

Abstract
Intracranial myxoid mesenchymal tumor harboring EWSR1 fusions with CREB family of genes was recently described, and it resembles the myxoid variant of angiomatoid fibrous histiocytoma. We present three pediatric patients with intracranial EWSR1‐rearranged myxoid mesenchymal neoplasm and provide a molecular genetic characterization of these tumors. Clinical histories and imaging results were reviewed. Histology, immunohistochemistry, EWSR1, FUS, NR4A3 fluorescence in situ hybridization (FISH), and next‐generation sequencing (NGS) were performed. A 12‐year‐old male (case 1), 14‐year‐old female (case 2), and 18‐year‐old male (case 3), presented with headaches, emesis, and seizures, respectively. The magnetic resonance images demonstrated tumors abutting the dura (cases 1 and 3) and in the third ventricle (case 2). All tumors were vascular, with solid sheets of monomorphic oval cells in a prominent myxoid/microcystic matrix. A thin fibrous pseudocapsule was present in all lesions, but definitive lymphocytic cuffing was absent. Morphologically, they closely resembled myxoid variant of angiomatoid fibrous histiocytoma. Mitoses were rare, and necrosis was absent. All tumors expressed desmin and GLUT1, and focal EMA and CD99. The proliferation index was low. FISH and NGS showed EWSR1–CREB1 fusion (cases 1 and 2), and EWSR1–CREM fusion (case 3). There were no FUS (16p11.2) or NR4A3 (9q22.33) rearrangements in case 3. Gains of 5q (including KCNIP1) and 11q (including CCND1) were present in cases 1 and 2. There were no common pathogenic genomic changes other than EWSR1 rearrangements across cases. CNS myxoid mesenchymal neoplasms with histological and immunophenotypic similarities to myxoid variant of AFH are rare, diagnostically challenging, and harbor EWSR1–CREB1 and also a novel EWSR1–CREM fusion not yet described in AFH. Therefore, it is uncertain if these tumors represent variants of AFH or a new entity. The copy number and mutational changes presented here provide support for future studies to further clarify this issue.
Keywords: BRAF, CREB1, CREM, EWSR1, myxoid mesenchymal tumor, myxoid variant angiomatoid fibrous histiocytoma, next‐generation sequencing
Introduction
EWSR1 rearrangements with CREB family of genes (CREB1, ATF1, and CREM) characterize a spectrum of mesenchymal tumors occurring in a variety of sites and with a wide range of biological behavior. Among them, angiomatoid fibrous histiocytoma (AFH) 3, 8, 10, 11, 23, 25, a tumor of low‐to‐intermediate malignant potential shows most often EWSR1–CREB1 fusion as a result of t(2:22)(q33;q12), and less frequently EWSR1–ATF1 or FUS–ATF1 resulting respectively from t(12:22)(q13;q12) or t(12;16)(qq13;p11) 8, 9. EWSR1–CREB1 fusion is the only fusion described in primary pulmonary myxoid sarcoma to date 14, 20, 27, 28, while EWSR1–ATF1 fusion has been described in clear cell sarcoma 5, 13; and hyalinizing clear cell carcinoma of salivary gland 4. The myxoid variant of AFH 15, 16, 24 has identical EWSR1 rearrangements with the classic AFH and is histologically characterized by a rich vascular network and vascular dilatation, epithelioid cells, and a prominent myxoid background. A pseudocapsule and lymphoid aggregates are usually present.
Myxoid AFH had not been reported in the central nervous system, although recently, Kao et al 17 described a small series of myxoid mesenchymal tumors that harbor EWSR1 rearrangements with CREB family of genes, including CREM in two cases. The authors hypothesized that these tumors, which show a predilection for intracranial location (4 out of 5 cases), but lack the typical capsular lymphocytic infiltrate and prominent vascular component of AFH myxoid variant, may represent a potentially novel myxoid mesenchymal tumor rather than AFH, myxoid variant. Additional cases with a comprehensive genomic analysis might be needed to support this observation.
This article presents three patients with myxoid mesenchymal intracranial tumors that we encountered in our practice and provides their detailed molecular characterization.
Materials and Methods
Clinical history
This study was performed with the approval of the Institutional Review Board at the Boston Children's Hospital and Brigham and Women's Hospital. Case 1 was identified in the archives of Boston Children's Hospital; cases 2 and 3 were encountered in the pathology practice at IWK Health Center, Halifax, NS, and Mayo Clinic, Rochester, MN, respectively. Cases 1 and 2 were presented at the 57th Diagnostic Slide Seminar of the American Association of Neuropathologists in June 2016 (http://neuro.pathology.pitt.edu/DSSFiles/Current DSS.htm). A summary of the clinical features of all three cases is presented in Table 1.
Table 1.
