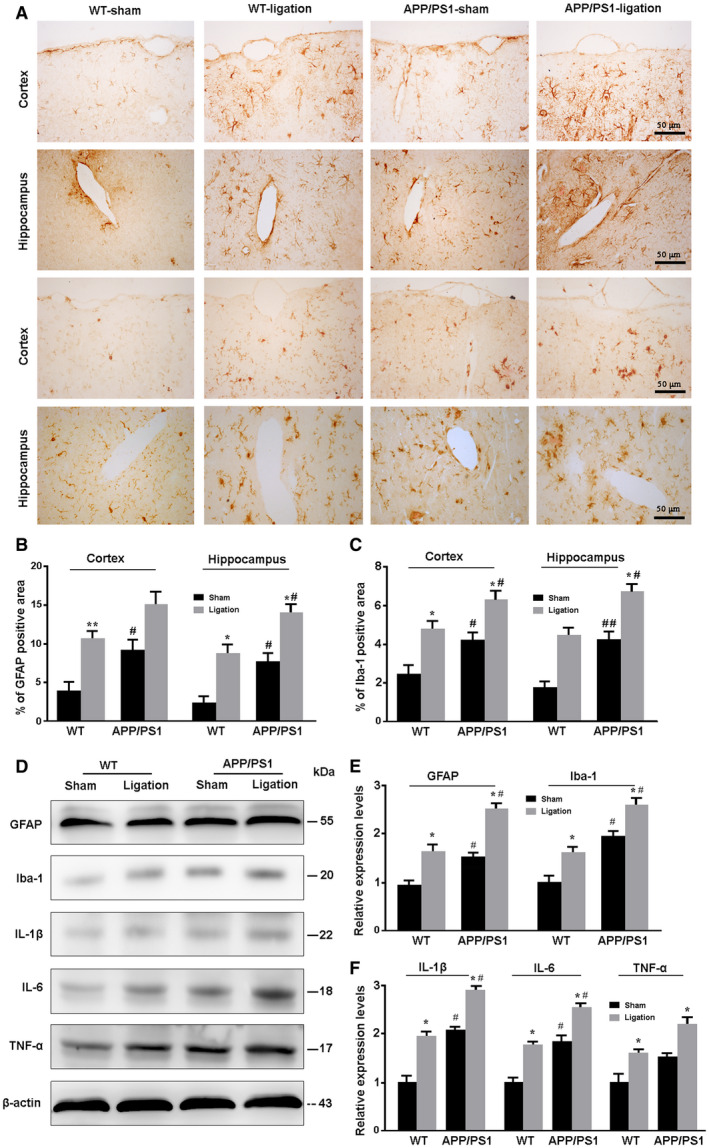
Analysis of reactive gliosis and neuroinflammatory response in the brain. A. Representative images showing expression and distribution of GFAP‐positive astrocytes and Iba‐1 positive microglia in the cortex and hippocampus of mice. B, C. The percentage of GFAP (B) and Iba‐1 (C) positive area in the cortex and hippocampus, respectively. D. Representative Western blot bands for GFAP, Iba‐1, IL‐1β, IL‐6 and TNF‐α in the brain of mice. E, F. Densitometry analysis of the expression levels of GFAP and Iba‐1 (E), and IL‐1β, IL‐6 and TNF‐α (F). Data represent mean ± SEM from four mice per group and analyzed by the two‐way ANOVA with Tukey’s post hoc test. Figure 4B: Ligation: cortex, F1,12 = 14.871, P = 0.002; hippocampus, F1,12 = 23.441, P = 0.000; Genotype: cortex, F1,12 = 8.336, P = 0.014; hippocampus, F1,12 = 15.396, P = 0.002; Interaction: cortex, F1,12 = 0.028, P = 0.871; hippocampus, F1,12 = 0.073, P = 0.790. Figure 4C: Ligation: cortex, F1,12 = 23.369, P = 0.000; hippocampus, F1,12 = 21.708, P = 0.001; Genotype: cortex, F1,12 = 12.752, P = 0.004; hippocampus, F1,12 = 19.618, P = 0.001; Interaction: cortex, F1,12 = 0.003, P = 0.955; hippocampus, F1,12 = 0.208, P = 0.656. Figure 4E: GFAP: Ligation (F1,12 = 14.982, P = 0.002); Genotype: (F1,12 = 10.334, P = 0.007); Interaction: (F1,12 = 0.413, P = 0.533); Iba‐1: Ligation (F1,12 = 12.281, P = 0.004); Genotype: (F1,12 = 24.940, P = 0.000); Interaction: (F1,12 = 0.004, P = 0.952). Figure 4F: IL‐1β: Ligation (F1,12 = 16.947, P = 0.001); Genotype: (F1,12 = 21.495, P = 0.001); Interaction: (F1,12 = 0.096, P = 0.762); IL‐6: Ligation (F1,12 = 14.939, P = 0.002); Genotype: (F1,12 = 20.409, P = 0.001); Interaction: (F1,12 = 0.072, P = 0.793); TNF‐α: Ligation (F1,12 = 14.141, P = 0.003); Genotype: (F1,12 = 11.330, P = 0.006); Interaction: (F1,12 = 0.018, P = 0.897). *P < 0.05, sham vs. LdcLNs;#P < 0.05, ##P < 0.01, WT vs. APP/PS1.
