

Abstract
Multiple Sclerosis is an autoimmune disorder causing neurodegeneration mostly in young adults. Thereby, myelin is lost in the inflammatory lesions leaving unmyelinated axons at a high risk to degenerate. Oligodendrocyte precursor cells maintain their regenerative capacity into adulthood and are able to remyelinate axons if they are properly activated and differentiate. Neuronal activity influences the success of myelination indicating a close interplay between neurons and oligodendroglia. The myelination profile determines the distribution of voltage‐gated ion channels along the axon. Here, we analyze the distribution of the sodium channel subunit Nav1.6 and the ultrastructure of axons after cuprizone‐induced demyelination in transgenic mice expressing GFP in oligodendroglial cells. Using this mouse model, we found an increased number of GFP‐expressing oligodendroglial cells compared to untreated mice. Analyzing the axons, we found an increase in the number of nodes of Ranvier in mice that had received cuprizone. Furthermore, we found an enhanced portion of unmyelinated axons showing vesicles in the cytoplasm. These vesicles were labeled with VGlut1, indicating that they are involved in axonal signaling. Our results highlight the flexibility of axons towards changes in the glial compartment and depict the structural changes they undergo upon myelin removal. These findings might be considered if searching for new neuroprotective therapies that aim at blocking neuronal activity in order to avoid interfering with the process of remyelination.
Keywords: nodes of Ranvier, oligodendrocyte precursor cells, remyelination, sodium channels, ultrastructure of axons, VGlut1‐positive vesicles
Introduction
Myelination in the central nervous system (CNS) is a highly dynamic process, bringing plasticity to the brain and neuronal circuits throughout life 49, 63. Myelin remodeling takes place in the healthy adult CNS, with internodes produced in adults being shorter compared to those generated during development 74, possibly adjusting conduction speed with age. During multiple sclerosis inflammatory damage causes loss of myelin sheaths which can be restored if remyelination occurs 23. Thereby, shorter internodes were observed after remyelination of lysolecithin‐induced demyelination 6 meaning internodes are shorter after remyelination. Remyelination is important because it restores nerve conduction 18, 56, 64 and protects axons from degeneration 33, 39, 45, 54.
Myelination is carried out by oligodendrocytes. Lineage tracing of NG2 cells 51 or oligodendrocyte precursor cells (OPCs) has revealed that these cells divide and differentiate into oligodendrocytes in postnatal brains 77. The fate of oligodendrocyte precursor cells (OPCs) can be followed by inducing genetically encoded fluorescent proteins in these cells and their offspring 76. OPCs persist into adulthood 21, and could even be detected in MS lesions in humans 9. Lineage tracing of OPCs was recently performed after toxin‐induced demyelination, revealing different response dynamics of OPCs dependent on the brain region that was analyzed 4. Thereby, OPCs give rise to newly developing oligodendrocytes capable of remyelinating axons in different models of demyelination 28, 65, 68, 75. Axonal activity seems to regulate myelin plasticity 22, 48.
Myelination and maturation of axons is influenced by the distribution and spacing of sodium channel subunits along central axons 24. During development, expression of the Nav1.2 subunit of sodium channels along axons is gradually replaced by Nav1.6 expression as myelination proceeds 7. Nav1.6 will then cluster to form the node of Ranvier 8, 34. Demyelinated axons show altered sodium channel expression and distribution along the naked axon 12, 13.
Thus, action potential propagation is determined by the myelination status of an axon as well as its distribution of sodium channels. Under pathological conditions of myelin removal, axons will adapt to these changes in order to maintain their ability to propagate action potentials. Most likely, there will be several gradations ranging from healthy myelinated axons up to neurodegeneration.
The cuprizone model is commonly used in studies focusing on de‐ and remyelination, although with varying protocols 36, 44, 55, 66 and is associated with toxin‐induced oligodendrocyte apoptosis 1, 38 and new generation of mature oligodendrocyte 44.
In the present study, we applied the Cuprizone‐model by feeding pellets to transgenic mice expressing GFP in cells of the oligodendrocyte lineage. Using those mice, we were able to detect changes in the oligodendroglial compartment after cuprizone treatment and relate them to physiological functions of axon bundles. As we observed an increase in generation of oligodendroglia, we assessed changes in the internode/node ratio by quantifying the number of nodes in all the experimental groups. Finally, ultrastructural analysis revealed an enhanced expression of vesicles that stained positive for VGlut1 in unmyelinated axons in the corpus callosum, as a reaction to the changes induced in the oligodendroglial compartment. Our results describe the structural changes in axons upon the reduction of mature oligodendrocyte, but before complete demyelination and functional loss. We conclude that axons are able to adapt to demyelination if it is not complete, and that remyelination at this stage is possible and prevents loss of axons and function.
These finely tuned processes may also occur at the onset of demyelinating diseases such as multiple sclerosis. Characterizing axonal changes as a response to myelin removal may reveal new processes that could be promoted in order to achieve remyelination.
Materials and Methods
Mice
The progeny of B6.129(Cg)‐Gt(ROSA)26Sor tm4(ACTB‐tdTomato,‐EGFP)Luo/J (ROSAmT/mG) 50 and B6.Cg‐Tg(Cspg4‐cre/Esr1*)Bakik/J (NG2CreERTM) 77 lines was used in all experiments. Breeding pairs were obtained from The Jackson Laboratory (stocks: 007676 and 008538, respectively) and bred in house under 12/12‐h light/dark conditions with water and food available ad libitum.
Ten‐week‐old mice were fed with either 0.2% cuprizone mixed into standard rodent chow in pellets (Ssniff) for 5 weeks or with normal chow. The caudal part of the corpus callosum was subsequently analyzed at different time points, namely after 5 weeks of cuprizone feeding, 5 weeks of cuprizone plus one additional week of remyelination (normal food), 5 weeks of cuprizone plus five additional weeks of remyelination and control (no cuprizone feeding at all) (experimental scheme, Figure 1).
Figure 1.
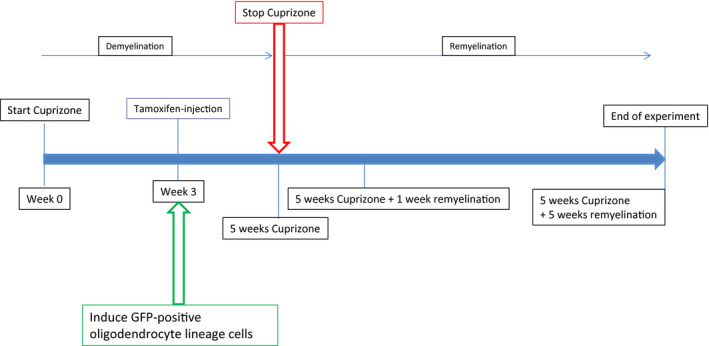
Time course of the experiments. Mice that were double transgenic for the NG2 Cre construct and the mT/mG reporter construct were fed with 0.2% cuprizone for 5 weeks. After this period remyelination was allowed for either 1 or 5 weeks. At week 3 after induction of demyelination by cuprizone, expression of membrane‐bound GFP in oligodendrocyte lineage cells was initiated by tamoxifen injection in all animals (cuprizone‐treated animals as well as age matched controls). Tissue was analyzed at indicated time points.
Cre activity in postnatal ROSAmT/mG:NG2‐CreERTM double‐transgenic mice was induced by intraperitoneal injection of 4‐hydroxytamoxifen (4‐OHT, Sigma). A 10 mg/mL stock solution was prepared by dissolving 4‐OHT in 19:1 vegetable oil:ethanol. Mice were injected with 1 mg of 4‐OHT per gram of body weight intraperitoneally 3 weeks after the start of cuprizone feeding or in age‐matched controls.
All experiments were performed in accordance with the German Animal Welfare Law and the allowance of the (local) authorities (Regierungspräsidium Tübingen).
Immunohistochemistry
At the end of each treatment, mice were anesthetized with a mixture of Ketamine 120 mg/kg/Xylazine 8 mg/kg and transcardially perfused with 4% paraformaldehyde (PFA) in 0.01 M phosphate‐buffered saline (PBS). Brains were removed, coronally cut into three pieces, embedded into TissueTek (OCT compound, Sakura), frozen and kept at −80°C. Ten‐micrometers‐thick cryo sections were cut with a Leica CM3050S Cryotome and Leica 819 Microtome Blades. Sections were air‐dried before staining, postfixed for 10 minutes with Ethanol at 4°C followed by 1 minute in acetone at room temperature. Sections were then rehydrated with with 0.1 M Tris‐buffered saline (TBS, pH 7.6) and blocked with blocking solution containing 5% (w/v) skimmed milk, 0.3% (v/v) Triton X‐100 (Roth) for 30 minutes. Primary antibodies were diluted in TBS containing skimmed milk over night at 4°C. Following washes with TBS, sections were incubated with secondary antibodies diluted in TBS/5% skimmed milk for 1 h at room temperature. Sections were washed, cell nuclei were counterstained by 10‐minute incubation with 0.2‐µg 4′6‐diamidino‐2‐phenylindole (DAPI) in TBS and mounted in VECTASHIELD hardset mounting medium (Vector Laboratories). Sections were imaged with a Zeiss LSM 710 Meta Confocal microscope.
Primary antibodies: OPCs were stained for the NG2 proteoglycan with rabbit anti‐mouse NG2 antibody [Dr. William Stallcup, Sanford Burnham Prebys Medical Discovery Institute (SBP), La Jolla, CA, USA], sodium channels were stained with rabbit anti‐Nav1.6 (Alomone labs), chicken anti‐GFP antibody was obtained from Abcam, myelin was stained with rabbit anti‐MBP (Cell Signaling). Secondary antibodies coupled to Alexa 488 and 633 were purchased from Invitrogen. Image analysis was done with ZEN, Image J or Adobe Photoshop.
For MBP, CC1 and VGlut1 staining, mice were anesthetized at the indicated time points with a mixture of isofluoran and oxygen (3%v/v) and decapitated. The brain was dissected in an ice‐cold N‐methyl‐D‐glucamine (NMDG)‐based solution (135 mM NMDG, 1 mM KCl, 1.2 mM KH2PO4, 20 mM choline bicarbonate, 10 mM glucose, 1.5 mM MgCl2, 0.5 mM CaCl2, pH 7.4, 310 mOsm) that was gassed with carbogen (95%O2 and 5%CO2). Four hundred‐micrometers‐thick coronal brain slices were cut with a Leica VT 1200S vibratome and transferred to 4% paraformaldehyde in 10 mM phosphate‐buffered saline for fixation overnight. Slices were embedded in agar for further sectioning (35‐µm‐thick slices) using a microtome HM650V, Thermo Scientific. Free‐floating sections were incubated with 10 mM citric acid (pH 6.0) for 1 h at 37°C for antigen retrieval. After washing, slices were incubated with blocking solution [3% Albumin Fraction V and 0.2% Triton‐X (both from Roth) in 0.1 mM Tris‐buffered saline (TBS)] for 1 h at 37°C. Slices were incubated with mouse anti‐APC (1:250, Ab‐7, Calbiochem), rat anti‐MBP (1:125, Abcam) or guinea pig anti‐VGlut1 (1:100, synaptic systems), diluted in blocking solution overnight at 4°C. After washing, secondary antibodies goat anti‐rat Alexa Fluor 633, goat anti‐mouse Alexa 633 or goat anti‐guinea pig Alexa Fluor 633 (1:250, Invitrogen) were applied for 3 h at room temperature. Sections were washed, cell nuclei were counterstained by 10 minutes of incubation with 0.2‐µg 4′6‐diamidino‐2‐phenylindole (DAPI) in TBS and mounted in VECTASHIELD hardset mounting medium (Vector Laboratories). Sections were imaged with a Zeiss LSM 710 Meta Confocal microscope.