Clinical and radiological characteristics of the three patients.
| Case | Age | Sex | Symptoms | MRI—location of lesion | MRI—tumor size (greatest dimension) | Other MRI findings |
|---|---|---|---|---|---|---|
| 1 | 12 | M | Headache | Posterior fossa/cerebellar, abutting dura | 2.5 cm × 2.3 × cm 1.0 cm | Lobulated, heterogeneous appearance, contrast enhancing, no tail‐sign present |
| 2 | 14 | F | Headache, nausea, vomiting, diplopia | Intraventricular (left lateral) | 3.8 cm × 3.6 cm × 3 cm | Heterogeneous appearance, surrounding vasogenic edema and mass effect |
| 3 | 18 | M | Generalized tonic‐clonic seizures | Midline/right frontal (falx area), surrounded by a thin rim of brain parenchyma | 3.0 cm × 2.0 cm × 1.5 cm | Contrast enhancing, surrounding vasogenic edema, no definitive dural attachment |
Case 1: a 12‐year‐old male presented with mild headache for approximately one month. MRI studies demonstrated a 2.5 cm × 2.3 cm × 1.0 cm, well‐defined extra‐axial lobulated mass in the left cerebellar hemisphere, abutting the dura (Figure 1a). The past medical history was notable for stage IV adrenal neuroblastoma, diagnosed at age 7 years, for which he was treated with chemotherapy, surgical resection, stem cell transplant and local radiotherapy. There was good response to therapy with no evidence of disease recurrence prior to this presentation. There was no prior cranial radiation. He underwent resection of the cerebellar mass, which was a tan, firm nodule attached to the dura.
Figure 1.
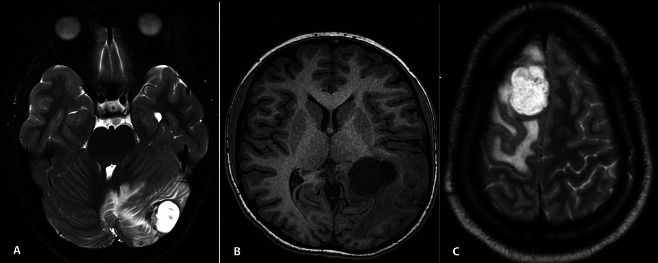
a. Case 1—axial T2‐hyperintense well‐demarcated tumor in right cerebellar hemisphere with surrounding edema, abutting the dura. b. Axial T1 MRI of case 2 shows well‐demarcated tumor involving left posterior horn of lateral ventricle. c. Case 3—axial T2‐hyperintense tumor in left frontal region, having similar features to those in case 1, but no definitive attachment to dura.
Case 2: a 14‐year‐old female presented with intermittent early morning headache, diplopia, nausea and vomiting. MRI revealed an intra‐axial 3.8 cm × 3.6 cm × 3 cm complex solid and cystic contrast‐enhancing mass of the posterior left ventricle with surrounding vasogenic edema and slight mass effect (Figure 1b). The resected lesional tissue was firm and tan‐pink.
Case 3: an 18‐year‐old male, presented with a seizure episode. A 3.0 cm × 2.0 cm × 1.5 cm frontal intra‐axial circumscribed, lobular mass in the proximity of the superior sagittal sinus, with associated vasogenic edema was found on MRI (Figure 1c). At surgery, the mass was intra‐axial well‐delineated, firm, tan, and surrounded by a rim of brain parenchyma.
Whole body imaging did not show other lesions in any of the patients.
Histopathology and immunohistochemistry
Hematoxylin–eosin (H&E) stained slides and immunostains were performed according to standard protocols on 4‐μm formalin‐fixed paraffin‐embedded sections using the following primary antibodies and conditions: INI1 (1:100; Ventana Medical Systems, Tuscon, AZ), somatostatin receptor 2A (Epitomics, UMB1, monoclonal, 1:2000); the remainder of the immunohistochemical stains were performed using automated staining with commercially available prediluted antibodies (EMA, S100, desmin, Glut‐1, GFAP, Olig2, SOX10, synaptophysin, myogenin, smooth muscle actin [SMA], CD68, keratin, CD34, CD99; Leica Biosystems, Buffalo Grove, IL). Counterstaining for nuclei was performed using Mayer's hematoxylin and coverslips were mounted with Permount (Fisher Scientific). Photographs were taken using an Olympus BX41 microscope and an Olympus DP25 camera.
Fluorescence in situ hybridization (FISH) analysis
FISH was performed on 4‐μm paraffin sections according to standard protocols 29. EWSR1 (22q12) rearrangement was evaluated using the Vysis LSI EWSR1 break‐apart Probe kit (Abbott Molecular, Des Plaines, IL). Also, FISH for FUS (16p11.2) and NR4A3 (9q22.33) were pursued for case 3 using commercially available dual‐color break‐apart probes (BAP).