Ligation of dcLNs impairs the polarized expression of AQP4 in the mPFC and hippocampus
Serving as the most abundant aquaporin in brain, AQP4 is selectively expressed on the paravascular endfeet of astrocytes. The polarized expression pattern of AQP4 is impaired due to reactive astroglosis, subsequently resulting in glymphatic clearance malfunction 31, 68. Therefore, we investigated effects of transgenic APP/PS1 and ligation of dcLNs on expression and polarization of AQP4. Consistent with the previous studies including from our laboratory 66, impaired AQP4 polarity was observed in the cerebral cortex and hippocampus of APP/PS1 mice (Figure 5A). Ligation of dcLNs increased the percentage of positive AQP4 area in the hippocampus of WT mice (P = 0.0149, vs. WT‐sham mice) and APP/PS1 mice (P = 0.0325, vs. APP/PS1‐sham) and the cortex of WT mice (P = 0.0133, vs. WT‐sham mice; Figure 5B). Blocking dural mater lymphatic drainage in both WT and APP/PS1 mice resulted in depolarization of AQP4 in the cortex (P = 0.0233, WT‐ligation vs. WT‐sham; P = 0.0113, APP/PS1‐ligation vs. APP/PS1‐sham) and hippocampus (P = 0.0264, WT‐ligation vs. WT‐sham; P = 0.0229, APP/PS1‐ligation vs. APP/PS1‐sham; Figure 5C). Notably, AQP4 expression was markedly increased at parenchymal domains surrounding Aβ plaques, as revealed by Congo red counterstain (P = 0.0215, APP/PS1‐sham vs. WT‐sham; P = 0.0406, APP/PS1‐ligation vs. APP/PS1‐sham; Figure 5A,D). Western blot consistently confirmed a synergistic effect of transferring of APP/PS1 gene and ligation of dcLNs on upregulation of AQP4 expression in the forebrain (P = 0.0256, WT‐sham vs. APP/PS1‐sham; P = 0.0342, WT‐ligation vs. APP/PS1‐ligation) (Figure 5E,F).
Figure 5.
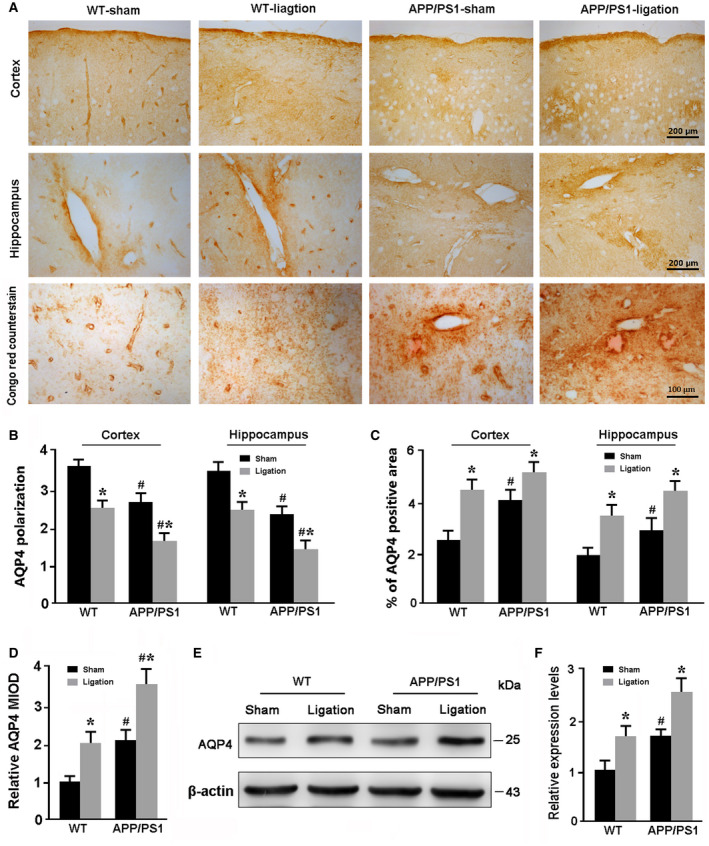
Analysis of AQP4 polarity and expression in the brain. A. Representative immunohistochemical images showing expression and distribution of AQP4 in the cerebral cortex and hippocampus. Ligation of dcLNs resulted in abnormal localization of AQP4 at parenchymal domains, especially surrounding Congo red‐positive Aβ plaques. B. Quantitative analyses of AQP4 polarization in the cerebral cortex and hippocampus. C. The percentage of AQP4 positive area in the hippocampus and cerebral cortex. D. The relative mean integrated optical density (MIOD) of AQP4 immunoreactivty at the parenchymal domains. E, F. Representative Western blot bands and densitometry analysis of Aβ1‐40, Aβ1‐42, Tau and PHF‐1 from the forebrain samples. Data represent mean ± SEM from four mice per group. The two‐way ANOVA with Tukey’s post hoc test. Figure 5B: Ligation: cortex: F1,12 = 21.317, P = 0.001; hippocampus: F1,12 = 17.706, P = 0.001; Genotype: cortex: F1,12 = 14.232, P = 0.003; hippocampus: F1,12 = 21.007, P = 0.001; Interaction: cortex: F1,12 = 0.007, P = 0.933; hippocampus: F1,12 = 0.019, P = 0.892. Figure 5C: Ligation: cortex: F1,12 = 13.991, P = 0.003; hippocampus: F1,12 = 18.548, P = 0.001; Genotype: cortex: F1,12 = 5.060, P = 0.044; hippocampus: F1,12 = 6.149, P = 0.029; Interaction: cortex: F1,12 = 1.324, P = 0.272; hippocampus: F1,12 = 0.018, P = 0.894. Figure 5D: Ligation: F1,12 = 9.198, P = 0.004; Genotype: F1,12 = 12.362, P = 0.001; Interaction: F1,12 = 0.044, P = 0.835. Figure 5F: Ligation: F1,12 = 12.913, P = 0.004; Genotype: F1,12 = 16.026, P = 0.002; Interaction: F1,12 = 0.004, P = 0.948. #P < 0.05, WT vs. APP/PS1; *P < 0.05, sham vs. LdcLNs.
