

Abstract
Clear cell meningioma (CCM) is a rare grade II histopathological subtype that usually occurs in young patients and displays high recurrence rate. Germline SMARCE1 mutations have been described in hereditary forms of this disease and more recently in small syndromic and sporadic CCM series. The diagnostic value of SMARCE1 in distinguishing between CCM and other meningioma variants has not been yet established. The aim of our study was to investigate the status of SMARCE1 in a series of CCMs and its morphological mimickers. We compared the performance of an anti‐SMARCE1 antibody and the molecular analysis of the SMARCE1 gene in a retrospective multicenter series of CCMs. All CCMs lossed SMARCE1 immunoexpression. Bi‐allelic inactivating events were found by NGS‐based sequencing in all of these cases, except for one, which was incompletely explored, but had a wild‐type sequence. We then validated the anti‐SMARCE1 antibody specificity by analyzing additional 305 pediatric and adult meningiomas of various subtypes and 15 non‐meningioma clear cell tumors by SMARCE1 immunohistochemistry. A nuclear immunostaining was preserved in all other meningioma variants, as well as non‐meningioma clear cell tumors. In conclusion, our series showed, for the first time, that SMARCE1 immunostaining is a highly sensitive biomarker for CCM, useful as a routine diagnostic biomarker.
Keywords: clear cell meningioma, microcystic meningioma, SMARCE1
INTRODUCTION
Meningiomas are the most frequent tumors of the central nervous system (CNS), representing 35.5% of all primary CNS tumors 12. They exhibit 13 different histopathological variants and are divided into three grades (grade I, II, and III) 23. Among them, CCM is a rare aggressive variant corresponding to grade II 23. Histopathologically, CCM may be difficult to distinguish from other morphological subtypes, especially grade I microcystic meningioma (MM) or other clear cell tumors 23.
The tumorigenesis of meningiomas, derived from arachnoid cells, depends on various germline and somatic gene mutations. The most common are germline NF2 mutations, responsible for neurofibromatosis type 2 (NF2) 24, 38. Two genes (SMARCB1 and SMARCE1), encoding two proteins of the SWI/SNF complex (BAF47 and SMARCE1), have also been recently implicated in hereditary meningiomatosis (1, 2, 4, 5, 7, 8, 9, 10, 12, 13, 14, 16, cf. review in 48). Over the last three years, germline mutations in the SMARCE1 gene have been described in 20 unrelated families (4, 12, 13, 14, cf. review in 48). These familial forms are linked to inherited germline mutations, mainly in exon 6 38, with inactivation of the wild‐type allele in the tumor cells, according to the Knudson hypothesis 20. SMARCE1‐deficient meningiomas are of only the clear cell subtype (4, 13, 14, 15, cf. review in 48). In contrast, tumors of NF2 and SMARCB1‐related meningiomas are morphologically heterogeneous with exceptional CCMs carrying NF2 mutations (1, 2, 3, 9, 10, 13, cf. review in 48). The role of the loss of SMARCE1 function in sporadic CCM has not yet been studied, but other SWI/SNF subunits (encoded by SMARCA2, ARID1A, PBRM1) have already been shown to be associated with other clear cell tumors, for example ovarian clear cell carcinoma 19, 33, 47.
In the literature, SMARCE1‐deficient CCMs have been particularly described in children (approximately 36% of reported CCMs, from 8 months to 17 years of age) (2, 4, 6, 8, 9, 11, 14, 17, 20, 23, 26, 27, 28, 29, 30, 32, 45, cf. review in 48) and young adults (approximately 21% of reported CCMs, from 18 to 30 years of age) (21, 27, 28, 29, 49, cf. review in 48). A previous series of 72 pediatric meningiomas has also showed that clear cell subtype (7% of related cases) is much more common in children than adults proportionately 44. This is consistent with the low mean (30.9 years) and median (26 years) age of patients with CCM (from 8 months to 88 years) relative to the median age of meningioma in general (65 years) 23. In the literature, the female‐to‐male ratio of patients with CCM was 1.1 (124 females and 110 males) (2, 4, 5, 7, 9, 10, 11, 13, 14, 17, 18, 21, 22, 26, 27, 30, 32, 34, 35, 36, 37, 38, 39, 41, 46, cf. review in 48). This characteristic is in contrast with classical meningioma epidemiology, showing a high frequency in females, particularly after menopause 23.
The existence of bi‐allelic alterations of SMARCE1 in some cases of clear cell meningiomatosis suggests that these alterations are oncogenic drivers in CCM, but this has not been tested in a large cohort of diverse histopathological variants of sporadic meningioma. Smith et al. tested 8 cases and identified 6 bi‐allelic alterations including 3 new mutations in exons 3 and 10 39. In this retrospective monocentric Asian cohort, a good correlation was found between immunohistochemical loss of SMARCE1 and SMARCE1 molecular alterations.
The aim of our study was to perform a comparative immunohistochemical and molecular analysis of the status of SMARCB1 and SMARCE1, as well as a molecular investigation of the NF2 gene, in a retrospective Caucasian multicenter series of CCMs to determine whether the status of SMARCE1 is the same in adult or pediatric CCMs, and therefore, whether it represents a specific diagnostic biomarker of CCM.
MATERIALS AND METHODS
Clear cell and microcystic meningioma study cohort
Patients
A total of 27 cases (all surgical specimens) were retrieved from the consultation archive database (1982–2016) of the Sainte‐Anne and Lariboisière Hospital pathology departments.