Transmission electron microscopy
For electron microscopy, mice were anesthetized with a mixture of ketamine 120 mg/kg/Xylazine 8 mg/kg and transcardially perfused with a mixture of 4%PFA/4% glutaraldehyde (GA) in PBS. Brains were removed, corpus callosum dissected and postfixed in the same fixative for 4 h. After washing in 0.1M cacodylate buffer (pH7.4), samples were postfixed in 1% OsO4, dehydrated in an ethanol series (50, 70, 96, and 100%) during which the 70% ethanol was saturated with uranyl acetate for contrast enhancement. Dehydration was completed by propylene oxide and specimens were embedded in Araldite (Serva, Heidelberg, Germany), hardened at 60°C for 48 h. Semi‐ and ultra‐thin sections were produced on a FCR Reichert Ultracut ultramicrotome (Leica, Bensheim, Germany). The semi‐thin sections were used to localize the correct region of the corpus callosum. After drying on glass slides, sections were stained for 1 min with Richardson's solution at 70°C and rinsed with aqua dest. For imaging, sections were mounted with DEPEX (Serva Electrophoresis GmbH, Heidelberg). From the selected region, ultra‐thin sections were made, mounted on Pioloform‐coated copper grids and contrasted with lead citrate and saturated uranyl acetate. Images were acquired with an EM10A electron microscope (Carl Zeiss, Oberkochen, Germany) and a digital camera (Tröndle, Germany).
Immunogold labeling
Four hundred‐micrometer‐thick coronal vibratome slices were prepared as described above. Slices were fixed with 4% paraformaldehyde in 10 mM phosphate‐buffered saline containing 0.1% glutaraldehyde (Electron Microscopy Sciences) for 2 h. After washing with PBS, slices were re‐sectioned to 50 µm using a microtome and transferred to TBS. Slices were incubated for 30 minutes with 0.05% Triton‐X and then blocked for 1 h using Aurion Blocking Solution and an additional hour with 3%BSA/0.2%Triton‐X in TBS. Slices were incubated with guinea pig anti‐VGlut1 antibody, diluted 1:13 in blocking solution, overnight at 4°C. After extensive washing with TBS, goat anti‐guinea pig antibody coupled to 15‐nm gold (Aurion), diluted 1:5 in blocking solution was added overnight at 4°C. After extensive washing, slices were postfixed with 2% glutaraldehyde in PBS, washed, corpos callosum was dissected and processed for conventional embedding in Araldite as described.
Image acquisition and counting
For the countings of GFP‐positive and NG2‐positive cells, images were obtained with a 40× Plan Apochromat NA 1.3 oil immersive (tile scan of the corpus callosum, z‐stacks collected at 1‐µm distance) with a Zeiss LSM710 Meta confocal microscope. All GFP+, NG2+ and DAPI‐positive cells in the corpus callosum of a 4‐µm‐thick slice were counted (maximum intensity projection) using the Image J Cell counter plugin (NIH, USA) and the ratio between GFP‐ or NG2‐positive cells/all DAPI+ cells was calculated. For the GFP+ cell counts, up to six slices were counted per mouse, averaged and then the average of n = 4 mice for all groups was assessed. For the NG2 counting, n = 3 or 4 for every group. One slice per mouse was counted.
Images from Nav1.6 stainings were obtained with a 63× Plan Apochromat NA 1.4 oil immersive. Subsequently, every field of view (134 µm2) was processed for automated counting of Nav1.6‐positive nodes of Ranvier by the Image J Analyze Particles function (NIH, USA). The clusters were size filtered to only include particles of 0.5–3 µm, circularity set to 0.00–1.00. For the cuprizone 5 weeks group, n = 6 mice were analyzed, for the 5 weeks cuprizone plus 1 week of remyelination, n = 5 mice were analyzed, for the 5 weeks cuprizone plus 5 weeks of remyelination, n = 6 mice were analyzed and for the control group, n = 7 mice were analyzed. Several slices per mouse were stained, and three images were taken per slice.
For the counting of CC1 cells, images were obtained with a 40× Plan Apochromat NA 1.3 oil immersive (tile scan of the corpus callosum, z‐stacks collected at 1‐µm distance) with a Zeiss LSM710 Meta confocal microscope. All CC1‐ and DAPI‐positive cells in the corpus callosum of a 15‐µm‐thick slice were counted (maximum intensity projection) using the Image J Cell counter plugin (NIH, USA) and the ratio between CC1‐positive cells/all DAPI+ cells was calculated. n = 3 mice for each group.
All images were acquired with 16 bit color depth and 4× averaging and were saved in Zeiss.lsm format.
Ultrastructural analysis was performed on 2.3 µm × 2.3 µm images acquired with 20.000× magnification. Approximately 16–22 images were taken per animal in sections of the corpus callosum in three animals per group. The number of axons containing vesicles was counted in each image and the average calculated per animal. On the same images, g‐ratios were assessed. Thereby, d = axon diameter, D = fiber diameter and g = d/D.
Compound action potentials
Mice were anesthetized with a mixture of isofluorane and oxygen (3%v/v) and decapitated. The brain was removed and dissected in ice cold N‐methyl‐D‐glucamine (NMDG)‐based solution (135 mM NMDG, 1 mM KCl, 1.2 mM KH2PO4, 20 mM choline bicarbonate, 10 mM glucose, 1.5 mM MgCl2, 0.5 mM CaCl2), pH 7.4, 310 mOsm, gassed with carbogen (95% O2, 5% CO2). Four hundred‐micrometers‐thick coronal brain slices were cut in the same solution using a Leica VT1200S vibratome. The slices were then transferred to a 32‐C Haas‐type interface incubation chamber and perfused with Ringer solution (124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4*H2O, 2 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, 10 mM glucose at pH 7.4, 300 mOsm, gassed with carbogen) and gradually cooled down to room temperature, allowing the slices to equilibrate.
After cooling down, individual slices were transferred to the recording chamber mounted on a stage of an upright microscope (FN‐1, Nikon, Japan). The slices were kept at room temperature and superfused continuously (about 2 ml/min) with gassed Ringer solution.
Pipettes were pulled from borosilicate glass capillaries (Science Products, Germany) with a vertical puller (Model PC10, Narishige, Japan) to an opening of 2–3 µm and were filled with Ringer solution. The recording and the stimulation pipette were placed at the same distance from the midline in fiber bundles of the caudal part of the corpus callosum, at a total distance of 1–1.4 mm distance from each other.
Per sweep, one pair of pulses (with an interpulse interval of 40 msec) was applied every 10 s, each pulse at a duration of 100–250 µsec and the indicated strength with an isolated pulse stimulator (A‐M Systems, Model 2100, Science Products, Germany).
CAPs were recorded with an extracellular amplifier (EXT‐10‐2F; NPI electronic), high‐pass filtered at 03.Hz and low‐pass filtered at 2 kHz and a sampling frequency of 25 kHZ. Data acquisition was performed using the Recording Artist (written by Rick Gerkin, Arizona State University, USA) running under Igor Pro 6.3 (Wave Metrics, Lake Oswego, USA). Controls were recorded in the presence of 1 µM tetrodotoxin citrate (TTX, Abcam) dissolved in Ringer solution.
For the analysis, 18–20 sweeps were averaged for each stimulation strength, and the amplitude of N1 (if present) and N2 of the first response was measured manually in Igor Pro.
Statistics
All statistical analyses were performed in IBM SPSS 23.0.0 software. Normality of data distributions was verified with Shapiro–Wilk's test. Homogeneity of variance between tested groups was validated with Levene's test. If data were normally distributed t‐tests were used for comparisons. If data were not normally distributed but variance was homogeneous between the groups, then Mann–Whitney U tests were used for comparison. Bar graphs were generated by using Igor Pro 6.3 (WaveMetrics, Lake Oswego, USA). Each bar represents group average while error bars represent standard error of the mean (SEM) in all figures.
Results
GFP‐expression in oligodendroglial cells after de‐ and remyelination
First, we assessed the changes in GFP expression in oligodendrocyte lineage cells in the different experimental groups (Figure 2). GFP expression was induced by tamoxifen injection 3 weeks after the start of cuprizone feeding (Figure 1). This schedule created a number of GFP cells that was large enough to be counted and compared between the different groups, while single cells were still distinguishable from each other. We analyzed the caudal part of the corpus callosum, since cuprizone‐induced demyelination is most pronounced in this part. Cuprizone treatment enhanced the expression of GFP in oligodendroglial cells compared to control mice that had not received cuprizone (Figure 2). Two weeks after tamoxifen injection, 6.15% (±0.59) of the cells were GFP positive in cuprizone‐treated mice (Figure 2A) compared to 3.78% (±0.34) in control mice of the same age and identical time after tamoxifen injection (Figure 2B) (P = 0.006). After 1 week of additional remyelination (and 3 weeks after tamoxifen injection), cuprizone‐treated mice had an increase from 6.15% to 8.85% (±1.37) of GFP‐positive cells (Figure 2C), while control mice of the same age and 3 weeks after tamoxifen injection had a lower increase from 3.78% to 4.56% (±0.32) GFP‐positive cells (Figure 2D) (P = 0.029). Finally, after 5 weeks of cuprizone treatment followed by 5 weeks of recovery (and in total 7 weeks after tamoxifen injection) 8.36% (±1.14) of the cells in caudal corpus callosum were GFP‐positive (Figure 2E), while only 4.93% (±0.90) of cells in the control mice of the same age and 7 weeks after tamoxifen injection were GFP positive (Figure 2F) (P = 0.047). Thus, there is an increase in GFP‐expressing cells over time that is strikingly more pronounced upon cuprizone treatment (Figure 2G).
Figure 2.