Next‐generation sequencing
DNA was isolated from tissue containing at least 20% tumor nuclei and analyzed by massively parallel sequencing using a solution‐phase Agilent SureSelect hybrid capture kit using methods previously described 19. Somatic mutations in tumor DNA were detected using the exome‐sequencing platform OncoPanel (Illumina HiSeq) in a CLIA‐certified laboratory. The OncoPanel assay detects mutations in 300 different cancer genes and 113 introns across 35 genes for rearrangement detection. Common single nucleotide polymorphisms (SNP) were accounted for using the following informatic steps: any SNP present at >0.1% in Exome Variant Server (NHLBI GO Exome Sequencing Project [ESP], Seattle, WA; URL: http://evs.gs.washington.edu/EVS/) or present in dbSNP was filtered. Variants also present in the COSMIC mutation database were rescued for manual review. Individual variants present at <10% allele fraction or in regions with <50× coverage were flagged for manual review. In an attempt to identify potential damaging effects of missense mutations, computational predictions of the impact of protein sequence variants were made using the software tool PolyPhen‐2 (2). Structural variants were detected using the BreaKmer algorithm, which identifies rearrangements and nucleotide‐level breakpoints by realigning variant contigs generated from assembling all misaligned reads within the targeted NGS data 1.
Results
Light microscopic features
The H&E‐stained sections of all tumors demonstrated a myxoid neoplasm of moderate cellularity (Figure 2a) with focal vague nodular architecture and hypocellular areas rich in collagen (Figure 2b). The cells were monomorphic and mildly atypical, growing in irregular trabecular (Figure 2c). A thin fibrous capsule was seen in all tumors (Figure 2d). The cells had oval‐to‐spindled nuclei, speckled chromatin, and moderate amounts of eosinophilic cytoplasm and were occasionally arranged in whorls (Figure 2e). Although the tumors had foci demonstrating a rich capillary network (Figure 2f), rare thin‐walled dilated vessels resembling lakes of blood were seen only in case 3 (Figure 2b). A discrete angiocentric pattern of growth was present particularly in case 1 (Figure 2g). Invasion of the brain and necrosis were not present, and mitoses were rare in cases 2 and 3. Case 1 had a single focus with up to 6 mitoses/10 high power fields. Well‐developed lymphocytic cuffing was not present in any of the cases. Occasional collagenous fibers similar to amianthoid fibers were present in every tumor, but without rosetting or sunburst appearance (Figure 2h).
Figure 2.
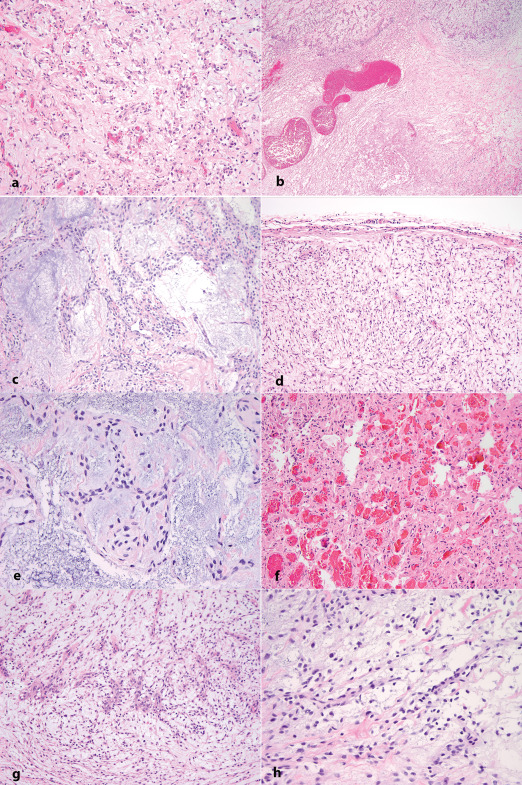
Histological characteristics. a. Hematoxylin–eosin‐stained section representative of the three tumors, showing epithelioid neoplasm composed of round‐to‐oval cells growing in short cords in a prominent myxoid background. b. A vague nodular pattern admixed with hypocellular areas rich in collagen, and occasional prominent vessels. c. Short cords and trabecula of monomorphic cells in a prominent myxoid background with scattered collagen fibers. d. The tumors were surrounded by a thin fibrous capsule. e. Cords and occasional whorls composed of monomorphic oval‐to‐spindle cells with minimal atypia in a prominent myxoid background. f. Focus of rich capillary network (more prominent in case 2). g. Discrete angiocentric pattern (more prominent in case 1). h. Spindle and stellate cells with collagen fibers resembling amianthoid fibers.