This retrospective and multicenter study included 27 tumors from 26 patients who underwent surgery at the Sainte‐Anne (n = 8), Lariboisière (n = 8), Mondor (n = 2), Necker (n = 3), Val‐de‐Grâce Hospitals (n = 1) or the Rothschild foundation (n = 3). One patient, originated from Nancy, was part of the national pediatric meningioma study cohort. One patient (patient 5), presenting multiple meningiomas, underwent surgery for two meningiomas. Patient characteristics and clinical data were retrieved from hospital records. The location of tumors was based on operative reports.
Histopathological review
The central pathology review was performed by two neuropathologists (ATE and PV). The tumors were classified and graded according to the 2016 WHO classification and were subtyped into MMs and CCMs 23.
Immunohistochemical study
A representative section was selected for each case. Unstained 3‐μm‐thick slides of formalin‐fixed, paraffin‐embedded tissues were obtained and submitted for immunostaining. Somatostatin receptor 2a (SSTR2) was used as a diagnostic marker for meningioma. SSTR2 (Reference ab134152, clone UMB1, dilution 1:200, monoclonal, rabbit, Abcam) was evaluated as positive (moderate or dark diffuse staining) or negative (no staining or weak staining) 28. SMARCE1 (Reference HPA003916, dilution 1:800, polyclonal, rabbit, Sigma‐Aldrich) and BAF47 (INI1) (Reference 612110, clone25/BAF47, dilution 1:200, mouse, BD Biosciences) were evaluated as positive when all nuclei were stained. Endothelial cells and lymphocytes were used as positive internal controls.
Targeted NGS genotyping of SMARCE1, SMARCB1 and NF2 genes
DNA extraction from 25 FFPE tumor samples was performed with the Maxwell® 16 FFPE Tissue LEV DNA Kit. DNA concentrations were determined using a Quant‐iT dsDNA HS assay kit and a Qubit 2.0 Fluorometer (Life Technologies, Saint‐Aubin, France).
Genotyping experiments were performed at the NGS facility of the Cochin hospital, Paris (Assistance Publique ‐ Hôpitaux de Paris, France). A custom Ampliseq panel targeting SMARCE1, NF2, SMARCB1, and SUFU genes was developed (reference IAD51599_119, ThermoFisherScientific). Genomic DNA was amplified to generate the library using the Ion AmpliSeq Library Kit 2.0 (Life Technologies). NGS library preparation, followed by amplification and purification, emulsion PCR, enrichment, loading on Ion 318™ chips, sequencing with an Ion Personal Genome Machine® (PGM™) System (Life Technologies), and data collection were performed as already described 31.
Sequence alignment was performed using the Torrent Mapping Alignment Program (TMAP, https://github.com/iontorrent/TMAP, Ion Torrent for Life Technologies), which was specifically developed to analyze Ion Torrent data. Aligned reads from .bam files were visualized using the tool Integrative Genomics Viewer v2.3 (IGV, https://www.broadinstitute.org/igv/) from the Broad Institute (Cambridge, MA).
Single nucleotide variants (SNVs) and short insertion and/or deletion detection from the bam files was performed using the Torrent Suite Variant Caller (TSVC) plugin from the Torrent Suite Software v5.0.4. (https://ioncommunity.thermofisher.com/community/products/software/torrent_suite, Life Technologies). Major calling parameters were chosen as follows to avoid false negative results: minimum sequencing depth ≥ 5X for SNVs and multiple nucleotide or complex variants and ≥ 10X for short insertions and/or deletions, minimum allele frequency (MAF) ≥ 1% for all using the TSVC.
Gene copy number analysis was performed using quantitative values (number of reads for each amplicon of each sample) of the Coverage Analysis plugin on the Ion Torrent Browser 5.0.4.0 (Life Technologies). Amplicon reads were first internally normalized for each sample: reads of target gene amplicons were individually divided by the total number of reads of the control gene amplicons. SUFU was used as the control gene for SMARCE1, NF2, and SMARCB1 copy number analysis. Normalized reads obtained for each amplicon of a sample were then divided by the average number of normalized reads of control samples for the corresponding amplicon. Copy number ratios of <0.75 were defined as deleted.
In case which blood DNA was available a constitutional mutation in SMARCE1 was looked for in peripheral blood lymphocytes.
Statistical analysis
Statistical analysis was performed using SPSS (version 20). Mean values and frequencies were used for the description of continuous and categorical variables, respectively. The degree of concordance between the results of SMARCE1 immunostaining and SMARCE1 genotyping was assessed using the Cohen's kappa coefficient. The categorization values suggested by Altman were used for interpretation of the kappa value 2.
Meningioma validation cohort
We retrieved a cohort of 305 meningiomas of various subtypes, graded according to the 2016 WHO classification, consisting of 120 large surgical meningioma samples and a TMA of 185 tumors (kindly provided by the APHM tumor bank AC‐2013‐1786/CRB number BB‐0033‐00097) 23. Twenty patients of large surgical meningioma samples were children (ages 1–16). In total, 169 WHO grade I, 95 WHO grade II, and 41 WHO grade III meningiomas were studied. Among them, the following rare histopathological variants were included: angiomatous (n = 2), secretory (n = 11), psammomatous (n = 9), metaplastic (n = 2), microcystic (n = 2), chordoid (n = 2), lymphoplasmacyte‐rich (n = 1) and rhabdoid (n = 2). The remaining meningiomas represented more common subtypes, such as meningothelial (n = 73), fibrous (n = 28), transitional (n = 54), atypical (n = 80) and anaplastic (n = 39) forms. No CCM was included in this cohort. A representative section (3‐μm‐thick slide) was selected for SMARCE1 immunostaining for each case.