Ligation of dcLNs exacerbates CD3+ T cells accumulation in the brain and dura mater of WT and APP/PS1 mice
It is now accepted that T cells routinely enter the central nervous system for immunosurveillance 12, 13, 26. The newly discovered lymphatic vessels in the dura mater offers a pathway for these immune cells to be transported out of the brain parenchyma and into the peripheral lymphatic system. In this study, we investigated the consequence of blocking dcLNs on excretion of CNS CD3+ T cells. Results showed that the percentage and absolute number of CD3+ T cells in the brain samples was no different between WT‐sham mice and APP/PS1‐sham mice (P = 0.6987, P = 0.2764, respectively). After ligation of dcLNs in both WT and APP/PS1 mice, accumulation of brain CD3+ T cells was observed in percentage and absolute number of T cells, respectively, (P = 0.0021, P = 0.0019, respectively, WT‐ligation vs. WT‐sham; P = 0.0002, P = 0.0005, respectively, APP/PS1‐ligation vs. APP/PS1‐sham), although more significant in APP/PS1 mice (P = 0.0035, P = 0.0134, respectively, APP/PS1‐ligation vs. WT‐ligation; Figure 6A,B,E). In addition, both WT and APP/PS1 mice, following ligation of dcLNs, showed accumulation of CD3+ T cells within the dura mater (P = 0.0004, P = 0.0144, respectively, WT‐ligation vs. WT‐sham; P = 0.0008; P = 0.0142, respectively, APP/PS1‐ligation vs. APP/PS1‐sham; Figure 6A,C,F), while a decrease in the percentage and absolute number of CD3+ T cells within the dcLNs was seen (P = 0.0016, P = 0.0086, respectively, WT‐ligation vs. WT‐sham; P = 0.0131, P = 0.0141, respectively, APP/PS1‐ligation vs. APP/PS1‐sham; Figure 6A,D,G).
Figure 6.
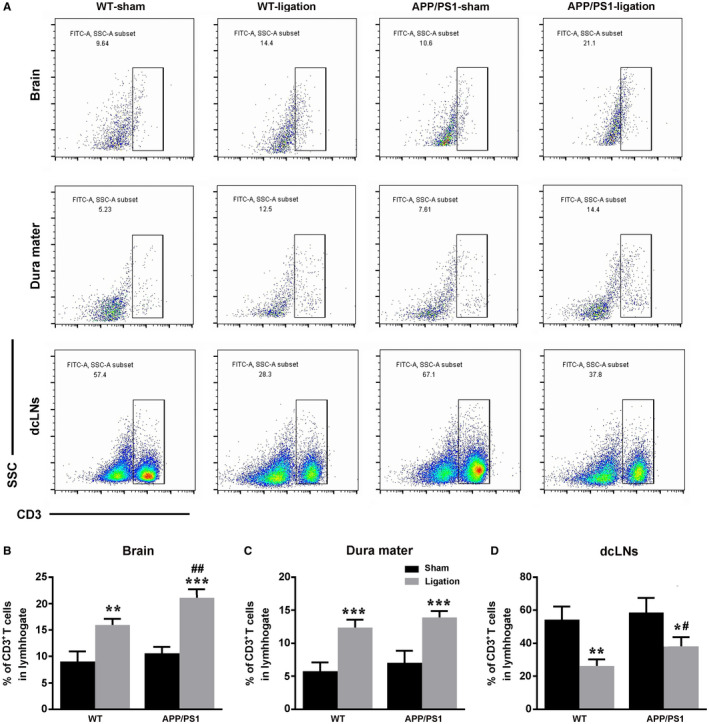
Analysis of CD3+ T cells distribution in the brain, dura mater and dcLNs of WT and APP/PS1 mice. A. Gating strategy and representative dot plots for CD3+ T cells in the brain, dura mater and dcLNs. B–D. Quantification of percentage of CD3+ T cells in the brain (B), dura mater (C) and dcLNs (D). E–G. Total number of CD3+ T cells per 10000 cells in the brain (E), dura mater (F) and dcLNs (G). All data represent mean ± SEM from four mice per group. The two‐way ANOVA with Tukey’s post hoc test. (B‐D) Brain, Ligation: F1,12 = 105.769, P = 0.000; Genotype: F1,12 = 19.259, P = 0.001; Interaction: F1,12 = 4.532, P = 0.055; Dura mater, Ligation: F1,12 = 86.871, P = 0.000; Genotype: F1,12 = 4.303, P = 0.060; Interaction: F1,12 = 0.008, P = 0.929; dcLNs, Ligation: F1,12 = 38.936, P = 0.000; Genotype: F1,12 = 5.456, P = 0.038; Interaction: F1,12 = 1.278, P = 0.280. (E‐G) Brain, Ligation: F1,12 = 73.394, P = 0.000; Genotype: F1,12 = 11.760, P = 0.005; Interaction: F1,12 = 3.595, P = 0.082; Dura mater, Ligation: F1,12 = 23.236, P = 0.000; Genotype: F1,12 = 0.795, P = 0.390; Interaction: F1,12 = 0.012, p = 0.914; dcLNs, Ligation: F1,12 = 26.388, P = 0.000; Genotype: F1,12 = 0.465, P = 0.508; Interaction: F1,12 = 0.677, P = 0.427. #P < 0.05, ##P < 0.01, ###P < 0.001, WT vs. APP/PS1; *P < 0.05, **P < 0.01, ***P < 0.001, sham vs. LdcLNs.
Ligation of dcLNs exacerbates neuronal apoptosis and synaptic protein loss in the hippocampus of APP/PS1 mice
Caspase‐3 is the key factor in the downstream cell apoptotic pathway and its expression level reflects apoptosis of neurons 34. We used immunohistochemical staining and western blot to detect and compare apoptosis of hippocampal neuron among different groups. The ratio of caspase‐3 positive apoptotic neurons in the hippocampus was no different in WT‐sham mice vs. WT‐ligation mice or APP/PS1‐sham mice, but significantly increased in APP/PS1‐ligation mice when compared to APP/PS1‐sham mice (P = 0.0393) or WT‐ligation mice (P = 0.0376; Figure 7A,B).Western blot also showed that both ligation and APP/PS1 genotype had a synergistic effect on upregulation of cleaved caspase‐3 levels (P = 0.0490, APP/PS1‐ligation vs. APP/PS1‐sham; P = 0.0217, APP/PS1‐ligation vs. WT‐ligation; Figure 7C,D).
Figure 7.