Immunohistochemistry
All three tumors demonstrated extensive (more than 90% tumor cells) and strong immunopositivity for desmin (Figure 3a) and GLUT‐1 (Figure 3b). The EMA (Figure 3c) and CD99 (Figure 3d) immunostains were focally positive in a cytoplasmic and membranous pattern, respectively. The S100, SOX10, CD34, SSTR2A, myogenin, GFAP, OLIG2, synaptophysin, and CAM5.2 immunostains were negative. Pale, variable and focal SMA expression was present in all three tumors in a cytoplasmic and membranous pattern that was considered nonspecific (not illustrated). Focal CD68 expression was present only in case 2. A CD68 immunostain was not performed in case 3. The proliferation labeling index, as demonstrated by the Ki67 immunostain, was low (less than 5%) with the exception of the focus in case 1, where the rate was up to 12%; however, due to the number of macrophages and occasional inflammatory cells, this value might be an overestimate. INI1 (BAF47) nuclear immunopositivity was retained in all three tumors.
Figure 3.
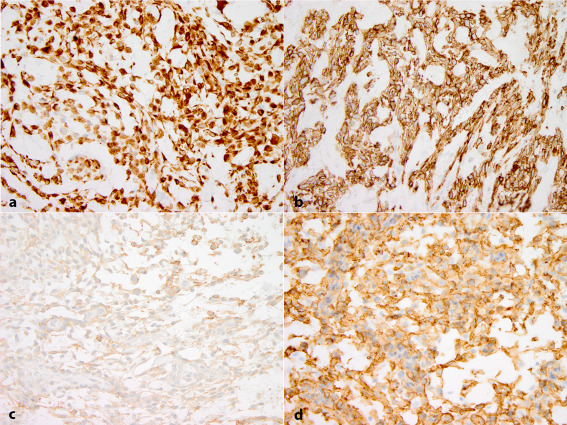
Immunohistochemical profile. Desmin (a) and Glut‐1 (b) immunostains diffusely and strongly positive, EMA (c) expressed in subset of tumor cells, CD99 immunostain (d) positive in a membranous pattern in large number of lesional cells.
Molecular genetic analysis
FISH demonstrated EWSR1 (22q12) rearrangement in 88% of nuclei in case 1, 93% of nuclei in case 2, and 63% of nuclei in case 3. FISH for FUS (16p11,2) and NR4A3 (9q22.33) was negative for rearrangements in case 3.
Targeted exome sequencing with subsequent structural variation analysis using the BreaKmer algorithm detected a reciprocal chromosomal rearrangement between chromosome 2 (mapping to intron 6 in case 1 and intron 7 in case 2) of CREB1 and chromosome 22 (mapping to intron 8 in cases 1 and 2) of EWSRI in cases 1 and 2. In case 3, BreaKmer identified a fusion between EWSR1 (mapping to intron 9) and CREM (mapping to intron 3) on chromosome 10 (Figure 4).
Figure 4.
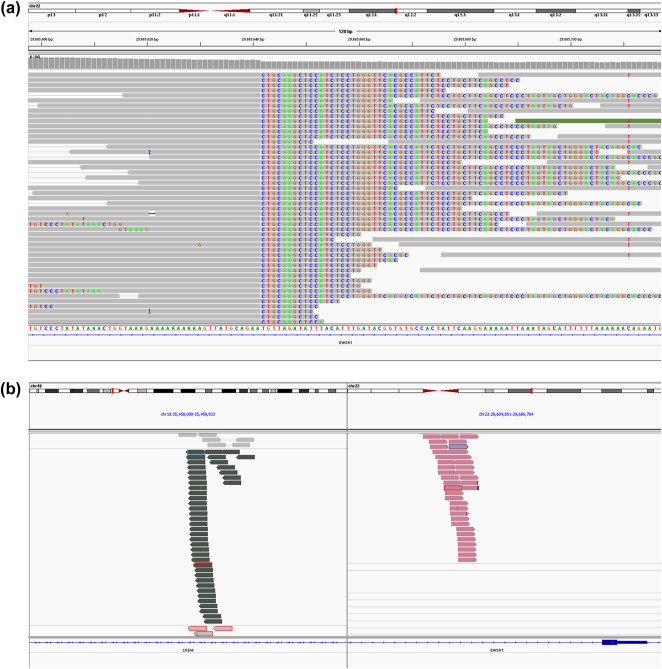
The EWSR1–CREM fusion in case 3 is supported by discordant read pairs indicating a break in EWSR1 (a) and alignment of split reads to EWSR1 and CREM (b) as predicted by the translocation analysis tool BreaKmer.
Copy number alterations detected by NGS demonstrated low‐level gains of 5q (including KCNIP1, 5q35.1) and 11q (including CCND1) in cases 1 and 2. The 5q gains in case 1 also involved 5q32 (including CSF1R and PDGFRB). Case 1 also demonstrated single copy deletions of 12p (including CCND2) and 19p (ZNF708). Additional alterations in case 2 included low copy number gains involving 15q (including NTRK3) and 18q (including GATA6). With the exception of the rearrangement in EWSR1, no additional copy number alterations were identified in case 3.