Non‐meningioma clear cell CNS tumor validation cohort
We retrieved an adult and pediatric cohort of 15 clear cell mimickers from the consultation archive database of the Sainte‐Anne Hospital pathology department. The following histopathological entities were included: metastases of renal clear cell carcinoma (n = 4), hemangioblastomas (n = 4), clear cell ependymomas (n = 4), Ewing sarcoma (n = 1), choroid plexus carcinoma (n = 1) and schwannoma (n = 1). A representative section (3‐μm‐thick slide) was selected for SMARCE1 immunostaining for each case.
The study design is summarized in Figure 1.
Figure 1.
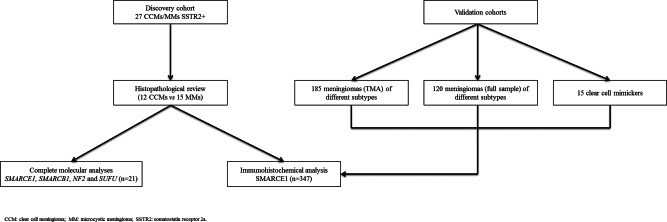
Workflow of the study.
RESULTS
Clinical data of the discovery cohort (CCM and MM)
The cohort included 22 adults (84.6%) and four children (15.4%). The mean age at surgery was 26.2 years (range, 7–72 years) for CCM and 55.2 years (range, 31–74 years) for MM. The female‐to‐male ratio was 0.8 for the CCM cohort and 6.5 (13 females and 2 males) for the MM cohort. The clinical data of patients with CCM are summarized in Table 1. Ten meningiomas (45.5%) were supratentorial, nine (40.9%) were located in skull‐base region, two (9.1%) in the posterior fossa, and one (4.5%) in the spine. Multiple meningiomas were identified in four patients, three with MMs and one with CCMs. They were found for two of the patients at diagnosis and for two during follow‐up. We obtained follow‐up data for 24 patients. Total surgery was performed on 15 patients, who had no evidence of the disease at the end of the follow‐up. The surgery was incomplete for two patients who then received adjuvant radiotherapy and were alive at the end of the follow‐up. Two patients with CCM, both children, died of the disease. No familial history was mentioned in medical records except in one patient (case 3) which had a sister with a spinal ependymoma but no more clinical and histopathological data were available.
Table 1.
Clinical data of the 11 patients with clear cell meningioma.
| Case | Age of onset (years), sex | Tumor location(s) | Resection type | Adjuvant treatment | Recurrence/progression | Recurrence interval (years) | Status at last follow‐up | OS (years) | Meningiomatosis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 13, F | Cervical C4‐C5 | GTR | 0 | 0 | NA | A | 60.0 | 0 |
| 2 | 6, M | Petrous and clivus | GTR | 0 | 1 | 1.9 | D | 5.6 | 0 |
| 3 | 18, F | Right temporal convexity | GTR | 0 | 0 | NA | A | 1.7 | 0 |
| 4 | 7, F | Petrous | STR | 0 | 1 | 2.8 | D | 6.9 | 0 |
| 5 | 23, F | Left jugum | STR | 0 | 0 | NA | A, stable residue | 15.9 | 1 |
| 5 | 26, F | Lumbar spine | GTR | 0 | 0 | NA | A | 15.9 | 1 |
| 6 | 72, M | Right trigeminal nerve | STR | 0 | 1 | 3.2 | A, stable residue | 3.2 | 0 |
| 7 | 20, F | Right pontocerebellar angle | NK | NK | NK | NK | NK | NK | NK |
| 8 | 41, M | Left pontocerebellar angle | STR | RT | 1 | 2.1 | A | 10.4 | 0 |
| 9 | 61, M | Right cavernous sinus | GTR | 0 | 0 | NA | NK | NK | 0 |
| 10 | 45, M | Right trigeminal nerve | GTR | 0 | 1 | 3.5 | A | 3.5 | 0 |
| 11 | 13, M | Petrous and cavernous sinus | STR | 0 | 1 | 1.3 | A, stable residue | 14.6 | 0 |
A: alive; D: died of progression of the disease; F: female; GTR: gross total resection; M: male; NA: not applicable; NK: not known; OS: overall survival; RT: radiotherapy; STR: subtotal resection.
Histopathological findings
The histopathological review classified 12 tumors as CCMs from 11 patients and 14 as MMs from 14 patients (Figure 2A,D). In one case, the extent of electrocoagulation artifacts did not allow discrimination between CCM and MM. One of the 14 MMs (case 21) was grade II because of high mitotic activity. All 12 CCMs were grade II, three with brain invasion (cases 3, 5 and 8), including one with high mitotic activity (case 3).
Figure 2.
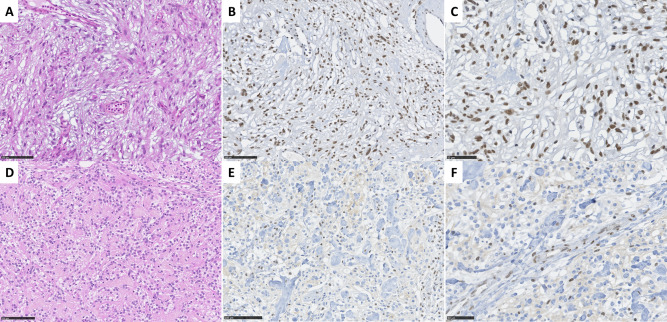
Histopathological and immunohistochemical findings. A–C a microcystic meningioma characterized by thin, elongated processes with ovoid nuclei (HES, 270×); preserved SMARCE1 nuclear staining in all tumor cells (280× and 400×). D–F a clear cell meningioma characterized by round to polygonal monotonous cells with clear cytoplasm, intermingled with thick interstitial collagen fibers (270×); loss of SMARCE1 nuclear staining in all tumor cells, note the preserved staining in endothelial cells (280× and 400×).