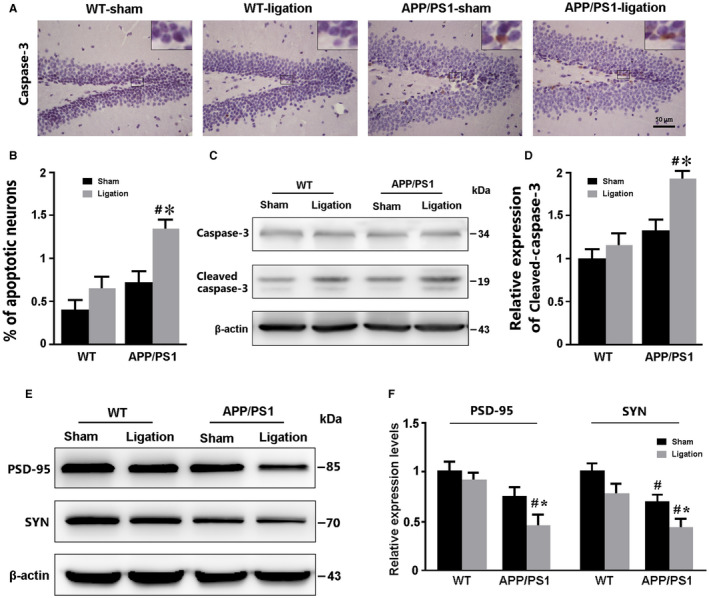
Analysis of neuron apoptosis and synaptic protein expression in the forebrain. A. Immunohistochemical staining for caspase‐3 in the DG region of the hippocampus. B. The percentages of caspase‐3‐positive apoptotic cells in the DG. C, D. Representative bands of Western blot and densitometry analysis of caspase‐3 protein levels in the forebrain samples. E, F. Representative bands of Western blot and densitometry analysis of PSD‐95 and SYN caspase‐3 protein levels in the forebrain samples. Data represent mean ± SEM from 4 mice per group. The two‐way ANOVA with Tukey’s post hoc test. Figure 6B: Ligation, F1,12 = 6.393, P = 0.026; Genotype, F1,12 = 9.095, P = 0.011; Interaction, F1,12 = 1.318, P = 0.273. Figure 7D: Ligation, F1,12 = 5.752, P = 0.034; Genotype, F1,12 = 10.925, P = 0.006; Interaction, F1,12 = 1.892, P = 0.194. Figure 7F: SYN, Ligation: F1,12 = 6.949, P = 0.022; Genotype, F1,12 = 17.290, P = 0.001; Interaction, F1,12 = 0.226, P = 0.643; PSD‐95: Ligation, F1,12 = 4.840, P = 0.048; Genotype, F1,12 = 13.798, P = 0.003; Interaction, F1,12 = 1.697, P = 0.217. #P < 0.05, WT vs. APP/PS1; *P < 0.05, sham vs. LdcLNs. #P < 0.05, WT vs. APP/PS1; *P < 0.05, sham vs. LdcLNs.
Previous studies have demonstrated that synaptic protein expression is decreased in APP/PS1 mice, and synaptic loss is highly correlated with cognitive impairment 23, 44. We further determined whether ligation of dcLNs would aggravate synaptic protein loss in APP/PS1 mice. As expected, APP/PS1‐ligation mice had low expression level of SYN and PSD‐95 in the forebrain, when compared to APP/PS1‐sham mice (P = 0.0366; P = 0.0236, respectively) and WT‐ligation mice (P = 0.0134; P = 0.0176, respectively). Additionally, APP/PS1‐sham mice showed decreases in SYN expression (P = 0.0479), but not PSD‐95 expression (P = 0.1056), relative to WT‐sham controls (Figure 7E,F).
Ligation of dcLNs impairs exploratory behavior and short‐term working memory in APP/PS1 mice
We assessed the exploratory behavior of mice using the open field test. Results revealed that APP/PS1 genotype had a significant negative effect on exploratory behavior. Ligation of dcLNs in both genotype mice exhibited decreased number of entrances into the center area (P = 0.0429, WT‐sham vs. WT‐ligation; P = 0.0066, APP/PS1‐sham vs. APP/PS1‐ligation; Figure 8A). Meanwhile, for the percentage of time spent in the center area, the decreasing effect of dcLN ligation was only observed in APP/PS1 mice (P = 0.0476, APP/PS1‐sham vs. APP/PS1‐ligation) rather than WT mice (P = 0.1201, WT‐sham vs. WT‐ligation; Figure 8B). Similarly, in the Y‐maze test that used for evaluating short‐term memory ability, APP/PS1 mice were more vulnerable to ligation of dcLN than WT mice, as revealed by the percentage of time spent in and the number of entering into and the novel arm (P = 0.0034, P = 0.0290, respectively, APP/PS1‐ligation vs. APP/PS1‐sham; P = 0.3658, P = 0.2104, respectively, WT‐ligation vs. WT‐sham; Figure 8C,D).
Figure 8.