No single nucleotide variants previously reported in the COSMIC cancer database (http://cancer.sanger.ac.uk/cosmic) were found in these three cases (Table 2). Several novel variants were however noted, but the majority of these occurred at allelic fractions consistent with normal germline variants. There were no common single nucleotide variants identified in the three cases. Of interest is the BRAF c.2128‐5dupT mutation observed in 8% of 325 reads in case 2, as BRAF mutations have not been described in EWSR1‐rearranged tumors, and such finding might have therapeutic implications.
Table 2.
Variants detected by targeted NGS (OncoPanel).
| Gene | DNA variant | Amino acid change | Variant type | Allelic fraction | Functional effect | PolyPhen‐2 functional effect prediction score | |
|---|---|---|---|---|---|---|---|
| Case 1 | PRF1 | c.529C>T | p.R177C | Missense | 0.40 | Probably damaging | 0.999 |
| MLH1 | c.409G>C | p.A137P | Missense | 0.43 | Benign | 0.169 | |
| MECOM | C.332G>C | p.G111A | Missense | 0.45 | Probably damaging | 1 | |
| FANCD2 | c.2309A>G | p.K770R | Missense | 0.46 | Benign | 0.171 | |
| ROS1 | c.6074T>A | p.I2025N | Missense | 0.46 | Probably damaging | 1 | |
| ATM | c.1726A>G | p.I576V | Missense | 0.48 | Possibly damaging | 0.675 | |
| CDKN2C | c.80A>G | p.N27S | Missense | 0.48 | Benign | 0.001 | |
| SDHA | c.728C>T | p.S243F | Missense | 0.49 | Benign | 0.146 | |
| Case 2 | BRAF | c.2128–5dupT | – | Frameshift | 0.08 | Disrupts protein tyrosine kinase domain at p.457‐714 | – |
| GLI2 | c.178G>C | p.A60P | Missense | 0.43 | Benign | 0.003 | |
| KMT2D | c.787C>T | p.R263C | Missense | 0.53 | Probably damaging | 0.99 | |
| Case 3 | NF1 | c.3160A>C | p.N1054H | Missense | 0.03 | Benign | 0.017 |
| TCF3 | c.1080C>A | p.P360P | Silent | 0.37 | – | – | |
| BRCA1 | c.2779G>T | p.A927S | Missense | 0.49 | Benign | 0.367 | |
| ERCC5 | c.1810G>T | p.V604L | Missense | 0.49 | Benign | 0.001 | |
| ERBB3 | c.3334delG | p.V1112Cfs*29 | Frameshift | 0.50 | Disrupts sequence distal to the protein tyrosine kinase domain | – | |
| CUX1 | c.1653G>A | p.M551I | Missense | 0.51 | Probably damaging | 0.966 | |
| STAT6 | c.1213–8C>G | – | Frameshift | 0.54 | – | – | |
| PRAME | c.21 + 889G>A | – | Frameshift | 0.64 | – | – |
Missense mutational effects are assessed by PolyPhen‐2 prediction of functional effects of human nsSNPs 2. Frameshift mutational effects are determined by gene analysis using COSMIC and Pfam (proteins family database). Potentially deleterious variants are indicated in bold.
To explore if these copy number changes and nucleotide variants might overlap with those seen in classic AFH, which has not been sequenced at this level, we identified in the archives at Boston Children's Hospital four cases of AFH in the 2012–2014 interval, of which one had available NGS. The patient was an 8‐year‐old boy with a classic AFH of the scalp. Aside from an EWSR1–ATF1 fusion demonstrated by BreaKmer analysis, there were no significant copy number changes. The nucleotide variants identified in this case are included in Table 3. None of the mutations found in this classic AFH were known to be pathogenic and none were reported previously in COSMIC. There was no significant overlap with the genomic findings of the three myxoid mesenchymal tumors.
Table 3.
Mutations identified by targeted NGS (OncoPanel) in one case of a classic AFH in the scalp of an 8‐year‐old male.
| Gene | DNA variant | Amino acid change | Variant type | Allelic fraction |
|---|---|---|---|---|
| ESR1 | c.227C>G | p.T76S | Missense | 0.48 |
| FANCA | c.893 + 101G>A | – | Intronic | 0.49 |
| KMT2D | c.6235–6C>G | – | Intronic | 0.5 |
| PRPF8 | c.*72C>G | – | Intronic | 0.48 |
| ROS1 | c.1757C>G | p.A586G | Missense | 0.42 |
| STAT3 | c.714G>C | p.E238D | Missense | 0.41 |
At 1‐year follow‐up, all three patients were well, without evidence of recurrence.