Immunohistochemical findings
Immunohistochemical analyses were performed on the entire cohort (27 tumors).
All meningiomas exhibited strong and diffuse SSTR2 staining with a preserved nuclear BAF47 staining. Preserved nuclear staining of SMARCE1 was present in all MMs (Figure 2B,C). In contrast, we observed a diffuse loss of nuclear SMARCE1 staining in all CCMs, which all included a strong vascular internal positive control (Figure 2E,F). To note, tumor cells often presented a weak cytoplasmic staining. The tumor with an inconclusive histopathological subtype was SMARCE1 immunoreactive (sufficient material was not available for molecular analysis). The histopathological and immunohistochemical results are summarized in Table 2.
Table 2.
Histopathological, immunohistochemical, and molecular findings of the CCM and MM study cohort.
| Case | Histopathological subtype, grade | Mitotic index (/1.6 mm2) | Loss of SMARCE1 immunoexpression | SMARCE1(NM_003079) sequence | Bi‐allelic loss of function | SMARCB1 (NM_003073) sequence | NF2 (NM_000268) sequence | SUFU (NM_016169) sequence |
|---|---|---|---|---|---|---|---|---|
| 1 | CCM, II | 1 | 1 |
Hit 1: Intron7 3′ acceptor site c.542–2A>G, p.? Hit 2: Exon 9 c.715C>T, p.(Arg239*) |
1 | WT | WT | WT |
| 2 | CCM, II | 1 | 1 | WT (CDS)/CNV: NI | NI | WT (CDS)/CNV: NI | WT (CDS)/CNV: NI | WT (CDS)/CNV: NI |
| 3 | CCM, II | 7 | 1 |
Hit 1: Exon7 c.472C>T, p.(Arg158*) Hit 2: Exon10 c.925_929delGAGCA, p.(Glu309Serfs*2) |
1 | WT | WT | WT |
| 4 | CCM, II | 1 | 1 |
Hit 1: Intron 6 5′ donor site c.369 + 1G>A, p.? Hit 2: LOH by whole gene deletion |
1 | WT | WT | WT |
| 5 | CCM, II | 0 | 1 |
Hit 1: Exon 5 c.197delC, p.(Pro66Glnfs*5) Hit 2: LOH by isodisomy |
1 |
WT (CDS) CNV: deletion |
WT (CDS) CNV: deletion |
WT |
| 5 | CCM, II | 0 | 1 |
Hit 1: Exon 5 c.197delC, p.(Pro66Glnfs*5) Hit 2: Exon 8 c.624_627delTGAG, p.(Ser208Argfs*26) |
1 | WT | WT | WT |
| 6 | CCM, II | 0 | 1 |
Hit 1: Intron 7 3′ acceptor site = c.542–1G>A, p.? Hit 2: Exon 8 c.547_548delGA, p.(Asp183*) |
1 | WT | WT | WT |
| 7 | CCM, II | 0 | 1 |
Hit 1: Exon 6 c.313C>T, p.(Arg105*) Hit 2: Intron 6 c.369 + 1G>T, p.? |
1 | WT | WT | WT |
| 8 | CCM, II | 3 | 1 |
Hit 1: Intron 6 5′ donor site c.369 + 1G>T, p.? Hit 2: Exon 7 c.458T>G, p.(Leu153*) CNV: NI |
1 | WT (CDS)/CNV: NI | WT (CDS)/CNV: NI | WT (CDS)/CNV: NI |
| 9 | CCM, II | 3 | 1 |
Hit 1: Exon 8 c.633_659delGGTGCCAGACGTTCGGTCAGTTGTCACinsT, p.(Val212Asnfs*3) Hit 2: LOH by whole gene deletion |
1 | WT |
WT (CDS) CNV: deletion |
WT |
| 10 | CCM, II | 0 | 1 |
Hit 1: Intron 6 5′ donor site c.369 + 2_+5delTAGG, p.? Hit 2: Intron 6 3′ acceptor site c.370–4delTT, p.? |
1 | WT | WT | WT |
| 12 | MM, I | 2 | 1 | WT (CDS)/CNV: NI | 0 | WT | WT | WT |
| 13 | MM, I | 0 | 0 | WT | 0 |
WT (CDS) CNV: deletion |
WT (CDS) CNV: deletion |
WT |
| 14 | MM, I | 0 | 0 | WT | 0 | WT | WT | WT |
| 15 | MM, I | 0 | 0 | WT | 0 | WT | WT | WT |
| 16 | MM, I | 1 | 0 | WT | 0 | WT | WT | WT |
| 17 | MM, I | 1 | 0 | WT | 0 | WT | WT | WT |
| 18 | MM, I | 1 | 0 | WT (CDS)/CNV: NI | 0 |
WT (CDS) CNV: deletion |
WT (CDS) CNV: deletion |
WT |
| 19 | MM, I | 0 | 0 | WT | 0 |
WT (CDS) CNV: deletion |
Hit 1: Intron 11 c.1122 + 1G>C Hit 2: LOH by isodisomy (absence of deletion) |
WT (CDS) CNV: deletion |
| 20 | MM, I | 0 | 0 | WT | 0 |
WT (CDS) CNV: deletion |
WT (CDS) CNV: deletion |
WT (CDS) CNV: deletion |
| 21 | MM, II | 8 | 0 | WT | 0 | WT | WT | WT |
| 22 | MM, I | 2 | 0 | WT | 0 | WT | WT | WT |
| 23 | MM, I | 1 | 0 | WT | 0 |
WT (CDS) CNV: deletion |
WT (CDS) CNV: deletion |
WT |
| 24 | MM, I | 2 | 0 | WT | 0 | WT | WT | WT |
CCM: clear cell meningioma; CDS: coding sequence; CNV: cony number variation analysis; MM: microcystic meningioma; NI: noninterpretable; WT: wild‐type (coding sequence and copy number).