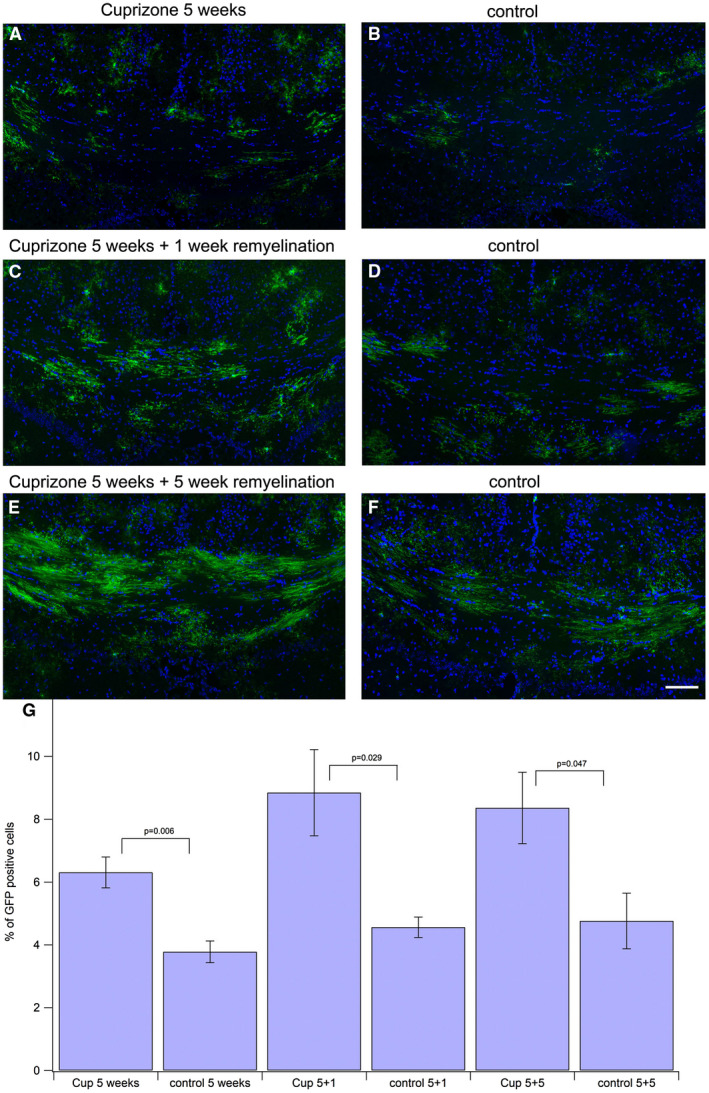
Amount of GFP‐positive oligodendroglial cells in corpus callosum is enhanced after cuprizone treatment. The GFP‐signal was enhanced by incubating with anti‐GFP antibody and Alexa488 labeled secondary antibody (green). Nuclei were stained with DAPI (blue). All DAPI‐positive nuclei were counted to assess the total number of cells in the corpus callosum. All GFP‐positive oligodendroglial cells were counted to assess the number of newly generated oligodendroglial cells. The ratio of GFP‐positive cells among all cells was calculated and compared to the corresponding controls (tissue taken at same period after tamoxifeninjection). The difference between cuprizone‐treated animals and controls was significant for all groups. Scale bar represents 100 μm. A. 5 weeks of cuprizone feeding, 2 weeks after tamoxifen injection. B. control, 2 weeks after tamoxifen injection. C. 5 weeks of cuprizone feeding plus 1 week of remyelination. Three weeks after tamoxifen injection. D. control, 3 weeks after tamoxifen injection. E. 5 weeks of cuprizone feeding plus 5 weeks of remyelination. Seven weeks after tamoxifen injection F: control, 7 weeks after tamoxifen injection. G. statistical comparison between the treatment groups and the corresponding controls.
After finding that feeding cuprizone pellets had an effect on GFP‐expressing oligodendrocyte lineage cells, we assessed the changes in myelin expression and function (Figure 3). We saw a reduction in MBP staining after 5 weeks of cuprizone feeding (Figure 3B1) compared to control (Figure 3A1). During the recovery phase, myelin expression was increased again at 5 weeks of cuprizone feeding plus 1 week of normal food (Figure 3C1) and 5 weeks of cuprizone feeding plus 5 weeks of normal food (Figure 3D1). Simultaneously, we measured compound action potentials (CAPs) in all groups to test the functionality of callosal fibers. It is generally assumed that the two peaks that are usually measured when recording CAPs in fibers of the corpus callosum belong to myelinated and unmyelinated fibers 52, which are both present in this brain region. Thereby, we noticed regional differences in CAP distribution within the caudal part of the corpus callosum as follows: CAPs recorded in control mice in the upper fiber bundles that appear “light” in the bright field microscope generally show a smaller first peak representing faster and myelinated fibers and a larger second peak representing slower and unmyelinated fibers (Figure 3A2). By contrast, CAPs recorded in the lower bundles that appear “dark” in the bright field microscope show a larger first peak and a smaller second peak, indicating that there are more myelinated fibers present in the “dark” bundle (Figure 3A3). Concomitantly, there is more MBP staining visible in this bundle (Figure 3A1). After 5 weeks of cuprizone feeding, MBP staining was clearly reduced (Figure 3B1). The first peak was completely lost in the “light” bundle, while the second peak persisted (Figure 3B2). The “dark” bundle seemed to be more resistant to cuprizone, as there was still a first and a second peak detectable, although in different proportions when compared to the control (Figure 3B3). At 5 weeks of cuprizone and 1 week of remyelination, we observed many cells in the corpus callosum that stained positive for MBP (Figure 3C1), representing newly forming MBP‐expressing cells. The myelin sheaths are not yet functional, since the first peak of the CAPs was still absent in the “light” bundle (Figure 3C2) and was weak in the “dark” bundle (Figure 3C3), whereas the second peak was weakly visible in both bundles. After 5 weeks of remyelination, myelin sheaths reappear in immunohistological stainings (Figure 3D1) and CAP measurements showed both peaks in the “light” (Figure 3D2) and “dark” (Figure 3D3) bundles of the corpus callosum, notably at the same proportions than in control mice. Thus, we could link immunohistochemistry to functionality of callosal fibers with this approach and describe the changes after cuprizone‐induced demyelination. We confirmed the presence of “light and dark” bundles in the corpus callosum in all experimental groups by performing light microscopy on semi‐thin sections. “Dark” bundles appear at higher density with more myelin rings, absorbing more of the basic dye as compared to “light” bundles (supporting Figure 1).
Figure 3.
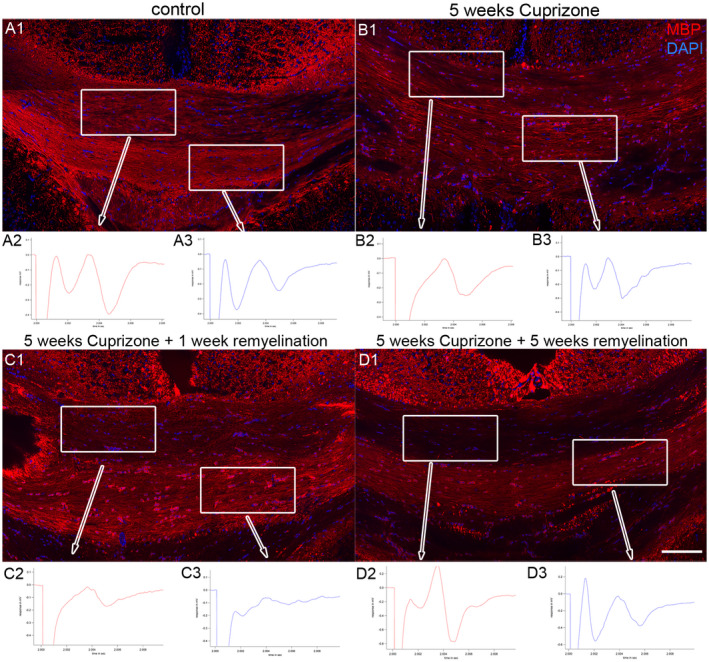
Effects of cuprizone‐feeding on myelin function. Staining for myelin basic protein MBP (red) is shown in control conditions (A1), after 5 weeks of cuprizone treatment (B1), at 5 weeks of cuprizone feeding and 1 week of remyelination (C1) and after 5 weeks of cuprizone and 5 weeks of remyelination (D1). DAPI staining is shown in blue. Scale bar represents 100 μm. Corresponding compound action potentials (CAPs) are depicted below each time point. Control: A2. CAP in upper “light” bundles, A3. CAP in lower “dark” bundles. 5 weeks cuprizone: B2. CAP in upper “light” bundles, B3. CAP in lower “dark” bundles. 5 weeks of cuprizone plus 1 week of remylination: C2. CAP in upper “light” bundles, C3. CAP in lower “dark” bundles. 5 weeks of cuprizone plus 5 weeks of remyelination: D2. CAP in upper “light” bundles, D3. CAP in lower “dark” bundles.
Changes in the number of NG2‐positive cells during de‐ and remyelination
To further evaluate the maturation state of the GFP‐expressing oligodendroglial cells, we analyzed the number of NG2‐positive oligodendrocyte precursor cells in the different treatment groups (Figure 4). In the 5 weeks of cuprizone‐feeding group (Figure 4A), we did not find an enhanced occurrence of NG2‐positive cells when compared to control (Figure 4B). The total number of NG2 cells was significantly enhanced only after 5 weeks of cuprizone treatment followed by 1 week of remyelination 12.83% (±1.35) (Figure 4C) compared to the control group (Figure 4D), where 6.67% (±1.48) of all cells were NG2 positive (P = 0.028). In the 5 weeks cuprizone and 5 weeks plus 5 weeks of remyelination group (Figure 4E), the enhancement in NG2‐positive cells between treatment and control group (Figure 4F) was slightly enhanced, but not significant. Figure 4G shows statistics for all groups. Of note, when we calculated the percentage of GFP‐positive cells that were at the same time positive for NG2, these values were on average about 25% for the control groups, but raised to 34.35% in the 5 weeks cuprizone group and to 37.88% in the 5 weeks cuprizone plus 1 week of remyelination group, the group in which the total number of NG2 cells was significantly enhanced. The amount of NG2‐positive cells among the GFP‐positive cells went back to 19.34% in the 5 weeks cuprizone plus 5 weeks of remyelination group. The rest of the GFP‐positive cells (80%–62%, depending on the group) must have further differentiated and lost the NG2 proteoglycan, which is a marker specifically found on glial precursors 57.
Figure 4.
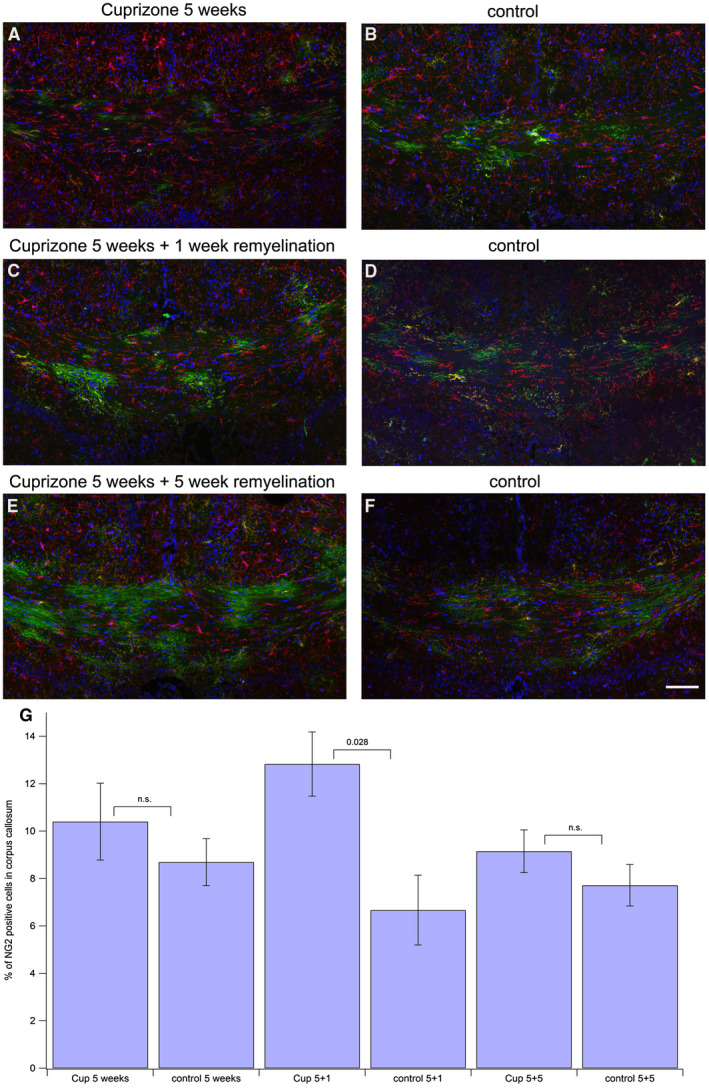
Assessing NG2‐positive portion of GFP‐expressing oligodendroglial cells. Tissue processing was the same as for Figure 2, except staining for NG2‐positive OPCs was performed with an anti‐NG2 antibody followed by an Alexa633‐labeled secondary antibody (shown in red), GFP (green), DAPI (blue). Scale bar represents 100 μm. A. 5 weeks of cuprizone feeding, 2 weeks after tamoxifen injection. B. control, 2 weeks after tamoxifen injection. C. 5 weeks of cuprizone feeding plus 1 week of remyelination. Three weeks after tamoxifen injection. D. control, 3 weeks after tamoxifen injection. E. 5 weeks of cuprizone feeding plus 5 weeks of remyelination. Seven weeks after tamoxifen injection. F. control, 7 weeks after tamoxifen injection. G. statistical comparison between the treatment groups and the corresponding controls.