Discussion
We describe three unusual myxoid mesenchymal tumors occurring intracranially in young patients, characterized by a peculiar myxoid morphology and showing EWSR1 rearrangements, closely resembling the cases recently described by Kao et al 17. Because of the histological features and chromosomal rearrangements seen in our three cases, the initial interpretation was of myxoid variant of AFH. Myxoid variant of AFH 15, 16, 24, 25 carries low‐to‐intermediate malignant potential, with reportedly only rare patients developing local recurrence; no distant metastases are reported. As in the classic AFH, the majority of the EWSR1 rearrangements in the myxoid variant involve fusion of EWSR1–CREB1. Less commonly, the EWSR1 rearrangement results from t(12;22)(q13;q12) leading to EWSR1–ATF1 7, 16. Rare examples of intracranial classic AFH, extra‐axial and intra‐axial parieto‐occipital, have been reported in the literature 6, 9, 12, 22, albeit none of these cases had myxoid features on histology. In the series of myxoid variant of AFHs described in the literature, none occurred intracranially. Of note, 1 of the 21 cases in the series of Schaefer et al. lacked a fibrous capsule and lymphocytic pseudocapsular cuffing, and blood‐filled cystic spaces were not present in 4 of the 21 cases. In a more recent series of 21 cases of AFH, Shi et al 25 found eight tumors that lacked pseudoangiomatoid spaces; two of these had prominent myxoid stroma, and one of these had prominent scleroting stroma. These series provide supportive evidence for a relatively wide histologic spectrum of AFH, which might challenge the hypothesis that these tumors represent a new entity.
The differential diagnosis of such myxoid mesenchymal tumors is wide; in our series, other entities with similar histology and EWSR1 rearrangements were largely excluded by immunohistochemical studies: myoepithelial tumors—negative cytokeratin, GFAP, equivocal SMA; microcystic meningioma with myxoid changes—negative somatostatin receptor 2A; an unusual glial or glioneuronal neoplasm—negative OLIG2, synaptophysin; extraskeletal myxoid chondrosarcoma—negative S100, and also by additional FISH studies in case 3 (no NR4A3 rearrangements for extraskeletal myxoid chondrosarcoma and FUS rearrangements for low grade fibromyxoid sarcoma) before NGS was performed. Myxoid AFH and primary pulmonary myxoid sarcoma (PPMS) have similar morphology, immunoprofile, and chromosomal rearrangements, and Smith et al documented these features in a brief three case series 26. Desmin immunostain is usually used as a method to distinguish between myxoid AFH (diffuse positivity) and PPMS (usually negative) 29. However, Smith et al describes one case of EWSR1‐rearranged potential PPMS with desmin expression. Although in this particular case, the EWSR1 fusion partner was not ATF1 or CREB1, the immunoprofile overlap raises the possibility that these two entities might be part of a single diagnostic spectrum; a more comprehensive molecular characterization of cases of PPMS and AFH might provide further evidence in that direction.
A curious finding was that all three tumors demonstrated extensive strong positivity for GLUT1 (Figure 3b), which has not been reported in AFH or in the recent series of five cases of myxoid mesenchymal tumor with EWSR1 rearrangements and predilection for CNS.
The tumors in our study are similar to those described by Kao et al. Notably, the similarities include occurrence in adolescents, intra‐ and extra‐axial locations (posterior fossa, intraventricular, frontal‐midline areas), and histological and immunohistochemical profile. In contrast, however, the tumors in our series demonstrated focal histologic features of classic AFH, within the prominent myxoid background: rich vascular meshwork and occasional dilated vascular spaces, thin fibrous pseudocapsule, and minimal lobular architecture. In addition, the immunoprofile of the cases in our series is identical to that of AFH (EMA and desmin expression and focal membranous CD99 positivity), and therefore, we are not entirely sure that they represent a new entity that is biologically distinct of the myxoid variant of AFH.
Two of our cases demonstrated the more common EWSR1–CREB1 fusion of AFH. The EWSR1–CREM fusion observed in case 3 has not been described in AFH, but was present in one intracranial case described by Kao et al. CREB1 and CREM, along with ATF1, are part of a related family of transcription factors with similar structure and function, but the significance of CREM as a fusion partner is unclear 18, 21; it is not unique to CNS located tumors as it was present in the index perirectal myxoid mesenchymal neoplasm.
In addition to the characterization of the fusions in our tumors, we also encountered low‐level copy number gains involving chromosomes 5q (including KCNIP1, 5q35.1) and 11q (including CCND1) in both cases 1 and 2. Also, in case 1, the 5q gains involved 5q32 (including CSF1R and PDGFRB), and a single copy deletion of 12p (including CCND2) and 19p (ZNF708) were also present. Additional alterations in case 2 included low copy number gains of 15q (including NTRK3) and 18q (including GATA6). There were no copy number changes present in case 3. Copy number alterations have not been previously reported in AFH and were not present in the classic AFH with available NGS results included in this study.