Targeted next generation sequencing (NGS) screening
Sufficient material for NGS‐based sequencing was available for 25 meningiomas. Among them, we were able to perform a complete gene copy number analysis on 21 meningiomas (10 CCMs and and 11 MMs) due to the expected inconsistent quality of DNA extracted from the FFPE samples. The genotyping results are summarized in Figure 3 and Table 2.
Figure 3.
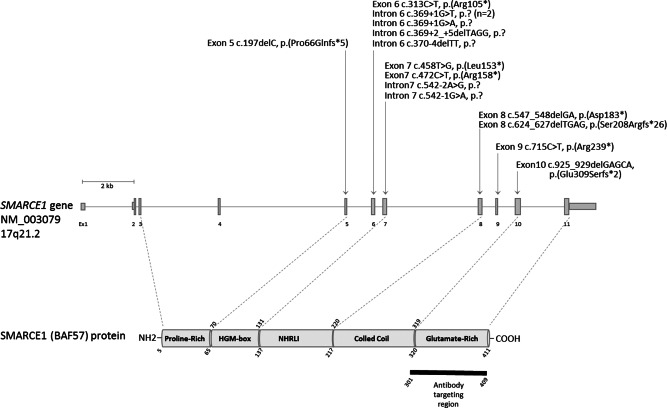
Schematic representation of the SMARCE1 gene and the corresponding encoded protein. Exons are depicted as vertical bars and numbered from exon 1 to exon 11. The position of the SMARCE1 variants identified in this study are indicated above the gene.
We observed no mutational SMARCE1 hits in any of the 11/11 MMs tested, whereas there were two losses of function SMARCE1 hits in all SMARCE1‐negative CCMs (10/10). By sequencing, SMARCE1 alterations were predicted to induce loss of function of the protein, with three nonsense, six frameshift and six splice variants. We also observed LOH by gene deletion for only two CCMs and copy neutral LOH for one tumor. SMARCE1 mutations did not always occur to the exclusion of NF2/SMARCB1 alterations. We observed the co‐occurrence of a NF2 deletion alone, or combined with a SMARCB1 deletion, in two SMARCE1‐mutated CCMs. We also identified NF2 whole gene deletions in conjunction with a SMARCB1 deletion in 4/11 MMs. Of note, we were able to identify an exon‐intron boundary NF2 mutation c.1122 + 1G > C in one of these four MMs. Only one patient (case 5) could be analyze for germline DNA: germline mutation of SMARCE1 was evidenced.
We did not observe point mutation of SUFU gene but mono‐allelic deletions were identified in two SMARCB1‐NF2‐deleted MMs.
Statistical analyses
All SMARCE1 negative CCMs presented a bi‐allelic SMARCE1 alteration. The correlation coefficient (Cohen's kappa coefficient) for these methods was excellent (1.00).
Meningioma and clear cell non‐meningioma validation cohorts
All 305 other various subtypes of meningiomas (100%) and 15 (100%) clear cell mimicking tumors were diffusely immunoreactive for SMARCE1.
DISCUSSION
Four meningioma variants, such as clear cell, chordoid (grade II), and papillary and rhabdoid (grade III) recur more frequently than others. The distinction between CCM from other variants is thus important in deciding on patient management and establishing a prognosis. Based on our extensive literature review of 234 CCMs (cf. Table S1 Supporting Information), we estimate that 45% of patients with CCM had tumor recurrence (local in 84%, local and other distant locations in 11%, and only distant location in 5% of reported cases) during the mean follow‐up of 45 months (3, 5, 6, 8, 10, 11, 12, 14, 15, 18, 19, 22, 23, 27, 29, 32, 34, 36, 37, 38, 39, 40, 41, 43, cf. review in 48). The locations of recurrence were distant intracranial in 40% of cases, distant neuraxis in 53% of cases and systemic (pulmonary) in 7% of cases (18, 26, cf. review in 48). This strongly contrasts with the recurrence rate of about 7–25% for benign meningiomas 23. Moreover, a retrospective literature analysis showed only 20 cases of multiple CCMs at diagnosis or during follow‐up (20/234, 8.5%; 8.3% in our series). This proportion did not differ from the other histopathological subtypes (less than 10%) 23. These data suggest that the pejorative prognosis of CCM is not due to a higher frequency of meningiomatosis but rather to an intrinsic aggressivity. In light of the literature data, the proportion of hereditary forms of CCM versus sporadic cases still needs to be elucidated. Smith et al. have showed over the past years the presence of SMARCE1 alterations in familial cases of CCM 39, 40, 41. Moreover, given the paucity of adjuvant treatment administered in our retrospectively discovery cohort, strong conclusions about prognosis cannot be made.