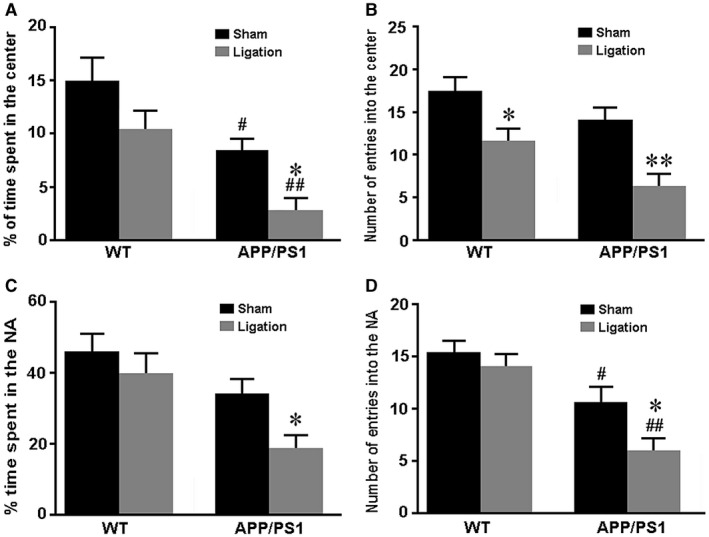
Analysis of exploratory behavior and short‐term working memory. A–C. The open field test. (A) Tracing of mouse movement during the 5‐minutes test period. (B) The percentage of time spent in the center area after tracing of mouse movement during the 10‐minutes test period. (C) The number of entries into the center area. D, E. The Y‐maze test. The percentage of time spent in the novel arm (D). The number of entries into the novel arm (E). Data represent mean ± SEM from 16 mice per group. The two‐way ANOVA with Tukey’s post hoc test. Figure 8A: Ligation, F1,60 = 5.388, P = 0.024; Genotype, F1,60 = 12.068, P = 0.001; Interaction , F1,60 = 0.029, P = 0.865; Figure 8B: Ligation, F1,60 = 5.918, P = 0.018; Genotype, F1,60 = 13.504, P = 0.001; Interaction, F1,60 = 0.112, P = 0.739; Figure 8C: Ligation, F1,60 = 4.534, P = 0.037; Genotype, F1,60 = 7.035, P = 0.010; Interaction, F1,60 = 0.511, P = 0.477; Figure 8D: Ligation, F1,60 = 5.673, P = 0.020; Genotype, F1,60 = 12.313, P = 0.001; Interaction, F1,60 = 0.096, P = 0.758. #P < 0.05; ##P < 0.01, WT vs. APP/PS1; *P < 0.05, sham vs. LdcLNs.
Discussion
Aβ and Tau aggregation triggers a series of pathological cascades in the early stages of AD. Finding effective clearance mechanisms to eliminate aggregated Aβ and Tau is thus far the fundamental strategy to combat AD 21, 22, 65. In the present study, we systematically investigated an involvement of the cerebral lymphatic system in AD‐like pathology of APP/PS1 mice. The results revealed that ligation of dcLNs, a terminal site of the central lymphatic drainage, aggravates brain Aβ and Tau accumulation, and further disturbs AQP4 polarity due to reactive astrocytes, which in turn damages glymphatic clearance function (Figure 9). This vicious cycle exacerbates secondary pathological cascades, including neuroinflammation, neuronal apoptosis, and synaptic protein loss in the hippocampus and cerebral cortex, worsening exploratory activity and memory deficits in APP/PS1 mice.
Figure 9.
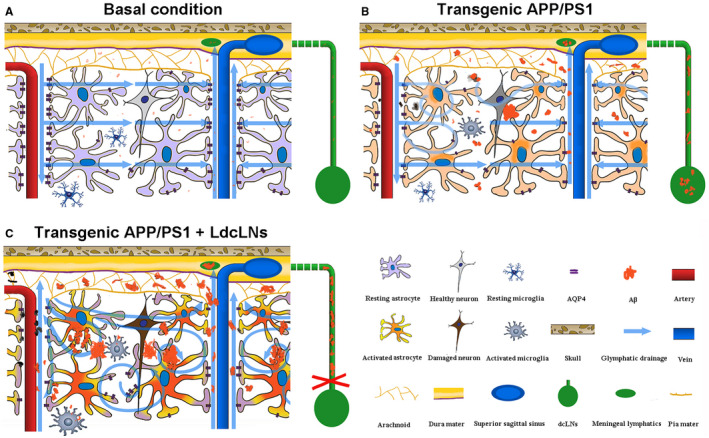
Schematic diagram of aggravated brain Aβ accumulation of APP/PS1 mice after LdcLNs. A. Under basal condition, perivascular AQP4 mediates active fluid transport from para‐arterial to para‐venous spaces facilitating drainage of wastes including Aβ from the brain parenchyma to the subarachnoid space and subsequently into the meningeal lymphatic vessels, eventually reaching the peripheral lymphatic system. B. In young APP/PS1 mice, depolarization of AQP4 caused by activated astrocytes impairs the interstitial fluid drainage, resulting in glymphatic clearance dysfunction and brain Aβ accumulation. C. Ligation of dcLNs blocks meningeal lymphatic drainage and accelerates Aβ plaque deposit within the brain, in turn causing more extensive reactive astroglosis, glymphatic dysfunction and neuron degeneration.
It is now known that the glymphatic system mediates the hydrostatic pressure driven clearance of metabolic macromolecules, including Aβ and Tau, from the brain parenchyma 30, 31, 61. ISF solutes within para‐arterial space are transported into the parenchyma and para‐venous space, then drained into the meningeal lymphatic vessels, eventually reaching the peripheral lymphatic system 31, 32, 41. In the present experiments, reduced glymphatic influx of intracisternal TR‐d3 is observed in 6‐month‐old APP/PS1 mice. Fluorescein transported into the dcLNs of APP/PS1 mice also significantly decreases, compared with that of WT mice. Furthermore, Aβ aggregation is observed within dural lymphatic microvessels of APP/PS1 mice, and blocking its excretion further increases its deposition in the brain parenchyma and dural lymphatics. These results demonstrate that impaired brain lymphatic drainage is involved in AD‐like pathology of APP/PS1 mice.
In agreement with the current results, previous experiments have reported that the decline of glymphatic clearance function already exists in the brain of young APP/PS1 mice, even before the formation of the Aβ plaques 24, 50. Previous studies have also indicated that glymphatic malfunction is associated with sustained exposure to Aβ. Aβ1‐40 microinjected into the frontal cortex causes vasoconstriction, and a decrease in the cerebral blood flow in the injected site 49. Furthermore, Aβ1‐40 injected into the cisterna magna also decreases the metabolic rate of the glymphatic system significantly 50.