A single mutational signature was not identified in these three cases (Table 2). Moreover, no variants of definitive significance in cancer were detected. As the tissue was analyzed in the absence of paired non‐tumor controls from the same patient, the possibility that these variants represent normal germline variants cannot be entirely excluded. However, the frameshift mutation in BRAF present in case 2, although not reported in cancer, is of interest, given the importance of BRAF alterations in the pathogenesis of many tumors, including pediatric brain tumors. An extensive molecular characterization of classic and myxoid variant AFH and of myxoid mesenchymal tumor described recently has not been published to date. In the classic AFH of the scalp reviewed by us, although there were no nucleotide variants reported in cancer, a ROS1 missense mutation (c.1757C>G, pA586G) was present. A ROS1 (c.6074T>A, pI576V) missense mutation was also present in case 1, and was absent in the other two tumors. Also a KMT2D intronic variant (c.6235‐6C>G) was seen in the classic AFH, and a missense KMT2D mutation (c.789C>T, p.R263C) was seen in case 2. Although these mutations are not identical, and their effect in cancer is not described, their occurrence in both classic AFH and in an intracranial myxoid mesenchymal tumor with similar biology to myxoid variant of AFH, is worthy of consideration in future studies, as they might suggest biological overlap.
In conclusion, we report three cases of intracranial myxoid mesenchymal tumors with EWSR1 fusions with CREB1 and CREM, and provide further insight in the genomics of these tumors. While signature copy number changes and single nucleotide variants were not identified, this extensive molecular characterization was not reported in these tumors or in AFH. The presence of a BRAF mutation in one of the cases might be of therapeutic interest. This is the second series of such tumors, the first one containing only four (out of five) intracranial cases; therefore, our study provides further promising support for this entity with a challenging differential diagnosis for the neuropathologists.
Acknowledgments
This study could not have been possible without the financial and technical support provided by the Department of Pathology at Boston Children's Hospital, Histology Laboratory at Boston Children's Hospital, and by the technicians in the Center for Advanced Molecular Diagnosis at the Brigham and Women Hospital. The authors would also like to thank the following pathologists for their input and expertise during the clinical evaluation of these cases: Dr. J.L. Hornick (Brigham and Women's Hospital, Boston, MA), Dr. A. Bahrami (St. Jude's Children's Research Hospital, Memphis, TN), and Dr. Aimee Popp (WellStar Medical Group, Marietta, GA).
References
- 1. Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas‐Rudilla V, Lin L et al (2015) BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res 43:e19. doi: 10.1093/nar/gku1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet Chapter 7;Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antonescu CR, Dal CP, Nafa K, Teot LA, Surti U, Fletcher CD, Ladanyi M (2007) EWSR1–CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46:1051–1060. [DOI] [PubMed] [Google Scholar]
- 4. Antonescu CR, Katabi N, Zhang L, Sung YS, Seethala RR, < Jordan RC, Perez‐Ordonez B et al, (2011) EWSR1ATF1 fusion is a novel and consistent finding in hyalinizing clear‐cell carcinoma of salivary gland. Genes Chromosomes Cancer 50:559–570. [DOI] [PubMed] [Google Scholar]
- 5. Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyl MEWS‐CREB1 (2006) a recurrent variant fusion in clear cell sarcoma—association with gastrointenstinal location and absence of melanocytic differentiation. Clin Cancer Res 12:5356–5362. [DOI] [PubMed] [Google Scholar]
- 6. Ashareef MA, Almadidy Z, Baker T, Perry A, Welsh CT, Vandergrift WA 3rd. (2016) Intracranial angiomatoid fibrous histiocytoma: case report and literature review. World Neurosurg 96:403–409. [DOI] [PubMed] [Google Scholar]
- 7. Chen G, Folpe AL, Colby TV, Sittampalam K, Patey M, Chen MG, Chan JK (2011) Angiomatoid fibrous histiocytoma: unusual sites and unusual morphology. Mod Pathol 24:1560–1570. [DOI] [PubMed] [Google Scholar]
- 8. Costa MJ, Weiss SW (1990) Angiomatoid malignant fibrous histiocytoma. A follow‐up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol 14:1126–1132. [PubMed] [Google Scholar]