In our study, two thirds of the identified SMARCE1 molecular variants are predicted to cause the truncation or absence of SMARCE1 protein, well explaining the negative SMARCE1 staining, targeting the COOH terminal amino acids 301––409. We were unable to investigate the functional consequences of the remaining third of SMARCE1 variants, corresponding to splice variants. However, immunohistochemical analysis clearly showed that the corresponding proteins are likely truncated or absent. The loss of SMARCE1 protein expression appears to be a sensitive and specific test for the diagnosis of CCM, as SMARCE1 immunostaining was lost in 12/12 (100%) of the CCMs in this study. These results confirm the data of Smith et al. in a smaller series of syndromic and sporadic CCMs 39. Our study constitutes the first proof of the diagnostic value of the loss of SMARCE1 expression by validation on a cohort of 320 adult and pediatric meningiomas of various grades and subtypes. SMARCE1 immunostaining could be routinely used to aid diagnosis as it is easy to interpret with a positive internal nuclear control in endothelial and inflammatory cells. As the expression was homogeneous and easy to interpret, it was lawful to analyze this staining on TMA. In case of artifacts (as one case of our study) or if meningioma subtyping problems due to tumor heterogeneity (as two tumors of Smith's study), SMARCE1 immunoexpression permit to reclassified the meningioma variant 39. Furthermore, none of the morphological mimickers of CCMs tested displayed a loss of SMARCE1 expression. Moreover, the loss of expression correlates well with the molecular status of SMARCE1 (Cohen's kappa coefficient was 1.00). However, we were unable to identify the SMARCE1 gene mutations in a single case of SMARCE1‐negative CCM. This discrepancy may due to the low quality of the DNA due to long‐term storage of FFPE samples (more than 30 years), making it impossible to assess the gene copy number. This may also be explained by the presence of a genetic alteration in non‐coding sequences or a post‐translational alteration, underlining the usefulness of protein expression testing in the diagnosis.
The epidemiological characteristics of our series are in agreement with the literature data, showing the lower CCM patients mean age (26.2 years) than patients with “non clear cell” meningiomas (53.1 years) (P < 0.001). The female‐to‐male ratio (= 3.7) was also significantly higher than CCM patients (P = 0.019). The literature review showed that CCM is mainly located in the spine (26% of reported pediatric cases) (8, 10, 11, 14, 15, 18, 19, 22, 23, 25, 27, 29, 34, 36, 37, 38, 40, 43, cf. review in 48) or posterior fossa (49% of reported pediatric cases) (5, 6, 32, 36, 37, 39, 43, cf. review in 48). The anatomical distribution of reported CCM cases according to the age of patients is summarized in Figure S1 (Supporting Information). Our series is not consistent with the literature concerning the location of the CCMs, with a high proportion at the base of the skull (58%). This discrepancy is possibly due to the fact that some of our neurosurgical teams are more implicated in skull‐base and cranial surgery than surgery of the spine.
In light of these data, we suggest genetic SMARCE1 testing and counseling for patients with a familial history of CCM or CCM occurring in young patients (<26 years), particularly if the tumor is located in the posterior fossa or the spine. Although this tumor is rarely fatal (only 5% of the reported patients died of their disease and 8% in our series, all children) (6, cf. review in 48), the presence of this alteration may alter the management of these patients, with close radiological monitoring (magnetic resonance imaging –MRI‐ of the brain and the spine) to prevent the apparition of further tumors. In our series, a germline mutation of SMARCE1 was identified in a young patient (case 5) with multiple meningiomas.
Somatic misactivation of the Hedgehog pathway is reported in meningioma 10, 42. Moreover, a germline SUFU gene variant has been reported in a large family presenting multiple meningiomas 1. Hence, SUFU gene was considered in our NGS custom design. No SUFU point mutation was identified in CCMs. We identified only copy number alterations in a subset of SMARCB1 and NF2‐deleted MMs. The consequence of the mono‐allelic loss of SUFU gene in the tumorigenesis of these MMs remains to be studied. Likewise, we have showed that SMARCE1 mutations identified in CCMs were not exclusive of NF2/SMARCB1 alterations as co‐occurrence of NF2 solely or combined to a SMARCB1 deletion was observed in two SMARCE1‐mutated CCMs. How mono‐allelic inactivation of NF2 (and SMARCB1) contribute to the CCM tumorigenesis remains to be investigated.
In summary, our study showed that the loss of SMARCE1 expression is a useful tool, sensitive and specific for diagnosing CCM.
DECLARATION OF INTERESTS
All authors declare they have no conflicts of interest related to this work.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Clinical and molecular data of reported cases of clear cell meningiomas in the literature.
Figure S1. Anatomical distribution of reported clear cell meningiomas (CCMs) in the literature according to age: most pediatric tumors are located in the posterior fossa and the spine, whereas adult tumors are mainly cranial and in the posterior fossa.
ACKNOWLEDGEMENTS
The authors thank all of the laboratory technicians in the Department of Pathology, Sainte‐Anne and Lariboisière Hospitals and the biobank of brain tumors of Necker Hospital. The authors thank the pathologist Dr Guillaume Gauchotte from Nancy Hospital who provided a case of clear cell meningioma.