These findings indicate that the demyelination increased the generation of OPCs during the first week of recovery, which then went back to normal rates.
Changes in the number of CC1‐positive cells during de‐ and remyelination
Mature oligodendrocytes in the corpus callosum of all treatment groups were counted after staining them with CC1 antibody. As shown in Figure 5, there is a significant decrease in the number of CC1‐positive oligodendrocytes after 5 weeks of cuprizone treatment (30.63% ±1.15), when compared to the controls (39.43% ±1.15). The number of CC1‐positive oligodendrocytes increases when cuprizone feeding is stopped, but there is a high variability in these groups, possibly due to the ongoing process of remodeling.
Figure 5.
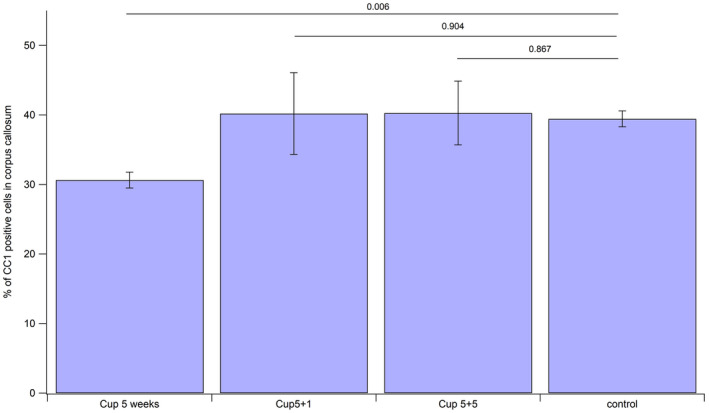
Numbers of CC1 cells in corpus callosum after 5 weeks cuprizone, 5 weeks cuprizone followed by 1 week of remyelination and 5 weeks cuprizone followed by 5 weeks of remyelination.
Distribution of Nav1.6‐positive nodes of Ranvier after remyelination
Since generation of new oligodendrocytes will eventually influence axonal organization, we next addressed the question whether the distribution of Nav1.6‐positive nodes of Ranvier in our model is altered. Mature nodes of Ranvier were stained by anti‐Nav1.6 antibodies. The nodal staining represents mature nodes of Ranvier, because most of them were flanked by Caspr staining (supporting Figure 2). Since the immunofluorescence data gave the impression, that nodal staining for Nav1.6 was enhanced along the time course of the experiments (Figure 6A–D), we quantified the number of nodal structures and compared these numbers between the different treatment groups (Figure 6E). For the analysis, we compared age matched control mice that had never received cuprizone (control group, pooled for all groups) with mice that had been fed with cuprizone pellets for 5 weeks, for 5 weeks plus an additional week with normal food and for 5 weeks plus additional 5 weeks of normal food representing the recovery phase. Indeed, quantification of mature nodes between the different groups revealed, that the number of nodes in the caudal corpus callosum was enhanced in all three treatment groups compared to untreated tissue. Whereas in the control, on average 384.40 (±32.6545) nodes were counted per field of view (Figure 6D), this number was enhanced to 513.17 (±9.2665) in the 5 weeks cuprizone group (Figure 6A) (P = 0.084 when compared to the control group), to 574.87 (±41.0244) in the 5 weeks of cuprizone plus 1 week of remyelination group (Figure 6B) (P = 0.084 when compared to the control group) and to 555.87 (±15.4733) in the 5 weeks of cuprizone plus 5 weeks of remyelination group (Figure 6C) (P = 0.036 when compared to the control group). The data show, that the enhanced generation of oligodendrocyte lineage cells is reflected in remodeling of axonal organization, namely the increase in the number of nodes of Ranvier. The difference was statistically significant between the control group and the 5 weeks of cuprizone feeding plus 5 weeks of remyelination group. This finding indicates, that the enhanced generation of oligodendroglial cells we observed after cuprizone treatment results in the generation of new myelin sheaths that are shorter compared to age matched controls, a process that is most pronounced at the end of the remyelination process at 5 weeks of remyelination. It should be mentioned that we also saw Nav1.6‐positive clusters that were not flanked by Caspr (Supporting Figure 2). These structures presumably represent prenodal clusters demonstrating nodal rearrangement, as they appear to be more frequent during the recovery phase.
Figure 6.

Distribution of Nav1.6 during de‐ and remyelination. Nodes of Ranvier in controls (no cuprizone treatment), 5 weeks cuprizone, 5 weeks cuprizone followed by 1 week of remyelination and 5 weeks cuprizone followed by 5 weeks of remyelination groups were visualized by staining for Nav1.6 followed by Alexa 633 labeled secondary antibody (shown in red). Scale bar represents 20 μm. Quantification of nodes is shown in (e).
GFP‐positive membrane protrusions are shortened after cuprizone treatment
To evaluate whether an increase in the number of nodes of Ranvier is linked to the generation of shorter internodes, the length of oligodendroglial protrusions stained by transgenic expression of membrane‐tagged GFP in this model were measured (Figure 7). Indeed, after 5 weeks of cuprizone treatment and 2 weeks after tamoxifen injection, the length of the GFP‐positive membrane protrusions was shortened from an average of 33.41 µm (±1.41) in control mice to an average of 25.69 µm (±1.08) (P = 0.005). A similar shortage could be observed after 5 weeks of cuprizone treatment plus 1 week of remyelination and 3 weeks after tamoxifen injection from 32.18 µm (±1.47) in controls to 23.46 µm (±1.83) (P = 0.004). After 5 weeks of cuprizone treatment and an additional 5 weeks of remyelination and 7 weeks after tamoxifen injection, the average length of GFP‐positive protrusions was shortened from 34.59 µm (±2.10) in the control group to 23.90 µm (±0.57) (P = 0.001). This finding indicates, that oligodendroglial cells tend to generate shorter membrane protrusions after cuprizone‐induced reduction of mature oligodendrocytes and myelin, presumably leading to shorter internodes.
Figure 7.
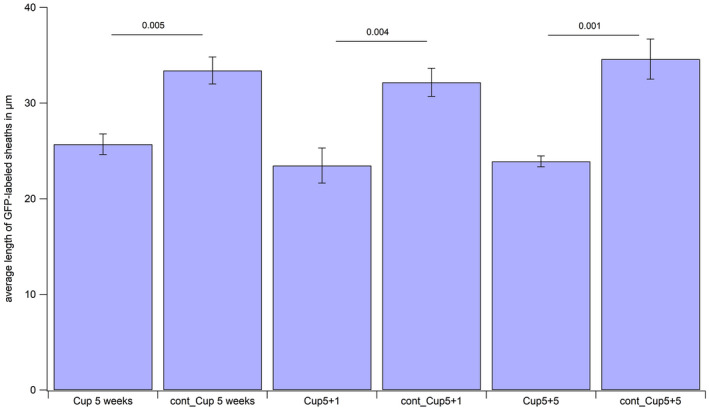
Length of GFP‐positive membrane protrusions. The average length of GFP‐expressing protrusions generated by oligodendroglia was assessed in all experimental groups and age‐matched controls.
Ultrastructural changes in axons during de‐ and remyelination
In order to address the question, whether enhanced generation of oligodendroglial cells and increase in Nav1.6‐positive nodes of Ranvier affects the ultrastructure of axons, we performed an electron microscopic study of the caudal part of corpus callosum in all four experimental groups (5 weeks of cuprizone treatment, 5 weeks of cuprizone plus 1 week of remyelination, 5 weeks of cuprizone plus 5 weeks of remyelination and untreated control). In control mice, we found myelinated as well as unmyelinated axons (Figure 8A). In addition, we saw small diameter axons (Figure 8B, example image taken from the 5 weeks of cuprizone treatment plus 1 week of remyelination group) that were most likely never myelinated before 62, 70. After cuprizone treatment and during remyelination, we frequently detected the presence of vesicles in unmyelinated axons (Figure 8C and D). Often, those axons showing vesicles were located in close proximity to protrusions belonging to an oligodendroglial cell (Figure 8C and D). From electron microscopic pictures, it was not possible to distinguish oligodendrocyte precursor cells vs. premyelinating oligodendrocytes vs. mature oligodendrocytes. To assess the incidence of axons that show vesicles, all axons per field of view were counted. On average, 1.25 (±0.13) axons per field of view contained vesicles in control mice, while this number raised to 2.66 (±0.35) after 5 weeks of cuprizone feeding, to 2.34 (±0.27) after 5 weeks of cuprizone feeding plus one week of remyelination and to 2.40 (±0.14) after 5 weeks of cuprizone feeding plus 5 weeks of remylination. The distribution of axons containing vesicles for all groups is shown in supporting Figure 3. The occurrence of vesicles reached the highest numbers directly after the cuprizone treatment, when the damage to myelin is at its peak. The number of vesicles remained elevated in the remyelination groups throughout the study when compared to age matched controls that had not received cuprizone.
Figure 8.
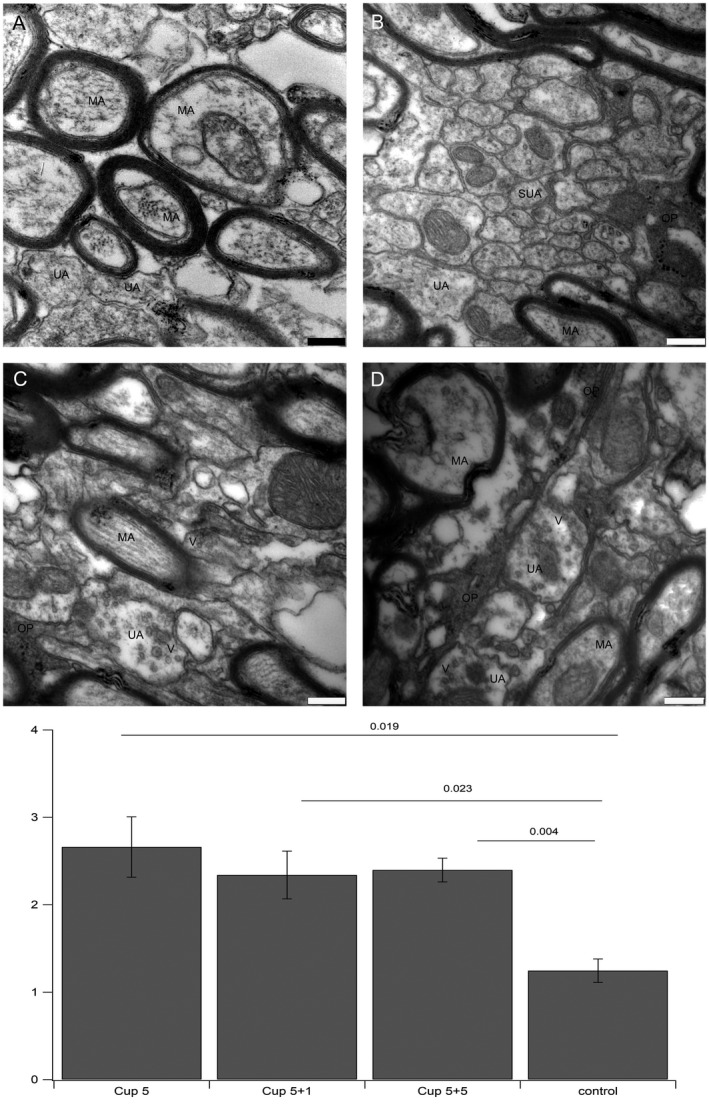
Ultrastructure of axons. A. myelinated axons in control conditions do not show vesicles. B. small diameter axons after cuprizone treatment do also not show vesicles. C and D. examples for enhanced expression of vesicles in demyelinated axons after cuprizone treatment. E. Statistics: The number of unmyelinated axons containing vesicles per image taken at the same magnification was counted. The differences in all three treatment groups were significantly different from controls. All images were taken at 20.000× fold magnification, bar represents 250 nm. V: vesicle, OP: oligodendroglial protrusion, MA: myelinated axon, UA: unmyelinated axon, SUA: small unmyelinated axon.