- 9. Dunham C, Hussong J, Seiff M, Pfeifer J, Perry A (2008) Primary intracerebral angiomatoid fibrous histiocytoma: report of a case with a t(12;22)(q13;q12) causing type 1 fusion of the EWS and ATF‐1 genes. Am J Surg Pathol 32:478–484. [DOI] [PubMed] [Google Scholar]
- 10. Enzinger FM (1979) Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 44:2147–2157. [DOI] [PubMed] [Google Scholar]
- 11. Fanburg‐Smith JC, Miettinen M (1999) Angiomatoid “malignant” fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol 30:1336–1343. [DOI] [PubMed] [Google Scholar]
- 12. Hansen JM, Larsen VA, Scheie D, Perry A, Skjoth‐Rasmussen J (2015) Primary intracranial angiomatoid fibrous histiocytoma presenting with anaemia and migraine‐like headaches and aura as early clinical features. Cephalalgia 35:1334–1336. [DOI] [PubMed] [Google Scholar]
- 13. Hisaoka M, Ishida T, Kuo TT, Matsuyama A, Imamura T, Nishida K et al (2008) Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 32:452–460. [DOI] [PubMed] [Google Scholar]
- 14. Jeon YK, Moon KC, Park SH, Chung DH (2014) Primary pulmonary myxoid sarcomas with EWSR1CREB1 translocation might originate from primitive peribronchial mesenchymal cells undergoing myofibroblastic differentiation. Virchows Arch 465:456–461. [DOI] [PubMed] [Google Scholar]
- 15. Justin Wong SB, Wee A, Puhaindran ME, Pang B, Lee VK (2015) Angiomatoid fibroys histiocytoma with prominent myxoid stroma: a case report and review of the literature. Am J Dermatopathol 37:623–631. [DOI] [PubMed] [Google Scholar]
- 16. Kao YC, Lan J, Tai HC, Li CF, Liu KW, Tsai JW et al (2014) Angiomatoid fibrous histiocytoma: clinicopathological and molecular characterization with emphasis on variant histomorphology. J Clin Pathol 67:210–215. [DOI] [PubMed] [Google Scholar]
- 17. Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, Rosenblum MK, Antonescu CR (2016) EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol 41:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neurib 35:605–623. [DOI] [PubMed] [Google Scholar]
- 19. MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L et al (2009) Profiling critical cancer gene mutations in clinical tumor samples. PLoS ONE 4:e7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsukuma S, Hisaoka M, Obar K, Kono T, Takeo H, Sato K, Hata Y (2012) Primary pulmonary myxoid sarcoma with EWSR1CREB1 fusion, resembling extraskeletal myxoid chondrosarcoma: case report with a review of literature. Pathol Int 62:817–822. [DOI] [PubMed] [Google Scholar]
- 21. Mayr B, Montmini M (2001) Transcritional regulation by the phosphorylation‐dependent factor CREB. Nat Rev Mol Cell Biol 2:599–609. [DOI] [PubMed] [Google Scholar]
- 22. Ochalski PG, Edinger JT, Horowitz MB, Stetler WR, Murdock GH, Kassam AB, Engh JA (2010) Intracranial angiomatoid fibrous histiocytoma presenting as recurrent multifocal intraparenchymal hemorrhage. J Neurosurg 112:978–982. [DOI] [PubMed] [Google Scholar]
- 23. Rossi S, Szuhai K, Ijszenga M, Tanke HJ, Zanatta L, Sciot R et al (2007) EWSR1CREB1 and EWSR1ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res 13:7322–7328. [DOI] [PubMed] [Google Scholar]
- 24. Schaefer IM, Fletcher CD (2014) Myxoid variant of so‐called angiomatoid “malignant fibrous histiocytoma”: clinicopathologic characterization. A series of 21 cases. Am J Surg Pathol 38:816–823. [DOI] [PubMed] [Google Scholar]
- 25. Shi H, Li H, Zhen T, Zhang F, Dong Y, Zhang W, Han A (2015) Clinicopathological features of angiomatoid fibrous histiocytoma: a series of 21 cases with variant morphology. Int J Clin Exp Pathol 8:772–778. [PMC free article] [PubMed] [Google Scholar]
- 26. Smith SC, Palanisamy N, Betz BL, Tomilins SA, Mehra R, Schmidt LA et al (2014) At the intersection of primary pulmonary myxoid sarcoma and pulmonary angiomatoid fibrous histiocytoma: observations from three new cases. Histopathology 65:144–146. [DOI] [PubMed] [Google Scholar]
- 27. Thway K, Fisher C (2012) Tumors with EWSR1CREB1 and EWSR1ATF1 fusions: the current status. Am J Surg Pathol 36:e1–e11. [DOI] [PubMed] [Google Scholar]
- 28. Thway K, Nicholson AG, Lawson K, Gonzalez D, Rice A, Balzer B et al (2011) Primary pulmonary myxoid sarcoma with EWSR1‐CREB1 fusion: a new tumor entity. Am J Surg Pathol 35:1722–1732. [DOI] [PubMed] [Google Scholar]
- 29. Weremowicz S, Schofield DE (2007) Preparation of cells from formalin‐fixed, paraffin‐embedded tissue for use in fluorescence in situ hybridization (FISH) experiments. Curr Protoc Hum Genet Chapter 8:Unit 8.8. [DOI] [PubMed] [Google Scholar]