REFERENCES
- 1. Aavikko M, Li S‐P, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E et al (2012) Loss of SUFU function in familial multiple meningioma. Am J Hum Genet 91:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altman D (1991) Practical Statistics for Medical Research. Chapman, London. [Google Scholar]
- 3. Antinheimo J, Haapasalo H, Haltia M, Tatagiba M, Thomas S, Brandis A et al (1997) Proliferation potential and histological features in neurofibromatosis 2‐associated and sporadic meningiomas. J Neurosurg 87:610–614. [DOI] [PubMed] [Google Scholar]
- 4. Anunobi CC, Bankole O, Ikeri NZ, Adeleke NA (2016) Suprasellar clear cell meningioma in an infant. Sultan Qaboos Univ Med J 16:e364–e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bacci C, Sestini R, Provenzano A, Paganini I, Mancini I, Porfirio B et al (2010) Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics 11:73–80. [DOI] [PubMed] [Google Scholar]
- 6. Ben Nsir A, Ben Hamouda K, Hammedi F, Kilani M, Hattab N (2016) Osteolytic clear cell meningioma of the petrous bone occurring 36 years after posterior cranial fossa irradiation: Case report. Neurol Neurochir Pol 50:297–302. [DOI] [PubMed] [Google Scholar]
- 7. Benchetritt M, Hofman V, Long E, Odin G, Basc E, Pasquier B et al (2008) Primary clear cell meningioma of the orbit mimicking a metastatic carcinoma: usefulness of immunohistochemistry and cytogenetic analysis. Virchows Arch Int J Pathol 452:209–213. [DOI] [PubMed] [Google Scholar]
- 8. Carrà S, Drigo P, Gardiman M, Perilongo G, Rigobello L (2003) Clear cell meningioma in a 22‐month‐old male: update after five years. Pediatr Neurosurg 38:162–163. [DOI] [PubMed] [Google Scholar]
- 9. Christiaans I, Kenter SB, Brink HC, van Os T. a M, Baas F, van den Munckhof P et al (2011) Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet 48:93–97. [DOI] [PubMed] [Google Scholar]
- 10. Clark VE, Erson‐Omay EZ, Serin A, Yin J, Cotney J, Ozduman K et al (2013) Genomic analysis of non‐NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339:1077–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Colen CB, Rayes M, McClendon J, Rabah R, Ham SD (2009) Pediatric spinal clear cell meningioma. Case report. J Neurosurg Pediatr 3:57–60. [DOI] [PubMed] [Google Scholar]
- 12. Dolecek TA, Propp JM, Stroup NE, Kruchko C (2012) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro‐Oncol 14:v1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dubois A, Sévely A, Boetto S, Delisle MB, Manelfe C (1998) Clear‐cell meningioma of the cauda equina. Neuroradiology 40:743–747. [DOI] [PubMed] [Google Scholar]
- 14. Evans LT, Van Hoff J, Hickey WF, Smith MJ, Evans DG, Newman WG, Bauer DF (2015) SMARCE1 mutations in pediatric clear cell meningioma: case report. J Neurosurg Pediatr 16:296–300. [DOI] [PubMed] [Google Scholar]
- 15. Foster S, Simonson WT, Duckert LG, Upton MP, Anzai Y (2012) Clear‐cell meningioma presenting as an infiltrative external auditory canal mass. Radiol Case Rep 7:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hadfield KD, Smith MJ, Trump D, Newman WG, Evans DG (2010) SMARCB1 mutations are not a common cause of multiple meningiomas. J Med Genet 47:567–568. [DOI] [PubMed] [Google Scholar]
- 17. Holtzman RN, Jormark SC (1996) Nondural‐based lumbar clear cell meningioma. Case report. J Neurosurg 84:264–266. [DOI] [PubMed] [Google Scholar]
- 18. Jallo GI, Kothbauer KF, Silvera VM, Epstein FJ (2001) Intraspinal clear cell meningioma: diagnosis and management: report of two cases. Neurosurgery 48:218–222. [DOI] [PubMed] [Google Scholar]
- 19. Jones S, Wang T‐L, Shih I‐M, Mao T‐L, Nakayama K, Roden R et al (2010) Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330:228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knudson AG (2001) Two genetic hits (more or less) to cancer. Nat Rev Cancer 1:157–162. [DOI] [PubMed] [Google Scholar]
- 21. Ko JK, Choi BK, Cho WH, Choi CH (2011) Non‐dura based intaspinal clear cell meningioma. J Korean Neurosurg Soc 49:71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu P‐I, Liu G‐C, Tsai K‐B, Lin C‐L, Hsu J‐S (2005) Intraspinal clear‐cell meningioma: case report and review of literature. Surg Neurol 63:285–289. [DOI] [PubMed] [Google Scholar]
- 23. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella‐Branger D, Perry A, Reifenberger G, von Deimling A (2016) World Health Organization Classification of Tumors of the Central Nervous System. IARC: Lyon, pp. 232–245. [DOI] [PubMed] [Google Scholar]
- 24. Louis DN, Ramesh V, Gusella JF (1995) Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol Zurich Switz 5:163–172. [DOI] [PubMed] [Google Scholar]
- 25. Mallya V, Singh A, Sharma K (2012) Clear cell meningioma of the cauda equina in an adult. Indian J Pathol Microbiol 55:262–264. [DOI] [PubMed] [Google Scholar]
- 26. Matsui H, Kanamori M, Abe Y, Sakai T, Wakaki K (1998) Multifocal clear cell meningioma in the spine: a case report. Neurosurg Rev 21:171–173. [DOI] [PubMed] [Google Scholar]
- 27. Melean G, Velasco A, Hernández‐Imaz E, Rodríguez‐Álvarez FJ, Martín Y, Valero A, Hernández‐Chico C (2012) RNA‐based analysis of two SMARCB1 mutations associated with familial schwannomatosis with meningiomas. Neurogenetics 13:267–274. [DOI] [PubMed] [Google Scholar]