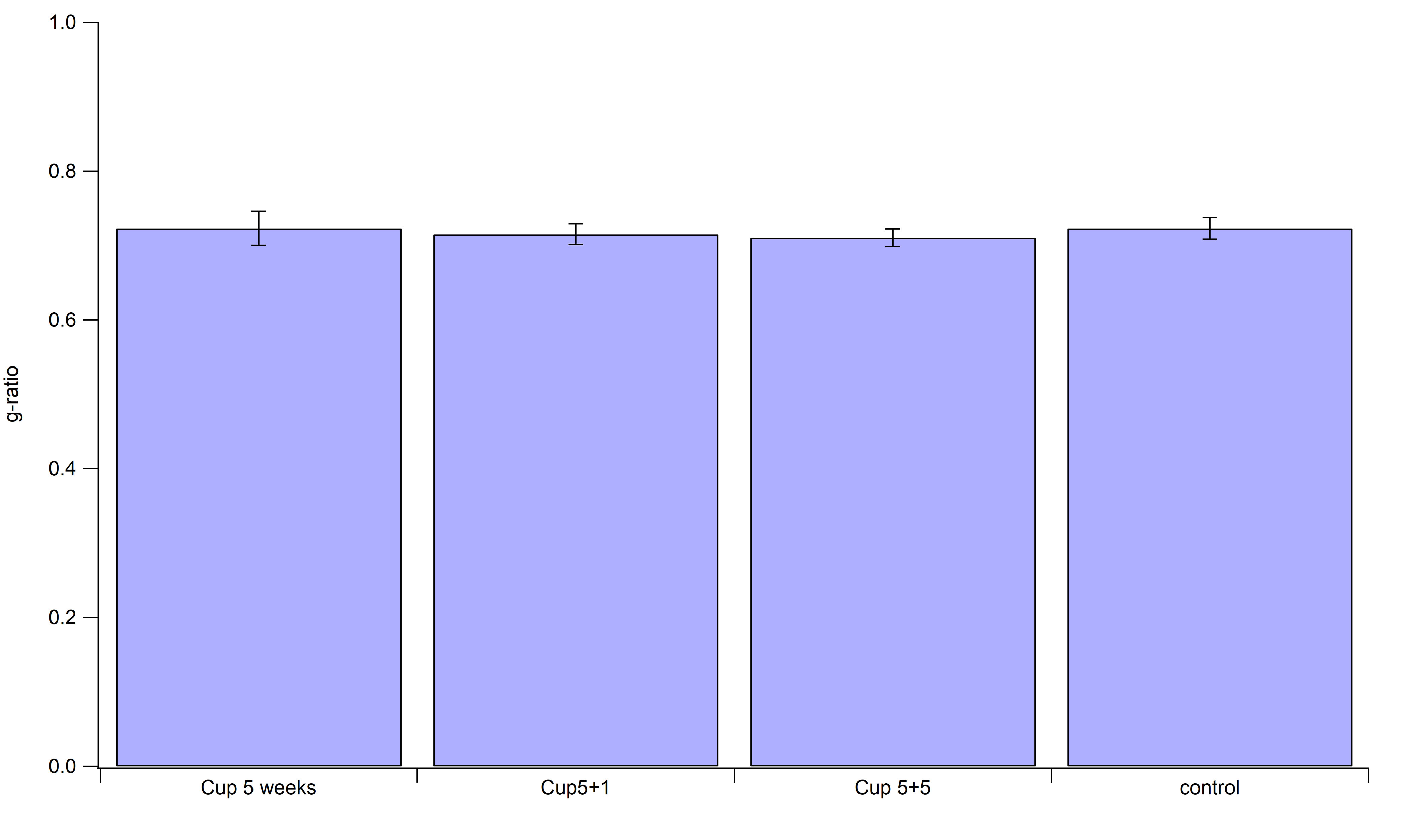
Additionally, we determined the g‐ratios in the same data set for all four groups, but did not find significant differences between them (Supporting Figure 4). As the g‐ratio is commonly used to asses myelin thickness, we can conclude that the thickness of myelin sheaths was not altered in our model.
Presence of synaptic vesicles increases after cuprizone treatment
To test the possibility, that the vesicles we observed in cuprizone‐treated animals are synaptic vesicles, we performed stainings for VGlut1, a marker for synaptic vesicles. Cuprizone‐treated animals (Figure 9A) showed more pronounced labeling for VGlut1 compared to controls (Figure 9B) in the corpus callosum, indicating that callosal fibers exposed to cuprizone‐induced reduction of oligodendrocytes expressed more synaptic vesicles. To confirm that VGlut1 indeed co‐localizes with synaptic vesicles, we set up a pre‐embedding immunogold staining procedure for VGlut1 in corpus callosum. As shown in Figure 10B, VGlut1 was labeled with 15‐nm gold particles on synaptic vesicles in the presynapse, confirming that VGlut1 was specifically labeled in synaptic vesicles. In Figure 10A, several vesicles expressed along axons that were not organized in a proper synapse, were also labeled with 15‐nm gold particles, confirming that axons did indeed upregulate synaptic vesicles.
Figure 9.
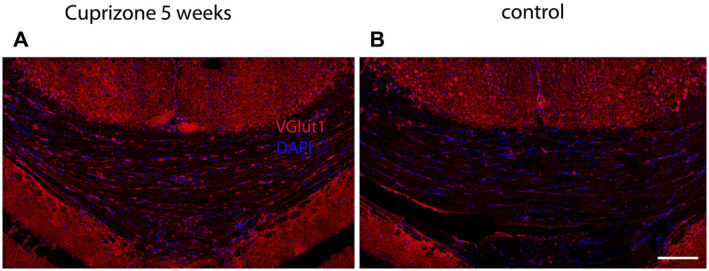
VGlut1 expression in Corpus Callosum. A. VGlut1 (in red) after 5 weeks of cuprizone feeding. B. VGlut1 in control DAPI is shown in blue. Scale bar represents 100 μm.
Figure 10.
Immunogold labeling for VGlut1. A. Cuprizone tissue shows vesicles in axons that are labeled with 15‐nm gold (arrows). B. In control tissue, vesicles in a synapse are labeled with 15‐nm gold (arrow). Scale bar represents 100 nm.
Discussion
In this study, we set up a model that is suitable to analyze and detect minor changes in the oligodendroglial compartment as well as in axons in the corpus callosum after treating transgenic mice with cuprizone pellets.
Although demyelination was not complete in our model and we could not observe differences in g‐ratios, the function of axon bundles was affected and there was a clear and significant increase in GFP‐expressing oligodendroglial cells, reflecting increased generation of oligodendroglial cells. The observed changes in the oligodendroglial compartment in turn induced an increase in the number of nodes of Ranvier, representing alterations in the distances between nodes and the lengths of internodes, and ultrastructural changes in axons, reflected by an increased occurrence of vesicles in unmyelinated axons of a certain diameter. These vesicles stained positive for VGlut1 by immunogold labeling, indicating an increase in axonal signaling. The observed changes follow a specific time schedule, as summarized in Table 1. GFP expression in oligodendroglial cells was enhanced in all treatment groups, when compared to the control, showing enhanced generation of newly forming oligodendrocytes after reduction of mature oligodendrocytes by feeding the toxin cuprizone. Since expression of GFP is stable after activation in NG2 cells in this model, it stays elevated during the course of recovery. MBP staining was reduced in all treatment groups as compared to the control and there was a significant reduction of mature CC1‐positive oligodendrocytes directly after cuprizone treatment. This finding is consistent with the functional readout applied, the measuring of compound action potentials. While it is generally assumed that the two peaks that are usually measured when recording CAPs in fibers of the corpus callosum belong to myelinated and unmyelinated fibers 52, the ratio between these two peaks is highly depending on positioning of the electrodes [summarized in 41]. After complete demyelination in the corpus callosum, the first of the two peaks is lost 15, 16. We could detect two peaks in all experimental groups as well as in the controls, but peaks were altered in a very specific and regionally distinct way in our model. Thereby, we identified differences between the anatomically superior “light” bundle and the anatomically inferior “dark” bundle in the caudal part of the corpus callosum. The first peak corresponding to the faster, myelinated fibers was larger in the “dark” bundle and smaller in the “light” bundle as compared to the second peak corresponding to the slower, unmyelinated fibers. The first peak disappeared only in the “light” bundle after 5 weeks of feeding cuprizone, and in the animals measured after 5 weeks of cuprizone and 1 week of remyelination. In the “dark” bundle, this peak was only reduced compared to the second peak but never lost. These findings indicate a regionally distinct susceptibility to cuprizone, even in the same anterior–posterior position within the brain. After 5 weeks of cuprizone feeding and additional 5 weeks of remyelination, all peaks reappeared and the ratios between first and second peak resembled control conditions, showing a reversible loss of myelin without affecting axonal function after a period of recovery. The number of NG2‐positive oligodendrocyte precursor cells was significantly enhanced only in one group, namely the 5 weeks of cuprizone treatment plus 1 week of remyelination, which is concomitant with an enhanced proliferation of OPCs in the early remyelinating phase. It is now accepted that OPCs differentiate into mature oligodendrocytes that can perform myelination 65, 75. There are no hints that mature oligodendrocytes do contribute to remyelination during regeneration after toxin‐induced demyelination 14. Since we saw enhanced numbers of GFP‐expressing cells after 5 weeks of cuprizone treatment and subsequent recovery, we assume that we induced proliferation of oligodendrocyte precursor cells and also differentiation, since the percentage of cells that stained positive for NG2 was only slightly enhanced and only significant in the 5 weeks cuprizone plus 1 week of remyelination group. This is in line with the common assumption that oligodendrocytes die and are replaced by dividing OPC that subsequently maturate when cuprizone treatment is stopped 44.
Table 1.