Suppressive effects of Aβ on glymphatic fluid transport could be associated with delocalization of AQP4 from the endfeet to the soma of reactive astrocytes. AQP4 is selectively localized on the endfeet of astrocytes around brain microvessels. The AQP4 polarity facilitates ISF in the parenchyma to form bulk flow 31, 47. Mislocalization of AQP4 disrupts the directional transport of ISF in the brain parenchyma, subsequently resulting in macromolecules not be expelled from the brain parenchyma in time 68. It is also reported that impaired perivascular AQP4 polarity occurs in either aged or APP/PS1 mouse brains and is closely related to damages of glymphatic clearance function 24, 50. Previous studies from our laboratory have found that deletion of the AQP4 gene in APP/PS1 mice aggravates Aβ deposition and vascular amyloidosis in the brain 66. In this experiment, we observed reactive astrocytes with impaired AQP4 polarity in the cortex and hippocampus of 6‐month‐old APP/PS1 mice. These evidences have collectively revealed an involvement of impaired cerebral lymphatic drainage in the pathogenesis of AD. Further study is necessary to investigate the extent of impairment in this clearance route of human patients with different stages of AD, which would help to establish a new method for the early diagnosis of AD.
In the present study, we found that in WT mice blockage of the central lymphatic pathway via dcLN ligation causes increases in the total Tau protein and phosphorylated Tau protein in the forebrain. Previous literature reports that K14‐VEGFR3‐Ig TG transgenic mice exhibit defective phenotypes of meningeal lymphatic vessels 1. The scavenging rate of injected fluorescent macromolecule tracer from the brain parenchyma significantly decreases in the transgenic mice, while the fluorescence tracer is completely absent in the dcLNs, suggesting an important role of the meningeal lymphatic vessels in brain macromolecular clearance 1. However, dcLN ligation does not result in an increase in Aβ content in the WT mouse brain. This indicates that Tau and its phosphorylated derivatives are more dependent on the lymphatic clearance pathway, while Aβ has other clearance pathways, such as transporting through the brain–blood barrier or intracellular enzymolysis 9, 45. Previous studies have stated that impairment of the glymphatic pathway causes increases in phosphorylated Tau levels after traumatic brain injury (changes in Aβ content have not been reported), and Tau pathology become more serious in AQP4 knockout mice 30, 70.
Moreover, we demonstrated that dcLN ligation in WT mice causes activation of glial cells, accumulation of CD3+ T cells within the brain, and increases inflammatory factors. The recently identified meningeal lymphatic vessels offers a pathway for immune cells out of the brain parenchyma through the peripheral lymphatic system 42, 43, acting as a potential site for beneficial T‐cell interaction within the CNS 10. Several studies have highlighted key roles for T cells in healthy brain functions. For example, T cells have been shown to deliver a neuroprotective function via the production of neurotrophins 46, 58. T cells area also capable of modulating glutamate release by astrocytes and microglia 15, 59, protecting neurons from degeneration 37. Conversely, its accumulation may cause adverse effects. It is reported that removal of the dcLNs would interrupt the normal flow of meningeal T cells, thereby resulting in cognitive malfunction, including spatial learning difficulties and memory impairment in mice 55. Consistently, our experimental results show that chronic blockage of dcLNs causes accumulation of CD3+ T cells within the brain and dura mater, while reduced CD3+ T cell numbers within the dcLNs. Moreover, we have found that ligation of dcLNs mildly impairs the exploratory behavior of WT mice, revealed by decreased number of entering into the central area during open field testing. It has been reported that a loss of T cells in immunocompromised mice would likely influence other immune system cells that reside within the meninges 55. Meningeal myeloid cells acquire a pro‐inflammatory phenotype in the absence of T cells, and these myeloid cells may produce cytokines such as IL‐1β, IL‐12 and TNF‐α 8, 35. The specific mechanism resulting in a negative effect of accumulated T cells in the brain still needs further investigation.
Apart from maintaining healthy brain functions, T cells seem to be involved in AD pathogenesis. Interestingly, early literature reported that AD patients with severe dementia showed improvement following transposition of the omentum to their brain 16, 17, 18. Physiological characteristics of the omentum include restoration of lymphatic continuity, improving blood vessels growth and secretion of biological substances, could result in beneficial effects on AD, although the exact mechanisms is not clear 17, 18. Subsequent studies reported an increase in CD8+ cytotoxic T‐cells and CD4+ T helper cells in the brain parenchyma of patients with AD 60. Furthermore, there is increasing evidence for a potential involvement of peripheral T cell subsets in AD 2, 7, 36, 39, 57. A decrease of CD4+CD25 high T cells, an increase in CD4+Foxp3+ Tregs, but no changes in CD8+ T cell subsets, have been observed in Alzheimer’s dementia 39, 57. Recently, a case control study reported an increase in circulating T helper 17 cells in the early stages of AD and an association of CD4+ CD127 low CD25+ regulatory T cells (Trges) with neurodegeneration marker Tau 52. In animal experiments, Tregs are shown to slow the AD‐like progression and restore cognitive function of APP/PS1 mice 7. By contrast, in 5×FAD transgenic mice, the depletion of Foxp3+ Tregs increases Aβ clearance and greatly ameliorates cognitive decline 36. In the present study, we found that the percentage and absolute number of CD3+ T cells in brain samples was no different between WT mice and APP/PS1 mice under intact condition, but increased significantly in APP/PS1 mice compared to WT controls after ligation of dcLNs. This indicates that in AD pathology, T cells flow back from the meninges to the parenchyma of the brain follow blockage of their drainage path into dcLNs. Disrupted BBB integrity is assumed to be a critical factor for peripheral T cells into the CNS parenchyma under certain pathological conditions, including AD 51. However, a recent study reported that chronic, partial microglia depletion is sufficient for peripheral macrophages readily engrafting the brain, independent of the BBB opening after irradiation 6. Taken together, further studies are necessary to precisely disclose the interplay between the CNS environment and the peripheral immune system, which could provide an effective new strategy to treat AD by targeting peripheral immune cell subsets.