- 28. Menke JR, Raleigh DR, Gown AM, Thomas S, Perry A, Tihan T (2015) Somatostatin receptor 2a is a more sensitive diagnostic marker of meningioma than epithelial membrane antigen. Acta Neuropathol 130:441–443. [DOI] [PubMed] [Google Scholar]
- 29. van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJM (2012) Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics 13:7. [DOI] [PubMed] [Google Scholar]
- 30. Park S‐H, Hwang S‐K, Park Y‐M (2006) Intramedullary clear cell meningioma. Acta Neurochir 148:463–466. [DOI] [PubMed] [Google Scholar]
- 31. Pasmant E, Parfait B, Luscan A, Goussard P, Briand‐Suleau A, Laurendeau I et al (2015) Neurofibromatosis type 1 molecular diagnosis: what can NGS do for you when you have a large gene with loss of function mutations?. Eur J Hum Genet EJHG 23:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Payano M, Kondo Y, Kashima K, Daa T, Yatsuka T, Kida H et al (2004) Two cases of nondura‐based clear cell meningioma of the cauda equina. APMIS Acta Pathol Microbiol Immunol Scand 112:141–147. [DOI] [PubMed] [Google Scholar]
- 33. Piva F, Santoni M, Matrana MR, Satti S, Giulietti M, Occhipinti G et al (2015) BAP1, PBRM1 and SETD2 in clear‐cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert Rev Mol Diagn 15:1201–1210. [DOI] [PubMed] [Google Scholar]
- 34. Rieske P, Zakrzewska M, Piaskowski S, Jaskólski D, Sikorska B, Papierz W et al (2003) Molecular heterogeneity of meningioma with INI1 mutation. Mol Pathol MP 56:299–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salunke P, Pal BK, Vyas S, Radotra BD (2012) Clear cell meningioma masquerading as trigeminal schwannoma. Surg Neurol Int 3:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmitz U, Mueller W, Weber M, Sévenet N, Delattre O, von Deimling A (2001) INI1 mutations in meningiomas at a potential hotspot in exon 9. Br J Cancer 84:199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schollenberg E, Easton AS (2013) A case of clear cell meningioma with tyrosine‐rich crystals. Int J Surg Pathol 21:411–412. [DOI] [PubMed] [Google Scholar]
- 38. Smith MJ (2015) Germline and somatic mutations in meningiomas. Cancer Genet 208:107–114. [DOI] [PubMed] [Google Scholar]
- 39. Smith MJ, Ahn S, Lee J‐I, Bulman M, du Plessis D, Suh Y‐L (2016) SMARCE1 mutation screening in classification of clear cell meningiomas. Histopathology. 70:814–820. doi: 10.1111/his.13135 [DOI] [PubMed] [Google Scholar]
- 40. Smith MJ, O'Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J et al (2013) Loss‐of‐function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet 45:295–298. [DOI] [PubMed] [Google Scholar]
- 41. Smith MJ, Wallace AJ, Bennett C, Hasselblatt M, Elert‐Dobkowska E, Evans LT et al (2014) Germline SMARCE1 mutations predispose to both spinal and cranial clear cell meningiomas. J Pathol 234:436–440. [DOI] [PubMed] [Google Scholar]
- 42. Strickland MR, Gill CM, Nayyar N, D'Andrea MR, Thiede C, Juratli TA, Schackert G, Borger DR, Santagata S, Frosch MP, Cahill DP, Brastianos PK, Barker FG (2016) Targeted sequencing of SMO and AKT1 in anterior skull base meningiomas. J Neurosurg 25:1–7. [DOI] [PubMed] [Google Scholar]
- 43. Teo JG, Goh KY, Rosenblum MK, Muszynski CA, Epstein FJ (1998) Intraparenchymal clear cell meningioma of the brainstem in a 2‐year‐old child. Case report and literature review. Pediatr Neurosurg 28:27–30. [DOI] [PubMed] [Google Scholar]
- 44. Thuijs NB, Uitdehaag BMJ, Van Ouwerkerk WJR, van der Valk P, Vandertop WP, Peerdeman SM (2012) Pediatric meningiomas in The Netherlands 1974–2010: a descriptive epidemiological case study. Childs Nerv Syst ChNS off J Int Soc Pediatr Neurosurg 28:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vural M, Arslantaş A, Ciftçi E, Artan S, Atasoy MA (2007) An unusual case of cervical clear‐cell meningioma in pediatric age. Childs Nerv Syst ChNS off J Int Soc Pediatr Neurosurg 23:225–229. [DOI] [PubMed] [Google Scholar]
- 46. Wang R, Al‐Khawaja D, Chiribao‐Negri C (2016) Clear cell meningioma: A case report with review of the literature. Pathology 48:S85. [Google Scholar]
- 47. Xia Q‐Y, Zhan X‐M, Fan X‐S, Ye S‐B, Shi S‐S, Li R et al (2016) BRM/SMARCA2‐negative clear cell renal cell carcinoma is associated with a high percentage of BRM somatic mutations, deletions and promoter methylation. Histopathology. 70:711–721. doi: 10.1111/his.13120 [DOI] [PubMed] [Google Scholar]
- 48. Yu KB, Lim MK, Kim HJ, Suh CH, Park HC, Kim EY, Han HS (2002) Clear‐cell meningioma: CT and MR imaging findings in two cases involving the spinal canal and cerebellopontine angle. Korean J Radiol 3:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H, Ma L, Wang Y‐B, Shu C, Kuang W, Huang Y‐A et al (2017) Intracranial Clear Cell Meningiomas: Study on Clinical Features and Predictors of Recurrence. World Neurosurg 97:693–700.e11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Table S1. Clinical and molecular data of reported cases of clear cell meningiomas in the literature.
Figure S1. Anatomical distribution of reported clear cell meningiomas (CCMs) in the literature according to age: most pediatric tumors are located in the posterior fossa and the spine, whereas adult tumors are mainly cranial and in the posterior fossa.