Summarizes all parameters that were assessed and the changes that occurred in the experimental groups compared to the control are indicated. GFP expression was significantly enhanced in all 3 experimental groups. Staining for MBP (myelin basic protein) was decreased in all 3 experimental groups. The number of NG2 cells was significantly enhanced only in the 5 weeks of Cuprizone plus 1 week of remyelination group, whereas the number was not significantly altered in the other groups. The number of CC1 positive cells was significantly decreased in the 5 weeks of Cuprizone feeding group only. The number of Nav1.6 positive nodes was increased in all experimental groups, although the difference was only significant in the 5 weeks of Cuprizone plus 5 weeks of remyelination. By contrast, the number of axons displaying vesicles was significantly enhanced in all three Cuprizone‐treated groups, while it was highest in the 5 weeks of Cuprizone group.
| Group | GFP expression | MBP staining | Number of NG2 cells | Number of CC1 cells | Number of Nav1.6‐positive nodes of Ranvier | Number of axons containing vesicles |
|---|---|---|---|---|---|---|
| 5 weeks of cuprizone feeding | ↑ | ↓ | ~ | ↓ | ~ | ↑↑ |
| 5 weeks of cuprizone feeding + 1 week of remyelination | ↑ | ↓ | ↑ | ~ | ~ | ↑ |
| 5 weeks of cuprizone feeding + 5 week of remyelination | ↑ | ↓ | ~ | ~ | ↑ | ↑ |
As remyelination proceeds, after 5 weeks of cuprizone treatment plus 5 weeks of remyelination, the increase in number of nodes of Ranvier was significant. This finding can be explained by the generation of new myelin sheaths with shorter internodes, as it has been already reported earlier for remyelinated internodes after lysolecithin‐induced demyelination 6 or naturally occurring myelin turnover in aging mice 74. Accordingly, when measuring GFP‐positive membrane protrusions after tamoxifen induced membrane‐tagged GFP‐expression in oligodendroglial cells, these protrusions were significantly shorter in cuprizone‐treated mice at all time points when compared to controls in our model. Shorter internodes after remyelination in MS patients have been predicted earlier 71 and could finally be shown by immunohistochemistry 10. By toxin‐enhanced generation of new oligodendroglia in our model, we observed the same effect. To our knowledge, this is the first study showing shorter internodes after remyelination in the cuprizone model of demyelination.
Finally, we found an increase in unmyelinated axons displaying vesicles in all treatment groups when compared to the untreated control, with the highest number directly after the cuprizone treatment. We suggest that the enhancement of vesicle expression occurs in demyelinated fibers. Under normal conditions, we detected only a small number of unmyelinated fibers showing vesicles, representing unmyelinated fibers. The enhanced number of unmyelinated axons displaying vesicles after cuprizone‐induced reduction of mature oligodendrocytes is thus caused by adding demyelinated axons expressing vesicles under these conditions. This finding points to the fact that axons become active and produce more synaptic vesicles upon cuprizone‐induced oligodendrocyte removal, and it is tempting to speculate that signaling towards newly developing oligodendroglia is increased in order to enhance axon‐glia communication and remyelination of affected axons. Our findings that these vesicles stain positive for VGlut1 at the ultrastructural level and that overall expression of VGlut1 is enhanced after cuprizone treatment strengthen this hypothesis
The cuprizone model is commonly used in studies focusing on de‐ and remyelination, although with varying protocols 36, 44, 55, 66 and is associated with oligodendrocyte apoptosis 1, 38. If cuprizone feeding is stopped the latest after 6 weeks, many axons get remyelinated, depending on the diameter of the axon 43. Nevertheless, the extent of demyelination is highly variable across the different areas of the brain and even within corpus callosum itself, with the caudal part of the corpus callosum being most vulnerable to demyelination 59, 60, 72. In addition to differences along the anterior–posterior axis of the corpus callosum, we describe differences in the inferior–superior position of the caudal part with respect to susceptibility to cuprizone. In the meantime, another study could show, that feeding cuprizone pellets is less potent in inducing demyelination compared to feeding freshly prepared rodent chow that was mixed with cuprizone powder daily 32. This is consistent with our observations, that feeding with cuprizone pellets, as used in this study, results in incomplete demyelination, but nevertheless induces changes in the generation of oligodendrocyte lineage cells that is probably closer to physiological turnover. Along these lines, another group showed changes in the protein structure of myelin after cuprizone treatment. Although the amount of myelin basic protein (MBP) in cuprizone‐treated mice was not reduced, the protein aggregated in an insoluble form in the pellet of brain homogenates from cuprizone‐treated mice, which was not observed in control brains or prion‐infected brain 26. This finding could explain the susceptibility of cuprizone‐treated tissue to detergents (own observations).
Alteration of numbers of the nodes of Ranvier have already been described under demyelinating conditions 37, 61. In studies where complete demyelination was induced, clustering of sodium channel subunit Nav1.6 was lost or greatly reduced 19 and did not return back to control levels, even after 6 weeks of remyelination 16. In this study, even the compound action potential could not be restored to normal levels, due to the damage of axons after complete demyelination, as shown by increase of ß‐APP 16. Despite these findings, nodes of Ranvier can be reestablished if remyelination is successful 42, 73 highlighting the capacity for regeneration of CNS. There seems to be a threshold at which level of demyelination, irreversible axonal loss, disruption of sodium channel clusters and reduction of sodium channels occur. In our study, the amount of nodes of Ranvier was even enhanced after cuprizone treatment. This can be explained by modest demyelination without loss of axons, followed by increased generation of oligodendroglial cells. The newly generated myelin sheaths are shorter, resembling those observed in aged healthy mice. Thus, the new generation of oligodendroglial cells on the one hand and axonal remodeling on the other hand seems to be a finely tuned and interdepending response to demyelination.
The electrical activity of axons triggers the onset of myelination 3, 17, 47. Myelination in turn influences the distribution of sodium channels 69 and excitability of neurons 29, 30, but does the distribution of sodium channels and nodes in turn alter the signaling to glial cells and possibly influence myelination? Induction of clustering of sodium channels by oligodendroglial‐secreted factors without existing myelin sheath could be shown in retinal ganglion cells 34, 35 and in hippocampal GABAergic neurons, influencing conduction velocity in these neurons 25. The existence of sodium channel clusters on naked axons was shown in demyelinated lesions of MS patients, suggesting that aggregation of nodal components preceded remyelination 11. Thus, the clustering of sodium channels at the node can also precede myelination. Changes in nodal structures, for example, an increase in length, have been described in MS, EAE and other CNS pathologies [reviewed in 2] and will finally alter action potential propagation. It remains to be determined how this will influence myelination.
In our ultrastructural analysis, we show a significant enhancement of vesicles in unmyelinated axons after cuprizone treatment. These vesicles occurring after demyelination could reflect enhanced signaling activities of demyelinated axons, as it was shown before that synaptic vesicles secreted by axons were involved in axon selection for myelination 31. It was shown previously by electrophysiological measurements, that OPCs receive glutamatergic synaptic input after lysolecithin‐induced demyelination in corpus callosum, which is lost as OPCs differentiate into oligodendrocytes 20. Later studies were able to link electrical activity in demyelinated axons to enhanced signaling towards OPCs that enhanced remyelination, most likely transmitted by synaptic input and glutamate release 27. Other investigators saw initial downregulation of synaptic contacts after demyelination while OPCs proliferate, with subsequent gain of synaptic input 53. Thus, the exact sequence of events between axons and OPCs after demyelination is not totally clear yet. Release of synaptic vesicles has been linked to regulation of myelination during development in zebrafish before 46. Vesicles at synaptic contacts were described in the initial studies about vesicular glutamate release onto OPCs during developmental myelination 5, 40, 78 and could be shown in cell culture systems of dorsal root ganglion (DRG) neurons and OPCs in the context of myelination 67. Thus, it is plausible that this type of communication is reestablished during remyelination in the adult brain. Our study now is the first one to demonstrate enhanced numbers of axons displaying vesicles during experimentally induced remyelination. The nature of these observed vesicles and the structures where they occur still needs further investigation. It is clear though, that the morphology of axon‐myelin interactions changes in demyelinating diseases 58.
Remodeling of myelin as it occurs during remyelination after a demyelinating event will change electrical properties of axons. How will this change in turn influence action potential propagation and signaling of demyelinated axons toward OPCs? We need to learn more about axon‐glia interactions during the different stages of neuronal damage during Multiple Sclerosis in order to develop therapies aiming at either protecting axons from over‐excitation and degeneration or promoting remyelination. These findings might also be taken into account when considering blocking neuronal activity, in order to avoid interfering with signaling of axons and the process of remyelination.
To our knowledge, this is one of the first studies investigating changes at the axon–oligodendroglia interface before functional loss becomes prominent. The alterations that are described may also play a role at the onset of demyelinating diseases such as multiple sclerosis, before neurodegeneration and disability impairs the patients. They may help understand the mechanisms of remyelination, and ultimately find ways to enhance these repair processes in order to prevent further damage.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
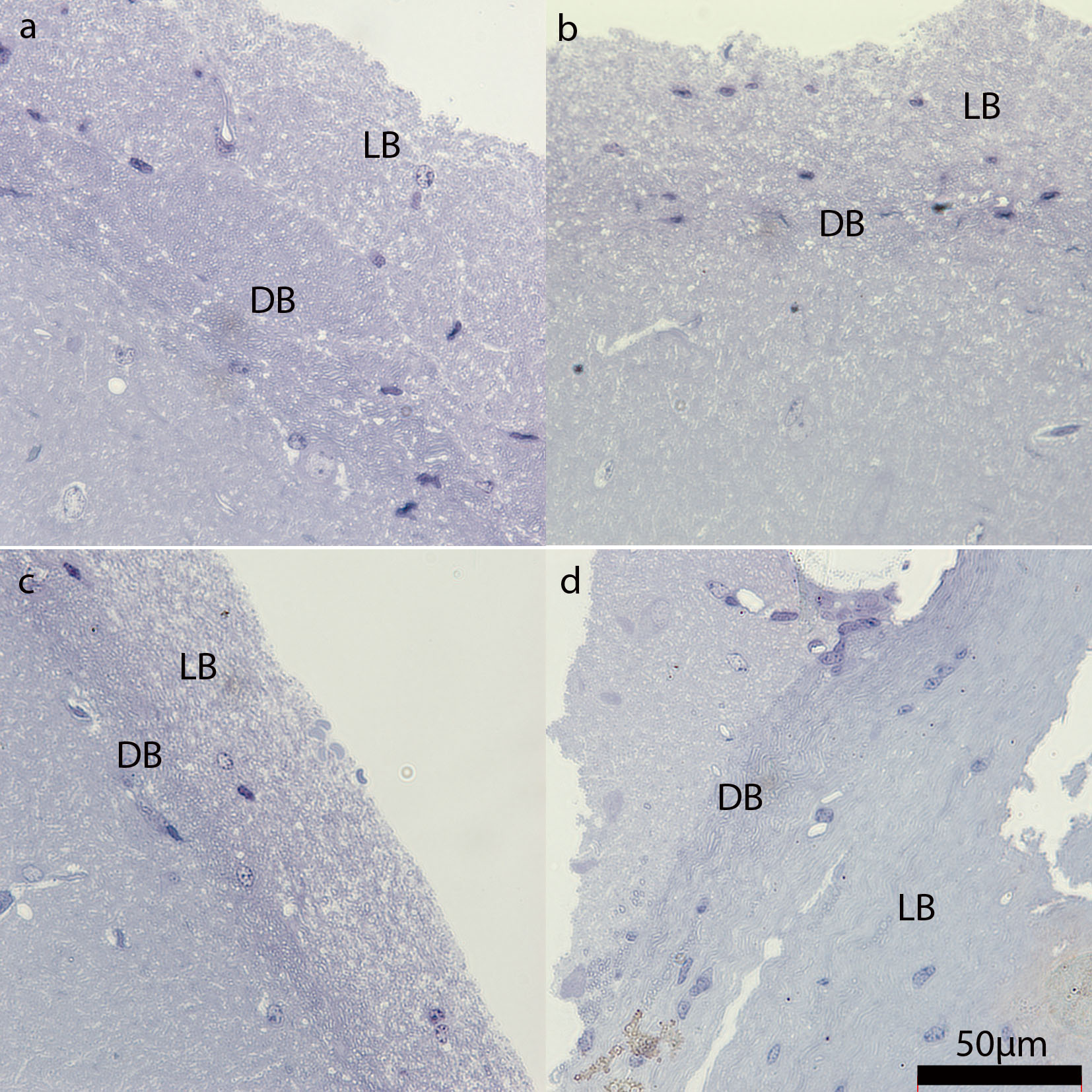
Figure S1. “Light” and “dark” bundles of the caudal part of mouse corpus callosum. Semi‐thin sections from electron microscopic preparations stained with Richardson's solution. Note the clear separation of the corpus callosum into light appearing bundles (LB) on the cortical side and dark appearing bundles (DB) on the hippocampal side in all experimental groups. A. control. B. 5 weeks of cuprizone treatment. C. 5 weeks of cuprizone treatment + 1 week of remyelination. D. 5 weeks of cuprizone treatment + 5 weeks of remyelination. Scale bar represents 50 µm.