In the present study, we systematically analyzed effects of blocking cerebral lymphatic drainage on the AD‐like pathological process in APP/PS1 mice. The results show that accumulation of Aβ in both meningeal lymphatic vessels and brain parenchyma is more serious in dcLN ligated APP/PS1 mice, resulting with various pathophysiological changes, including aggregated neuroinflammatory reaction, loss of synaptic protein and impairment of exploratory behavior and working memory. We observed that after ligation of dcLNs, the percentage of positive area for GFAP increased, along with impaired AQP4 polarity in the cortex and hippocampus of APP/PS1 mice. As mentioned above, accumulated Aβ and phosphorylated Tau disrupts selective expression pattern of AQP4 in microvascular astrocyte processes 30, 50, 70, which in turn hampers clearance of Aβ and phosphorylated Tau via the glymphatic system. The vicious cycle between abnormal aggregation of neurotoxic macromolecule and impairment of glymphatic clearance undoubtedly facilitates AD progression.
In summary, these results demonstrate an interaction of impairments between intracranial and extracranial lymphatic clearance components in promoting AD‐like pathology of APP/PS1 mice. These findings suggest protecting brain lymphatic clearance function will delay, and even prevent, abnormal aggregation of brain Aβ and Tau, thus serving a novel strategy for prevention and treatment of AD.
Supporting information
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81671070 and 81772454). All authors have no conflict of interest to declare.
References
- 1. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M et al (2015) A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A et al (2015) Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun 6:7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beeton C, Chandy KG (2007) Isolation of mononuclear cells from the central nervous system of rats with EAE. J Vis Exp 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao M, Pu T, Wang L, Marshall C, He H, Hu G et al (2017) Early enriched physical environment reverses impairments of the hippocampus, but not medial prefrontal cortex, of socially‐isolated mice. Brain Behav Immun 64:232–243. [DOI] [PubMed] [Google Scholar]
- 5. Chun H, Lee CJ (2018) Reactive astrocytes in Alzheimer’s disease: a double‐edged sword. Neurosci Res 126:44–52. [DOI] [PubMed] [Google Scholar]
- 6. Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E et al (2018) Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med 215:1627–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dansokho C, Ait Ahmed D, Aid S, Toly‐Ndour C, Chaigneau T, Calle V et al (2016) Regulatory T cells delay disease progression in Alzheimer‐like pathology. Brain 139:1237–1251. [DOI] [PubMed] [Google Scholar]
- 8. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deane R, Bell RD, Sagare A, Zlokovic BV (2009) Clearance of amyloid‐beta peptide across the blood‐brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets 8:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR et al (2010) Regulation of learning and memory by meningeal immunity: a key role for IL‐4. J Exp Med 207:1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DosSantos Picanco LC, Ozela PF, de Fatima de Brito Brito M, Pinheiro AA, Padilha EC, Braga FS et al (2018) Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr Med Chem 25:3141–3159. [DOI] [PubMed] [Google Scholar]
- 12. Engelhardt B, Ransohoff RM (2005) The ins and outs of T‐lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol 26:485–495. [DOI] [PubMed] [Google Scholar]
- 13. Engelhardt B, Ransohoff RM (2012) Capture, crawl, cross: the T cell code to breach the blood‐brain barriers. Trends Immunol 33:579–589. [DOI] [PubMed] [Google Scholar]
- 14. Gadani SP, Smirnov I, Smith AT, Overall CC, Kipnis J (2017) Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med 214:285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garg SK, Banerjee R, Kipnis J (2008) Neuroprotective immunity: T cell‐derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol 180:3866–3873. [DOI] [PubMed] [Google Scholar]
- 16. Goldsmith HS, Wu W, Zhong J, Edgar M (2003) Omental transposition to the brain as a surgical method for treating Alzheimer’s disease. Neurol Res 25:625–634. [DOI] [PubMed] [Google Scholar]
- 17. Goldsmith HS (2004) The evolution of omentum transposition: from lymphedema to spinal cord, stroke and Alzheimer’s disease. Neurol Res 26:586–593. [DOI] [PubMed] [Google Scholar]
- 18. Goldsmith HS (2014) Benefit of omental blood flow in Alzheimer’s disease: effect on deteriorating neurons. J Alzheimers Dis 42(Suppl 3):S277–S280. [DOI] [PubMed] [Google Scholar]
- 19. Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI et al (1998) Glial‐neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol 8:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Zimmer VC, Hartmann T (2013) Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer’s disease. Front Aging Neurosci 5:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardy J (2006) Amyloid double trouble. Nat Genet 38:11–12. [DOI] [PubMed] [Google Scholar]
- 22. Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- 23. Haroutunian V, Katsel P, Roussos P, Davis KL, Altshuler LL, Bartzokis G (2014) Myelination, oligodendrocytes, and serious mental illness. Glia 62:1856–1877. [DOI] [PubMed] [Google Scholar]
- 24. Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA et al (2011) Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 121:431–443. [DOI] [PubMed] [Google Scholar]
- 25. Heneka MT, O'Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184:69–91. [DOI] [PubMed] [Google Scholar]
- 26. Hickey WF (2001) Basic principles of immunological surveillance of the normal central nervous system. Glia 36:118–124. [DOI] [PubMed] [Google Scholar]
- 27. Hillemeyer P, White MD, Pascual DW (2002) Development of a transient CD4(+)CD8(+) T cell subset in the cervical lymph nodes following intratracheal instillation with an adenovirus vector. Cell Immunol 215:173–185. [DOI] [PubMed] [Google Scholar]
- 28. Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M et al (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378:321–330. [DOI] [PubMed] [Google Scholar]
- 29. Huang H, Wang L, Cao M, Marshall C, Gao J, Xiao N et al (2015) Isolation housing exacerbates Alzheimer’s disease‐like pathophysiology in aged APP/PS1 mice. Int J Neuropsychopharmacol 18:pyu116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L et al (2014) Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34:16180–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jessen NA, Munk AS, Lundgaard I, Nedergaard M (2015) The glymphatic system: a beginner’s guide. Neurochem Res 40:2583–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jimenez S, Baglietto‐Vargas D, Caballero C, Moreno‐Gonzalez I, Torres M, Sanchez‐Varo R et al (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age‐dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28:11650–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Juraver‐Geslin HA, Durand BC (2015) Early development of the neural plate: new roles for apoptosis and for one of its main effectors caspase‐3. Genesis 53:203–224. [DOI] [PubMed] [Google Scholar]