{kind=link}
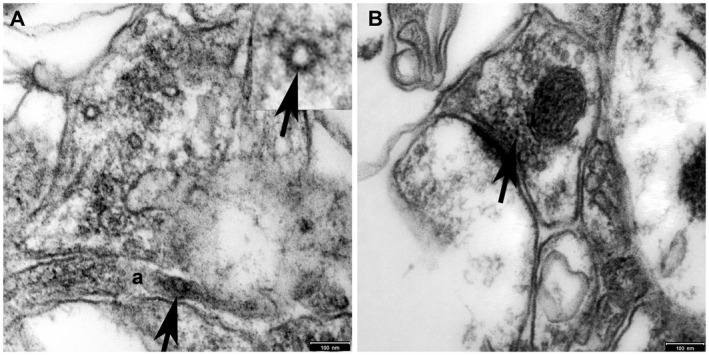
Figure S2. Nav1.6‐positive nodes are flanked by Caspr. Presence of mature nodes of Ranvier was verified by staining for the axonal, paranodal protein Caspr/contactin (1–4), which flanks the mature nodes of Ranvier, before counting the nodes. Mature nodes of Ranvier expressing Nav1.6 (red) are flanked by the paranodal protein Caspr (green) in all three experimental groups and control. Note that not all of the nodes are flanked by Caspr. These structures seem to be increased after cuprizone treatment and during recovery as compared to the control. Scale bars represent 20 µm.
{kind=link}
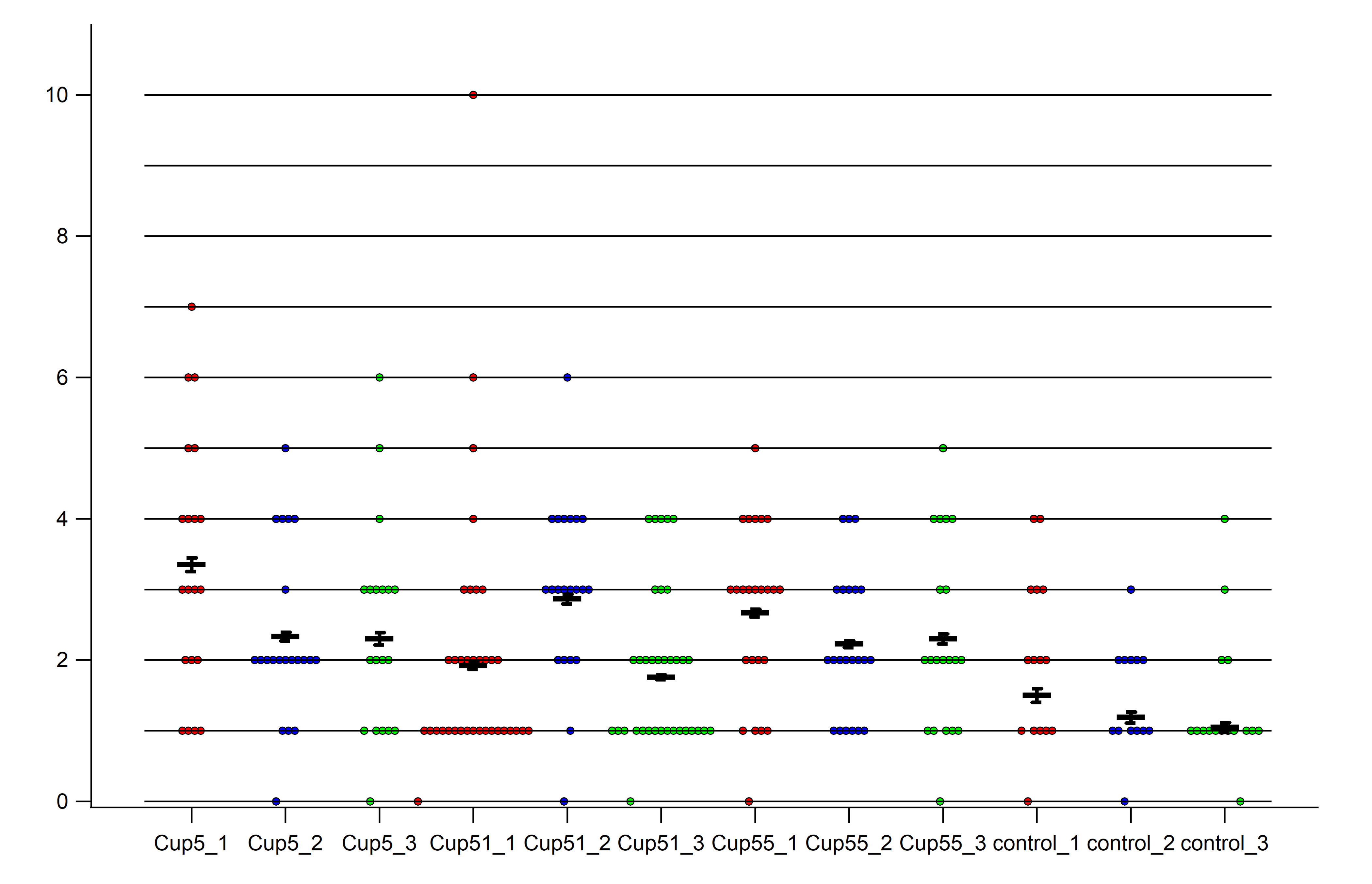
Figure S3. Distribution of axons containing vesicles per image shown in a scatter plot. The distribution of the number of axons containing vesicles per image is shown for all images and for all samples (n = 3 for all groups) in a scatter plot. The scatter plot was generated using Igor Pro 6.3 (WaveMetrics, Lake Oswego, USA). Cup5_1, Cup5_2 and Cup5_3 represent the samples for the 5 weeks of cuprizone treatment group. Cup51_1, Cup51_2 and Cup51_3 represent the samples for the 5 weeks of cuprizone plus 1 week of remyelination group. Cup55_1, Cup55_2 and Cup55_3 represent the 5 weeks of cuprizone plus 5 weeks of remyelination group. Control_1, control_2 and control_3 are the samples for the control group.
{kind=link}
Figure S4. Determination of g‐ratios. The EM‐data set was used to measure g‐ratios. Thereby, d=axon diameter was divided by D=fiber diameter in order to obtain an indication of myelin sheath thickness. No statistical difference could be detected between all three experimental groups and the control.
{kind=link}
Acknowledgments
We thank Dr. Maria Kukley and the members of her laboratory Dr. Ting‐Jiun Chen, Bartosz Kula and Balint Nagy for helpful discussions and support. We thank Dr. Axinia Döring and Dr. Julia Fitzgerald for helpful comments on the manuscript. We thank Dr. William Stallcup, SBP Institute, La Jolla, USA, for the NG2 antibody. We thank Kristin Mounts for help with assessing the g‐ratios. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grant PF574/5‐1 to FP and the Werner Reichardt Centre for Integrative Neuroscience (EXC307).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Acs P, Komoly S (2012) Selective ultrastructural vulnerability in the cuprizone‐induced experimental demyelination. Ideggyogy Szemle 65:266–270. [PubMed] [Google Scholar]
- 2. Arancibia‐Carcamo IL, Attwell D (2014) The node of Ranvier in CNS pathology. Acta Neuropathol 128:161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barres BA, Raff MC (1993) Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361:258–260. [DOI] [PubMed] [Google Scholar]
- 4. Baxi EG, DeBruin J, Jin J, Strasburger HJ, Smith MD, Orthmann‐Murphy JL, et al (2017) Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone‐induced demyelination. Glia 65:2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergles DE, Roberts JD, Somogyi P, Jahr CE (2000) Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405:187–191. [DOI] [PubMed] [Google Scholar]
- 6. Blakemore WF, Eames RA, Smith KJ, McDonald WI (1977) Remyelination in the spinal cord of the cat following intraspinal injections of lysolecithin. J Neurol Sci 33:31–43. [DOI] [PubMed] [Google Scholar]
- 7. Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G (2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30:91–104. [DOI] [PubMed] [Google Scholar]
- 8. Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR (2000) Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA 97:5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD (2000) NG2‐positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20:6404–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, Chomyk AM, et al (2012) Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol 72:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coman I, Aigrot MS, Seilhean D, Reynolds R, Girault JA, Zalc B, Lubetzki C (2006) Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 129:3186–3195. [DOI] [PubMed] [Google Scholar]
- 12. Craner MJ, Lo AC, Black JA, Waxman SG (2003) Abnormal sodium channel distribution in optic nerve axons in a model of inflammatory demyelination. Brain 126:1552–1561. [DOI] [PubMed] [Google Scholar]
- 13. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG (2004) Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA 101:8168–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crawford AH, Tripathi RB, Foerster S, McKenzie I, Kougioumtzidou E, Grist M, et al (2016) Pre‐existing mature oligodendrocytes do not contribute to remyelination following toxin‐induced spinal cord demyelination. Am J Pathol 186:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crawford DK, Mangiardi M, Tiwari‐Woodruff SK (2009) Assaying the functional effects of demyelination and remyelination: revisiting field potential recordings. J Neurosci Methods 182:25–33. [DOI] [PubMed] [Google Scholar]
- 16. Crawford DK, Mangiardi M, Xia X, Lopez‐Valdes HE, Tiwari‐Woodruff SK (2009) Functional recovery of callosal axons following demyelination: a critical window. Neuroscience 164:1407–1421. [DOI] [PubMed] [Google Scholar]
- 17. Demerens C, Stankoff B, Logak M, Anglade P, Allinquant B, Couraud F, et al (1996) Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci USA 93:9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duncan ID, Brower A, Kondo Y, Curlee JF Jr, Schultz RD (2009) Extensive remyelination of the CNS leads to functional recovery. Proc Natl Acad Sci USA 106:6832–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dupree JL, Mason JL, Marcus JR, Stull M, Levinson R, Matsushima GK, Popko B (2004) Oligodendrocytes assist in the maintenance of sodium channel clusters independent of the myelin sheath. Neuron Glia Biology 1:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Etxeberria A, Mangin JM, Aguirre A, Gallo V (2010) Adult‐born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat Neurosci 13:287–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ffrench‐Constant C, Raff MC (1986) The oligodendrocyte‐type‐2 astrocyte cell lineage is specialized for myelination. Nature 323:335–338. [DOI] [PubMed] [Google Scholar]
- 22. Fields RD (2015) A new mechanism of nervous system plasticity: activity‐dependent myelination. Nat Rev Neurosci 16:756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franklin RJ, Ffrench‐Constant C (2008) Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci 9:839–855. [DOI] [PubMed] [Google Scholar]
- 24. Freeman SA, Desmazieres A, Fricker D, Lubetzki C, Sol‐Foulon N (2016) Mechanisms of sodium channel clustering and its influence on axonal impulse conduction. Cell Mol Life Sci 73:723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Freeman SA, Desmazieres A, Simonnet J, Gatta M, Pfeiffer F, Aigrot MS, et al (2015) Acceleration of conduction velocity linked to clustering of nodal components precedes myelination. Proc Natl Acad Sci USA 112:E321–E328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frid K, Einstein O, Friedman‐Levi Y, Binyamin O, Ben‐Hur T, Gabizon R (2015) Aggregation of MBP in chronic demyelination. Ann Clin Transl Neurol 2:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I, et al (2015) Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun 6:8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gensert JM, Goldman JE (1997) Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 19:197–203. [DOI] [PubMed] [Google Scholar]
- 29. Hamada MS, Kole MH (2015) Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. J Neurosci 35:7272–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamada MS, Popovic MA, Kole MH (2017) Loss of saltation and presynaptic action potential failure in demyelinated axons. Front Cell Neurosci 11:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hines JH, Ravanelli AM, Schwindt R, Scott EK, Appel B (2015) Neuronal activity biases axon selection for myelination in vivo . Nat Neurosci 18:683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hochstrasser T, Exner GL, Nyamoya S, Schmitz C, Kipp M (2017) Cuprizone‐containing pellets are less potent to induce consistent demyelination in the corpus callosum of C57BL/6 mice. J Mol Neurosci 61:617–624. [DOI] [PubMed] [Google Scholar]
- 33. Irvine KA, Blakemore WF (2008) Remyelination protects axons from demyelination‐associated axon degeneration. Brain 131:1464–1477. [DOI] [PubMed] [Google Scholar]
- 34. Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA (2001) Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 30:105–119. [DOI] [PubMed] [Google Scholar]
- 35. Kaplan MR, Meyer‐Franke A, Lambert S, Bennett V, Duncan ID, Levinson SR, Barres BA (1997) Induction of sodium channel clustering by oligodendrocytes. Nature 386:724–728. [DOI] [PubMed] [Google Scholar]
- 36. Kipp M, Clarner T, Dang J, Copray S, Beyer C (2009) The cuprizone animal model: new insights into an old story. Acta Neuropathol 118:723–736. [DOI] [PubMed] [Google Scholar]
- 37. Kojima W, Hayashi K (2018) Changes in the axo‐glial junctions of the optic nerves of cuprizone‐treated mice. Histochem Cell Biol 149:529–536. [DOI] [PubMed] [Google Scholar]
- 38. Komoly S (2005) Experimental demyelination caused by primary oligodendrocyte dystrophy. Regional distribution of the lesions in the nervous system of mice [corrected]. Ideggyogy Szemle 58:40–43. [PubMed] [Google Scholar]
- 39. Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, et al (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 157:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kukley M, Capetillo‐Zarate E, Dietrich D (2007) Vesicular glutamate release from axons in white matter. Nat Neurosci 10:311–320. [DOI] [PubMed] [Google Scholar]
- 41. Li L, Velumian AA, Samoilova M, Fehlings MG (2016) A novel approach for studying the physiology and pathophysiology of myelinated and non‐myelinated axons in the CNS white matter. PLoS One 11:e0165637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marin MA, de Lima S, Gilbert HY, Giger RJ, Benowitz L, Rasband MN (2016) Reassembly of excitable domains after CNS axon regeneration. J Neurosci 36:9148–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mason JL, Langaman C, Morell P, Suzuki K, Matsushima GK (2001) Episodic demyelination and subsequent remyelination within the murine central nervous system: changes in axonal calibre. Neuropathol Appl Neurobiol 27:50–58. [DOI] [PubMed] [Google Scholar]
- 44. Matsushima GK, Morell P (2001) The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 11:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mei F, Lehmann‐Horn K, Shen YA, Rankin KA, Stebbins KJ, Lorrain DS et al (2016) Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A, Lyons DA (2015) Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo . Nat Neurosci 18:628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mitew S, Gobius I, Fenlon LR, McDougall SJ, Hawkes D, Xing YL et al (2018) Pharmacogenetic stimulation of neuronal activity increases myelination in an axon‐specific manner. Nat Commun 9:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mitew S, Xing YL, Merson TD (2016) Axonal activity‐dependent myelination in development: insights for myelin repair. J Chem Neuroanat 76:2–8. [DOI] [PubMed] [Google Scholar]
- 49. Mount CW, Monje M (2017) Wrapped to adapt: experience‐dependent myelination. Neuron 95:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double‐fluorescent Cre reporter mouse. Genesis 45:593–605. [DOI] [PubMed] [Google Scholar]
- 51. Nishiyama A, Chang A, Trapp BD (1999) NG2+ glial cells: a novel glial cell population in the adult brain. J Neuropathol Exp Neurol 58:1113–1124. [DOI] [PubMed] [Google Scholar]
- 52. Reeves TM, Phillips LL, Povlishock JT (2005) Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol 196:126–137. [DOI] [PubMed] [Google Scholar]
- 53. Sahel A, Ortiz FC, Kerninon C, Maldonado PP, Angulo MC, Nait‐Oumesmar B (2015) Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front Cell Neurosci 9:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schultz V, van der Meer F, Wrzos C, Scheidt U, Bahn E, Stadelmann C, et al (2017) Acutely damaged axons are remyelinated in multiple sclerosis and experimental models of demyelination. Glia 65:1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skripuletz T, Gudi V, Hackstette D, Stangel M (2011) De‐ and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected. Histol Histopathol 26:1585–1597. [DOI] [PubMed] [Google Scholar]
- 56. Smith KJ, Blakemore WF, McDonald WI (1979) Central remyelination restores secure conduction. Nature 280:395–396. [DOI] [PubMed] [Google Scholar]
- 57. Stallcup WB (2002) The NG2 proteoglycan: past insights and future prospects. J Neurocytol 31:423–435. [DOI] [PubMed] [Google Scholar]
- 58. Stassart RM, Mobius W, Nave KA, Edgar JM (2018) The axon‐myelin unit in development and degenerative disease. Front Neurosci 12:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Steelman AJ, Thompson JP, Li J (2012) Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci Res 72:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stidworthy MF, Genoud S, Suter U, Mantei N, Franklin RJ (2003) Quantifying the early stages of remyelination following cuprizone‐induced demyelination. Brain Pathol 13:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stojic A, Bojcevski J, Williams SK, Diem R, Fairless R (2018) Early nodal and paranodal disruption in autoimmune optic neuritis. J Neuropathol Exp Neurol 77:361–373. [DOI] [PubMed] [Google Scholar]
- 62. Sturrock RR (1980) Myelination of the mouse corpus callosum. Neuropathol Appl Neurobiol 6:415–420. [DOI] [PubMed] [Google Scholar]
- 63. Tomassy GS, Dershowitz LB, Arlotta P (2016) Diversity matters: a revised guide to myelination. Trends Cell Biol 26:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Trapp BD, Stys PK (2009) Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol 8:280–291. [DOI] [PubMed] [Google Scholar]
- 65. Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD (2010) NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci 30:16383–16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van der Star BJ, Vogel DY, Kipp M, Puentes F, Baker D, Amor S (2012) In vitro and in vivo models of multiple sclerosis. CNS Neurol Disord Drug Targets 11:570–588. [DOI] [PubMed] [Google Scholar]
- 67. Wake H, Ortiz FC, Woo DH, Lee PR, Angulo MC, Fields RD (2015) Nonsynaptic junctions on myelinating glia promote preferential myelination of electrically active axons. Nat Commun 6:7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Watanabe M, Toyama Y, Nishiyama A (2002) Differentiation of proliferated NG2‐positive glial progenitor cells in a remyelinating lesion. J Neurosci Res 69:826–836. [DOI] [PubMed] [Google Scholar]
- 69. Waxman SG (2006) Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 7:932–941. [DOI] [PubMed] [Google Scholar]
- 70. Waxman SG, Bennett MV (1972) Relative conduction velocities of small myelinated and non‐myelinated fibres in the central nervous system. Nature New Biol 238:217–219. [DOI] [PubMed] [Google Scholar]
- 71. Waxman SG, Brill MH (1978) Conduction through demyelinated plaques in multiple sclerosis: computer simulations of facilitation by short internodes. J Neurol Neurosurg Psychiatry 41:408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xie M, Tobin JE, Budde MD, Chen CI, Trinkaus K, Cross AH, et al (2010) Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol 69:704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xing YL, Roth PT, Stratton JA, Chuang BH, Danne J, Ellis SL, et al (2014) Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J Neurosci 34:14128–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, et al (2013) Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zawadzka M, Rivers LE, Fancy SP, Zhao C, Tripathi R, Jamen F, et al (2010) CNS‐resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 6:578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhu X, Bergles DE, Nishiyama A (2008) NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135:145–157. [DOI] [PubMed] [Google Scholar]
- 77. Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A (2011) Age‐dependent fate and lineage restriction of single NG2 cells. Development 138:745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE (2007) Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci 10:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. “Light” and “dark” bundles of the caudal part of mouse corpus callosum. Semi‐thin sections from electron microscopic preparations stained with Richardson's solution. Note the clear separation of the corpus callosum into light appearing bundles (LB) on the cortical side and dark appearing bundles (DB) on the hippocampal side in all experimental groups. A. control. B. 5 weeks of cuprizone treatment. C. 5 weeks of cuprizone treatment + 1 week of remyelination. D. 5 weeks of cuprizone treatment + 5 weeks of remyelination. Scale bar represents 50 µm.
Figure S2. Nav1.6‐positive nodes are flanked by Caspr. Presence of mature nodes of Ranvier was verified by staining for the axonal, paranodal protein Caspr/contactin (1–4), which flanks the mature nodes of Ranvier, before counting the nodes. Mature nodes of Ranvier expressing Nav1.6 (red) are flanked by the paranodal protein Caspr (green) in all three experimental groups and control. Note that not all of the nodes are flanked by Caspr. These structures seem to be increased after cuprizone treatment and during recovery as compared to the control. Scale bars represent 20 µm.
Figure S3. Distribution of axons containing vesicles per image shown in a scatter plot. The distribution of the number of axons containing vesicles per image is shown for all images and for all samples (n = 3 for all groups) in a scatter plot. The scatter plot was generated using Igor Pro 6.3 (WaveMetrics, Lake Oswego, USA). Cup5_1, Cup5_2 and Cup5_3 represent the samples for the 5 weeks of cuprizone treatment group. Cup51_1, Cup51_2 and Cup51_3 represent the samples for the 5 weeks of cuprizone plus 1 week of remyelination group. Cup55_1, Cup55_2 and Cup55_3 represent the 5 weeks of cuprizone plus 5 weeks of remyelination group. Control_1, control_2 and control_3 are the samples for the control group.
Figure S4. Determination of g‐ratios. The EM‐data set was used to measure g‐ratios. Thereby, d=axon diameter was divided by D=fiber diameter in order to obtain an indication of myelin sheath thickness. No statistical difference could be detected between all three experimental groups and the control.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.