- 35. Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW et al (2003) Cytokine‐induced sickness behavior. Brain Behav Immun 17(Suppl 1):S112–S118. [DOI] [PubMed] [Google Scholar]
- 36. Kipnis J, Avidan H, Caspi RR, Schwartz M (2004) Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci U S A 101:14663–14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kipnis J, Gadani S, Derecki NC (2012) Pro‐cognitive properties of T cells. Nat Rev Immunol 12:663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D et al (2014) Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76:845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D et al (2009) Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis 17:91–103. [DOI] [PubMed] [Google Scholar]
- 40. Louveau A, Da Mesquita S, Kipnis J (2016) Lymphatics in neurological disorders: a neuro‐lympho‐vascular component of multiple sclerosis and Alzheimer’s disease? Neuron 91:957–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J (2017) Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest 127:3210–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al (2010) Decreased clearance of CNS beta‐amyloid in Alzheimer’s disease. Science 330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McClean PL, Holscher C (2014) Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 76(Pt A):57–67. [DOI] [PubMed] [Google Scholar]
- 45. Miners JS, Barua N, Kehoe PG, Gill S, Love S (2011) Abeta‐degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol 70:944–959. [DOI] [PubMed] [Google Scholar]
- 46. Moalem G, Gdalyahu A, Shani Y, Otten U, Lazarovici P, Cohen IR et al (2000) Production of neurotrophins by activated T cells: implications for neuroprotective autoimmunity. J Autoimmun 15:331–345. [DOI] [PubMed] [Google Scholar]
- 47. Nakada T (2014) Virchow‐Robin space and aquaporin‐4: new insights on an old friend. Croat Med J 55:328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nishitsuji K, Hosono T, Uchimura K, Michikawa M (2011) Lipoprotein lipase is a novel amyloid beta (Abeta)‐binding protein that promotes glycosaminoglycan‐dependent cellular uptake of Abeta in astrocytes. J Biol Chem 286:6393–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C (2001) A beta‐peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 281:H2417–2424. [DOI] [PubMed] [Google Scholar]
- 50. Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E et al (2016) Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol Dis 93:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15:300–312. [DOI] [PubMed] [Google Scholar]
- 52. Oberstein TJ, Taha L, Spitzer P, Hellstern J, Herrmann M, Kornhuber J et al (2018) Imbalance of circulating T(h)17 and regulatory T cells in Alzheimer’s disease: a case control study. Front Immunol 9:1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prox J, Rittger A, Saftig P (2012) Physiological functions of the amyloid precursor protein secretases ADAM10, BACE1, and presenilin. Exp Brain Res 217:331–341. [DOI] [PubMed] [Google Scholar]
- 54. Qiu WQ, Folstein MF (2006) Insulin, insulin‐degrading enzyme and amyloid‐beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging 27:190–198. [DOI] [PubMed] [Google Scholar]
- 55. Radjavi A, Smirnov I, Derecki N, Kipnis J (2014) Dynamics of the meningeal CD4(+) T‐cell repertoire are defined by the cervical lymph nodes and facilitate cognitive task performance in mice. Mol Psychiatry 19:531–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sagare AP, Bell RD, Zlokovic BV (2012) Neurovascular dysfunction and faulty amyloid beta‐peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG et al (2010) PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 21:927–938. [DOI] [PubMed] [Google Scholar]
- 58. Serpe CJ, Byram SC, Sanders VM, Jones KJ (2005) Brain‐derived neurotrophic factor supports facial motoneuron survival after facial nerve transection in immunodeficient mice. Brain Behav Immun 19:173–180. [DOI] [PubMed] [Google Scholar]
- 59. Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X et al (2005) Protective autoimmunity: interferon‐gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem 92:997–1009. [DOI] [PubMed] [Google Scholar]
- 60. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M et al (2002) Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol 124:83–92. [DOI] [PubMed] [Google Scholar]
- 61. Weller RO, Djuanda E, Yow HY, Carare RO (2009) Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol 117:1–14. [DOI] [PubMed] [Google Scholar]
- 62. Wu HY, Kuo PC, Wang YT, Lin HT, Roe AD, Wang BY (2018) β‐Amyloid induces pathology‐related patterns of tau hyperphosphorylation at synaptic terminals. J Neuropathol Exp Neurol 77:814–826. [DOI] [PubMed] [Google Scholar]
- 63. Wyss‐Coray T (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 12:1005–1015. [DOI] [PubMed] [Google Scholar]
- 64. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M et al (2013) Sleep drives metabolite clearance from the adult brain. Science 342:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xin SH, Tan L, Cao X, Yu JT, Tan L (2018) Clearance of amyloid beta and tau in Alzheimer’s disease: from mechanisms to therapy. Neurotox Res. 34:733–748. [DOI] [PubMed] [Google Scholar]
- 66. Xu Z, Xiao N, Chen Y, Huang H, Marshall C, Gao J et al (2015) Deletion of aquaporin‐4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol Neurodegener 10:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang C, Huang X, Huang X, Mai H, Li J, Jiang T et al (2016) Aquaporin‐4 and Alzheimer’s disease. J Alzheimers Dis 52:391–402. [DOI] [PubMed] [Google Scholar]
- 68. Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LM, Nilsson LN et al (2011) Loss of astrocyte polarization in the Tg‐ArcSwe mouse model of Alzheimer’s disease. J Alzheimers Dis 27:711–722. [DOI] [PubMed] [Google Scholar]
- 69. Yoon SS, Jo SA (2012) Mechanisms of amyloid‐beta peptide clearance: potential therapeutic targets for Alzheimer’s disease. Biomol Ther (Seoul) 20:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhao ZA, Li P, Ye SY, Ning YL, Wang H, Peng Y et al (2017) Perivascular AQP4 dysregulation in the hippocampal CA1 area after traumatic brain injury is alleviated by adenosine A2A receptor inactivation. Sci Rep 7:2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials